

Abstract
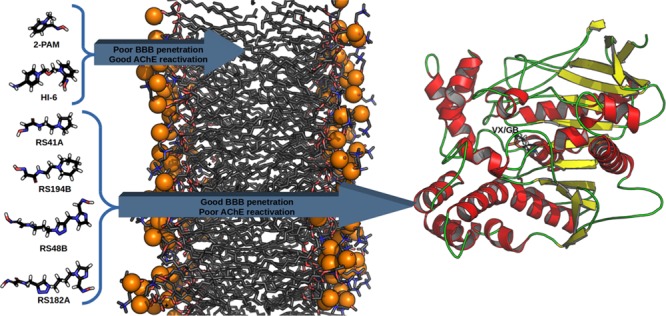
The deleterious effects of nerve agents over the enzymes acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) turned these compounds into the most dangerous chemical weapons known. Among the antidotes in use today against these agents, oximes in combination with other drugs are the only treatment with any action. HI-6 and 2-PAM are cationic oximes proved to be effective for the reactivation of AChE inhibited by the nerve agents VX and sarin (GB). However, when it comes to reactivation of AChE inside the central or peripheral nervous systems, charged molecules present low diffusion due to low penetration through the blood–brain barrier. Uncharged oximes appear as an interesting alternative to solve this problem, but the development and enhancement of more efficient uncharged oximes capable of reactivating human AChE is still necessary. Given the limitations for in vivo and in vitro experimental studies with nerve agents, modeling is an important tool that can contribute to a better understanding of factors that may affect the efficiency of uncharged oximes. In order to investigate the interaction and behavior of cationic and uncharged oximes, we performed here molecular docking, molecular dynamics simulations, and binding energies calculations of the known cationic oximes HI-6 and 2-PAM plus four uncharged oximes found in the literature, complexed with human AChE (HssACHE) conjugated with the nerve agents VX and GB. The uncharged oximes showed different behaviors, especially RS194B, which presented stability inside AChE-VX, but presented free binding energy lower than cationic oximes, suggesting that structural alterations could favor its interactions with these complexes. In contrast, HI-6 and 2-PAM showed higher affinities with more negative binding energy values and larger contribution of the amino acid Asp74, demonstrating the importance of the quaternary nitrogen to the affinity and interaction of oximes with AChE-GB and AChE-VX conjugates.
Introduction
Despite being toxic pesticides and/or insecticides, the major risk represented by the organophosphorus (OP) compounds to human health comes from their use as chemical warfare agents.1−5 OP compounds are defined as nerve agents because their main target is the human central nervous system (CNS), where they are capable of inhibiting the hydrolysis, usually performed by the enzyme acetylcholinesterase (AChE),2,4−6 of the neurotransmitter acetylcholine (ACh) in post-synaptic membranes and neuromuscular junctions. The inhibition mechanism of the OP is analogous to the reaction of AChE with ACh7,8 and is well described in the literature,6,9−17 except for the step of the leaving group of the OP, resulting in the phosphorylation of AChE which triggers the cholinergic syndrome, defined as an overstimulation of structures innervated by cholinergic fibers.17−21
The literature reports that the reversal of the inhibition process is possible by reactivating AChE using oximes, which are nucleophiles capable of displacing the OP by dephosphorylating the residue Ser203 (in the human case), that is part of the catalytic triad with His447 and Glu334 of the active site.9,11,22−28 However, the oxime should be administered as soon as possible to prevent the process called “aging” of AChE, which is characterized by the elimination of an alkyl moiety (R1) from the OP.11,22−26,28 Among the oximes found in the literature, 1-[[2-[(E)-hydroxyiminomethyl]pyridin-1-ium-1-yl]methoxymethyl]pyridin-1-ium-4-carboxamide (Asoxime chloride, or most commonly HI-6) (Figure 1) and (NE)-N-[(1-methylpyridin-1-ium-2-yl)methylidene]hydroxylamine (pralidoxime, or most commonly 2-PAM) (Figure 1) stand out for their efficiency in the AChE reactivation process.11,22−26,29−35 Even with studies suggesting the importance of the quaternary nitrogen in AChE reactivation, some studies in the literature argue that the cationic oximes, which present great reactivating potential in in vitro and in vivo tests, show a significant reduction in their efficiency.36−39 This is attributed to the fact that such oximes do not present good diffusion through the blood–brain barrier (BBB) and diffuse through the blood and peripheral tissues.36−40 Therefore, in this work, we decided to analyze, through molecular modeling, the behavior and interactions of the neutral oximes RS41A, RS194B, RS48B, and RS182A (Figure 1) found in the literature,36−39 which presented good diffusion through the BBB in comparison to the commercial oximes HI-6 and 2-PAM, in order to determine some factors, besides the quaternary nitrogen, that may influence the interaction of the complexes of AChE with sarin (GB) and VX, which are two of the most lethal chemical warfare agents.2
Figure 1.

Structures of HI-6 and 2-PAM and uncharged oximes RS41A, RS194B, RS48B, and RS182.
Methodology
The oximes had their aqueous ionization constants (pKa) evaluated at biological pH (pH = 7.4) in the chemicalize server (www.chemicalize.com) by the prediction of a dissociation constant method, using micro constants.41 Oximes HI-6, RS41A, and RS194B were predicted to present ionization at this pH (see Figure S1 of Supporting Information) but not the other oximes.
The three-dimensional structures of each oxime, guided by previous results, had the partial atomic charge calculation performed in Spartan 08 Suite42 using the semiempirical method RM1.43−47
The three-dimensional model structures of AChE inhibited by OPs (AChE-OP) complexed with HI-6 were constructed through mutations on the crystallographic structure of Mus musculus AChE complexed with HI-6 and inhibited by GB (AChE-GB) and VX (AChE-VX), available in the RCSB-Protein Data Bank48 under the codes 2WHP(49) and 2Y2U, respectively, using the software SPDBViewer.50,51 The target and the template presented more than 85% of homological identity. The models obtained were further used to perform docking studies using the software Molegro Virtual Docker (MVD) with MolDock52 as the docking algorithm. Water molecules from the crystal were conserved in the calculations in order to investigate eventual interactions with the solvent. To calculate the interaction energy, the MolD score uses the spatial configuration of the atoms contained in the study systems. In our specific case, this spatial configuration is presented by the crystallographic structure. The re-docking of HI-6 in the active site of crystallographic AChE was performed in order to validate the docking protocol. A sphere with 11 Å of radius, centered in the binding pocket, was the selected docking region. Because of the stochastic nature of the docking algorithm, about 20 runs were performed for each compound, resulting in 30 poses (conformation and orientation of the oxime) generated for each run. The criteria adopted to select the best poses were the most negative value (suggesting greater stability and affinity) of the scoring function, hydrogen bonds, and the distance between the O atom of the oxime and the P atom of the OP. The poses selected were used as initial configurations for the molecular dynamics (MD) simulations.
