

Abstract
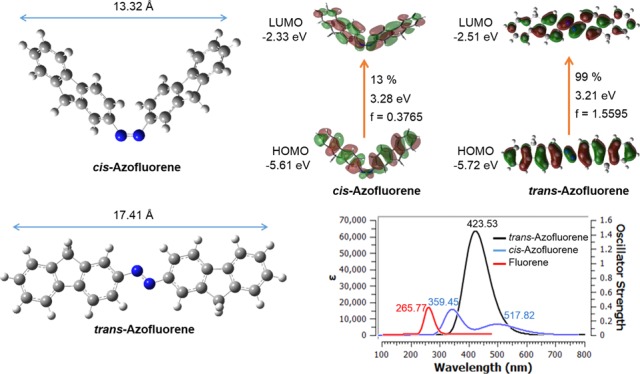
In the present work, a theoretical study was carried out to study the molecular structure, harmonic vibrational frequencies, normal force field calculations, and Raman scattering activities for fluorene π-conjugation spacer containing azo-based dye named trans- and cis-bis(9H-fluoren-2-yl)diazene (AzoFL) at density functional theory using B3LYP (Becke-3–Lee–Yang–Parr) functional and 6-31+G(d,p) basis set. The theoretical calculations have also been performed with fluorene and the trans- and cis-isomers of diazene, difluorodiazene by the same method DFT-B3LYP/6-31+G(d,p) and basis set. The present DFT calculation shows that the trans-AzoFL is more stable than the cis-AzoFL by 16.33 kcal/mol. We also report the results of new assignments of vibrational frequencies obtained on the basis of the present calculations. Time-dependent DFT (TD-DFT) and ZIndo calculations have been performed to study the UV–vis absorption behavior and frontier molecular orbitals for the above-mentioned compounds. The UV–vis spectrum from TD-DFT calculation shows the π–π* transition bands at λmax 423.53 nm (εmax 6.0 × 104 M–1 cm–1) and at λmax 359.45 nm (εmax 1.7 × 104 M–1 cm–1), respectively, for trans- and cis-AzoFL. Compared to parent trans-diazene (λmax 178.97 nm), a significant variation to longer wavelength (∼245 nm) is observed due to the incorporation of the fluorene (FL) ring into the −N=N– backbone. The co-planarity of the two FL rings with the longer N=N bond length compared to the unsubstituted parent diazene indicates the effective red shift due to the extended π-conjugation in trans-AzoFL. The nonplanarity of cis-AzoFL (48.1° tilted about the C–N bond relative to the planar N=N–C bond) reflects its ∼64 nm blue shift compared to that of trans-counterpart.
1. Introduction
Azo compounds represent one of the oldest and largest class of synthesized organic compounds used not only in dye industry1 but also in analytical chemistry as indicators in acid–base, redox, and complexometric titration.2,3 In addition, azo compounds were reported to exhibit biological activities such as antibacterial, antifungal, pesticides, antiviral, and anti-inflammatory properties.4−10 Beyond their dyeing properties and biological activities, azo compounds exhibit interesting electronic and geometrical features relating to their application for reversible optical data storage.11−17 The storage process makes use of the light-induced trans–cis–trans isomerization of the azo moiety, thereby utilizing the local variation of the refractive index of the medium.11 Because of its ability to induce a molecular motion and a significant geometric change upon trans ⇆ cis photoisomerization, azo compounds can be utilized for the construction of light-driven molecular devices.18,19
The light induced changes in the molecular structure and physical properties of azo moiety associated with E ⇆ Z photoisomerization have led to the incorporation of azobenzene into a wide variety of molecular architectures including polymers, dendrimers, liquid crystals, self-assembled monolayers, and biomaterials.20−25 Because trans-azobenzene shows intense π–π* absorption in the UV region, the rapid trans-to-cis isomerization can be induced by noncoherent UV light. The cis isomer has an enhanced n−π* absorption in the visible region; the cis-to-trans isomerization is triggered through visible-light irradiation.20 The light-driven structural changes of the azobenzene unit incorporated into a larger compound affect the properties of azo-functionalized molecular systems.18 Emerging applications of azo compounds require the extension of π-conjugated systems of azo derivatives to design visible-light-driven switches.26−30 The increasing π-conjugated length allows for more obvious red shift of azo π → π* transition bands.26 Therefore, the synthesis of azo-containing π-conjugated compounds attracts considerable attention because of the possible red shifts of azo π → π* transition bands and novel optoelectrical properties.26 On the basis of these fascinating features and properties of azo-containing π-conjugated compounds, our aim in this present work is to provide a purely theoretical perspective on the optimized geometries, orbital energies (HOMO, LUMO), IR, Raman activity, and UV–vis spectra of trans and cis-bis(9H-fluoren-2-yl)diazene (AzoFL) (Figure 1).
Figure 1.

Chemical structures of trans-and cis-bis(9H-fluoren-2-yl)diazene (AzoFL).
The valuable electronic properties of fluorene-based compounds, characterized by extensive π conjugation together with their photochemical features, make them promising candidates for use in organic light-emitting diodes,31,32 solar cells,33,34 and field-effect transistors.35,36 In an effort to gain a better understanding of the structure and electronic properties of difluorene-substituted diazene, in this paper we have investigated the optimized geometries and vibrational and absorption spectra of cis and trans-bis(9H-fluoren-2-yl)diazene (Figure 1) and compared them with those of fluorene, cis, and trans isomers of diazene and difluorodiazene (Figure 2) using the same method and basis set.
Figure 2.

Structures of fluorene and cis and trans isomers of diazene and difluorodiazene.
All of the calculations for the above-mentioned three pairs of azo compounds and fluorene are calculated by the same widely used density functional theory37 (DFT) which has capability to produce different results with high accuracy and better consistency.11,37−44 In spite of the vast literature on the studies of photoisomerization and other photophysical properties of azo dyes by means of various spectroscopic and photochemical methods, the chemistry of azo compounds are not well understood completely yet. This is probably due to the fact that sometimes it is difficult to isolate cis- and trans-isomers of the azo compounds in pure form possibly due to reversible cis–trans isomerization of the compounds. Thereby, the determination of the different properties of a pure isomer of azo compound is not straightforward. In this paper, our target is to investigate the different properties of the cis- and trans-isomers of azo fluorene individually and compare them with those of the parent diazene, fluorine, and difluorodiazene using the same DFT method and same basis set by theoretical aspect. The findings of this study might be important to understand the chemistry of π-conjugated azo compounds.
2. Results and Discussion
2.1. Geometrical Structures
The atom numbering of the trans- and cis-isomers of the model compound, bis(9H-fluoren-2-yl)diazene (AzoFL) is shown in Figure 1. The optimized geometries of both the cis- and trans-isomers of AzoFL calculated at the B3LYP/6-31+G(d,p) level are shown in Figure 3. The B3LYP/6-31+G(d,p) optimized molecular geometries of fluorene (FL) and cis- and trans-isomers of parent diazene (DZ) and difluorodiazene (DFDZ) are presented in Figure 4. The most relevant optimized geometric parameters of trans- and cis-AzoFL and fluorene (FL) are summarized in Table 1. The geometric parameters of other azo compounds are listed in Table 2 and shown in Figure 4.
Figure 3.
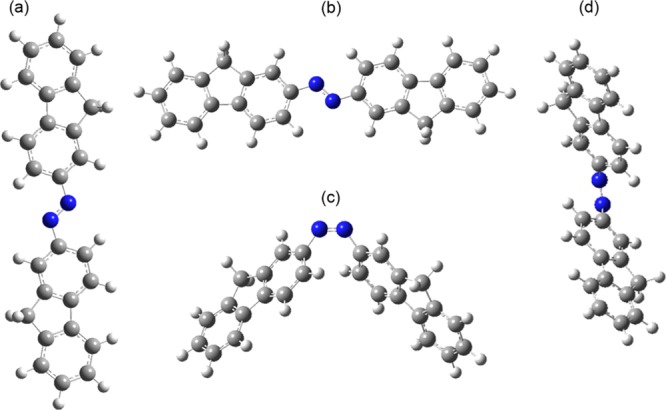
Optimized geometries of trans-AzoFL (a,b) and cis-AzoFL (c,d) calculated at B3LYP/6-31+G(d,p) method. Deep blue: N, ash: C, cyano: H.
Figure 4.
