

Abstract

Poly(aspartic acid) (PAA) is a biodegradable water-soluble anionic polymer that can potentially replace poly(acrylic acid) for industrial applications and has shown promise for regenerative medicine and drug delivery. This paper describes an efficient and sustainable route that uses protease catalysis to convert l-aspartate diethyl ester (Et2-Asp) to oligo(β-ethyl-α-aspartate), oligo(β-Et-α-Asp). Comparative studies of protease activity for oligo(β-Et-α-Asp) synthesis revealed α-chymotrypsin to be the most efficient. Papain, which is highly active for l-glutamic acid diethyl ester (Et2-Glu) oligomerization, is inactive for Et2-Asp oligomerization. The assignment of α-linkages between aspartate repeat units formed by α-chymotrypsin catalysis is based on nuclear magnetic resonance (NMR) trifluoacetic acid titration, circular dichroism, and NMR structural analysis. The influence of reaction conditions (pH, temperature, reaction time, and buffer/monomer/α-chymotrypsin concentrations) on oligopeptide yield and average degree of polymerization (DPavg) was determined. Under preferred reaction conditions (pH 8.5, 40 °C, 0.5 M Et2-Asp, 3 mg/mL α-chymotrypsin), Et2-Asp oligomerizations reached maximum oligo(β-Et-α-Asp) yields of ∼60% with a DPavg of ∼12 (Mn 1762) in just 5 min. Computational modeling using Rosetta software gave relative energies of substrate docking to papain and α-chymotrypsin active sites. The substrate preference calculated by Rosetta modeling of α-chymotrypsin and papain for Et2-Asp and Et2-Glu oligomerizations, respectively, is consistent with experimental results.
Introduction
Polycarboxylates such as poly(malic acid),1−6 poly(glutamic acid) (PGA),7,8 and poly(aspartic acid) (PAA)9−14 are alternatives to traditional nonbiodegradable polyanionic materials (e.g., polyacrylic acid) that are used in a wide variety of products. The poor hydrolytic stability of ester-linked poly(malic acid) is problematic for commodity applications. In contrast, PAA and PGA have amide-linked backbones with sufficient hydrolytic stability that enable their use in a wide range of industrial applications.
Poly(α,β-dl-aspartate)s, T-PAA, from high-temperature condensation polymerization (see below), are of substantial interest for industrial processes. T-PAA inhibits deposition of various calcium (e.g., calcium carbonate, calcium sulfate, and calcium phosphate) and barium salts and, therefore, is a potential substitute for PAA in water-treatment applications.15,16 The T-PAA used for such applications has an Mw, Mn, and Mw/Mn of 5200, 1800, and 2.9, respectively.16 T-PAA hydrogels are pH-, salt-, and temperature-responsive.17 These hydrogels can function in a variety of applications such as personal care and biomedical systems.17 Other potential industrial applications for T-PAA include as ingredients in cleaning formulations and as oil field treatment additives.18 PAA is under study for biomedical applications such as hydrogels,19 gene delivery assemblies,20 polyelectrolyte films,21 nanoparticle drug-delivery systems,22 scaffolding for tissue growth,23 artificial skin, and so forth.24
There are several different synthetic routes to PAAs. For large-scale industrial applications, T-PAA is synthesized at high temperature (∼180 and 230 °C) such that acrylic acid is converted to polysuccinimide which is subsequently hydrolyzed under alkaline conditions to give T-PAA.9 Another route to T-PAA is via polymerization of maleic acid produced from reaction of ammonia and maleic anhydride.25,26 However, T-PAA synthesis is energy-intensive and gives polymers with irregular structures. The latter is due to racemization of stereocenters and ring-opening of succinimide units that leads to about 30% α-linkages and 70% β-linkages. Furthermore, branching and cross-linking can occur during the polymerization process.27
PAA is also synthesized via formation and subsequent ring-opening polymerization of N-carboxyanhydride (NCA) monomers.24,28 The corresponding NCA–PAA is normally linear and enantiopure. NCA-ROP also allows for the preparation of polypeptides in high molecular weight, controlled dispersity, and with unnatural and [D]-enantiomer repeat units.29 Habraken et al.28 reported the synthesis of γ-benzyl-l-glutamate with an Mn up to 22 200 and Mw/Mn as low as 1.09. However, NCA-ROP requires protection–deprotection chemistry to avoid initiation by nucleophilic amino acid side chain23,30 toxic phosgene-based chemicals to cyclize amino acids forming NCA monomers, organic solvents for synthesis, exhaustive purification of NCA monomers, and strictly anhydrous and often inert atmospheric conditions.22,24−31
This paper aims at developing alternative environmentally friendly (“green”) routes to PAA. For this, we turn to protease-catalyzed peptide synthesis (PCPS) routes to PAA (p-PAA). Kinetically driven PCPS occurs under mild conditions (e.g., 30–40 °C) and uses amino acids in their ethyl ester (Et-AA) form. Enzyme selectivity often results in the formation of peptides with uniform α-linked AAs while mild reaction conditions avoid racemization during enzymatic polymerizations.32−34
Thus far, reports of PCPS routes to p-PAA are sparse and lack information on how the enzyme structure results in differences in substrate specificity. Uemura et al. reported that polyethylene glycol 10 000-modified papain catalyzes the oligomerization of Et2-Asp in benzene giving an oligomer mixture with chain lengths from a heptamer to decamer. However, when the oligomerization reaction was performed in aqueous media, a switch in substrate specificity occurred such that oligo(Asp) was not formed.35 Matsumura et al. reported an alkalophilic proteinase (AP) from Streptomyces sp-catalyzed Et2-Asp oligomerization occurring after 3 days, with 88% α-linkages and an average molecular weight (Mw) up to 2500 determined by size exclusion chromatography.36 Soeda et al. reported that Et2-Asp is oligomerized by a bacterial protease from Bacillus subtilis BS in organic media (e.g., acetonitrile) that contains about 4.5% water.37 Reactions conducted for 2 days gave α-linked oligo(β-ethyl l-aspartate) and oligo(β-Et-α-Asp), in yields up to 85%, Mw by gel permeation chromatography up to 3700 and a chain length distribution from mass-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) of 5–20 (the highest peak intensity at 10 units, Mn = 1500).37 However, these previous studies are limited in their scientific value because (i) the activity of proteases used is not defined by a standard assay, (ii) an alkalophilic protease from a Streptomyces sp and a bacterial protease from B. subtilis BS are not sufficiently classified and identified to determine the structure of the protease used. For (i) and (ii), the abovementioned information on the protease activity, source, and structure is essential to enable others to repeat the published methods and investigate mechanisms on a molecular level. It is also important to avoid organic solvents during PCPS oligomerizations and to complete reactions within a few hours and not days for potential commercial adoption.
Although the potential of many common commercial proteases to catalyze Et2-Asp oligomerizations remains uninvestigated, there are a substantial number of publications that describe the use of readily available proteases to convert Et2-Glu to oligo(Glu).32,38,39 Uyama et al. reported that papain, bromelain, and α-chymotrypsin are all active for Et2-Glu oligomerization giving α-linked oligo(γ-Et-Glu). Oligomer chain lengths ranged from 5 to 9 residues, and the highest yield was 80% after a 24 h reaction.39 Aso et al. reported papain-catalyzed oligomerization of Et2-Glu that gave a 65% yield of oligo(γ-Et-Glu) in 3 h with a DPavg of 7–9.38 Later, Li et al. reported papain-catalyzed Et2-Glu oligomerization in phosphate buffer (40 °C, 1 h) to give oligo(γ-Et-Glu) in 81% yield with a DPavg of 8–9.32 Papain also proved to be an effective catalyst for co-oligomerizations of Et2-Glu with Et-Tyr and Et-Leu.39,40
Computational studies have provided molecular insights into peptide hydrolysis reactions by papain41−44 and α-chymotrypsin45,46 and, to a lesser extent, peptide bond synthesis via aminolysis.47 Modeling protease substrate interactions in the papain system using Quantum Mechanics/Molecular Mechanics (QM/MM) approaches provided insights into the stereopreference of papain-catalyzed oligomerization of l-alanine over d-alanine via aminolysis reactions.47 However, previous studies have not conducted a comparative analysis of different enzyme–substrate pairs to obtain insights into the molecular bases for experimentally observed substrate preferences, possibly because of the large computational expense associated with QM/MM approaches. We reasoned that modeling the molecular interactions between substrates and enzymes using the enzyme design framework we developed with Rosetta software would allow us to rapidly and efficiently identify the steric and energetic factors that determine the exactitude of fit for different substrates within papain and chymotrypsin active sites. These calculations align well with our experimental observations and thus inform a molecular-level understanding of specificity differences observed herein.