For the MD simulations, the complexes AChE-OP were constructed using the GROMACS 4.6.7 Suite53−56 and confined inside of octahedral boxes with around 1000 nm3 under periodic boundary conditions.57 The boxes were filled with around 24,000 water molecules using the model TIP3P58−60 to reproduce the effect of the solvent. The force field used was OPLS-AA,61 which presents excellent results for protein systems but has no parameters for small organic molecules, making a parameterization step for the oximes necessary. The topology and coordinate files were created, from the docking poses, with the AcPype62 software, and the atomic charges restrained electrostatic potential method63−66 calculated by semi-empirical AM1-BCC,67 parameterized to reproduce the method Hartree–Fock and basis set 6-31G*, which reproduces the experimental data of the oxime structure very well.68 The energy minimization of the systems was achieved in four steps: first, using steepest descent algorithm, with position restrained (PR) and convergence criterion of 100.00 kJ mol–1 nm–1; second, using steepest descent again with the same convergence criterion but without PR; third, using conjugate gradients, with a convergence criterion of 20.00 kJ mol–1 nm–1; and the last step, using the low-memory Broyden–Fletcher–Goldfarb–Shanno69 algorithm approach, with a convergence criterion of 10.00 kJ mol–1 nm–1. In order to balance temperature and pressure, the minimized complexes were submitted to thermalization steps. The equilibration of the temperature was achieved using the canonical ensemble (NVT),70 keeping the number of particles, volume, and temperature constant, with a time of 100 ps. The pressure equilibration was achieved continuing the simulation from the NVT equilibration phase and using isothermal–isobaric ensemble (NPT),70 keeping the number of particles, pressure, and temperature constant. The equilibration step was followed by a production step of 40 ns at 310 K and 1 bar, using 2 fs of the integration time with the lists of pairs being updated at every 5 steps. The cut-off for Lennard Jones and Coulomb interactions was between 0 and 1.2 nm. The leap-frog algorithm was used in the production step with the Nosé–Hoover thermostat71 (τ = 0.5 ps) at 310 K and the Parrinello–Rahman barostat72 (τ = 2.0 ps) at 1 bar. All Arg and Lys residues were assigned with positive charges, and all Glu and Asp residues were assigned with negative charges. The VMD73 software was used to visualize the simulation trajectories.
The molecular mechanics Poisson–Boltzmann (PB) surface area method has been widely used for free energy calculations in biological systems and has shown good affinity for proteins and ligands.42,45−47,74−79 The PB equation is used to calculate the contributions to the interaction energy between a ligand and a protein.80,81 To calculate the free binding energies between the studied oximes and the AChE-OP complexes, we used the g_mmpbsa80,82 software, which uses the trajectories resulting from MD simulations to determine the interactions between two pre-established groups, in our case, AChE-OP and oxime, previously separated into an index file. In this method, the free binding energy is defined as follows
| 1 |
Fitting the eq 1 to the cases under study, we have45
| 2 |
where GComplex represents the free energy of the complex AChE/GA/oxime, GAChE+OP represents the free energy of the protein, and GOxime represents the free energy of the oxime.80
Each term in the eq 2 above must be calculated in the following way
| 3 |
where “⟨ ⟩” represents mean values and EMM is the mechanics potential energy in vacuum. Temperature and entropy are depicted by the terms T and S, respectively, which together represent the entropic contribution to the free energy in a vacuum.80 The energy necessary to transfer a solute from the vacuum to the solvent (GSolvation) is called the free energy of solvation and is obtained from the sum of two terms
| 4 |
Gpolar arises from the electrostatic interaction between the solute and solvent, and it is obtained by solving the PB equation using the implicit (continuum) approximation.80
To calculate the binding energy components, the systems were subjected to three steps: the calculation of the potential energy in vacuum, the calculation of polar solvation energy, and the calculation of nonpolar solvation energy. Among the several models available for the nonpolar component, we used the SASA model83 with a surface tension of 0.0226778 (kJ/mol2) and a probe radius of 1.4 Å. For the calculations, dielectric constants of 6, 80, and 1 were used for the protein, water, and void, respectively. Subsequently, two python scripts (available at https://rashmikumari.github.io/g_mmpbsa/) were used. The scripts compile the energy component calculation files into two output files containing ΔGBindind and the contribution of each amino acid.
Analysis Methods
From the obtained trajectory, analyses were performed using tools offered by the GROMACS package53−56 and plotted with the Origin software.84 The distance between the centers of mass (COM) of the OP and the oxime as a function of time was obtained by the g_dist tool, which determines the distance between the COM of two system components from the x, y, and z coordinates. The g_rms tool provided root-mean-square deviation (RMSD) by comparing each structure from the trajectory to a reference structure; in this case, the initial system structure. The hydrogen bonds (H-bond) were determined based on cutoffs for the angle and distance of the H-bond donor and the H-bond acceptor through the g_hbond tool, where OH and NH groups are considered the donors, while O and N are the acceptors.54,56
In order to analyze and compare the interaction sites of AChE-VX and AChE-GB, the contribution of each amino acid to the binding energies and the volume of the interaction site were calculated. The contributions were determined by the g_mmpbsa tool, which decomposes the total binding energy into the contribution made by each residue, enabling a comparison of the relative contribution of residues to the overall binding energy.80 To determine the volume of the interaction site, the grid-based cavity prediction algorithm was used, which determines a region as a possible cavity and its volume, creating a discrete grid with a resolution of 0.5 Å, covering the protein and, at each grid point a sphere of radius 1.4 Å is placed.52 The grid points will be referred to as part of the inaccessible volume where clashes occur with the protein atom spheres and all other points are referred to as accessible.52 Each accessible grid point is checked to see if it is part of a cavity. Starting from a given grid point, the algorithm chooses a random direction, and this direction (and the opposite direction) is followed until the grid boundaries are hit, checking if an inaccessible grid point is hit on the way, and this is repeated a number of times. If the percentage of lines hitting an inaccessible volume is larger than a given threshold, the point is marked as being part of a cavity.52 The threshold is adjusted according to the location of the cavities found, where a value of 0% is only possible away from the protein, as opposed to a value of 100%, corresponding to a binding site deeply buried in the protein.52 In order to evaluate the influence of the OP on the conformation of inhibited AChE, g_rmsf was used to compute the root-mean-square fluctuation (RMSF) of backbone amino acid positions, in the trajectory of the AChE-OP simulation, using the initial configuration as refs (54) and (56). The SuperPose85 server was used to perform the AChE-VX and AChE-GB superposition and calculate the RMSD between structures. To generate the image, we used UCSF Chimera86 (http://www.rbvi.ucsf.edu/chimera) with the PyChimera87 script.
Results and Discussion
An analysis of the protonation state of oximes as a function of pH was performed by chemicalize, through the prediction of the dissociation constant method, using micro constants. The protonation state chosen was the one with the highest prevalence in the physiological pH of human blood and compared to the experimental data provided.37 HI-6 shows a deprotonation of oxime OH, with a prevalence of 93%, while RS41A and RS194B shows the cyclic N atom protonated, with prevalence of 96 and 98%, respectively (see Figure S1). The calculations were performed with the most abundant species of each oxime.