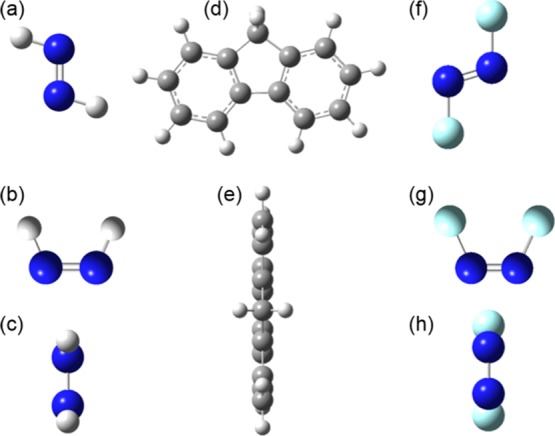
B3LYP/6-31+G(d,p) optimized geometries of (a) trans-DZ; (b,c) cis-DZ; (d,e) FL; (f) trans-DFDZ; and (g,h) cis-DFDZ,. Deep blue: N; cyano: H; ash: C; sky blue: F.
Table 1. Optimized Geometric Parametersa of Fluorene (FL), trans-AzoFL, and cis-AzoFL in the Ground State Calculated at B3LYP/6-31+G(d,p) and AM1 Methods.
| FL |
trans-AzoFL |
cis-AzoFL |
||||
|---|---|---|---|---|---|---|
| parametersa | AM1 | DFTb | AM1 | DFTb | AM1 | DFTb |
| N=N | 1.231 | 1.262 | 1.204 | 1.252 | ||
| C–N | 1.436 | 1.414 | 1.442 | 1.433 | ||
| C1–C2 | 1.403 | 1.401 | 1.421 | 1.407 | 1.417 | 1.406 |
| C2–C3 | 1.392 | 1.401 | 1.407 | 1.412 | 1.405 | 1.408 |
| C3–C4 | 1.402 | 1.398 | 1.399 | 1.390 | 1.398 | 1.394 |
| C4–C11 | 1.385 | 1.398 | 1.383 | 1.403 | 1.384 | 1.400 |
| C5–C12 | 1.385 | 1.398 | 1.385 | 1.399 | 1.385 | 1.399 |
| C5–C6 | 1.402 | 1.398 | 1.402 | 1.397 | 1.402 | 1.397 |
| C6–C7 | 1.392 | 1.401 | 1.392 | 1.402 | 1.392 | 1.401 |
| C7–C8 | 1.403 | 1.401 | 1.403 | 1.401 | 1.408 | 1.401 |
| C8–C13 | 1.382 | 1.392 | 1.382 | 1.392 | 1.382 | 1.392 |
| C1–C10 | 1.429 | 1.392 | 1.378 | 1.388 | 1.379 | 1.388 |
| C11–C12 | 1.461 | 1.470 | 1.460 | 1.466 | 1.461 | 1.468 |
| C12–C13 | 1.429 | 1.411 | 1.429 | 1.413 | 1.429 | 1.412 |
| C9–C10 | 1.504 | 1.516 | 1.505 | 1.515 | 1.505 | 1.516 |
| C9–C13 | 1.504 | 1.516 | 1.504 | 1.516 | 1.504 | 1.516 |
| C10–C11 | 1.429 | 1.411 | 1.429 | 1.411 | 1.428 | 1.413 |
| C1–H | 1.099 | 1.087 | 1.100 | 1.086 | 1.101 | 1.087 |
| C2–H | 1.100 | 1.086 | ||||
| C3–H | 1.100 | 1.086 | 1.102 | 1.084 | 1.102 | 1.086 |
| C4–H | 1.110 | 1.086 | 1.100 | 1.087 | 1.099 | 1.086 |
| C5–H | 1.110 | 1.086 | 1.099 | 1.086 | 1.099 | 1.086 |
| C6–H | 1.100 | 1.086 | 1.100 | 1.086 | 1.100 | 1.086 |
| C7–H | 1.100 | 1.086 | 1.100 | 1.086 | 1.100 | 1.086 |
| C8–H | 1.099 | 1.087 | 1.099 | 1.087 | 1.098 | 1.087 |
| C9–H | 1.119 | 1.098 | 1.120 | 1.098 | 1.119 | 1.098 |
| C9–H | 1.119 | 1.098 | 1.120 | 1.098 | 1.119 | 1.098 |
| N1–C2–C3 | 124.9 | 124.6 | 122.5 | 123.0 | ||
| C2–N1=N2 | 119.7 | 115.5 | 129.4 | 124.4 | ||
| N1=N2–C2′ | 119.7 | 115.5 | 129.4 | 124.4 | ||
| C2–C3–C4 | 120.9 | 120.6 | 121.1 | 120.3 | 121.1 | 120.3 |
| C1–C2–C3 | 120.9 | 120.5 | 119.8 | 120.2 | 120.0 | 120.3 |
| C10–C1–C2 | 118.7 | 119.1 | 118.7 | 119.3 | 118.6 | 119.1 |
| C3–C4–C11 | 118.6 | 118.9 | 119.1 | 119.4 | 118.9 | 119.4 |
| C4–C11–C10 | 120.5 | 120.4 | 120.2 | 120.3 | 120.2 | 120.1 |
| C1–C10–C11 | 120.5 | 120.5 | 121.1 | 120.4 | 110.0 | 120.6 |
| C9–C10–C11 | 110.5 | 110.0 | 110.0 | 110.0 | 110.0 | 110.0 |
| C9–C13–C12 | 110.5 | 110.0 | 110.1 | 110.0 | 110.1 | 110.1 |
| C10–C9–C13 | 103.3 | 102.8 | 103.3 | 102.7 | 103.3 | 102.7 |
| C10–C11–C12 | 108.3 | 108.6 | 108.4 | 108.7 | 108.4 | 108.6 |
| C13–C12–C11 | 108.3 | 108.6 | 108.3 | 108.5 | 108.3 | 108.5 |
| C12–C5–C6 | 118.6 | 118.9 | 118.6 | 118.8 | 118.6 | 118.9 |
| C5–C6–C7 | 120.9 | 120.6 | 120.9 | 120.6 | 120.9 | 120.7 |
| C6–C7–C8 | 120.9 | 120.5 | 120.9 | 120.6 | 120.9 | 120.6 |
| C7–C8–C13 | 118.7 | 119.1 | 118.7 | 119.0 | 118.7 | 119.0 |
| C8–C13–C12 | 120.5 | 120.5 | 120.4 | 120.4 | 120.4 | 120.4 |
| C13–C12–C5 | 120.5 | 120.4 | 120.5 | 120.5 | 120.5 | 120.5 |
| C1–C2–N1 | 115.3 | 115.2 | 117.3 | 116.1 | ||
| C3–C2–N1 | 124.9 | 124.6 | 122.5 | 123.0 | ||
| C2N1N2C2′ | 179.3 | –179.99 | 2.3 | 10.9 | ||
| C3C2N1N2 | –15.7 | 0.01 | 46.9 | 48.1 | ||
Bond lengths in angstroms and bond angles and dihedral angles in degrees.
B3LYP/6-31+G(d,p).
Table 2. Calculated Optimized Geometric Parameters of trans-Diazene (DZ), cis-Diazene (DZ), trans-Difluoro Diazene (DFDZ), and cis-Difluoro Diazene (DFDZ).
|
trans-DZ |
trans-DFDZ |
|||||||
|---|---|---|---|---|---|---|---|---|
| parametersa | AM1 | DFTb | AM1 | HFc | HFd | HFe | DFTb | expf |
| N=N | 1.212 | 1.244 | 1.244 | 1.192 | 1.192 | 1.188 | 1.225 | 1.224 |
| dN1–H1 | 1.018 | 1.036 | ||||||
| dN2–H2 | 1.018 | 1.036 | ||||||
| ∠H1N1N2 | 112.3 | 106.7 | ||||||
| ∠N1N2H2 | 112.3 | 106.7 | ||||||
| ∠HNNH | 180.0 | 180.0 | ||||||
| dN1–F1 | 1.348 | 1.339 | 1.339 | 1.326 | 1.395 | 1.398 | ||
| dN2–F2 | 1.348 | 1.339 | 1.339 | 1.326 | 1.395 | |||
| ∠F1N1N2 | 113.0 | 106.9 | 106.9 | 107.5 | 105.1 | 115.5 | ||
| ∠N1N2F2 | 113.0 | 106.9 | 106.9 | 107.5 | 105.1 | |||
| ∠FNNF | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | |||
|
cis-DZ |
cis-DFDZ |
|||||||
|---|---|---|---|---|---|---|---|---|
| parametersa | AM1 | DFTb | AM1 | HFc | HFd | HFe | DFTb | expf |
| N=N | 1.197 | 1.242 | 1.220 | 1.193 | 1.193 | 1.190 | 1.217 | 1.209 |
| dN–H | 1.019 | 1.043 | ||||||
| dN–H | 1.019 | 1.043 | ||||||
| ∠H1N1N2 | 120.6 | 113.0 | ||||||
| ∠N1N2H2 | 120.6 | 113.0 | ||||||
| ∠HNNH | 0.0 | 0.0 | ||||||
| dN1–F1 | 1.356 | 1.337 | 1.337 | 1.327 | 1.399 | 1.409 | ||
| dN2–F2 | 1.356 | 1.337 | 1.337 | 1.327 | 1.399 | |||
| ∠F1N1N2 | 124.2 | 114.4 | 114.4 | 114.6 | 114.9 | 114.4 | ||
| ∠N1N2F2 | 124.2 | 114.4 | 114.4 | 114.6 | 114.9 | |||
| ∠FNNF | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |||
d, bond lengths in angstroms and ∠, bond angles, and dihedral angles in degrees.