The focus of this work is (i) determining differences in activity of common commercially available proteases (α-chymotrypsin, trypsin, papain, and bromelain) for oligomerization of Et2-Asp and Et2-Glu oligomerizations, (ii) based on this information, developing a well-defined and repeatable route to prepare oligo(Asp), and (iii) gaining a molecular-level understanding of observed differences in protease selectivity by computational methods. The results reveal that papain and α-chymotrypsin have remarkably different activities for these similar substrates. Because α-chymotrypsin was found to be an effective catalyst for oligo(Et2-Asp) synthesis, studies were then performed to elucidate the extent that reaction parameters (pH, reaction time, and concentrations of buffer, monomer, and protease) influence α-chymotrypsin-catalyzed Et2-Asp oligomerizations. The results of this work led to an efficient PCPS route to oligo(β-Et-Asp). We then used computational modeling with Rosetta software to interrogate on a molecular-level selectivity difference found between papain- and α-chymotrypsin-catalyzed Et2-Asp and Et2-Glu oligomerizations.
Experimental Section
Materials
High-performance liquid chromatography (HPLC) grade l-aspartic acid diethyl ester hydrochloride (Et2-Asp) with 99.8% purity and α-chymotrypsin from bovine pancreas with 94% protein were purchased from Chem-Impex International. l-Glutamic acid diethyl ester hydrochloride (Et2-Glu) was purchased from TCI. Crude papain from Carica papaya (30 000 USP units/mg of solid) was purchased from Calbiochem Co. Ltd. Trypsin from bovine pancreas and bromelain from pineapple stem were purchased from Sigma-Aldrich. Potassium phosphate dibasic trihydrate, casein, trichloroacetic acid, sodium carbonate anhydrous, sodium acetate trihydrate, calcium acetate, and l-tyrosine were purchased from Sigma-Aldrich. Folin & Ciocalteu’s phenol reagent was purchased from MP Biomedicals. All the abovementioned chemicals, reagents, and enzymes were obtained in the highest available purity and were used as received.
Methods
General Procedure for Protease-Catalyzed Synthesis of Oligo(β-Et-Asp)
Et2-Asp in the L-stereochemical configuration (1.128 g, 5 mmol) and 10 mL of phosphate buffer solution were transferred to a 50 mL Eppendorf tube. The solution was set to a predetermined pH, and protease (16 units/mL) was added to the reaction mixture. The tube was gently stirred in a water bath at 40 °C for a predetermined reaction time. The pH of the reaction media was controlled by automated titration as described below. Reactions were terminated by acidifying to pH 3 with 3 M HCl, cooled to room temperature, and 20 mL of distilled water was added. The insoluble product was separated by centrifugation (9000 rpm) and washed twice with cold distilled water. The resulting product was lyophilized giving a white powder. 1H NMR (500 MHz, DMSO-d6) spectrum of oligo(l-Asp): δ = 1.17 (t, 3H, CH3 of Et), δ = 2.5–2.8 (m, 2H, CH2), δ = 3.75 (m, 1H, CH end group), δ = 4.05 (q, 2H, CH2 of Et), δ = 4.56 (m, 1H, CH), δ = 8.0–8.3 (m, 1H, NH).
Procedure for Reduction of Oligo(β-Et-Asp)
To a suspension of 110 mg (0.82 mmol ethyl ester units) of oligo(β-Et-Asp) in chloroform (5 mL) at 0 °C, 2 M LiAlH4 in tetrahydrofuran (1.2 mL, 2.4 mmol) was added dropwise. The resulting mixture was warmed to room temperature and stirred for 18 h. The reaction was then quenched with 1 M HCl(aq) for 30 min and the mixture was dialyzed against distilled water for 24 h. The resulting reduced peptide was freeze-dried and analyzed by nuclear magnetic resonance (NMR) for linkage assignment.
Control of the Reaction pH
Because the pH decreases during oligomerizations, an automated pH control was employed. A tiamo titration control system and Metrohm CH9101 dosing unit were used. The dosing solution (1.0 M NaOH) was added at up to 0.1 mL/min, and the frequency at which the probe checked the pH was set to 1.0 s. The reaction medium pH was controlled within 0.05 units of the set value.
Protease Activity Assay
Protease activity was measured following the Universal Protease Activity Assay protocol using casein as the substrate as previously reported.48,49
Instrumental Methods
NMR Spectroscopy
1D and 2D proton (1H) NMR spectra were recorded on a Varian spectrometer at 500 MHz or a Bruker spectrometer at 600 MHz. 1D 1H and 13C NMR experiments were performed in DMSO-d6 at 10–30 mg/mL oligopeptides with a data acquisition delay of 1 s and a total of 64–128 scans. 2D 1H NMR experiments (1H–1H COSY) were performed in DMSO-d6 at 30 mg/mL with 128 scans. Data collection and analysis were performed using MestReNova software. Proton chemical shifts were referred to tetramethylsilane at 0.00 pm. The solvent-induced helix–coil transition was investigated by adding trifluoroacetic acid (TFA) that covered a range of concentrations from 0 to 10% (v/v) to peptide (50 mg) dissolved in 5 mL of CDCl3. All spectra were recorded at 25 °C.
Mass-Assisted Laser Desorption/Ionization-Time-of-Flight
MALDI-TOF spectra were recorded using a Bruker ultraflex III MALDI-TOF/TOF mass spectrometer. The instrument was operated in a positive ion linear mode with an accelerating potential of +20 kV. A saturated solution of α-cyano-4-hydroxycinnamic acid in 66% acetonitrile, 33% distilled water, and 1% TFA was used as the matrix.
Circular Dichroic Measurements
Circular dichroism (CD) spectra were recorded on a Jasco 815 Spectrometer using a 2 mm quartz cell at 25 °C. Peptides (0.2 mg/mL) in CHCl3 were prepared for measurements. All the spectra were obtained in the wavelength region between 180 and 260 nm.
Protease Active Site Modeling
Computational modeling of acyl–enzyme intermediates was performed with the RosettaScripts application utilizing the module enzdes (enzyme design) in the Rosetta software suite and scored by the talaris_2014 score function.50,51 Initial chemical structures of Et2-Glu and Et2-Asp and their respective hydrolyzed intermediates were created with Avagadro.52 These chemical structures generated a set of 200 conformers using the conformational generation tool in the program mercury, curated by the CCDC.53
The protein crystal structures for papain (9pap) and α-chymotrypsin (1yph) were used for enzyme modeling, with the acyl–enzyme intermediate geometry determined from chymotrypsin (1oxg) and papain (2bu3).54−57 Papain and α-chymotrypsin were prepared by a FastRelax protocol with heavy constraints for initial minimization. Using the geometry determined from the 1oxg and 2bu3 crystal structures, enzdes constraints were developed between the hydrolyzed acyl intermediate, the attacking diester nucleophile, and the catalytic triads of papain and α-chymotrypsin. The geometry of the acyl–enzyme complex was used to create starting placements of the hydrolyzed acyl intermediates and the attacking nucleophiles within the context of both papain and α-chymotrypsin. Because there are two orientations by which the nucleophile can dock in the enzyme to attack the acyl intermediate, two sets of geometries were created with flipped orientations of the diester, designated c1 and c2.
Sampling of the acyl–enzyme complex was performed through an initial two runs of repack on the enzyme active site (specifically on both the acyl intermediate and the attacking nucleophile) to alleviate clashes between the small molecules and the enzyme, followed by four minimization and repacking steps on the full acyl–enzyme complex pocket. The abovementioned small molecule conformers were used to simulate the flexibility of the diesters and hydrolyzed intermediates. Constraints were applied such that only the enzyme pocket and small molecules would be sampled. This sampling was repeated 50 times for each set of docked complexes for a total of 16 acyl–enzyme complexes: two sets for each enzyme (papain or α-chymotrypsin), two sets for each linkage modeled (α and β or γ), two sets for either the glutamic or aspartic acid diester oligomerization, and a final two sets for each potential orientation that the nucleophile can dock into the enzyme to attack the acyl intermediates (c1 and c2).
Results and Discussion
Schemes of kinetically driven PCPS are available in previous publications.32 In summary, prior to peptide bond formation, an enzyme–acyl intermediate is formed between the carbonyl of an activated amino acid and the cysteine hydroxyl or serine thiol functional groups at the protease active site. Then, the enzyme-activated substrate complex is de-acylated by the nucleophilic attack of the free amine of another amino acid monomer or oligomer (the preferred nucleophile) or water. The former case will result in peptide bond formation and chain propagation, whereas hydrolysis will result in a deactivated carboxylic acid group. When the peptide reaches a chain length where it is no longer soluble in the reaction medium, it precipitates, shifting the reaction equilibrium to peptide formation. During peptide bond formation, HCl is liberated into the reaction medium thereby decreasing its pH. In order to maintain optimal protease activity, a sodium hydroxide solution was added to the reaction mixture by an automated dosing unit. In all studies described below, the L-stereochemical configuration of Et2-Asp and Et2-Glu was used.