The main selection criteria of the selected docking poses were the shortest distance between the O atom of the oxime and P atom of the OP (which would favor the reactivation reaction) and higher affinity (more negative scoring function). Table 1 presents the values calculated by the MVD47 MolDock scoring function for interactions of oximes with the AChE-OP complexes. In the docking results, we highlight the behavior of the larger molecular structures (HI-6, RS48B, and RS182A), which presented MolDock scoring function more negative than the smaller ones (2-PAM, RS41A, and RS194B) for all systems, being directly related to the interaction energies and H-bonds formed with amino acids. It is important to highlight that this difference may be directly related to the fact that the crystallographic structure of the inhibited enzyme is complexed with HI-6, implying in a larger interaction site, favoring the interactions of larger oximes. The interaction modes of the best poses of each oxime inside each system are shown in Figures S2 and S3 of the Supporting Information.
Table 1. Values for MolDock Score Function, H-Bond, Distance OOxime–POP, and Amino Acid Interacting with the Oximes Inside the AChE-OP Complexes.
| oxime | MD score (kJ mol–1) | H-bond (kJ mol–1) | distance O–P (nm) | amino acids |
|---|---|---|---|---|
| AChE-VX | ||||
| HI-6 | –521.24 | –35.31 | 0.45 | Tyr124, Tyr133, Tyr337 |
| 2-PAM | –332.04 | –25.65 | 0.65 | Gly120, Ser125, Tyr133 |
| RS41A | –356.39 | –28.12 | 0.66 | Ser125, Tyr133 |
| RS194B | –328.78 | –24.89 | 0.66 | Ser125, Tyr133 |
| RS48B | –489.24 | –26.61 | 0.55 | Tyr124, Ser125, Tyr337 |
| RS182A | –499.07 | –28.07 | 0.51 | Ser125, Thr83, Tyr337 |
| AChE-GB | ||||
| HI-6 | –505.47 | –15.82 | 0.56 | Tyr124, Ser298, Tyr337 |
| 2-PAM | –256.19 | 0.00 | 0.32 | ____ |
| RS41A | –362.38 | –20.63 | 0.86 | Tyr341, Phe295 |
| RS194B | –391.58 | –10.54 | 0.84 | Tyr341 |
| RS48B | –613.83 | –40.38 | 0.41 | Ser293, Phe295, Arg296, Ser298 |
| RS182A | –536.10 | –53.35 | 0.38 | Tyr124, Glu285, Trp286, Phe295, Arg296, Ser298 |
The efficacy of oximes depends on their reactivity and affinity toward the AChE-OP complex, where the reactivity derived from the nucleophilic activity of the oxime moiety and the affinity is derived from physicochemical features (steric compatibility, electrostatic effects, and hydrophobic interactions).24,88 The graphs of RMSD vs time for the AChE-OP/oxime complexes (Figure 2) were plotted in order to analyze the stability and affinity of the systems.
Figure 2.

RMSD vs time for the AChE-VX (black)/oxime (dark cyan) and AChE-GB (green)/oxime (orange) complexed with (a) HI-6, (b) 2-PAM, (c) RS41A, (d) RS194B (e) RS48B, and (f) RS182A. The RMSD of AChE inhibited is from the amino acid backbone variations.
The results for 2-PAM (Figure 2b) stand out because it presents low position variation in both systems. This fact could be related to the affinity and the interactions performed with the enzymes, as observed in the binding energy values (Table 2).
Table 2. Binding Energies between AChE-VX and AChE-GB and the Studied Oximes, Kox Obtained in the Literature, Contribution of Asp74 to the Binding Energy, and the Volume of the Oxime Interaction Sitea.
| oxime | binding energies (kJ mol–1) | Kox (mM) | Asp74 interaction energies (kJ mol–1) | interaction site volume (nm3) |
|---|---|---|---|---|
| AChE-VX | ||||
| HI-6 | –2022.36 | –233.47 | 0.20 | |
| 2-PAM | –894.23 | 0.2538 | –45.62 | 0.19 |
| RS41A | –102.43 | 4.638 | 1.79 | 0.33 |
| RS194B | –150.80 | 2.138 | 4.12 | 0.18 |
| RS48B | –201.81 | ___ | –14.00 | 0.25 |
| RS182A | –204.16 | ___ | –7.41 | 0.25 |
| AChE-GB | ||||
| HI-6 | –2034.62 | –174.02 | 0.36 | |
| 2-PAM | –971.34 | 0.3438 | –61.24 | 0.26 |
| RS41A | –145.65 | 1138 | –5.62 | 0.41 |
| RS194B | –126.67 | 1.938 | –4.34 | 0.33 |
| RS48B | –254.06 | ___ | –14.16 | 0.38 |
| RS182A | –356.88 | ___ | –15.19 | 0.44 |
Empty cells are due to data not found in the literature.
HI-6, unlike 2-PAM, presented a larger position variation with AChE-GB (Figure 2a) but was restricted to the range of 0.17 and 0.22 nm, throughout the simulation, which can be considered stable, given its degree of freedom and frame size. The position changes of HI-6 inside AChE-VX (Figure 2a) are stabilized after 20 ns (20,000 ps) of simulation. This behavior can be explained by the larger volume of the interaction site of HI-6 inside the complex AChE-VX, related to AChE-GB. This causes the solvent molecules present in the site to take longer to settle, an effect evidenced by the H-bonds formed between HI-6 and the solvent (Figure 3a) during the simulation, which increases from one to three after 20 ns. Plots of RMSD for the apo enzymes shown in Figure S4 show that the systems stabilize before the 20 ns of MD simulation.
Figure 3.

H-bonds formed between oximes HI-6 (a) and RS48B (b) and solvent inside the complex AChE-VX, during the MD simulations.
In vivo experiments, the difference in the efficiency of HI-6 compared to 2-PAM against VX and GB poisoning is not evident or statistically significant, as previously described in the literature.23,31,89 However, in in vitro studies, HI-6 showed high affinity and higher velocity of reactivation than 2-PAM at low concentrations for both nerve agents. Meanwhile, 2-PAM was able to significantly reactivate AChE inhibited by both OPs, but concentrations that did not correspond to in vivo recommended doses were required.34,88,90,91 These data are evidenced in our studies by the results of binding energies (Table 2) of these oximes, where higher affinity of HI-6 for the AChE-OP complexes is evident, which can highlight here the influence of the two quaternary N atoms in electrostatic effects, which granted twice more affinity compared to 2-PAM. The values of Kox found in the literature38 for 2-PAM, RS41A, and RS194B inside AChE-GB and AChE-VX (see Table 2), corroborate our theoretical results once they are much lower for 2-PAM. As Kox refers to the dissociation of the oxime from the interaction site, the lower its value the higher the affinity of the oxime for the site, similar to the interaction energies.
Sit et al. and later Radić et al.,37,38 in their studies demonstrated that RS41A and RS194B penetrate CNS quickly and a favorable antidotal action was achieved. However, in our studies, the oximes RS41A and RS194B (except for the system AChE-VX/RS194B, which will be discussed later) showed a larger position variation throughout the MD simulation. Behavior reflected by their respective binding energies with the AChE-OP complexes, showing much lower affinity, compared to the cationic oximes. Importantly, the presence of a protonated cyclic amine in both oximes, given the result of the study of ionization constants, was not sufficient to provide a good affinity for the complex AChE-OP because when evaluating the variation of the distances between the COM of oxime and OP (Figure 4), we observed that RS41A (Figure 4e,f) and RS194B (Figure S3 of Supporting Information) were moving away from the OP and consequently from the catalytic site. This considerably impairs the reactivation process, given the importance of the oxime’s affinity for the interaction site and effectiveness of the kinetic process.24,49,88,92−94
Figure 4.