B3LYP/6-31+G(d,p).
HF/6-31+G(d,p); N=Ncis (1.19323 Å); N=Ntrans (1.19208 Å); N–Fcis (1.133918 Å); N–Ftrans (1.133745 Å).
HF/6-31++G(d,p); N=Ncis (1.19323 Å); N=Ntrans (1.19208 Å).
HF/6-311+G(d,p); N=Ncis (1.19043 Å); N=Ntrans (1.18799 Å); N–Fcis (1.132657 Å); N–Ftrans (1.132601 Å).
The optimized geometry parameters (Table 1) show that the trans-AzoFL is almost planar (central CNNC dihedral angle: 179.99°) according to our DFT calculation. Complete geometry optimization for cis-AzoFL in present work resulted in nonplanarity (central CNNC dihedral angle: 10.9°) of the molecule. The fluorene (FL) rings are rotated by 48.1° about the C–N bond relative to planar N=N–C arrangement to decrease the H–H non bonded interaction in cis-AzoFL. According to our DFT calculation the energy difference shows that the trans-AzoFL in its ground state is more stable than the cis-AzoFL by 16.33 kcal/mol (Table 3).
Table 3. Calculated Energies (Hartree), Energy Differences (kcal/mol) between the Cis- and Trans-Isomers of AzoFL, DFDZ, and DZ and Their Respective Dipole Moments (Debye), Respectively.
| compound | methoda | Etrans | Ecis | Ecis–transg | μ (trans) | μ (cis) |
|---|---|---|---|---|---|---|
| AzoFL | AM1b | 0.261988 | 0.254685 | –4.58 | 0.17 | 2.99 |
| DFTc | –1111.176069 | –1111.150053 | +16.33 | 0.00 | 3.12 | |
| DFDZ | AM1b | 0.049665 | 0.033056 | –10.4 | 0.00 | 0.66 |
| HFd | –307.595444 | –307.593029 | +1.52 | 0.00 | 0.17 | |
| HFe | –307.595444 | –307.593029 | +1.52 | 0.00 | 0.17 | |
| HFf | –307.673508 | –307.670734 | +1.74 | 0.00 | 0.18 | |
| DFTc | –309.033536 | –309.036420 | –1.81 | 0.00 | 0.22 | |
| DZ | AM1b | 0.050244 | 0.051651 | +0.88 | 0.00 | 2.70 |
| HFd | –110.006960 | –109.994657 | +7.72 | 0.00 | 3.37 | |
| DFTc | –110.651970 | –110.641101 | +6.82 | 0.00 | 3.20 |
The symmetry of trans-DZ and DFDZ in different methods are C2h, C2v for cis-DZ and DFDZ; C2 for both the trans- and cis-AzoFL.
Semiempirical AM1 method using predefined ZDO basis set.
B3LYP/6-31+G(d,p) basis set.
6-31+G(d,p) basis set.
6-31++G(d,p) basis set.
6-311+G(d,p) basis set.
The negative values of energy difference in respective cases indicate the cis-preference over trans-isomer.
The trans-AzoFL has no dipole moment, whereas the cis-AzoFL exhibits a dipole moment of 3.12 D. However, our semiempirical AM1 calculation shows that the cis-AzoFL is more stable by 4.18 kcal/mol (Table 3) compared to that of trans-AzoFL, which possess some deviation from planarity having a bit dipole moment (0.17 D).
Our calculated geometry parameters at B3LYP/6-31+G(d,p) for trans-DZ (Table 2) (NN: 1.244 Å, NH: 1.036 Å, ∠NNH: 106.7°) is well agreed with the earlier reported experimental value (NN: 1.247 Å, NH: 1.029 Å, ∠NNH: 106.3°)45 and theoretical work46 (NN: 1.238 Å, NH: 1.035 Å, ∠NNH: 107°) by B3LYP/6-311++G(d,p) method. The calculated work47 by CCSD(T)/CBS found (NN: 1.246 Å, NH: 1.029 Å, ∠NNH: 106.4°) which also has good agreement with our present work (Table 2). The geometric parameters (Table 2) for cis-diazene (NN: 1.242 Å, NH: 1.043 Å, ∠NNH: 113°) calculated by present B3LYP/6-31+G(d,p) method is also quite well agreed with the earlier reported (NN: 1.237 Å, NH: 1.041 Å), (∠NNH: 113°) by B3LYP/6-311++G(d,p) method.48 All of the ground-state geometries were verified by vibrational frequency analysis at the same level of theory and found as true minima because negative vibrational frequencies were absent in all cases.
The calculated energies (hartree), energy differences (kcal/mol) between the cis- and trans-isomers of AzoFL, DFDZ, and DZ and their respective dipole moments (debye) are summarized in Table 3. The trans-AzoFL was found as more stable than the cis-AzoFL by the calculation at B3LYP/6-31+G(d,p). Similarly trans-DZ was also found stable as compared to cis-DZ. Back et al.48 by near-ultraviolet absorption investigation of diazene in gas phase showed that the trans-DZ was the most stable isomer. However, the cis-DFDZ was found as more stable (Table 3) by 1.81 kcal/mol than the trans-DFDZ by B3LYP/6-31+G(d,p), which supports the preference of cis-DFDZ energetically by the earlier work.49 The trans- and cis-isomers of AzoFL in ground state adopted the C2 symmetry, whereas the trans-DZ and trans-DFDZ adopt the C2h point groups. The cis-DZ, cis-DFDZ, and FL possess C2v points group. We have made a comparative study of the N=N, N–H, H–F, C–N, C–C, and C–H bond lengths as well as C–N=N and C–C–N bond angles in DZ, FL, DFDZ, and AzoFL. As shown in Tables 1 and 2, we have found that the N=N bond lengths of trans-isomers of DZ, DFDZ, and AzoFL, are 1.244, 1.225, and 1.262 Å, respectively. The N=N bond length order among the three trans-isomers has been found as AzoFL > DZ > DFDZ by our DFT-B3LYP/6-31+G(d,p) calculation, and the same trend has been observed for the respective cis-isomers as well. Upon substitution in the parent trans-DZ molecule by two electron donor fluorene (FL) moiety causes an increase of the N=N bond distance from 1.244 to 1.262; an 0.018 Å increase of bond length is observed. This is due to the extensive π-bond conjugation of the N=N bond with the fluorene (FL) ring in trans-isomer of AzoFL. On the other hand, incorporation of the two F atoms in the parent DZ by replacing two H-atoms causes shortening of the N=N bond length from 1.244 to 1.225 Å (Table 2) in trans-DFDZ. Hence, an opposite trend, a decrease of 0.019 Å is observed in trans-DFDZ compared to that of trans-DZ. This effect is stronger in cis-DFDZ, a bit shorter of 0.025 Å N=N bond length in cis-DFDZ is found compared to cis-DZ (Table 2).
As aromatic fluorene (FL) moiety is the major structural unit of our target AzoFL, we have calculated FL for comparison even though there are detailed experimental50 as well as some theoretical works44 present in the literature. Our calculated structure of FL (Table 1) by B3LYP/6-31+G(d,p) is well agreed with the reported work done by Lee and Boo44 calculated at the B3LYP/6-31G* level. There is reasonable agreement found with the reported X-ray crystal structure.51 A minor deviation was observed with the X-ray crystal structure51 of bond angles, for example, ∠C1C10C11 by 1.43°.
Our DFT calculation shows that the FNN angle in the trans-DFDZ is 105.1° whereas the same angle in cis-form is 114.9°. Our HF calculation shows that the FNN angle in the trans-form is 106.9°, whereas the same angle in cis-form is 114.4°. This supports earlier work.49 As fluorine atoms are electronegative, they have stronger electron affinity relative to the nitrogen atoms and possibility to polarize the bonds. The cis-isomer has a small dipole moment (0.22 D), whereas the trans-DFDZ has no dipole moment according to our present B3LYP/6-31+G(d,p) calculation.