Comparative Activity of Proteases for Aspartate and Glutamate Diethyl Ester Oligomerization
In previous work by Li et al., papain was found to be an excellent catalyst for conversion of Et2-Glu to oligo(γ-Et-Glu) giving this product in ∼81% yield within about 15 min.32 Furthermore, α-chymotrypsin, trypsin, and bromelain are common commercial proteases with broad specificities for peptide synthesis.32,36,37,39 Because the structures of Et2-Asp and Et2-Glu differ by only one methylene group between the α-carbon and the β- and γ-carboxyl groups, respectively, we determined the relative ability of these catalysts to effectively prove the hypothesis that papain would also be an effective catalyst for oligo(β-Et-Asp) synthesis. Table 1 lists results of Et2-Asp and Et2-Glu oligomerizations using α-chymotrypsin, trypsin, papain, and bromelain. The activity of each protease was determined by the Universal Protease Activity Assay using casein as the substrate (see Experimental Section). For each protease, the quantity of the enzyme used is that which results in 16 units/mL activity where a unit is the release of 1 μmol of tyrosine from casein at 37 °C min–1 mL–1. Protease activities in unit mg–1 mL–1 are given as a footnote in Table 1. Hence, activity differences of the four proteases are normalized by adjusting the amount of protease to give 16 units/mL activity in reaction solutions. The results as shown in Table 1 reveal that papain and α-chymotrypsin have opposite specificities for Et2-Asp and Et2-Glu. The reaction times are based on monitoring the pH drop that corresponded to base addition by the pH-stat to maintain the pH at 8.5 (see Experimental Section). When base addition ceased, indicating completion of the reaction, the reaction was terminated. If reactions were continued beyond this point, reverse hydrolysis would likely occur. No precipitation was observed for papain-catalyzed Et2-Asp oligomerizations over a 1 h reaction time. During this reaction time, there was no evidence of oligomerization based on pH-stat monitoring. Furthermore, analysis of the supernatant by LC–mass spectrometry (MS) showed that only the monomer remained and no low chain length oligomers were formed. In contrast, by 5 min, α-chymotrypsin-catalyzed Et2-Asp oligomerization gave a ∼58% yield of oligo(Et-Asp), while the yield of oligo(Et-Glu) at 1 h was significantly less (16%). Trypsin also catalyzes the oligomerization of Et2-Asp and Et2-Glu giving yields of 15 and 5%, respectively. Furthermore, bromelain was a poor catalyst for oligomerizations of both Et2-Asp and Et2-Glu giving yields of 0 and 3%, respectively.
Table 1. α-Chymotrypsin, Trypsin, Papain, and Bromelain Catalysis of Et2-Asp and Et2-Glu Oligomerization Reactionsa.
| monomer | proteaseb | reaction timec | yield %d |
|---|---|---|---|
| Et2-Asp | α-chymotrypsin | 5 min | 58 ± 2 |
| trypsin | 30 min | 15 ± 3 | |
| papain | 1 h | no reactione | |
| bromelain | 1 h | no reactione | |
| Et2-Glu | α-chymotrypsin | 30 min | 16 ± 1 |
| trypsin | 30 min | 5 ± 1 | |
| papain | 1 h | 60 ± 5 | |
| bromelain | 1 h | 3 ± 1 |
Reactions were performed in 0.6 M phosphate buffer with 0.5 M monomer at pH 8.5 and 40 °C.
An activity unit for each protease is the quantity in mg that results in the release of 1 μmol of tyrosine min–1 mL–1 in buffer solution at 37 °C. The activity of α-chymotrypsin, trypsin, papain, and bromelain by the casein hydrolysis assay is 5.68, 3.31, 0.895, and 0.88 units mg–1 mL–1, respectively.48,49 The amount of protease was normalized so that, for each protease, the weight corresponding to 16 units mg–1 mL–1 was used for oligomerizations.
Reaction times were determined by monitoring the pH drop that corresponded to base addition by the pH-stat to maintain the pH at 8.5 (see Experimental Section). When base addition ceased, indicating completion of the reaction, the reaction was terminated.
Determined by the weight of the precipitated polymer over the theoretical weight of the product formed in 100% yield.
The assertion that no reaction occurred when papain and bromelain were catalysts for Et2-Asp oligomerization is based on both the absence of precipitate formation and analysis of the supernatant by LC–MS that showed only a monomer and no low chain length oligomers.
Time Course Study: α-Chymotrypsin-Catalyzed Et2-Asp Oligomerization
Considering the promising results of α-chymotrypsin-catalyzed Et2-Asp oligomerization, further studies were performed to determine how changes in the reaction time, temperature, pH, and concentrations of the buffer/enzyme/monomer affect the peptide yield and DPavg. Figure 1 displays the results of α-chymotrypsin-catalyzed oligo(Et-Asp) synthesis as a function of time at 25 and 40 °C (see Figure 1 legend for reaction conditions). The oligomerization yield, determined by gravimetric analysis of the precipitated product, proceeded rapidly. Analysis of the precipitated product by 1H NMR (see below) confirmed that it is in fact oligo(Et-Asp). For reactions at 25 and 40 °C, the product formed within the first 10 s of protease addition. The yield reached 22–28% at 30 s and increased rapidly during the first 2 min. By 5 min, the yields reached maximum values of 58 and 54% at 40 and 25 °C, respectively. Further increase in the reaction time at 25 °C shows that the yield plateaued at 54%. This is likely due to product formation during the 5 min reaction time along with a low molecular weight water-soluble coproduct (the latter was not experimentally verified). Furthermore, by 5 min, the reaction at 25 °C appeared highly viscous which results in diffusion constraints that will slow down product hydrolysis after precipitation. In contrast, at 40 °C, a decrease in % yield occurred during extended reaction times (Figure 1). This is explained by the relatively lower viscosity at 40 °C that enables protease-catalyzed hydrolysis of ester or amide groups of precipitated products converting them to the water-soluble coproduct. Given what appears to be a slightly more rapid oligomerization at 40 °C at 5 min, further investigations of α-chymotrypsin-catalyzed synthesis of oligo(Et-Asp) are performed at 40 °C.
Figure 1.
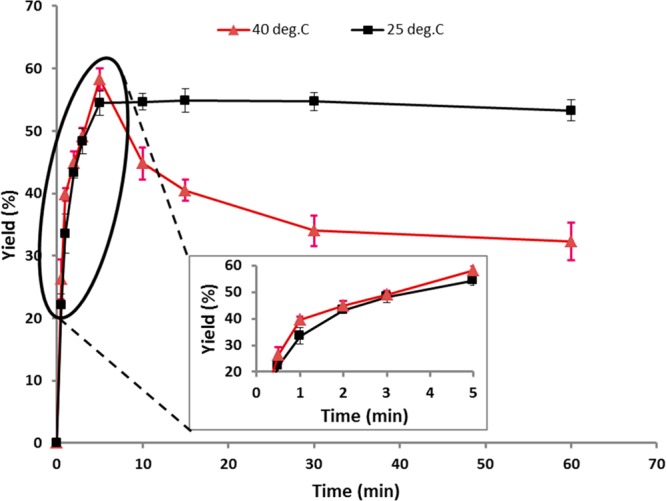
Time course of oligo(aspartate) synthesis based on the yield of the precipitated product for reactions performed at 25 and 40 °C consisting of 0.3 M Et2-Asp, 2 mg/mL α-chymotrypsin, and 0.6 M phosphate buffer at pH 8.5. The inserted graph shows the expansion of the 0.5–5 min region.
Structural Analysis
The 1D 1H NMR spectrum of Et2-Asp and the corresponding oligo(Et-Asp), synthesized by α-chymotrypsin catalysis for 15 min (reaction conditions are in the Figure 2 legend), is displayed in Figure 2 along with peak assignments. Confirmation of peak assignments was based on 2D 1H–1H COSY and 2D HSQC NMR experiments (see Figures S1 and S2, respectively). Further confirmation of peak assignments resulted from the comparison of the monomer and peptide peak signal positions and broadening. First, the amine proton signal of the monomer shifted from one peak at 8.7 ppm to a broad resonance with multiple peaks between 8.0 and 8.3 ppm because of peptide formation. Second, the α-CH methane proton of the monomer shifted downfield from 4.3 to 4.6 ppm. The β-CH2 also moved upfield from 3.0 to 2.5–2.8 ppm. The change of signal integration intensities between the monomer and peptide is also consistent with peptide formation. In the monomer spectrum, peak D (α-CH) and A (ethyl ester CH3) have an integration ratio of exactly 1:6, while in the oligomer product spectrum, this ratio changed to 1:3.8 because of the loss of ethyl ester groups during peptide bond formation. The oligomer peaks are assigned as follows, δ = 1.17 (t, 3H, CH3 of Et), δ = 2.5–2.8 (m, 2H, β-CH2), δ = 3.54 (m, 1H, CH end group, see the Supporting Information), δ = 4.05 (q, 2H, CH2 of Et), δ = 4.56 (m, 1H, α-CH), and δ = 8.0–8.3 (m, 1H, NH).