Population histogram of COM distances between OP and oximes presented during MD and determined by interactions between the oximes and the complexes AChE-OP. (a) VX/HI-6, (b) GB/HI-6, (c) VX/2-PAM, (d) GB/2-PAM, (e) VX/RS41A, and (f) GB/RS41A. Other histograms can be checked in Figures S4 of the Supporting Information
Specifically, RS194B inside the AChE-VX interaction site presented a small range of position variation as a function of time, different from the results presented inside AChE-GB. However, as previously mentioned, the graphs of distances between COM of oxime and OP show that RS194B moves slightly away from the catalytic site.
RS41A and RS194B presented low affinity in both systems, as can be seen through the values of binding energy, especially when compared to the cationic oximes, in agreement with the results reported by Sit et al. and Radić et al.37,38 Despite having binding energy values below the cationic oximes, RS194B has better stability and affinity than RS41A inside AChE-VX, even though both have similar structures with the only difference in the cyclic moiety. The stability of RS194B inside AChE-VX can be justified by the hydrophobic interactions between the cyclic moiety and amino acids of the interaction site, favored by its smaller volume (Table 2). Besides, we can add the fact that it does not form stable H-bonds with any of the AChE-OP complexes and, instead, formed more H-bonds with solvent molecules (3 H-bond with the solvent inside AChE-VX and 5 H-bond with the solvent inside AChE-GB).
Oxime RS48B presented a great variation of position up to 20 ns of simulation in both systems, as previously reported. Initially, there is a displacement toward the catalytic site (evidenced by the distance histogram of the COM, of Figure S5), but as can be observed in Figure 3b, like with HI-6, after 20 ns, the H-bonds formed with solvent molecules contributed to stabilize the molecule at the interaction site. RS182A, like RS48B, initially moves away from the catalytic site (see distance histogram of COM, at Figure S5) and then remains stable in both systems. This can be justified by the large number of H-bond formed (up to six in the AChE-VX/RS182A complex and up to eight in the AChE-GB/RS182A complex). However, both RS48B and RS182A, despite having the larger structures, higher number of substituents, and more H-bond formed, presented affinities for the AChE-OP complex far below the cationic oximes and close to the protonated ones, as can be seen by the binding energies values shown in Table 2. The position changes of one heterocycle, apparently did not change the affinity of these oximes for AChE-VX, unlike AChE-GB, where there is a higher affinity of RS182A, most likely due to interactions that favored greater stability.
Among the amino acids that contributed to the higher affinity of the cationic oximes, we can highlight Glu334 and Asp74. Glu334, being part of the catalytic triad of AChE, playing a fundamental role in the hydrolysis of ACh, as previously described.95,96 Asp74 is part of the so-called peripheral anionic site (PAS) located around the entrance to the active site gorge, which in addition to Asp74, consists of residues Tyr72, Tyr124, Trp286, and Tyr341.96,97 The PAS is a site where a ligand can be tethered to a drug candidate not only for increasing the overall binding affinity but also by enhancing the reactivation of OP-inhibited AChE.95,98 The effect of the PAS contribution on the affinity between oximes and the AChE-OP complex can be evidenced by the difference in the binding energies of the cationic oximes related to other oximes. Because the area of PAS is surrounded by acidic residues, there is a concentration of negative charges in this region, capable of binding cationic and aromatic oximes.95−97 This is probably the reason why HI-6 presents a higher binding energy than 2-PAM, once one of these two positively charged nitrogen docks closer to the PAS site region.
The difference in contribution from Asp74 to HI-6 inside the AChE-OP complexes can be explained by the conformational difference of the interaction sites (Figure 5) due to the smaller volume observed for the complex AChE-VX compared to AChE-GB. With a smaller volume at the interaction site, the PAS region can interact better with HI-6 inside the AChE-VX site (see Figure 5a and Table 2).
Figure 5.

Influence of the differences between the interaction sites of AChE-VX (a) and AChE-GB (b) on the amino acid contributions to the binding energies of HI-6. The different colors in the tertiary structure of the AChE-OP complexes represent (in kJ mol–1) the contributions of each amino acid and the regions to the binding energy between oxime and the complex AChE-OP.
According to the literature, the AChE inhibition reaction by VX induces a slight structural change in the side chain of His447, while the AChE-GB structure shows an apo-like conformation for His447,94 and phosphonylation causes a reduction in space in the cavity relative to apo-like AChE.88 However, when we analyze the fluctuation (Figure 6) of the amino acids of the AChE-OP complexes without the oxime, we find that the AChE-GB complex presents greater position variation from the initial system configuration when compared to AChE-VX. Amino acids from both systems started from the same initial configuration, showing that GB can influence the structure of the entire protein because of its interaction with the amino acids of the active site. This interaction gives the interaction site greater volume and directly affects the interactions between amino acids and oximes. It can be seen from the RMSF (Figure 6) that AChE-VX presents largest fluctuations in only three regions, between amino acids 72–98, 250–280, and 330–360, respectively. These fluctuations are very important because these regions contains all amino acids of the PAS, demonstrating the importance of this site for the interaction with the oximes and AChE reactivation.
Figure 6.
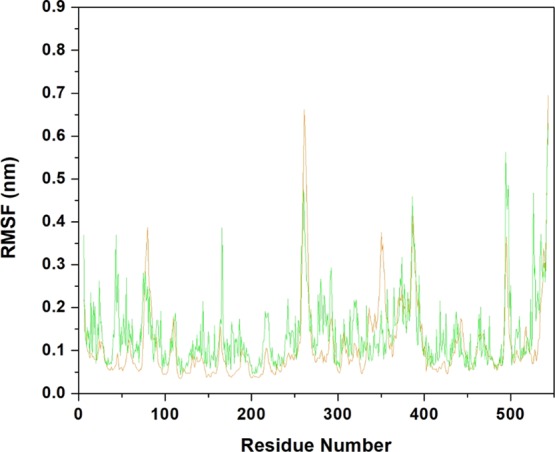
RMSF of the amino acids of AChE-VX (orange) and AChE-GB (green) fitting the initial structure of each system as a reference.
The protein superposition demonstrated that there is a conformational difference between the systems, as can be observed in Figure 7, where the backbones have an RMSD of 3.79 nm, showing that there are conformational differences in the two systems and, as commented before, this has great influence on the interaction with the oximes. It is important to mention that there are no reports yet of the experimental data proving the volume difference between the AChE-VX and AChE-GB interaction sites, only data suggesting that there is a difference between them.94
Figure 7.