The N=N bond length of cis-DFDZ is found to be shorter (Table 2) than that of the corresponding trans-DFDZ by our B3LYP-DFT/6-31+G(d,p) calculation. On the contrary, the N–F bond (1.399 Å) in cis-DFDZ is longer (0.004 Å) than its trans-counterpart (1.395 Å). It should be mentioned that the shortening of the N=N bond in conjunction with elongation of the N–F bond indicates the presence of negative hyperconjugation.49,55 This difference in geometrical parameters leads to a higher stability of the cis-DFDZ, which is nicely reflected in our DFT-B3LYP/6-31+G(d,p) calculation. In addition, a considerable widening of ∠NNF has been observed for cis-DFDZ (Table 2) compared to that of trans-DFDZ. The reason for such type of structural change is due to repulsion of the F atom lone pairs, the electrostatic repulsion of the N–F dipolar bonds, and steric effect.49 Such type of structural/geometrical change has also been observed by earlier work.49,55,56 In our DFT-B3LYP/6-31+G(d,p) calculation, the two C–N bonds in cis-AzoFL is also found to be longer by (0.019 Å) compared to that of trans-AzoFL, whereas the same bond is longer by only 0.006 Å in semiempirical AM1 (Table 1).
The N=N bond of DZ, DFDZ, and AzoFL (Tables 1 and 2) is shorter in cis-isomer over trans-isomer by 0.015, 0.024, and 0.027 Å in semiempirical AM1 method. Similar behavior, that is, shorter N=N bond in DZ, DFDZ, and AzoFL by 0.002, 0.008, and 0.01 Å by DFT/6-31+G(d,p) method.
Our result from semiempirical AM1 method shows the preference of cis-isomer over trans-isomer (Table 3) by 4.58 and 10.4 kcal/mol for AzoFL and DFDZ, respectively. On the other hand, the parent trans-DZ isomer is stable by 0.88 kcal/mol over cis-DZ. The preference of cis-AzoFL over the trans-isomer by AM1 method is not clear, but the preference of cis-isomer over trans-isomer for DFDZ due to cis-effect is known in the literature for dihalodiazenes.49,52,55,56 Different explanations were found for the cis-effect in the literature by different authors, viz., (i) the sum of the repulsive forces between the N lone pairs and between the two N–F bonds is less in cis-DFDZ compared to that of the trans-DFDZ,57 (ii) mutual interplay of various interactions, for example, antiperiplanar interaction, Coulombic interaction, and lone pair-lone pair interaction in diazene moiety.49 (iii) delocalization of the N lone pair over the antibonding orbital of the adjacent N–F bond along with the lone pair delocalization of F over the antibonding orbital of the N=N bond,58 and (iv) mutual interactions between the nitrogen lone pairs and the neighboring antibonding orbital of the N–X bond (X = F, Cl, Br).56 The shorter N=N bond length is also observed in the parent and unsubstituted cis-DZ along with longer N–H and wider NNH angle compared to that of trans-DZ. However, the parent trans-DZ isomer is stable by 0.88 kcal/mol over cis-DZ, and the cis-effect, that is, the stability of cis-DZ over trans-DZ was not observed in our both the DFT and semiempirical AM1 calculation in accordance with different previous work.49,55 Because DZ contains no F atoms, as a consequence there are no lone pair electrons for delocalization of halogen lone pairs into the antibonding orbitals of N=N bond. This could be the inability of parent cis-DZ to get any stabilizing energy via delocalization effects and causes preference of trans-isomer.55
An attempt were also taken to observe the cis effect by the HF method using three different basis sets, for example, 6-31+G(d,p), 6-31++G(d,p), and 6-311+G(d,p), respectively for DFDZ. The ab initio Hartree–Fock produces insignificant but somewhat longer N=N bond length by 0.00044 Å in cis-DZ (1.21530 Å) over trans-DZ (1.21486 Å) using 6-31+G(d,p) basis set. Similar insignificant longer N=N bond length is also observed in cis-DFDZ by 0.00115 and 0.00244 Å over trans-DFDZ by HF using 6-31+G(d,p) and 6-311+G(d,p) basis set, respectively. The N–F bond of cis-DFDZ is also found to be longer compared to trans-DFDZ by 0.00173 and 0.00056 Å in HF/6-31+G(d,p) and 6-311+G(d,p) basis sets. In HF both the 6-31+G(d,p) and 6-311+G(d,p) basis sets produces longer N=N bond and N–F bond. In both cases, they have larger FNN bond angles. The FNN bond angle of cis-DFDZ is 0.2° in wider by HF/6-311+G(d,p) basis set compared to HF/6-311+G(d,p) over trans-isomer. The HF calculation shows that 6-31+G(d,p) and 6-31++G(d,p) basis set produces the same geometric parameters and equal energy (Table 3). However, energetically preference of the cis-DFDZ was not found by all of the basis sets of HF methods by our present work. Earlier work by HF49,55 with small basis set and the SS-MRCCSD/aug-cc-pVDZ55 calculation was also unable to show the cis-effect.
2.2. Electronic Absorption Spectra
The photophysical properties of trans- and cis-AzoFL consisting of donor (fluorene ring) and acceptor (−N=N−) azo group have been investigated in gas phase by theoretical calculation. The UV–vis absorption spectra of parent trans- and cis-DZ, electron withdrawing F atom-containing difluorodiazene (DFDZ) and FL were calculated and made comparison with the model compound AzoFL. In the past decade, time-dependent DFT (TD-DFT) has become the leading method for the calculation of excitation energies and optical properties of organic molecules.59−62 Starting from the each fully optimized ground-state structures of B3LYP/6-31+G(d,p), TD-DFT excited-state calculations with the hybrid functional B3LYP and 6-31+G(d,P) basis set were calculated on the three lowest spin allowed singlet–singlet transitions for the above-mentioned azo compounds and fluorene in the gas phase. The calculated UV–vis spectra of those compounds are shown in Figure 5. The theoretical excitation energies (Eex), oscillator strengths (f), and absorption wavelengths (λmax) are listed in Tables 4–6. All of the transition probabilities of the different trans- and cis-azo compounds by TD-DFT calculation are given in Tables 4 and 5, respectively.
Figure 5.

UV–vis spectrum of (a) trans- and cis-AzoFL, (b) trans- and cis-DZ, (c) trans- and cis-DFDZ (inset: UV–vis peak of cis-DFDZ: half-width at half height 0.033 eV), and (d) FL (inset: UV–vis peak of FL UV–vis peak: peak half-width at half height 0.033 eV) obtained by TD-DFT/B3LYP/6-31+G(d,p) calculation. The calculated UV–vis spectra are represented with a Gaussian UV–vis peak half-width at half height 0.333 eV.
Table 4. Comparison of Electronic Absorption Wavelengths λMax (nm), Excitation Energies, Eex (eV), and Oscillator Strengths (f) Obtained by TD/DFT and ZIndo Calculation for the Model AzoFL and Other Compounds for π–π* Transition.
|
trans- |
cis- |
|||||||
|---|---|---|---|---|---|---|---|---|
| method | properties | DZ | DFDZ | AzoFL | DZ | DFDZ | AzoFL | FL |
| TD/DFTa,b | λmax | 178.97 | 189.32 | 423.53 | 205.43 | ∼190.00 | 359.45 | 265.77 |
| Eex | 6.9277 | 6.5490 | 2.9274 | 6.0355 | 6.4312 | 3.4492 | 4.6650 | |
| f | 0.0386 | 0.0111 | 1.5595 | 0.0277 | 0.0104 | 0.3765 | 0.2862 | |
| ZIndoc,d | λmax | 140.80 | 175.65 | 387.20 | 135.98 | 169.51 | 355.21 | 296.84 |
| Eex | 8.8057 | 7.0586 | 3.2021 | 9.1177 | 7.3142 | 3.4905 | 4.1767 | |
| f | 0.4735 | 0.4028 | 1.5678 | 0.5269 | 0.3670 | 0.7533 | 0.4446 | |
Using B3LYP/6-31+G(d,p).
From initial optimized geometry of B3LYP/6-31+G(d,p).
Using semi empirical ZIndo with predefined STO-3G basis set.
From initial optimized geometry of semi empirical AM1.
Table 6. Electronic Transition, Absorption Wavelengths λMax (nm), Excitation Energies, Eex (eV), and Oscillator Strengths (f) Obtained by TD-DFT/B3LYP/6-31+G(d,p) Calculation for all of the cis-Azo Compounds from the Optimized Initial Geometry at B3LYP/6-31+G(d,p)e.
| compound | electronic transition | λmax | f | Ex | MOa | MOb | symc | wave functionsd |
|---|---|---|---|---|---|---|---|---|
| cis-DZ | S0 → S1 | 371.78 | 0.0056 | 3.3348 | 8 → 9 | 0.70904 | B1 | H → L (100%) |
| S0 → S2 | 205.43 | 0.0277 | 6.0355 | 8 → 10 | 0.70584 | B2 | H → L + 1 (99%) | |
| S0 → S3 | 183.64 | 0.0000 | 6.7516 | 7 → 9 | 0.70622 | A2 | H – 1 → L (99%) | |
| cis-DFDZ | S0 → S1 | 194.49 | 0.0000 | 6.3748 | 14 → 17 | 0.70624 | A2 | H – 2 → L (99%) |
| S0 → S2 | 192.79 | 0.0104 | 6.4312 | 15 → 18 | 0.34743 | B1 | H – 1 → L + 1 (24%) | |
| 16 → 17 | 0.61543 | H → L (75%) | ||||||
| S0 → S3 | 180.82 | 0.0058 | 6.8569 | 15 → 18 | 0.61259 | B1 | H – 1 → L + 1 (75%) | |
| 16 → 17 | –0.34590 | H → L (23%) | ||||||
| cis-AzoFL | S0 → S1 | 517.82 | 0.1774 | 2.3944 | 92 → 95 | –0.24158 | B | H – 2 → L (11%) |
| 94 → 95 | 0.65138 | H → L (84%) | ||||||
| S0 → S2 | 359.45 | 0.3765 | 3.4492 | 92 → 95 | 0.63933 | B | H – 2 → L (81%) | |
| 94 → 95 | 0.25998 | H → L (13%) | ||||||
| S0 → S3 | 352.82 | 0.0486 | 3.5141 | 93 → 95 | 0.65249 | A | H – 1 → L (85%) | |
| 94 → 96 | –0.23956 | H → L + 1 (11%) |
Molecular orbitals involved in the transition.