Figure 2.

1H NMR (500 MHz, DMSO-d6) spectra of (A) the monomer (Et2-Asp) and (B) oligo(Et-Asp) synthesized using 0.3 M Et2-Asp, 2 mg/mL α-chymotrypsin, and 0.6 M phosphate buffer (pH 8.5), at 40 °C, for 15 min. The peptide structure is shown as α-linked, this will be discussed below.
The DPavg of oligo(Et-Asp) from the 40 °C 15 min reaction (see Figure 2 legend) was determined by 1H NMR peak integration of methine protons D at 4.56 ppm relative to methine proton F at 3.54 ppm (the internal and end group methine hydrogens, respectively) is 11.1 (Mn 1600 g/mol). The same peptide product was analyzed by MALDI-TOF and the spectrum is displayed in Figure 3. The adjacent series of peaks differ by 143 m/z, which corresponds to the molecular weight of an aspartic acid ethyl ester residue in the peptide chain. MALDI-TOF peaks (sodium adducts) corresponding to the oligopeptide range from DP 7 to DP 34, although the most intense peaks range from DP 8 to 13, consistent with the NMR-determined DPavg. The MALDI-TOF spectrum indicates that the oligo(Et-Asp) synthesized by α-chymotrypsin is a mixture of various DP oligomers. One of the peak series, expanded for clarity, shows the major peak and minor peaks. The higher signal at m/z = 1928.165 corresponds to oligo(Et-Asp) of DP = 13 with no loss of ethyl ester groups. The two signals at m/z = 1900.166 and 1922.139 correspond to peptides where hydrolysis of one ethyl ester pendant or the chain end group occurred, forming the corresponding carboxylic acid and salt forms, respectively.
Figure 3.

MALDI-TOF spectrum of oligo(Et-Asp) synthesized using 0.3 M Et2-Asp, 2 mg/mL α-chymotrypsin, and 0.6 M phosphate buffer (pH 8.5), at 40 °C, for 15 min.
Et2-Asp molecules have two ethyl ester groups attached to the α- and β-carbonyls, respectively. During the enzymatic reaction, either ester group can undergo nucleophilic attack by the active site serine (Ser-195) to form an acyl–enzyme complex. This could lead to two possibilities, formation of α-linked oligoaspartate, oligo(β-Et-α-Asp), or β-linked oligoaspartate, oligo(α-Et-β-Asp). If both α- and β-linkages exist in oligoaspartate, two α-methine proton signals (D) with different chemical shifts will be observed. A major α-proton signal at ∼4.5 ppm and a minor peak at ∼4.4 ppm are observed (Figure 2). From the COSY spectrum shown in Figure S1, no cross-peaks are observed for the signal at 4.4, although this may be due to its low intensity. The ratio of intensities between signals at 4.5 and 4.4 is about 25:1.
To determine whether oligoaspartate ethyl ester synthesized by α-chymotrypsin-catalysis is α- or β-linked, the effects of TFA titration on 1D 1H NMR spectra and CD measurements were studied. Poly(β-Et-α-l-Asp) adopts a right-handed α-helix conformation in chloroform and undergoes a helix–coil transition upon addition of TFA.58−60Figure 4A shows that the α-H (D) shifts downfield as TFA (0.3–10%) was added to the oligoaspartate/CDCl3 solution. This downfield shift is consistent with that observed for the helix–coil transition of poly(α-l-amino acids).60−62 The α-H in poly(α-Et-β-l-Asp) undergoes an upfield shift thus following the opposite trend under the same experimental conditions. The formation of α-linked polyaspartate ethyl ester was further confirmed by a CD spectrum conducted in pure chloroform (Figure 4b). A trough at 230 nm is observed in the CD spectrum at molar ellipticity of −5500, which is consistent with the result observed for poly(β-Et-α-l-Asp) in previous publications.58−60
Figure 4.

TFA-induced conformational changes in oligo(Et-Asp): (A) α-H signal (D) shifts in the presence of 0.3–10% TFA (v/v) in CDCl3; (B) CD spectrum in chloroform.
Because of the overlap of the two carbonyl groups (CO ester and CO amide) in the 13C NMR, direct assignment of the linkage chemistry by 2D-1H13C HMBC, as previously reported,62 was not possible. To obtain direct evidence for the linkage chemistry of oligo(β-Et-α-l-Asp) synthesized by α-chymotrypsin catalysis, the reduction of ester functionalities using LiAlH4 was performed to obtain the corresponding peptide with hydroxyl side chains. Analysis of the 2D-1H,1H COSY spectrum as shown in Figure S3 reveals a strong cross-peak between the β-CH2 protons at ∼1.90 ppm and the methylene protons of CH2OH at ∼3.70 ppm. This provides unambiguous and direct evidence that the peptide is α-linked.
Influence of Reaction Parameters on α-Chymotrypsin Catalysis of l-Aspartate Diethyl Ester (Et2-Asp)
A series of studies were performed to assess how the selected reaction parameters (pH and concentrations of the buffer, monomer, and protease) influence the yield and DPavg of α-chymotrypsin-catalyzed Et2-Asp oligomerizations. Figure S4 displays the results of how the reaction medium pH influences peptide formation (see Figure S4 legend for reaction conditions). Control of pH was achieved by using an automated pH-stat. To quench the reaction at 5 min, concentrated HCl was added that reduces the pH to 2–3. High enzyme activity was maintained between pH 7.0 and 8.5 with yields that varied from 51 to 60%. A decrease in the pH to 6.5 and an increase in the pH to 9.0 resulted in significant decreases in yield (34 and 44%, respectively). Oligopeptide DPavg tends toward increasing values (∼10–14) as the medium pH increases. There is a significant difference between the DPavg’s of peptides formed at pH 7.5 and 9.5 (12 and 14.5, respectively). It may be that under alkaline pH conditions, the extent of ester hydrolysis increases thereby increasing oligo(Et-Asp) solubility that enables continued chain growth prior to precipitation.
Figure 5 displays the strong influence of Et2-Asp concentration on product formation. Oligo(Et-Asp) yield increased from 8 to 33 and 50% by increasing the monomer concentration from 0.1 to 0.2 and 0.3 M, respectively. A small increase from 0.15 to 0.2 M Et2-Asp resulted in a 21% increase in product yield. Hence, a relatively low substrate concentration is sufficient to enable kinetic control of product formation as aminolysis is favored over hydrolysis reactions. A small increase in the product yield from 50 to 58% occurred over the relatively large substrate concentration range of 0.3–0.5 M. An increase in the substrate concentration above 0.5 M resulted in substantial decreases (33% from 0.5 to 0.8 M) in yield. We hypothesize that the increased reaction time is required to reach higher yields as the substrate concentration is increased. However, experiments to test this hypothesis were not performed. Figure 5 also shows that there is no significant change in DPavg for substrate concentrations between 0.1 and 0.5 M. Because the data at higher concentrations were from one measurement, the resulting absence of standard deviation data does not allow further discussion of DPavg in the 0.6 to 0.8 M region of the plot.
Figure 5.

Effect of substrate concentration on oligo(Et-Asp) yield and DPavg. Reactions were conducted using 2 mg/mL α-chymotrypsin, 0.6 M phosphate buffer, at 40 °C, for 5 min at pH 8. Values are the mean from triplicate experiments. Error bars define the standard deviation.