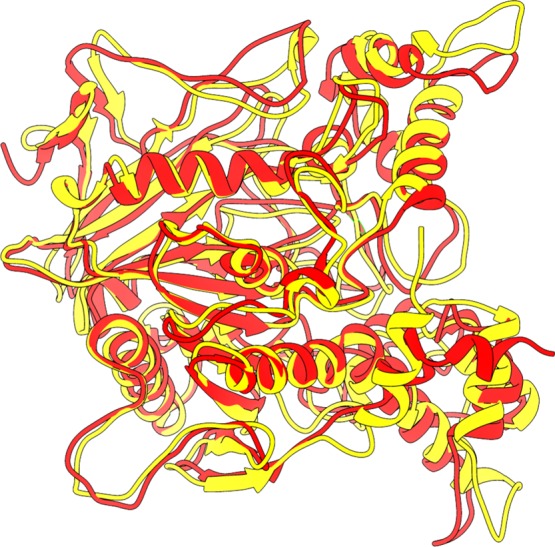
AChE-VX (red) and AChE-GB (yellow) structure superposition demonstrating the differences between their most stable conformations.
Conclusions
Our results were capable of showing that even being able to diffuse through the CNS, the uncharged oximes studied may not present good affinity for VX and GB-inhibited AChE when compared to the cationic commercial oximes HI-6 and 2-PAM. For the larger oximes, the H-bond formed with the solvent were crucial for their stability in the interaction site, as seen for HI-6, RS48B, and RS182A. The interaction of amino acids of PAS with the oximes demonstrated the great importance of the quaternary nitrogen to improve the affinity between the oxime and the complex AChE-OP but, also, that this interaction is not as effective for RS41A and RS194B. The simulations showed that the difference in the spatial configuration in the oxime interaction region inside AChE-VX and AChE-GB may represent a problem in the development of efficient oximes against intoxication with VX and GB because this may cause considerable difference in affinity with the oximes. In summary, these studies expand and extend the database on uncharged oximes and should be helpful in assessing their potential as an alternative antidote for nerve agent poisoning.
Acknowledgments
The authors wish to thank the Military Institute of Engineering and the Federal University of Lavras for the infrastructure and the Brazilian financial agencies CAPES, CNPq (grant no. 308225/2018-0) and FAPERJ (grant no. E-02/202.961/2017). This work was also supported by the excellence project UHK.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03737.
Oxime ionization constant; oxime interaction modes; and distance of COM (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ribeiro T. S.; Prates A.; Alves S. R.; Oliveira-Silva J. J.; Riehl C. A. S.; Figueroa-Villar J. D. The Effect of Neutral Oximes on the Reactivation of Human Acetylcholinesterase Inhibited with Paraoxon. J. Braz. Chem. Soc. 2012, 23, 1216–1225. 10.1590/s0103-50532012000700004. [DOI] [Google Scholar]
- Ganesan K.; Raza S.; Vijayaraghavan R. Chemical Warfare Agents. J. Pharm. Bioallied Sci. 2010, 2, 166–178. 10.4103/0975-7406.68498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- França T. C. C.; Silva G. R.; Castro A. T. Defesa Química: Uma Nova Disciplina No Ensino de Química. Rev. Virtual Química 2010, 2, 84–104. [Google Scholar]
- Chauhan S.; Chauhan S.; D’Cruz R.; Faruqi S.; Singh K. K.; Varma S.; Singh M.; Karthik V. Chemical Warfare Agents. Environ. Toxicol. Pharmacol. 2008, 26, 113–122. 10.1016/j.etap.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Szinicz L. History of Chemical and Biological Warfare Agents. Toxicology 2005, 214, 167–181. 10.1016/j.tox.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Kim K.K.; Tsay O. G.; Atwood D. A.; Churchill D. G. Destruction and Detection of Chemical Warfare Agents. Chem. Rev. 2015, 111, 5345–403. 10.1021/cr100193y. [DOI] [PubMed] [Google Scholar]
- Arias H. R.; Gu R.-X.; Feuerbach D.; Guo B.-B.; Ye Y.; Wei D.-Q. Novel Positive Allosteric Modulators of the Human Α7 Nicotinic Acetylcholine Receptor. Biochemistry 2011, 50, 5263–5278. 10.1021/bi102001m. [DOI] [PubMed] [Google Scholar]
- Gu R.-X.; Zhong Y.-Q.; Wei D.-Q. Structural Basis of Agonist Selectivity for Different NAChR Subtypes: Insights from Crystal Structures, Mutation Experiments and Molecular Simulations. Curr. Pharm. Des. 2011, 17, 1652–1662. 10.2174/138161211796355119. [DOI] [PubMed] [Google Scholar]
- Gonçalves A. d. S.; França T. C. C.; Figueroa-Villar J. D.; Pascutti P. G. Molecular Dynamics Simulations and QM/MM Studies of the Reactivation by 2-PAM of Tabun Inhibited Human Acethylcolinesterase. J. Braz. Chem. Soc. 2011, 22, 155–165. 10.1590/s0103-50532011000100021. [DOI] [Google Scholar]
- Gonçalves A. d. S.; França T. C. C.; Wilter A.; Figueroa-Villar J. D. Molecular Dynamics of the Interaction of Pralidoxime and Deazapralidoxime with Acetylcholinesterase Inhibited by the Neurotoxic Agent Tabun. J. Braz. Chem. Soc. 2006, 17, 968–975. 10.1590/s0103-50532006000500022. [DOI] [Google Scholar]
- Ajami D.; Rebek J. Chemical Approaches for Detection and Destruction of Nerve Agents. Org. Biomol. Chem. 2013, 11, 3936–4124. 10.1039/c3ob40324f. [DOI] [PubMed] [Google Scholar]
- Larini L.Toxicologia Dos Praguicidas; Editora Manole: São Paulo, 1999. [Google Scholar]
- Martin T.; Lobert S. Chemical Warfare. Toxicity of Nerve Agents. Crit. Care Nurse 2003, 23, 15–20. [PubMed] [Google Scholar]
- Sidell F. R.; Takafuji E. T.; Franz D. R.. Medical Aspects of Chemical Warfare, 1a Edição.; United States Government Printing: Washington, 1997. [Google Scholar]
- Marrs T. C.; Maynard R. L.; Sidell F. R. In Chemical Warfare Agents: Toxicology and Treatment, 2nd ed.; Marrs T. C., Maynard R. L., Sidell F. R., Eds.; John Wiley & Sons Ltd, 2007. [Google Scholar]
- Bosgra S.; van Eijkeren J. C. H.; van der Schans M. J.; Langenberg J. P.; Slob W. Toxicodynamic Analysis of the Inhibition of Isolated Human Acetylcholinesterase by Combinations of Methamidophos and Methomyl in Vitro. Toxicol. Appl. Pharmacol. 2009, 236, 1–8. 10.1016/j.taap.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Bajgar J. Complex View on Poisoning with Nerve Agents and Organophosphates. Acta medica (Hradec Kral. 2005, 48, 3–21. 10.14712/18059694.2018.23. [DOI] [PubMed] [Google Scholar]
- Delfino R. T.; Ribeiro T. S.; Figueroa-Villar J. D. Organophosphorus Compounds as Chemical Warfare Agents: A Review. J. Braz. Chem. Soc. 2009, 20, 407–428. 10.1590/s0103-50532009000300003. [DOI] [Google Scholar]
- Karalliedde L.; Baker D.; Marrs T. C. Organophosphate-Induced Intermediate Syndrome. Toxicol. Rev. 2006, 25, 1–14. 10.2165/00139709-200625010-00001. [DOI] [PubMed] [Google Scholar]