Molecular orbital coefficients.
sym, orbital symmetry-singlet.
The wave functions based on the eigenvectors predicted by TD-DFT. H and L are used to denote the HOMO and LUMO.
Percentage of contribution obtained by (100 × c × c × 2), where c is the coefficient.
Table 5. Absorption Wavelengths λMax (nm), Excitation Energies, Eex (eV), and Oscillator Strengths (f) Calculated by TD/DFT-B3LYP/6-31+G(d,p) Method for all of the trans-Azo Compounds and FL From the Initial Optimized Geometry at B3LYP/6-31+G(d,p).
| compound | electronic transition | λmax | f | Ex | MOa | MOb | symc | wave functionsd,e |
|---|---|---|---|---|---|---|---|---|
| trans-DZ | S0 → S1 | 387.78 | 0.0000 | 3.1972 | 8 → 9 | 0.70891 | BG | H → L (100%) |
| S0 → S2 | 184.08 | 0.0000 | 6.7354 | 8 → 10 | 0.70579 | AG | H → L + 1 (99%) | |
| S0 → S3 | 178.97 | 0.0386 | 6.9277 | 8 → 11 | 0.70527 | BU | H → L + 2 (99%) | |
| trans-DFDZ | S0 → S1 | 227.47 | 0.0000 | 5.4505 | 16 → 17 | 0.70544 | BG | H → L (99%) |
| S0 → S2 | 189.32 | 0.0111 | 6.5490 | 15 → 17 | 0.29661 | BU | H – 1 → L (17%) | |
| 16 → 18 | 0.63732 | H → L + 1 (81%) | ||||||
| S0 → S3 | 179.59 | 0.0000 | 6.9033 | 15 → 18 | 0.70238 | BG | H – 1 → L + 1 (98%) | |
| trans-AzoFL | S0 → S1 | 489.35 | 0.0000 | 2.5336 | 93 → 95 | 0.69879 | B | H – 1 → L (97%) |
| S0 → S2 | 423.53 | 1.5595 | 2.9274 | 94 → 95 | 0.70581 | B | H → L (99%) | |
| S0 → S3 | 344.48 | 0.0000 | 3.5992 | 92 → 95 | 0.68177 | A | H – 2 → L (92%) | |
| 94 → 96 | –0.13547 | H → L + 1 (3%) | ||||||
| FL | S0 → S1 | 276.39 | 0.1648 | 4.4858 | 42 → 45 | 0.22186 | B2 | H – 2 → L (9%) |
| 42 → 46 | 0.11652 | H – 2→L + 1 (2%) | ||||||
| 44 → 45 | 0.48597 | H → L (47%) | ||||||
| 44 → 46 | –0.43727 | H → L + 1 (38%) | ||||||
| S0 → S2 | 265.77 | 0.2862 | 4.6650 | 42 → 45 | –0.15521 | B2 | H – 2 → L (4%) | |
| 44 → 45 | 0.48553 | H → L (47%) | ||||||
| 44 → 46 | 0.48096 | H → L + 1 (46%) | ||||||
| S0 → S3 | 256.82 | 0.0072 | 4.8277 | 43 → 45 | 0.55822 | A1 | H – 1 → L (62%) | |
| 44 → 47 | –0.40501 | H → L + 2 (32%) |
Molecular orbitals involved in the transition.
Molecular orbital coefficients.
sym, orbital symmetry-singlet.
The wave functions based on the eigenvectors predicted by TD-DFT. H and L are used to denote the HOMO and LUMO.
Percentage of contribution obtained by (100 × c × c × 2), where c is the co-efficient.
The present TD-DFT calculations show that the model trans-AzoFL afforded characteristics broad and long-waved absorption band around 300–700 nm (Figure 5a). The band at λmax 423.53 nm is very high with a molar extinction coefficient εmax 6.0 × 104 M–1 cm–1, which is indicative of the π–π* transition63 (S0–S2) in trans-AzoFL. On the other hand, the band for n−π* transition was not observed in trans-AzoFL by TD-DFT calculation. The spectra (Figure 5a) of cis-AzoFL shows the disappearance of the band at λmax 423.53 nm, while a well resolved band at 359.45 nm (S0–S2) for π–π* and a second band at 517.82 nm (S0–S1) for n−π* transition, respectively, was observed (Figure 5a). The band at 359.45 nm (π–π*) is decreased in intensity (εmax 1.7 × 104 M–1 cm–1), whereas the n−π* transition band at 517 nm has strong εmax 7.0 × 103 M–1 cm–1 absorbance compared to that of other azo compounds under study. The absorption band for the π–π* transition in cis-AzoFL shifts to shorter wavelength at λmax 359 45 nm, a 64.08 nm blue shift is observed compared to that of trans-AzoFL. The broad band at λmax 517.82 nm (n−π*) transition for cis-AzoFL (Figure 5a) is shifted to longer wavelength compared to all other cis-azo compounds by present TD-DFT calculation.
Liu and co-workers26 investigated the UV–vis spectrum of 1,2-bis(9,9-dioctyl-9H-fluoren-2-yl)diazene in 1,2-dichloroethane (concentration of the compound is 0.02 g/L) and found the experimental absorption maxima (λmax) for π–π* transition at 394 nm and n−π* transition at 500 nm. They26 also performed TD-DFT calculation at the level of ONIOM (M06-2x/6-31G*: AM1), and the calculated absorption maximum (π–π* transition) of 1,2-bis(9,9-dioctyl-9H-fluoren-2-yl)diazene was found at 345 nm. These results supports our TD-DFT calculated UV–vis spectra of trans-AzoFL (π–π* transition band at λmax 423.53 nm) at the level of B3LYP/6-31+G(d,p) in gas phase.
Bagheri and Hashemianzadeh34 employed TD-DFT calculations with B3LYP/6-311+G** basis set, based on the optimized geometries of B3LYP/6-311+G** for azo dye-containing fluorene derivative at one end and 4-carboxyphenyl group at the other end of the azo group (−N=N−). The TD-DFT calculated maximum wavelengths (π–π* transition) of the azo dye34 are shown at 405.41 nm in gas phase and at 438.62 nm in THF in UV–vis absorption spectra. The steady-state UV–visible absorption spectrum of trans-azobenzene in n-hexane shows one weak band at 445 nm assigned for the n−π* transition (S1 state) and a stronger band at 315 nm for π–π* transition (S2 state) by Lednev et al.64 The n−π* transition is very weaker (ε ≈ 400 M–1 cm–1) and is not allowed in the trans-isomer of azobenzene compounds by symmetry rules. However, the electronic transition n−π* (380–520 nm) is allowed in cis-isomer, resulting in an increase in intensity with respect to the trans-isomer in azobenzene compounds.65,66
The present TD-DFT calculation performed by our group shows that the parent trans-DZ (Figure 5b) has λmax 178.97 nm (εmax 1.4 × 103 M–1 cm–1) for π–π* (S0–S3) transition. The n−π* transition band in the parent trans-DZ was also not observed similar to trans-AzoFL. The band at 178.97 nm in cis-DZ (Figure 5b) completely disappears and instead of that two new well-separated nice bands at λmax 205.43 nm (εmax 1.2 × 103 M–1 cm–1) for π–π* (S0–S2) and at λmax 371.78 nm (εmax 200 M–1 cm–1) for n−π* (S0–S1) transition, respectively, is found. It is also observed that in cis-DZ (Figure 5b), the λmax at 205.43 nm (εmax 1.2 × 103 M–1 cm–1) for π–π* transition is decreased in intensity compared to that of trans-DZ λmax 178.97 nm (εmax 1.4 × 103 M–1 cm–1) and shifts to longer wavelength.