Figure 6 provides data on how reaction protease concentration effects the yield and DPavg of α-chymotrypsin-catalyzed oligo(Et-Asp) synthesis. Details of reaction conditions are given in the Figure 6 legend. As discussed above, the catalytic activity of α-chymotrypsin is 5.68 units mg–1 mL–1. The experiments described in Figures 1, 5, and S4, S5 used 2 mg/mL α-chymotrypsin or 11.36 units/mL. The product yield increased from 27 to 40 and 58% with protease concentrations of 0.5, 1, and 2 mg/mL. Further increase in protease concentration from 2 to 3 and 4 mg/mL did not significantly change the product yield. However, increasing the α-chymotrypsin concentration from 4 to 5 mg/mL decreased the yield from 57 to 39%. Furthermore, dramatic decreases in DPavg of oligo(Et-Asp) occur with increased protease concentration. For example, at 2 and 5 mg/mL α-chymotrypsin, the DPavg decreases from 12.5 to 6.9. Thus, both oligopeptide yield and DPavg decrease with increased protease and constant (0.5 M) monomer concentration. This is explained by referring to Figure 1 where, at 40 °C, the yield of oligo(Et-Asp) is 28 and 58% at 30 s and 5 min, respectively. Further increase in the reaction time to 15 and 30 min resulted in a decrease in % yield to 40.5 and 34.0%, respectively. Increase in enzyme concentration is expected to accelerate the rate at which the oligopeptide is formed and subsequently degraded. In other words, we hypothesize that at enzyme concentrations >2.0 mg/mL, the peak yield and degradation moves to shorter times resulting in peptide degradation being the dominant reaction considerably before 5 min. This hypothesis remains to be tested in future studies.
Figure 6.

Effect of protease concentration on oligo(β-Et-α-Asp) ester yield and DPavg. Reactions were conducted using 0.5 M l-aspartic acid diethyl ester (Et2-l-Asp) hydrochloride salt, 0.6 M phosphate buffer, at 40 °C, for 5 min at pH 8. Values are the mean from triplicate experiments. Error bars define the standard deviation.
Figure S5 provides data on the relationship between buffer concentration and oligo(Et-Asp) yield and DPavg. After attaining a certain molecular weight, oligo(Et-Asp) precipitates from solution. By increasing the phosphate buffer concentration, the yield might increase because of “salting out” of the product. The highest yield is at 0.6 M buffer solution. Further increase of buffer concentration resulted in decreased yield, probably because of the negative impact of the high ionic strength on the ionic states of protease active site residues. The peptide yield remained at about 40% at buffer concentrations above 0.9 M. A large decrease in the DPavg occurs at buffer concentrations above 0.9 M. This is explained by the decreased solubility of oligo(Et-Asp) at high buffer concentrations that results in product precipitation at relatively lower average chain lengths.
Computational Modeling
To understand the molecular basis of the observed protease selectivity for oligomerizations of Et2-Glu and Et2-Asp in the L-stereochemical configuration, computational modeling was performed using Rosetta software. Simulations were used to determine the relative energies of Et2-Asp and Et2-Glu monomer–substrate binding to papain and α-chymotrypsin active sites in which the respective enzyme–acyl complexes of aspartate and glutamate monoethyl esters had been formed.
In this set of computational models, two pairs of models were constructed for the diethyl esters in the program Avogadro. The first pair with the α-ester hydrolyzed (α-AME for aspartic acid monoethyl ester and α-GME for glutamic acid monoethyl ester) and the second pair of monoethyl esters with the β-ester of Et2-Asp hydrolyzed (β-AME) and the γ-ester of Et2-Glu hydrolyzed (γ-GME). These were then modeled into the active site to generate the respective acyl–enzyme intermediates. The (attacking nucleophile) diethyl ester monomers Et2-Asp and Et2-Glu were also generated using Avogadro.
α-Chymotrypsin Models
In the reactive substrate-bound α-chymotrypsin models, a pocket near S195 (nucleophilic residue in the enzyme) fits each modeled substrate (Figure 7). In modeling the amine nucleophile into the catalytic site, the positioning and conformational preference of the ester gives insight into the preference for α-linkages observed in experiments. For the Et2-Asp nucleophile, there are two conformers of Et2-Asp observed, depending on the acylation position (α or β) (Figure 7A,B, respectively). Both conformers orient the amine group into a position optimal for attack on the carbonyl carbon while being activated by the H57 imidazole. However, for the β-AME-chymotrypsin models, intramolecular or intermolecular steric hindrances are observed for the Et2-Asp nucleophile. In contrast, the α-AME-α-chymotrypsin models lack these Et2-Asp clashes (Table S1), which may explain the preferential formation of oligo(β-Et-α-Asp) instead of oligo(β-Et-β-Asp).
Figure 7.

α-Chymotrypsin active site-docking models: Et2-Asp (green) forming an α-chymotrypsin (blue) acyl intermediate with (A) α-hydrolyzed aspartic acid (pink) and (B) β-hydrolyzed aspartic acid (red). Et2-Glu (orange) forms acyl intermediates with (C) α-chymotrypsin-α-hydrolyzed glutamic acid (teal) and (D) α-chymotrypsin-γ-glutamic acid (purple). Both α-hydrolyzed species demonstrate ester burial in the active site pocket with relaxed conformations and hydrogen bonding interactions between the α-hydrolyzed amine and the carboxylate oxygen.
For the α-GME-α-chymotrypsin acyl intermediate with Et2-Glu, conformations of the nucleophile and the hydrolyzed ester exhibit clashes with α-chymotrypsin (Figure 7C,D). For Et2-Glu to adopt a catalytically active orientation, unfavorable interactions of the Et2-Glu carbonyl oxygens (distance < 3.0 Å) with either the carbonyl oxygen of H57 or the carbonyl oxygen of F38 occur. The γ-hydrolyzed models (Figure 7D) are even more unfavorable as the Et2-Glu conformation has additional carbonyl oxygen interactions with S39, and the interaction between the F38 carbonyl oxygen and the Et2-Glu α carbonyl oxygen approaches 2.6 Å. Thus, conformational changes, which in turn may reduce the catalytic efficiency, may be required for polymerization. These observations are consistent with the reduced amount of the product obtained in the α-chymotrypsin-catalyzed Et2-Glu oligomerization reaction (Table 1).
Papain Models
In the case of papain catalysis, models of Et2-Glu attacking the α-GME acyl intermediate (Figure 8C) reveal that Et2-Glu is in an energetically favorable conformational state with the Et2-Glu amine in an optimal orientation and distance for attack. That is, in the simulations conducted, no steric hindrance was observed. In the γ-GME acyl intermediate models (Figure 8D), both Et2-Glu and γ-GME have energetically unfavorable conformational states with inter- and intramolecular clashes between the diethylester and the hydrolyzed intermediate.
Figure 8.

Papain active site-docking models: Et2-Asp (green) forming a papain (wheat) acyl intermediate with (A) α-hydrolyzed aspartic acid (pink) and (B) β-hydrolyzed aspartic acid (red), both demonstrating intramolecular steric clashes which inhibit catalysis. Contrarily, the (C) papain-α-hydrolyzed glutamic acid (teal) acyl intermediate with Et2-Glu (orange) substrates are in relaxed conformations, while the (D) papain-γ-glutamic acid (purple) acyl intermediate illustrates two intramolecular steric clashes.
For α-AME-acyl-papain intermediate models (Figure 8A), the orientation of the Et2-Asp amine is unfavorable for the formation of an amide bond in both conformations. Conformations adopted by Et2-Asp exhibit intramolecular steric clashes between the carbonyl oxygens in both conformations. In the β-hydrolyzed aspartic acid set of acyl intermediate models (Figure 8B), conformations of the Et2-Asp amine required for the effective nucleophilic attack at the β-AME-acyl intermediate are highly strained. These models are consistent with the observation that papain is unable to oligomerize Et2-Asp (Table 1).
The abovementioned observations of molecular sterics being consistent with experimental observations are also reflected in energies of docked molecules calculated from Rosetta (Table S1). In Rosetta modeling, the “total score” represents the overall energy of a specific protease–substrate model and the “constraint score” is a reflection of how favorable the catalytic geometry of a given model is. The ideal catalytic geometry is defined based on mechanistic studies. In both cases, a lower score is more favorable. The models demonstrate that total scores in all cases are similar (for a given enzyme) and constraints score (penalty), describing the fit of the model to a catalytically competent conformational state, agree well with experimental data (Table S1). Specifically, α-chymotrypsin has higher oligomerization yields for Et2-Asp relative to Et2-Glu, and the α-chymotrypsin models correspondingly have lower constraint scores in the aspartic acid acyl intermediates over the glutamic acid acyl intermediates. In papain models, the Et2-Glu models have better constraint scores than the Et2-Asp models, corresponding to papain’s ability to preferentially polymerize diethyl esters of glutamic acid but not aspartic acid.