- Hulse E. J.; Davies J. O. J.; Simpson A. J.; Sciuto A. M.; Eddleston M. Respiratory Complications of Organophosphorus Nerve Agent and Insecticide Poisoning. Implications for Respiratory and Critical Care. Am. J. Respir. Crit. Care Med. 2014, 190, 1342–1354. 10.1164/rccm.201406-1150ci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretto A. Experimental and Clinical Toxicology of Anticholinesterase Agents. Toxicol. Lett. 1998, 102–103, 509–513. 10.1016/s0378-4274(98)00245-8. [DOI] [PubMed] [Google Scholar]
- Mercey G.; Verdelet T.; Renou J.; Kliachyna M.; Baati R.; Nachon F.; Jean L.; Renard P.-Y. Reactivators of Acetylcholinesterase Inhibited by Organophosphorus Nerve Agents. Acc. Chem. Res. 2012, 45, 756–766. 10.1021/ar2002864. [DOI] [PubMed] [Google Scholar]
- Koplovitz I.; Stewart J. R. A Comparison of the Efficacy of HI6 and 2-PAM against Soman, Tabun, Sarin, and VX in the Rabbit. Toxicol. Lett. 1994, 70, 269–279. 10.1016/0378-4274(94)90121-x. [DOI] [PubMed] [Google Scholar]
- Kuca K.; Jun D.; Musilek K. Structural Requirements of Acetylcholinesterase Reactivators. Mini Rev. Med. Chem. 2006, 6, 269–277. 10.2174/138955706776073510. [DOI] [PubMed] [Google Scholar]
- Soukup O.; Tobin G.; Kumar U. K.; Binder J.; Proska J.; Jun D.; Fusek J.; Kuca K. Interaction of Nerve Agent Antidotes with Cholinergic Systems. Curr. Med. Chem. 2010, 17, 1708–1718. 10.2174/092986710791111260. [DOI] [PubMed] [Google Scholar]
- Kuca K.; Musilek K.; Jun D.; Karasova J.; Soukup O.; Pejchal J.; Hrabinova M. Structure-Activity Relationship for the Reactivators of Acetylcholinesterase Inhibited by Nerve Agent VX. Med. Chem. 2013, 9, 689–693. 10.2174/1573406411309050008. [DOI] [PubMed] [Google Scholar]
- Gonçalves A. d. S.; França T. C. C.; Caetano M. S.; Ramalho T. C. Reactivation Steps by 2-PAM of Tabun-Inhibited Human Acetylcholinesterase: Reducing the Computational Cost in Hybrid QM/MM Methods. J. Biomol. Struct. Dyn. 2014, 32, 301–307. 10.1080/07391102.2013.765361. [DOI] [PubMed] [Google Scholar]
- Bester S. M.; Guelta M. A.; Cheung J.; Winemiller M. D.; Bae S. Y.; Myslinski J.; Pegan S. D.; Height J. J. Structural Insights of Stereospecific Inhibition of Human Acetylcholinesterase by VX and Subsequent Reactivation by HI-6. Chem. Res. Toxicol. 2018, 31, 1405–1417. 10.1021/acs.chemrestox.8b00294. [DOI] [PubMed] [Google Scholar]
- Bajgar J.; Fusek J.; Kassa J.; Kuca K.; Jun D. Chemical Aspects of Pharmacological Prophylaxis against Nerve Agent Poisoning. Curr. Med. Chem. 2009, 16, 2977–2986. 10.2174/092986709788803088. [DOI] [PubMed] [Google Scholar]
- Whitmore C.; Cook A. R.; Mann T.; Price M. E.; Emery E.; Roughley N.; Flint D.; Stubbs S.; Armstrong S. J.; Rice H.; et al. The Efficacy of HI-6 DMS in a Sustained Infusion against Percutaneous VX Poisoning in the Guinea-Pig. Toxicol. Lett. 2018, 293, 207–215. 10.1016/j.toxlet.2017.11.007. [DOI] [PubMed] [Google Scholar]
- Reymond C.; Jaffré N.; Taudon N.; Menneteau M.; Chaussard H.; Denis J.; Castellarin C.; Dhote F.; Dorandeu F. Superior Efficacy of HI-6 Dimethanesulfonate over Pralidoxime Methylsulfate against Russian VX Poisoning in Cynomolgus Monkeys (Macaca Fascicularis). Toxicology 2018, 410, 96–105. 10.1016/j.tox.2018.09.005. [DOI] [PubMed] [Google Scholar]
- Antonijevic B.; Stojiljkovic M. P. Unequal Efficacy of Pyridinium Oximes in Acute Organophosphate Poisoning. Clin. Med. Res. 2007, 5, 71–82. 10.3121/cmr.2007.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassa J.; Musilek K.; Zdarova Karasova J.; Kuca K.; Bajgar J. Two Possibilities How to Increase the Efficacy of Antidotal Treatment of Nerve Agent Poisonings. Mini Rev. Med. Chem. 2012, 12, 24–34. 10.2174/138955712798869011. [DOI] [PubMed] [Google Scholar]
- Kuca K.; Cabal J.; Kassa J.; Jun D.; Hrabinová M. Comparison of in Vitro Potency of Oximes (Pralidoxime, Obidoxime, HI-6) to Reactivate Sarin-Inhibited Acetylcholinesterase in Various Parts of Pig Brain. J. Appl. Toxicol. 2005, 25, 271–276. 10.1002/jat.1053. [DOI] [PubMed] [Google Scholar]
- Kuča K.; Musílek K.; Jun D.; Pohanka M.; Žd′árová Karasová J.; Novotný L.; Musilová L. Could Oxime HI-6 Really Be Considered as “Broad-Spectrum” Antidote?. J. Appl. Biomed. 2009, 7, 143–149. 10.32725/jab.2009.016. [DOI] [Google Scholar]
- Radić Z.; Sit R. K.; Garcia E.; Zhang L.; Berend S.; Kovarik Z.; Amitai G.; Fokin V. V.; Sharpless K. B.; Taylor P. Mechanism of Interaction of Novel Uncharged, Centrally Active Reactivators with OP-HAChE Conjugates. Chem. Biol. Interact. 2013, 203, 67–71. 10.1016/j.cbi.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radić Z.; Sit R. K.; Kovarik Z.; Berend S.; Garcia E.; Zhang L.; Amitai G.; Green C.; Radić B.; Fokin V. V.; et al. Refinement of Structural Leads for Centrally Acting Oxime Reactivators of Phosphylated Cholinesterases. J. Biol. Chem. 2012, 287, 11798–11809. 10.1074/jbc.m111.333732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit R. K.; Radić Z.; Gerardi V.; Zhang L.; Garcia E.; Katalinić M.; Amitai G.; Kovarik Z.; Fokin V. V.; Sharpless K. B.; et al. New Structural Scaffolds for Centrally Acting Oxime Reactivators of Phosphylated Cholinesterases. J. Biol. Chem. 2011, 286, 19422–19430. 10.1074/jbc.m111.230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik Z.; Maček N.; Sit R. K.; Radić Z.; Fokin V. V.; Barry Sharpless K.; Taylor P. Centrally Acting Oximes in Reactivation of Tabun-Phosphoramidated AChE. Chem. Biol. Interact. 2013, 203, 77–80. 10.1016/j.cbi.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M.; Wang Z.; Zhou Y.; Xu W.; Li S.; Wang L.; Wei D.; Qiao Z. A Novel Drug Candidate for Alzheimer’s Disease Treatment: Gx-50 Derived from Zanthoxylum Bungeanum. J. Alzheimer’s Dis. 2013, 34, 203–213. 10.3233/jad-121831. [DOI] [PubMed] [Google Scholar]
- Szegezdi J.; Csizmadia F.. Prediction of Dissociation Constants Using Microconstants. In American Chemical Society National Meeting, 2004; pp 2–3.