Figure 5d shows a broad band around 200–350 nm for fluorene (FL). The three bands (Figure 5d inset, half-width at half height 0.033 eV) at 256.82 nm (S0–S3), 265.77 nm (S0–S2), and 276.39 nm (S0–S1) merge together at λmax 265.77 nm (εmax 1.6 × 103 M–1 cm–1) for the π–π* transition (S0–S2).
It is crystal like clear that a significant variation on the absorption spectra of AzoFL occurred by incorporation of the fluorene (FL) ring into the −N=N– backbone (Figure 5). The same trend in extinction-coefficient, that is, much higher extinction-coefficient and higher oscillator strength in trans-AzoFL in comparison with that of parent trans-DZ (Figure 5) is observed.
The results show that incorporation of the FL ring into the −N=N– back bone causes bathochromic shifts of both the trans- and cis-AzoFL and higher extinction-coefficient (Figure 5a,d). A 157.76 and 93.68 nm wavelength increment is observed compared to FL in trans- and cis-AzoFL, respectively, for π–π* transition band. The weak band for n−π* (S0–S1) transition at λmax 371.78 nm (εmax 200 M–1 cm–1) for cis-diazene shifts to λmax 517.82 nm (εmax 7.0 × 103 M–1 cm–1) in cis-AzoFL, a red shift of 146.04 nm is observed with higher intensity. On the other hand, the intensity of the π–π* band in both the cis-DZ and cis-AzoFL causes hypochromic effect by TD-DFT calculation compared to the corresponding trans-isomers.
In trans-AzoFL, the absorption maxima λmax 423.53 nm of π–π* transition showed an obvious red shift of ∼245 nm increment to longer wavelength compared to that of trans-diazene (λmax 178.97 nm). This effective red shift is attributed due to the extended π-conjugation length which reflects the longer N=N bond length of AzoFL (Table 1). Even a 154.02 nm of wavelength increment toward longer wave length is observed in cis-azoFL (λmax 359.45 nm) compared to that of cis-diazene (λmax 205.43 nm). Because of coplanarity of the two FL rings in trans-isomer, the π–π* transition band shifts to lower energy longer wavelength compared to that of cis-AzoFL.
Introducing two F atoms into the −N=N– backbone in DFDZ shows interesting results. The trans-DFDZ (Figure 5c) has a band at λmax 189.32 nm (S0–S2) with low absorbance. The molar absorptivity was found only εmax ≈ 420 M–1 cm–1 with low oscillator strength (0.0111). It is expected that π–π* transition should have high molar absorptivity usually at εmax ≈ 104 M–1 cm–1, but this unusual result is surprising. The π–π* transition band at λmax 189.32 nm of trans-DFDZ causes a red shift of 10.35 nm compared to that of trans-DZ (λmax 178.97 nm, εmax ≈ 1.4 × 103 M–1 cm–1).
In cis-DFDZ, a broad band appeared at λmax ≈ 190 nm with low molar absorptivity (εmax ≈ 500 M–1 cm–1) by Gaussian UV–vis peak half-width at half height (0.333 eV) in UV–vis spectra (Figure 5c). However, the band was found as separated bands at λmax 180.82 nm (S0–S3, f = 0.0058) and λmax 192.79 nm (S0–S2, f = 0.0104) (Figure 5c, inset) at UV–vis peak half-width at half height (0.033 eV). Compared to cis-DZ (λmax 205.43 nm, εmax 1.2 × 103 M–1 cm–1), cis-DFDZ (λmax ≈ 190 nm, εmax ≈ 500 M–1 cm–1) shows a blue shift of 15.43 nm with reduced molar absorptivity. The cis-DFDZ (λmax ≈ 190 nm, εmax ≈ 500 M–1 cm–1) and trans-DFDZ (λmax 189.32 nm, εmax ≈ 420 M–1 cm–1) shows a similar type of absorption behavior (Figure 5c).
In order to examine the TD-DFT excited-state behavior of the DZ and DFDZ, a further investigation was carried out (Table S1). TD-DFT//B3LYP/6-31+G(d,p) calculations by using different initial geometries obtained from HF/6-31+G(d,p) and HF/6-31++G(d,p) basis sets were done. The two initial geometries gave the similar results by TD-DFT//B3LYP/6-31+G(d,p) calculations. In trans-DFDZ, a band appeared at λmax ≈ 168 nm with low molar absorptivity (εmax ≈450 M–1 cm–1) by Gaussian UV–vis peak half-width at half height (0.333 eV) in UV–vis spectra (Figure S1a). However, the band was found as separated bands at λmax 161.62 nm (S0–S3, f = 0.0092) and λmax 172.36 nm (S0–S2, f = 0.0067) (Figure S1a, inset) at UV–vis peak half-width at half height (0.233 eV). By using HF/6-31+G(d,p) as initial geometry in TD-DFT//B3LYP/6-31+G(d,p) calculation, the absorptivity is enhanced in some extent and causes a ∼17 nm red shift in cis-DFDZ (λmax 185.82 nm, S0–S1,f = 0.0181, εmax ∼750 M–1 cm–1) compared to trans-DFDZ (λmax 168 nm, εmax ≈ 450 M–1 cm–1).
ZIndo excited-state calculations with the predefined STO-3G basis set by using optimized geometries of semiempirical AM1 as the initial structure were also calculated on the three lowest spin allowed singlet–singlet transitions for the above-mentioned azo compounds and FL in the gas phase. The electronic transition data, for example, the theoretical excitation energies (Eex), oscillator strengths (f), and absorption wavelengths (λmax) are listed in the Tables 4, S2 and S3. The calculated UV–vis spectra of the three pairs of azo compounds and FL by ZIndo are shown in Figure 6.
Figure 6.
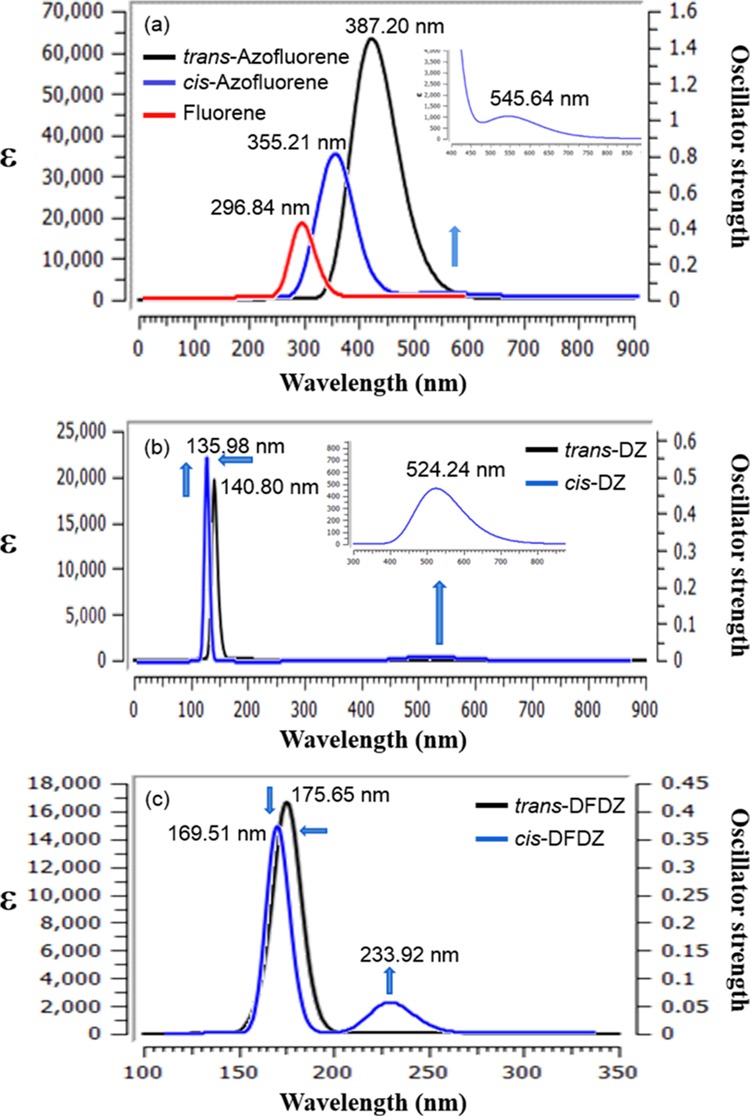
Calculated UV–vis spectra of (a) trans- and cis-AzoFL with FL (b) trans- and cis-DZ (c) trans- and cis-DFDZ by ZIndo. The calculated UV–vis spectra are represented with a Gaussian UV–vis peak half-width at half height 0.333 eV or 2685.83 cm–1.