To further investigate the positional preference for polymerization, we calculated energies of the acyl–enzyme moiety linked to the enzyme at different positions (α, β, and γ) and found that Rosetta’s fa_intra_rep score term, the term describing the Lennard-Jones repulsive energy between atoms within the same molecule (lower is more favorable), correlates well with α-chymotrypsin’s and papain’s preferred mode of polymerization at the α-position (Table S2). The fa_intra_rep score term is calculated by summing over the repulsive interactions between all atoms in the molecule. These values allow us to compare the relative strain in different molecules and different conformers of the monomer. However, as the values are not absolute energies, they are meaningful only in comparison with each other. Thus, the experimentally observed preferences of the enzymes for Et2-Asp relative to Et2-Glu polymerization can be qualitatively recapitulated using Rosetta modeling. The hydrolytic cleavage of amide/peptide bonds by proteases is mechanistically well-understood and has been investigated extensively by computational approaches.63−65 However, little attention has been paid to computationally investigating the underlying basis for substrate specificity in the peptide bond formation reaction catalyzed by proteases. Previous modeling efforts on papain-catalyzed dipeptide synthesis focused on the choice of the ester group or on rationalizing the l-amino acid versus d-amino acid preference (47, 66). Our results show that consideration of the energetics of interaction between the acylated enzyme and the amino acid nucleophile is also necessary to better recapitulate the substrate preference of protease enzymes in peptide synthesis. The qualitative agreement of Rosetta-calculated energetics with experimental observations of substrate specificity in peptide synthesis also indicates that a Rosetta-based approach should be applicable for the redesign of proteases for peptide synthesis which would require remodeling of both the S1 (acyl donor-binding) and S1’ (amine-binding) pockets on the protease to control specificity.
Conclusions
Four proteases were selected to assess their relative activities for oligomerization of Et2-Asp and Et2-Glu that differ by just one methylene unit between their α-carbon and corresponding β- and γ-carboxylate functionalities, respectively. This comparative study revealed that papain and α-chymotrypsin have remarkably different oligomerization activities for these similar substrates. Papain-catalyzed oligomerization of Et2-Glu and Et2-Asp gave yields of 60 and 0%, respectively. Computational modeling proved consistent with these results because Et2-Glu forms energetically favorable conformational states for the reaction with the papain-α-linked to γ-ethyl glutamate monoester (the acyl–enzyme complex). In contrast, when Et2-Asp is the incoming nucleophile attacking the acyl–enzyme complex (papain-α-linked to β-ethyl aspartate), computational modeling shows that conformations required for amide bond formation are highly strained. Because α-chymotrypsin proved to be most effective for Et2-Asp oligomerization, studies were performed to characterize the linkage chemistry (i.e., α- vs β) of oligo(Et-Asp). Titration of an oligo(Et-Asp) chloroform solution with TFA results in a downfield shift of the methine hydrogen with increasing TFA concentration, consistent with the fact that oligo(Et-Asp) is α-linked. This result is consistent with the CD spectrum where a trough at 230 nm was observed. Because NMR assignment of linkage chemistry proved inconclusive because of overlapping peaks, the ester side chains were reduced to hydroxyl moieties. NMR experiments on this product show unambiguously that aspartate units are connected by α-linked peptide bonds.
α-Chymotrypsin catalyzed oligomerization at 40 °C occurred rapidly such that in 30 s and 5 min, % yields reached ∼25 and 58%, respectively. A small increase in monomer (Et2-Asp) concentration from 0.15 to 0.2 M resulted in a 21% increase in the product yield demonstrating that a relatively low substrate concentration is sufficient to enable kinetically driven aminolysis reactions that build peptide chains. An α-chymotrypsin concentration of 2 mg/mL (0.5 M substrate) gave a 58% yield with a DPavg of 12. Finally, computational modeling of α-chymotrypsin led to lower energy structures that favor formation of α-linkages and a preference for oligomerization of aspartate over glutamate diethyl ester.
Although papain can have broad specificity for oligomerization of different amino acid ethyl esters such as phenyl alanine, leucine, alanine, and tyrosine, papain has high selectivity that discriminates between aspartate and glutamate diethyl esters. α-Chymotrypsin-catalyzed oligomerization of Et2-Asp occurs rapidly giving DPavg values between 12 and 14 (Mn values of 1762–2048). Such chain lengths, based on prior work with thermal polyaspartate, have proven useful as antiscalants for water processing and also hold promise as building blocks to construct block copolymers for drug delivery and hydrogel formation.
Acknowledgments
This work was supported by the National Science Foundation Award # 1067415 to R.A.G.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03290.
Rosetta scores of papain and α-chymotrypsin acyl intermediates with corresponding nucleophilic diesters; full atom intramolecular repulsive scores for enzyme–substrate systems under study; 2D 1H–1H COSY NMR (600 MHz, DMSO-d6) spectrum of oligo(l-aspartate) synthesized by α-chymotrypsin catalysis; 2D HSQC NMR (600 MHz, DMSO-d6) spectrum of oligo(l-aspartate) synthesized by α-chymotrypsin catalysis; 2D 1H,1H COSY spectrum in D2O of reduced oligoAsp; plot with data on the relationship between reaction pH and oligo(Et-Asp) yield and DPavg; and plot with data on how buffer concentration effects oligo(Et-Asp) yield and DPavg (PDF)
Author Contributions
R.A.G. conceived the idea for the studies and provided overall guidance on experiments and data interpretation. F.Y. planned and performed experiments to determine protease activity on aspartate and glutamate substrates. F.T. performed and interpreted 2-D NMR experiments. E.D. used Rosetta modeling of α-chymotrypsin and papain to determine substrate specificity. S.D.K. supervised and helped interpret computer simulations and calculations. The paper was written by F.Y. and E.D. and edited by F.T., S.D.K., and R.A.G. All authors accepted the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Fujishige S.; Morita R.; Brewer J. R.; Nagata N.; Nakahara T.; Tabuchi T. Auto-Catalytic Cleavage of Poly(β-L-Malic Acid) in Aqueous Solution. Macromol. Rapid Commun. 1993, 14, 163–166. 10.1002/marc.1993.030140303. [DOI] [Google Scholar]
- Guerin P.; Vert M.; Braud C.; Lenz R. W. Optically Active Poly (\-Malic-Acid). Polym. Bull. 1985, 14, 187–192. 10.1007/bf00708479. [DOI] [Google Scholar]
- Matsumura S.; Beppu H.; Nakamura K.; Osanai S.; Toshima K. Preparation of Poly(β-Malic Acid) by Enzymatic Ring-Opening Polymerization of Benzyl β-Malolactonate. Chem. Lett. 1996, 25, 795–796. 10.1246/cl.1996.795. [DOI] [Google Scholar]
- Arnold S. C.; Lenz R. W. Synthesis of Stereoregular Poly(Alkyl Malolactonates). Macromol. Symp. 1986, 6, 285–303. 10.1002/masy.19860060128. [DOI] [Google Scholar]