- Shao Y.; Molnar L. F.; Jung Y.; Kussmann J.; Ochsenfeld C.; Brown S. T.; Gilbert A. T. B.; Slipchenko L. V.; Levchenko S. V.; O’Neill D. P.; et al. Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- Rocha G. B.; Freire R. O.; Simas A. M.; Stewart J. J. P. RM1: A Reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. 10.1002/jcc.20425. [DOI] [PubMed] [Google Scholar]
- Gonçalves A. d. S.; França T. C. C.; Figueroa-Villar J. D.; Pascutti P. G. Conformational Analysis of Toxogonine, TMB-4 and HI-6 Using PM6 and RM1 Methods. J. Braz. Chem. Soc. 2010, 21, 179–184. 10.1590/s0103-50532010000100025. [DOI] [Google Scholar]
- de Souza F. R.; Garcia D. R.; Cuya T.; Kuca K.; Alencastro R. B.; França T. C. C. Behavior of Uncharged Oximes Compared to HI6 and 2-PAM in the Human AChE-Tabun Conjugate: A Molecular Modeling Approach. J. Biomol. Struct. Dyn. 2017, 36, 1430–1438. 10.1080/07391102.2017.1324322. [DOI] [PubMed] [Google Scholar]
- de Souza F. R.; Guimarães A. P.; Cuya T.; Freitas M. P.; Gonçalves A. da S.; Forgione P.; Costa França T. C. Analysis of Coxiela Burnetti Dihydrofolate Reductase via in Silico Docking with Inhibitors and Molecular Dynamics Simulation. J. Biomol. Struct. Dyn. 2016, 35, 2975–2986. 10.1080/07391102.2016.1239550. [DOI] [PubMed] [Google Scholar]
- Bastos L. d. C.; de Souza F. R.; Guimarães A. P.; Sirouspour M.; Guizado T. R. C.; Forgione P.; Ramalho T. C.; França T. C. C. Virtual Screening, Docking and Dynamics of Potential New Inhibitors of Dihydrofolate Reductase from Yersinia Pestis. J. Biomol. Struct. Dyn. 2015, 34, 2184–2198. 10.1080/07391102.2015.1110832. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekström F.; Hörnberg A.; Artursson E.; Hammarström L. G.; Schneider G.; Pang Y. P. Structure of HI-6-Sarin-Acetylcholinesterase Determined by x-Ray Crystallography and Molecular Dynamics Simulation: Reactivator Mechanism and Design. PLoS One 2009, 4, e5957 10.1371/journal.pone.0005957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N.; Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Schwede T.; Kopp J.; Guex N.; Peitsch M. C. SWISS-MODEL: An Automated Protein Homology-Modeling Server. Nucleic Acids Res 2003, 31, 3381–3385. 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R.; Christensen M. H. MolDock : A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; Van Der Spoel D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; Van Der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Van der Spoel D.; Van Drunen R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. 10.1016/0010-4655(95)00042-e. [DOI] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Martínez L.; Borin I. A.; Skaf M. S.. Fundamentos de Simulação Por Dinâmica Molecular. In Métodos de Química Teórica e Modelagem Molecular; Morgon N. H., Coutinho K., Eds.; Editora Livraria da Física, 2007; pp 413–452. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Harrach M. F.; Drossel B. Structure and Dynamics of TIP3P, TIP4P, and TIP5P Water near Smooth and Atomistic Walls of Different Hydroaffinity. J. Chem. Phys. 2014, 140, 174501. 10.1063/1.4872239. [DOI] [PubMed] [Google Scholar]
- Mark P.; Nilsson L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. 10.1021/jp003020w. [DOI] [Google Scholar]
- Kaminski G. A.; Friesner R. A.; Tirado-rives J.; Jorgensen W. L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. 10.1021/jp003919d. [DOI] [Google Scholar]
- Sousa da Silva A. W.; Vranken W. F. ACPYPE - AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro A. A. S. T.; Horta B. A. C.; Alencastro R. B. d. MKTOP: A Program for Automatic Construction of Molecular Topologies. J. Braz. Chem. Soc. 2008, 19, 1433–1435. 10.1590/s0103-50532008000700031. [DOI] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Kollman P. a.; Kollmann P. A. Application of RESP Charges To Calculate Conformational Energies, Hydrogen Bond Energies, aCrgies of Solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. 10.1021/ja00074a030. [DOI] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W.; Kollman P. a. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. 10.1021/j100142a004. [DOI] [Google Scholar]
- Wang J.; Cieplak P.; Kollman P. A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules?. J. Comput. Chem. 2000, 21, 1049–1074. . [DOI] [Google Scholar]
- Jakalian A.; Jack D. B.; Bayly C. I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- Gonewar N. R.; Jadhav V. B. J. K. D.; Sarawadekar R. G. Theoretical Calculations of Infrared, NMR and Electronic Spectra of 2-Nitroso-1, Naphthol or 1-2 Naphthoquinine-2 Oxime and Comparison with Experimental Data. Res. Pharm. 2012, 2, 18–25. [Google Scholar]
- Byrd R. H.; Lu P.; Nocedal J.; Zhu C. A Limited Memory Algorithm for Bound Constrained Optimization. SIAM J. Sci. Comput. 1995, 16, 1190–1208. 10.1137/0916069. [DOI] [Google Scholar]
- Bosko J. T.; Todd B. D.; Sadus R. J. Molecular Simulation of Dendrimers and Their Mixtures under Shear: Comparison of Isothermal-Isobaric (NpT) and Isothermal-Isochoric (NVT) Ensemble Systems. J. Chem. Phys. 2005, 123, 034905. 10.1063/1.1946749. [DOI] [PubMed] [Google Scholar]
- Evans D. J.; Holian B. L. The Nose-Hoover Thermostat. J. Chem. Phys. 1985, 83, 4069–4074. 10.1063/1.449071. [DOI] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VDM: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- de Almeida J. S. F. D.; Guizado T. R.; Guimaraes A. P.; Ramalho T. C.; Goncalves A. S.; de Koning M. C.; Franca T. C. Docking and Molecular Dynamics Studies of Peripheral Site Ligand-Oximes as Reactivators of Sarin-Inhibited Human Acetylcholinesterase. J. Biomol. Struct. Dyn. 2016, 34, 2632–2642. 10.1080/07391102.2015.1124807. [DOI] [PubMed] [Google Scholar]