ZIndo produces nice bands for π–π* and n−π* transitions for the three pairs of azo compounds. The π–π* transition band of trans- and cis-AzoFL were observed at λmax 387.20 nm and λmax 355.21 nm, respectively, by ZIndo. As shown in (Figures 5 and 6), similar behavior and same spectral pattern were observed by introducing FL ring into the backbone of −N=N– unit. A nice bathochromic shift (Figure 6a) of π–π* transition band of trans-AzoFL (λmax 387.20 nm, εmax 6.0 × 104 M–1 cm–1) and cis-AzoFL (λmax 355.21 nm, εmax 3.0 × 104 M–1 cm–1) compared to that of FL (λmax 296.84 nm, εmax 1.85 × 104 M–1 cm–1) were observed. A comparison of π–π* transition band of cis- and trans-AzoFL with parent trans-DZ (λmax 140.80 nm (S0 → S3), εmax 2.1 × 104 M–1 cm–1) and cis-DZ (λmax 135.98 (S0 → S3), εmax 2.0 × 104 M–1 cm–1) also shows that cis- and trans-AzoFL are red-shifted by ZIndo method.
The cis- and trans-DFDZ also shows some extent of red shift compared to that of corresponding isomers of DZ.
The assignment of n−π* transition band of the above-mentioned cis-compounds is straightforward. The transition bands (n−π*) are at λmax 545.64 nm (εmax 950 M–1 cm–1), λmax 524.24 nm (εmax 400 M–1 cm–1), and λmax 233.92 nm (εmax 2 × 103 M–1 cm–1) for cis-AzoFL, cis- DZ, and cis-DFDZ respectively. The n−π*transition band of both the cis-AzoFL and cis-DFDZ is red-shifted compared to that of cis-DZ.
Though the ZIndo produces n−π* transition band in trans-AzoFL at λmax 562.02 nm (εmax ≈ 450, f = 0.0105, Gaussian UV–vis peak half-width at half height 0.233 eV) but the n−π* bands were not seen in trans-DFDZ and parent trans-DZ in both the DFT and ZIndo method.
Unlike the spectral pattern obtained from TD-DFT method, ZIndo produces well-separated π–π* (S0 → S3) and n−π* (S0 → S2) transition bands at λmax 169.51 nm (εmax 1.4 × 104, f = 0.3671) and λmax 233.92 nm (εmax 1.4 × 104, f = 0.0543), respectively, for cis-DFDZ (Figure 6c). A slight blue shift and small hypochromic effect for π–π* transition were observed for the parent DZ and DFDZ compared to that of respective trans-isomers by ZIndo method. In trans-DFDZ, the transition of (S0 → S3) at 163.06 nm (f = 0.0524) is underneath the π–π* transition band (S0 → S2) at 175.65 nm (f = 0.4028).
It is noteworthy that using DFT/6-31+G(d,p) as initial geometry in TD-DFT//B3LYP/6-31+G(d,p) calculation, there is no significant differences were observed between absorption spectra of cis and trans-DFDZ (Figure 5c). However, with different initial geometry, HF/6-31+G(d,p) was used in TD-DFT//B3LYP/6-31+G(d,p) calculation, and the π–π* transition band of trans-DFDZ was blue-shifted (∼17 nm) compared to cis-DFDZ (Figure S1a). In the case of ZIndo method, trans-DFDZ was red-shifted (∼6 nm) compared to cis-DFDZ (Figure 6c).
2.3. Frontier Molecular Orbitals
The highest occupied molecular orbital (HOMO) and the lowest-lying unoccupied molecular orbital (LUMO) are known as frontier molecular orbital (FMO). The molecular orbital is a mathematical function that describes the behavior of an electron or a pair of electrons within a molecule.67 These functions are plotted as surfaces around the molecular structure. The HOMO represents the ability to donate an electron, on the other hand LUMO as an electron acceptor. The energy gap between the HOMO and LUMO determines not only the chemical reactivity and kinetic stability, but also optical and electrical properties of a molecule.68
The energies of six important molecular orbitals and the 3D plots of the third HOMO [HOMO – 2], second highest [HOMO – 1], and the highest HOMO, the lowest unoccupied MO [LUMO], second lowest unoccupied MOs [LUMO + 1], and the third lowest unoccupied MOs [LUMO + 2] of the model compound AzoFL calculated using B3LYP/6-31+G(d,p) basis set at DFT level of theory are shown in Figures 7 and 8. The energy values of HOMO, LUMO, and energy gap between them, Eg (HOMO–LUMO), and dipole moments of the ground and excited states of the AzoFL, DFDZ; parent DZ and FL are listed in Table 7.
Figure 7.
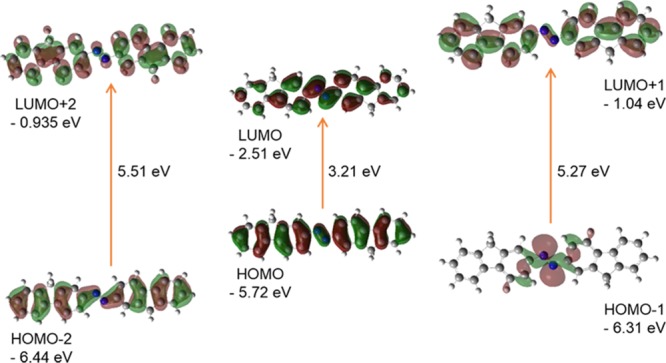
Diagram of FMO (isovalue: 0.02 [e bohr–3]1/2 of trans-AzoFL generated from TD/DFT calculation). Green and Maroon colors depict different phases.
Figure 8.
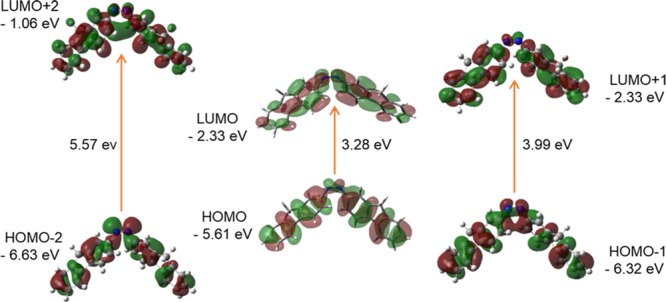
Diagram of FMO (isovalue: 0.02 [e bohr–3]1/2 of cis-AzoFL generated from TD/DFT calculation). Green and Maroon colors depict different phases.
Table 7. Energy Valuesa of HOMO, LUMO, and Energy Gap Between Them, Eg (HOMO–LUMO), Dipole Momentsb (μ) of the AzoFL, DFDZ; Parent DZ and FL.
| DFTc |
semiempiricald |
dipole
moment |
||||||
|---|---|---|---|---|---|---|---|---|
| compound | HOMO | LUMO | ΔEg | HOMO | LUMO | ΔEg | μgrounde,f | μexcitede,g |
| trans-AzoFL | –5.72 | –2.51 | 3.21 | –8.49 | –1.02 | 7.46 | 0.00 | 0.00 |
| 0.17 | 0.30 | |||||||
| cis-AzoFL | –5.61 | –2.33 | 3.28 | –8.70 | –0.81 | 7.89 | 3.12 | 3.12 |
| 2.99 | 3.24 | |||||||
| trans-DFDZ | –10.30 | –3.15 | 7.14 | –13.67 | –2.21 | 11.42 | 0.00 | 0.00 |
| 0.00 | 0.00 | |||||||
| cis-DFDZ | –10.77 | –2.93 | 7.84 | –13.85 | –2.02 | 11.83 | 0.22 | 0.22 |
| 0.66 | 0.57 | |||||||
| trans-DZ | –6.96 | –1.99 | 4.98 | –10.32 | 0.84 | 10.97 | 0.00 | 0.00 |
| 0.00 | 0.00 | |||||||
| cis-DZ | –7.07 | –2.06 | 5.01 | –0.86 | –10.56 | 11.42 | 3.20 | 3.20 |
| 2.70 | 3.99 | |||||||
| FL | –6.04 | –1.12 | 4.93 | –8.71 | –0.22 | 8.49 | 0.58 | 0.58 |
| 0.37 | 0.70 | |||||||
Energies are in electron volts (eV).
Dipole moments are in debye.
DFT calculation using B3LYP/6-31+G(d,p).
Semiempirical ZIndo.
Upper value: DFT.
Down value: AM1.
Down value: ZIndo.
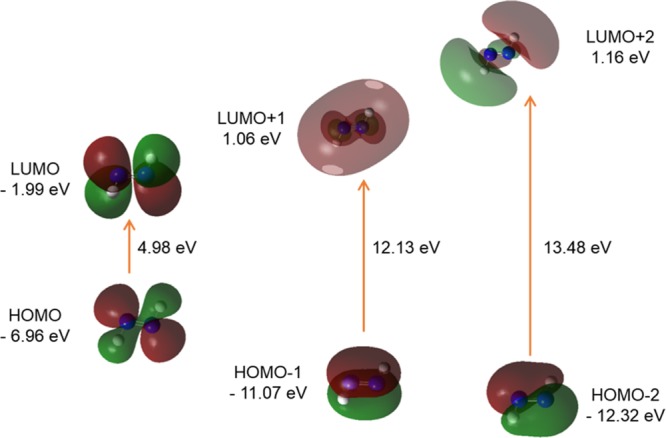
The model trans-AzoFL compound has a total of 610 alpha orbitals, out of which 94 are occupied and the remaining 516 are virtual orbitals. The orbital 94 represents HOMO, whereas orbital 95 represents LUMO orbitals. In our analyses, we found that the energy values of HOMO and LUMO are −5.72 and −2.51 eV, respectively, in trans-AzoFL (Figure 7, Table 7).