- Ouchi T.; Fujino A. Synthesis of Poly(α-Malic Acid) and Its Hydrolysis Behavior in Vitro. Macromol. Chem. Phys. 1989, 190, 1523–1530. 10.1002/macp.1989.021900703. [DOI] [Google Scholar]
- Benvenuti M.; Lenz R. W. Polymerization and Copolymerization of β-Butyrolactone and Benzyl-β-Malolactonate by Aluminoxane Catalysts. J. Polym. Sci., Part A: Polym. Chem. 1991, 29, 793–805. 10.1002/pola.1991.080290602. [DOI] [Google Scholar]
- Kunioka M.; Goto A. Biosynthesis of Poly(γ-Glutamic Acid) from L-Glutamic Acid, Citric Acid, and Ammonium Sulfate in Bacillus Subtilis Ifo3335. Appl. Microbiol. Biotechnol. 1994, 40, 867–872. 10.1007/bf00173990. [DOI] [Google Scholar]
- Cromwick A.-M.; Gross R. A. Effects of Manganese (Ii) on Bacillus Licheniformis Atcc 9945a Physiology and γ-Poly(Glutamic Acid) Formation. Int. J. Biol. Macromol. 1995, 17, 259–267. 10.1016/0141-8130(95)98153-p. [DOI] [PubMed] [Google Scholar]
- Roweton S.; Huang S. J.; Swift G. Poly(Aspartic Acid): Synthesis, Biodegradation, and Current Applications. J. Environ. Polym. Degrad. 1997, 5, 175–181. 10.1007/BF02763661. [DOI] [Google Scholar]
- Alford D. D.; Wheeler A. P.; Pettigrew C. A. Biodegradation of thermally synthesized polyaspartate. J. Environ. Polym. Degrad. 1994, 2, 225–236. 10.1007/bf02071970. [DOI] [Google Scholar]
- Swift G. Directions for Environmentally Biodegradable Polymer Research. Acc. Chem. Res. 1993, 26, 105–110. 10.1021/ar00027a005. [DOI] [Google Scholar]
- Soeda Y.; Toshima K.; Matsumura S. Sustainable Enzymatic Preparation of Polyaspartate Using a Bacterial Protease. Biomacromolecules 2003, 4, 196–203. 10.1021/bm0200534. [DOI] [PubMed] [Google Scholar]
- Low K. C.; Koskan L. P. Symthetic Polyaspartic Acid and Its Uses. Polym. Mater. Sci. Eng. 1993, 69, 253. [Google Scholar]
- Tomida M.; Nakato T.; Kuramochi M.; Shibata M.; Matsunami S.; Kakuchi T. Novel Method of Synthesizing Poly(Succinimide) and Its Copolymeric Derivatives by Acid-Catalysed Polycondensation of L-Aspartic Acid. Polymer 1996, 37, 4435–4437. 10.1016/0032-3861(96)00267-4. [DOI] [Google Scholar]
- Hasson D.; Shemer H.; Sher A. State of the Art of Friendly “Green” Scale Control Inhibitors: A Review Article. Ind. Eng. Chem. Res. 2011, 50, 7601–7607. 10.1021/ie200370v. [DOI] [Google Scholar]
- Low K. C.; Wheeler A. P.; Koskan L. P.. Commercial Poly(Aspartic Acid) and Its Uses. In Hydrophilic Polymers; Glass J. E., Ed.; Advances in Chemistry; American Chemical Society, 1996; Chapter 6, pp 99–111. [Google Scholar]
- Zohuriaan-Mehr M. J.; Pourjavadi A.; Salimi H.; Kurdtabar M. Protein- and Homo Poly(Amino Acid)-Based Hydrogels with Super-Swelling Properties. Polym. Adv. Technol. 2009, 20, 655–671. 10.1002/pat.1395. [DOI] [Google Scholar]
- Schwamborn M. Chemical Synthesis of Polyaspartates: A Biodegradable Alternative to Currently Used Polycarboxylate Homo- and Copolymers. Polym. Degrad. Stab. 1998, 59, 39–45. 10.1016/s0141-3910(97)00184-5. [DOI] [Google Scholar]
- Juriga D.; Nagy K.; Jedlovszky-Hajdú A.; Perczel-Kovách K.; Chen Y. M.; Varga G.; Zrínyi M. Biodegradation and Osteosarcoma Cell Cultivation on Poly(Aspartic Acid) Based Hydrogels. ACS Appl. Mater. Interfaces 2016, 8, 23463–23476. 10.1021/acsami.6b06489. [DOI] [PubMed] [Google Scholar]
- Nie J.-J.; Dou X.-B.; Hu H.; Yu B.; Chen D.-F.; Wang R.-X.; Xu F.-J. Poly(Aspartic Acid)-Based Degradable Assemblies for Highly Efficient Gene Delivery. ACS Appl. Mater. Interfaces 2015, 7, 553–562. 10.1021/am506730t. [DOI] [PubMed] [Google Scholar]
- Pilbat A.-M.; Ball V.; Schaaf P.; Voegel J.-C.; Szalontai B. Partial Poly(Glutamic Acid) - Poly(Aspartic Acid) Exchange in Layer-by-Layer Polyelectrolyte Films. Structural Alterations in the Three-Component Architectures. Langmuir 2006, 22, 5753–5759. 10.1021/la060454v. [DOI] [PubMed] [Google Scholar]
- Yao X.; Xie C.; Chen W.; Yang C.; Wu W.; Jiang X. Platinum-Incorporating Poly(N-Vinylpyrrolidone)-Poly(Aspartic Acid) Pseudoblock Copolymer Nanoparticles for Drug Delivery. Biomacromolecules 2015, 16, 2059–2071. 10.1021/acs.biomac.5b00479. [DOI] [PubMed] [Google Scholar]
- Kim H. J.; Kim U.-J.; Kim H. S.; Li C.; Wada M.; Leisk G. G.; Kaplan D. L. Bone Tissue Engineering with Premineralized Silk Scaffolds. Bone 2008, 42, 1226–1234. 10.1016/j.bone.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Iwatsuki M. Biodegradation of Copoly(L-Aspartic Acid/L-Glutamic Acid) in Vitro. Biopolymers 1990, 29, 549–557. 10.1002/bip.360290310. [DOI] [PubMed] [Google Scholar]
- Koskan L. P.; Meah A. R. Y.. Production of High-Molecular-Weight Polysuccinimide and High-Molecular-Weight Poly(Aspartic Acid) from Maleic Anhydride and Ammonia. U.S. Patent 5,219,952A, Jun 15, 1993.
- Boehmke G.; Schmitz G.. Process for the Preparation of Polysuccinimide, Polyaspartic Acid and Their Salts. U.S. Patent 5,468,838A, Nov 21, 1995.
- Thombre S. M.; Sarwade B. D. Synthesis and Biodegradability of Polyaspartic Acid: A Critical Review. J. Macromol. Sci., Part A: Pure Appl.Chem. 2005, 42, 1299–1315. 10.1080/10601320500189604. [DOI] [Google Scholar]
- Habraken G. J. M.; Wilsens K. H. R. M.; Koning C. E.; Heise A. Optimization of N-carboxyanhydride (NCA) polymerization by variation of reaction temperature and pressure. Polym. Chem. 2011, 2, 1322–1330. 10.1039/c1py00079a. [DOI] [Google Scholar]
- Hadjichristidis N.; Iatrou H.; Pitsikalis M.; Sakellariou G. Synthesis of Well-Defined Polypeptide-Based Materials via the Ring-Opening Polymerization of α-Amino Acid N-Carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. 10.1021/cr900049t. [DOI] [PubMed] [Google Scholar]
- Huang J.; Hastings C. L.; Duffy G. P.; Kelly H. M.; Raeburn J.; Adams D. J.; Heise A. Supramolecular Hydrogels with Reverse Thermal Gelation Properties from (Oligo)tyrosine Containing Block Copolymers. Biomacromolecules 2013, 14, 200–206. 10.1021/bm301629f. [DOI] [PubMed] [Google Scholar]
- Choi Y. Y.; Jang J. H.; Park M. H.; Choi B. G.; Chi B.; Jeong B. Block Length Affects Secondary Structure, Nanoassembly and Thermosensitivity of Poly(ethylene glycol)-poly(L-alanine) Block Copolymers. J. Mater. Chem. 2010, 20, 3416–3421. 10.1039/b922956f. [DOI] [Google Scholar]
- Li G.; Vaidya A.; Viswanathan K.; Cui J.; Xie W.; Gao W.; Gross R. A. Rapid Regioselective Oligomerization of L-Glutamic Acid Diethyl Ester Catalyzed by Papain. Macromolecules 2006, 39, 7915–7921. 10.1021/ma061419y. [DOI] [Google Scholar]
- Thormann M.; Thust S.; Hofmann H.-J.; Bordusa F. Protease-Catalyzed Hydrolysis of Substrate Mimetics (Inverse Substrates): A New Approach Reveals a New Mechanism. Biochemistry 1999, 38, 6056–6062. 10.1021/bi9828425. [DOI] [PubMed] [Google Scholar]
- Wehofsky N.; Koglin N.; Thust S.; Bordusa F. Reverse Proteolysis Promoted by in Situ Generated Peptide Ester Fragments. J. Am. Chem. Soc. 2003, 125, 6126–6133. 10.1021/ja0344213. [DOI] [PubMed] [Google Scholar]
- Uemura T.; Fujimori M.; Lee H.-H.; Ikeda S.; Aso K. Polyethylene Glycol-Modified Papain Catalyzed Oligopeptide Synthesis from the Esters of L-Aspartic and L-Glutamic Acids in Benzene. Agric. Biol. Chem. 1990, 54, 2277–2281. 10.1080/00021369.1990.10870303. [DOI] [Google Scholar]
- Matsumura S.; Tsushima Y.; Otozawa N.; Murakami S.; Toshima K.; Swift G. Enzyme-Catalyzed Polymerization of L-Aspartate. Macromol. Rapid Commun. 1999, 20, 7–11. 10.1002/(sici)1521-3927(19990101)20:1<7::aid-marc7>3.3.co;2-d. [DOI] [Google Scholar]