- Kar P.; Lipowsky R.; Knecht V. Importance of Polar Solvation and Configurational Entropy for Design of Antiretroviral Drugs Targeting HIV-1 Protease. J. Phys. Chem. B 2013, 117, 5793–5805. 10.1021/jp3085292. [DOI] [PubMed] [Google Scholar]
- Jayaram B.; Sprous D.; Young M. A.; Beveridge D. L. Free Energy Analysis of the Conformational Preferences of A and B Forms of DNA in Solution. J. Am. Chem. Soc. 1998, 120, 10629–10633. 10.1021/ja981307p. [DOI] [Google Scholar]
- Vorobjev Y. N.; Almagro J. C.; Hermans J. Discrimination between Native and Intentionally Misfolded Conformations of Proteins: ES/IS, a New Method for Calculating Conformational Free Energy That Uses Both Dynamics Simulations with an Explicit Solvent and an Implicit Solvent Continuum Model. Proteins Struct. Funct. Genet. 1998, 32, 399–413. . [DOI] [PubMed] [Google Scholar]
- Estévez J.; Rodrigues de Souza F.; Romo M.; Mangas I.; Costa Franca T. C.; Vilanova E. Interactions of Human Butyrylcholinesterase with Phenylvalerate and Acetylthiocholine as Substrates and Inhibitors: Kinetic and Molecular Modeling Approaches. Arch. Toxicol. 2019, 93, 1281–1296. 10.1007/s00204-019-02423-8. [DOI] [PubMed] [Google Scholar]
- Garcia D. R.; Souza F. R.; Paula Guimarães A.; Castro Ramalho T.; Palermo de Aguiar A.; Celmar Costa França T. Design of Inhibitors of Thymidylate Kinase from Variola Virus as New Selective Drugs against Smallpox: Part II. J. Biomol. Struct. Dyn. 2019, 37, 4569–4579. 10.1080/07391102.2018.1554510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R.; Kumar R.; Lynn A. g_mmpbsa-A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Homeyer N.; Gohlke H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10037–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitkoff D.; Sharp K. A.; Honig B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. 10.1021/j100058a043. [DOI] [Google Scholar]
- Edwards P. M. Origin 7.0: Scientific Graphing and Data Analysis Software. J. Chem. Inf. Comput. Sci. 2002, 42, 1270–1271. 10.1021/ci0255432. [DOI] [Google Scholar]
- Maiti R.; Van Domselaar G. H.; Zhang H.; Wishart D. S. SuperPose: A Simple Server for Sophisticated Structural Superposition. Nucleic Acids Res 2004, 32, 590–594. 10.1093/nar/gkh477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pedregal J. R.-G.; Maréchal J.-D. PyChimera: use UCSF Chimera modules in any Python 2.7 project. Bioinformatics 2018, 34, 1784–1785. 10.1093/bioinformatics/bty021. [DOI] [PubMed] [Google Scholar]
- Kuca K.; Cabal J.; Kassa J.; Jun D.; Hrabinova M. Vitro Potency of H Oximes (HI-6, HLö-7), the Oxime BI-6, and Currently Used Oximes (Pralidoxime, Obidoxime, Trimedoxime) to Reactivate Nerve Agent-Inhibited Rat Brain Acetylcholinesterase. J. Toxicol. Environ. Health. A 2006, 69, 1431–1440. 10.1080/15287390500364283. [DOI] [PubMed] [Google Scholar]
- Olson C. T.; Menton R. G.; Riser R. C.; Matthews M. C.; Stotts R. R.; Romano J. R.; Koplovitz I.; Hackley B. E.; Johnson J. B. Efficacies of Atropine/2-PAM and Atropine/HI-6 in Treating Monkeys Intoxicated with Organophosphonate Nerve Agents. Int. J. Toxicol. 1997, 16, 9–20. 10.1080/109158197227314. [DOI] [Google Scholar]
- Kassa J.; Jun D.; Kuca K. The Reactivating and Therapeutic Efficacy of Oximes to Counteract Russian VX Poisonings. Int. J. Toxicol. 2006, 25, 397–401. 10.1080/10915810600846971. [DOI] [PubMed] [Google Scholar]
- Kuca K.; Jun D.; Cabal J.; Hrabinova M.; Bartosova L.; Opletalova V. Russian VX: Inhibition and Reactivation of Acetylcholinesterase Compared with VX Agent. Basic Clin. Pharmacol. Toxicol. 2006, 98, 389–394. 10.1111/j.1742-7843.2006.pto_267.x. [DOI] [PubMed] [Google Scholar]
- Hörnberg A.; Tunemalm A.-K.; Ekström F. Crystal Structures of Acetylcholinesterase in Complex with Organophosphorus Compounds Suggest That the Acyl Pocket Modulates the Aging Reaction by Precluding the Formation of the Trigonal Bipyramidal Transition State. Biochemistry 2007, 46, 4815–4825. 10.1021/bi0621361. [DOI] [PubMed] [Google Scholar]
- Millard C. B.; Koellner G.; Ordentlich A.; Shafferman A.; Silman I.; Sussman J. L. Reaction Products of Acetylcholinesterase and VX Reveal a Mobile Histidine in the Catalytic Triad. J. Am. Chem. Soc. 1999, 121, 9883–9884. 10.1021/ja992704i. [DOI] [Google Scholar]
- Artursson E.; Andersson P. O.; Akfur C.; Linusson A.; Börjegren S.; Ekström F. Catalytic-Site Conformational Equilibrium in Nerve-Agent Adducts of Acetylcholinesterase: Possible Implications for the HI-6 Antidote Substrate Specificity. Biochem. Pharmacol. 2013, 85, 1389–1397. 10.1016/j.bcp.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Franjesevic A. J.; Sillart S. B.; Beck J. M.; Vyas S.; Callam C. S.; Hadad C. M. Resurrection and Reactivation of Acetylcholinesterase and Butyrylcholinesterase. Chem.—A Eur. J. 2019, 25, 5337–5371. 10.1002/chem.201805075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallender W. D.; Szegletes T.; Rosenberry T. L. Acetylthiocholine Binds to Asp74 at the Peripheral Site of Human Acetylcholinesterase as the First Step in the Catalytic Pathway. Biochemistry 2000, 39, 7753–7763. 10.1021/bi000210o. [DOI] [PubMed] [Google Scholar]
- Johnson G.; Moore S. The Peripheral Anionic Site of Acetylcholinesterase: Structure, Functions and Potential Role in Rational Drug Design. Curr. Pharm. Des. 2006, 12, 217–225. 10.2174/138161206775193127. [DOI] [PubMed] [Google Scholar]
- De Koning M. C.; Joosen M. J. A.; Noort D.; Van Zuylen A.; Tromp M. C. Peripheral Site Ligand-Oxime Conjugates: A Novel Concept towards Reactivation of Nerve Agent-Inhibited Human Acetylcholinesterase. Bioorganic Med. Chem. 2011, 19, 588–594. 10.1016/j.bmc.2010.10.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.