- Soeda Y.; Toshima K.; Matsumura S. Sustainable Enzymatic Preparation of Polyaspartate Using a Bacterial Protease. Biomacromolecules 2003, 4, 196–203. 10.1021/bm0200534. [DOI] [PubMed] [Google Scholar]
- Aso K.; Uemura T.; Shiokawa Y. Protease-Catalyzed Synthesis of Oligo-L-Glutamic Acid from L-Glutamic Acid Diethyl Ester. Agric. Biol. Chem. 1988, 52, 2443–2449. 10.1271/bbb1961.52.2443. [DOI] [Google Scholar]
- Uyama H.; Fukuoka T.; Komatsu I.; Watanabe T.; Kobayashi S. Protease-Catalyzed Regioselective Polymerization and Copolymerization of Glutamic Acid Diethyl Ester. Biomacromolecules 2002, 3, 318–323. 10.1021/bm010135c. [DOI] [PubMed] [Google Scholar]
- Li G.; Raman V. K.; Xie W.; Gross R. A. Protease-Catalyzed Co-Oligomerizations of L-Leucine Ethyl Ester with L-Glutamic Acid Diethyl Ester: Sequence and Chain Length Distributions. Macromolecules 2008, 41, 7003–7012. 10.1021/ma800946d. [DOI] [Google Scholar]
- Arafet K.; Ferrer S.; Moliner V. Computational Study of the Catalytic Mechanism of the Cruzain Cysteine Protease. ACS Catal. 2017, 7, 1207–1215. 10.1021/acscatal.6b03096. [DOI] [Google Scholar]
- Fekete A.; Komáromi I. Modeling the archetype cysteine protease reaction using dispersion corrected density functional methods in ONIOM-type hybrid QM/MM calculations; the proteolytic reaction of papain. Phys. Chem. Chem. Phys. 2016, 18, 32847–32861. 10.1039/c6cp06869c. [DOI] [PubMed] [Google Scholar]
- de Beer R. J. A. C.; Zarzycka B.; Amatdjais-Groenen H. I. V.; Jans S. C. B.; Nuijens T.; Quaedflieg P. J. L. M.; van Delft F. L.; Nabuurs S. B.; Rutjes F. P. J. T. Papain-Catalyzed Peptide Bond Formation: Enzyme-Specific Activation with Guanidinophenyl Esters. ChemBioChem 2011, 12, 2201–2207. 10.1002/cbic.201100267. [DOI] [PubMed] [Google Scholar]
- Wei D.; Huang X.; Liu J.; Tang M.; Zhan C.-G. Reaction Pathway and Free Energy Profile for Papain-Catalyzed Hydrolysis of N-Acetyl-Phe-Gly 4-Nitroanilide. Biochemistry 2013, 52, 5145–5154. 10.1021/bi400629r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. J. P. An investigation into the applicability of the semi-empirical method PM7 for modelling the catalytic mechanism in the enzyme chymotrypsin. J. Mol. Model. 2017, 23, 154. 10.1007/s00894-017-3326-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis L.; Di Renzo F.; Germani R.; Goracci L.; Spreti N.; Tiecco M. α-Chymotrypsin superactivity in quaternary ammonium salt solution: kinetic and computational studies. RSC Adv. 2016, 6, 46202–46211. 10.1039/c6ra07425a. [DOI] [Google Scholar]
- Gimenez-Dejoz J.; Tsuchiya K.; Numata K. Insights into the stereospecificity in Papain-Mediated Chemoenzymatic Polymerization from Quantum Mechanics/Molecular Mechanics Simulations. ACS Chem. Biol. 2019, 14, 1280–1292. 10.1021/acschembio.9b00259. [DOI] [PubMed] [Google Scholar]
- Anson M. L. The Estimation of Pepsin, Trypsin, Papain, and Cathepsin with Hemoglobin. J. Gen. Physiol. 1938, 22, 79–89. 10.1085/jgp.22.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folin O.; Ciocalteau V. On Tyrosine and Tryptophan Determinations in Proteins. J. Biol. Chem. 1929, 73, 627. [Google Scholar]
- Fleishman S. J.; Leaver-Fay A.; Corn J. E.; Strauch E.-M.; Khare S. D.; Koga N.; Ashworth J.; Murphy P.; Richter F.; Lemmon G.; Meiler J.; Baker D. Rosettascripts: A Scripting Language Interface to the Rosetta Macromolecular Modeling Suite. PLoS One 2011, 6, e20161 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara M. J.; Leaver-Fay A.; Tyka M. D.; Stein A.; Houlihan K.; DiMaio F.; Bradley P.; Kortemme T.; Baker D.; Snoeyink J.; Kuhlman B. Combined Covalent-Electrostatic Model of Hydrogen Bonding Improves Structure Prediction with Rosetta. J. Chem. Theory Comput. 2015, 11, 609–622. 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool. http://avogadro.cc/.
- Taylor R.; Cole J.; Korb O.; McCabe P. Knowledge-Based Libraries for Predicting the Geometric Preferences of Druglike Molecules. J. Chem. Inf. Model. 2014, 54, 2500–2514. 10.1021/ci500358p. [DOI] [PubMed] [Google Scholar]
- Kamphuis I. G.; Kalk K. H.; Swarte M. B. A.; Drenth J. Structure of Papain Refined at 1.65 Å Resolution. J. Mol. Biol. 1984, 179, 233–256. 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- Razeto A.; Galunsky B.; Kasche V.; Wilson K. S.; Lamzin V. S.. High Resolution Structure of Bovine Alpha-Chymotrypsin. n. pag. Crossref. Web; RSCB Protein Data Bank, 2006. DOI: 10.2210/pdb1YPH/pdb. [DOI] [Google Scholar]
- Singh N.; Jabeen T.; Sharma S.; Roy I.; Gupta M. N.; Bilgrami S.; Somvanshi R. K.; Dey S.; Perbandt M.; Betzel C.; Srinivasan A.; Singh T. P. Detection of Native Peptides as Potent Inhibitors of Enzymes. FEBS J. 2004, 272, 562–572. 10.1111/j.1742-4658.2004.04499.x. [DOI] [PubMed] [Google Scholar]
- Vivares D.; Arnoux P.; Pignol D. A Papain-Like Enzyme at Work: Native and Acyl-Enzyme Intermediate Structures in Phytochelatin Synthesis. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18848–18853. 10.1073/pnas.0505833102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury E. M.; Carpenter B. G.; Goldman H. Conformational Studies of Polymers and Copolymers of L-Aspartate Esters. I. Preparation and Solution Studies. Biopolymers 1968, 6, 837–850. 10.1002/bip.1968.360060607. [DOI] [PubMed] [Google Scholar]
- Zotti M. D.; Formaggio F.; Crisma M.; Peggion C.; Moretto A.; Toniolo C. Handedness Preference and Switching of Peptide Helices. Part I: Helices Based on Protein Amino Acids. J. Pept. Sci. 2014, 20, 307–322. 10.1002/psc.2638. [DOI] [PubMed] [Google Scholar]
- León S.; Alemán C.; Muñoz-Guerra S. Helix Sense in Poly(β-Alkyl α-L-Aspartate)S. Macromolecules 1997, 30, 6662–6667. 10.1021/ma970087h. [DOI] [Google Scholar]
- Bradbury E. M.; Cary P. D.; Crane-Robinson C.; Hartman P. G. Nuclear Magnetic Resonance of Synthetic Polypeptides. Pure Appl. Chem. 1973, 36, 53–92. 10.1351/pac197336010053. [DOI] [Google Scholar]
- Totsingan F.; Centore R.; Gross R. A. CAL-B Catalyzed Regioselective Bulk Polymerization of L-Aspartic Acid Diethyl Ester to α-Linked Polypeptides. Chem. Commun. 2017, 53, 4030–4033. 10.1039/c7cc01300k. [DOI] [PubMed] [Google Scholar]
- Topf M.; Várnai P.; Richards W. G. Ab Initio Qm/Mm Dynamics Simulation of the Tetrahedral Intermediate of Serine Proteases: Insights into the Active Site Hydrogen-Bonding Network. J. Am. Chem. Soc. 2002, 124, 14780–14788. 10.1021/ja026219q. [DOI] [PubMed] [Google Scholar]
- Kraut J. Serine Proteases: Structure and Mechanism of Catalysis. Annu. Rev. Biochem. 1977, 46, 331–358. 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- Martínez-González J. Á.; González M.; Masgrau L.; Martínez R. Theoretical Study of the Free Energy Surface and Kinetics of the Hepatitis C Virus Ns3/Ns4a Serine Protease Reaction with the Ns5a/5b Substrate. Does the Generally Accepted Tetrahedral Intermediate Really Exist?. ACS Catal. 2015, 5, 246–255. 10.1021/cs5011162. [DOI] [Google Scholar]
- de Beer R. J. A. C.; Zarzycka B.; Mariman M.; Amatdjais-Groenen H. I. V.; Mulders M. J.; Quaedflieg P. J. L. M.; van Delft F. L.; Nabuurs S. B.; Rutjes F. P. J. T. Papain-Specific Activating Esters in Aqueous Dipeptide Synthesis. ChemBioChem 2012, 13, 1319–1326. 10.1002/cbic.201200017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.