It is evident from Figures 7 and 8 that the HOMO and LUMO are localized on almost the whole molecule showing π- and π*-bonding MO, respectively. HOMO – 1 is localized on the N=N linkage, C2, C1, and C2′, C1′ atoms of the trans-AzoFL ring with almost no participation of the FL linker groups (Figure 7). The energy separation between the HOMO and the LUMO of trans-AzoFL is 3.21 eV, whereas the value is 3.28 eV for cis-AzoFL (Table 7). The HOMO (94a)–LUMO (95b) transition implies for π–π*(S0–S2) transition with 99% probability (Table 6).
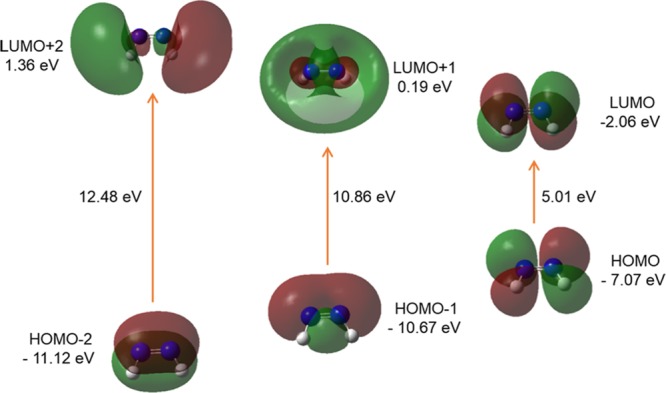
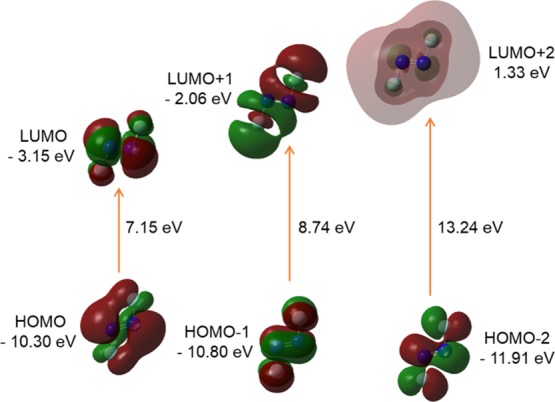
The 3D FMOs of FL, DZ, and DFDZ are shown in Figures 9–13, respectively. Both the trans-DZ and cis-DZ has a total of 48 alpha molecular orbitals, out of which 8 are occupied and the remaining 40 are virtual orbitals. The orbital 8 represents HOMO whereas 9 represents LUMO orbitals in DZ. In trans-DZ HOMO – 1 is π-bonding MO whereas in cis-DZ HOMO – 2 is π-bonding MO. LUMO is showing π*-antibonding MO. The LUMO + 2 in both the cis- and trans-diazene are showing similar behavior.
Figure 9.
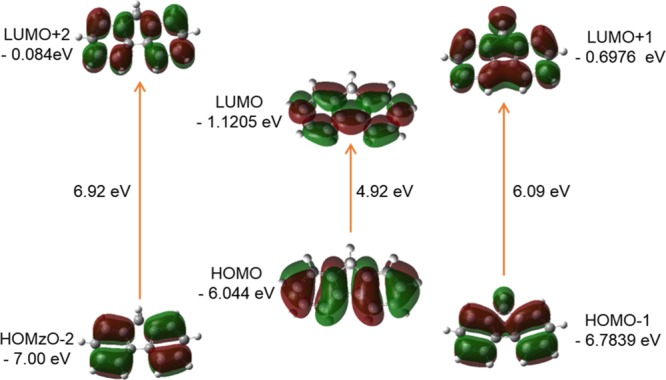
FMO orbitals (isovalue: 0.02 [e bohr–3]1/2 of FL generated from TD/DFT calculation). Green and Maroon colors depict different phases.
Figure 13.
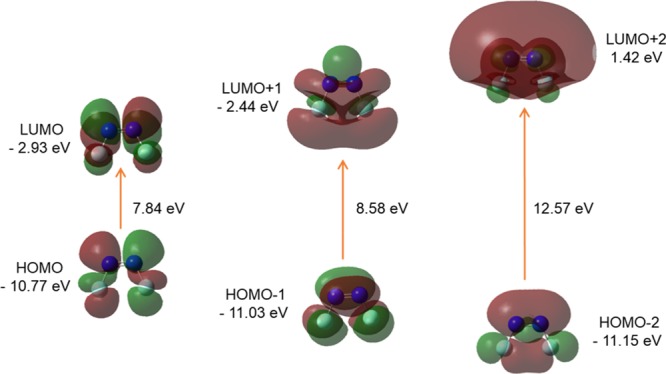
FMO orbitals (isovalue: 0.02 [e bohr–3]1/2 of cis-DFDZ generated from TD/DFT calculation). Green and Maroon colors depict different phases.
Figure 10.
FMO orbitals (isovalue: 0.02 [e bohr–3]1/2 of trans-DZ generated from TD/DFT calculation). Green and Maroon colors depict different phases.
Figure 11.
FMO orbitals (isovalue: 0.02 [e bohr–3]1/2 of cis-DZ generated from TD/DFT calculation). Green and Maroon colors depict different phases.
The orbitals 16 and 17 represent the HOMO and LUMO, respectively, in both the cis- and trans- DFDZ. The LUMO pattern of both the trans- and cis-DFDZ looks similar, whereas HOMO is different (Figures 12 and 13). The lone pairs on the nitrogen atoms are jot out in the plane of the molecule as seen in the HOMO of trans-DFDZ (Figure 12). The HOMO–LUMO energies and gap (Eg) between the HOMO–LUMO are given in the Table 7.
Figure 12.
FMO orbitals (isovalue: 0.02 [e bohr–3]1/2 of trans-DFDZ generated from TD/DFT calculation). Green and Maroon colors depict different phases.
From the HOMO and LUMO energies, global reactivity descriptor properties can be calculated.69−72 The ionization potential I and electron affinity A are equal to orbital energies of HOMO and LUMO as I = −EHOMO and A = −ELUMO. The ionization potential I and electron affinity A are found as 5.72 and 2.51 eV (Table 8), respectively, for trans-AzoFL. The electronegativity χ = (I + A)/2, chemical potential, μ = −χ, chemical hardness η = (I – A)/2, chemical softness, S = 1/η, electrophilicity index (ω) = μ2/2η, respectively, is calculated and tabulated in Table 8. The global reactivity descriptors of FL and trans- and cis-AzoFL, DZ, and DFDZ are summarized and given in the Table 8.
Table 8. Calculated Polarizabilitya (α) and Global Reactivity Descriptorsb by B3LYP/6-31+G(d,p) Basis Set at DFT Level of Theory.
| compound | α | I | A | χ | μ | η | S | ω |
|---|---|---|---|---|---|---|---|---|
| trans-AzoFL | 430.03 | 5.72 | 2.51 | 4.12 | –4.12 | 1.61 | 0.62 | 5.26 |
| cis-AzoFL | 365.23 | 5.61 | 2.33 | 3.97 | –3.97 | 1.64 | 0.61 | 4.80 |
| trans-DFDZ | 21.12 | 10.30 | 3.15 | 6.73 | –6.73 | 3.58 | 0.28 | 6.33 |
| cis-DFDZ | 20.69 | 10.77 | 2.93 | 6.85 | –6.85 | 3.92 | 0.26 | 5.98 |
| trans-DZ | 16.34 | 6.96 | 1.99 | 4.48 | –4.48 | 2.49 | 0.40 | 4.03 |
| cis-DZ | 16.72 | 7.07 | 2.06 | 4.57 | –4.57 | 2.51 | 0.40 | 4.16 |
| FL | 152.05 | 6.04 | 1.12 | 3.58 | –3.58 | 2.46 | 0.41 | 2.62 |
Polarizability, α in a.u.
I, ionization potential; A, electron affinity; χ, electronegativity; μ, chemical potential; η, chemical hardness; S, chemical softness and ω, electrophilicity index in eV.
2.4. Assignments of Vibrational Frequencies
Nowadays, description of theoretical vibrational spectra has attracted much attention not only for the identification of different compounds but also for spectrochemical investigation. There have been several theoretical reports on vibrational frequencies for trans-azobenzene in the ground state at the MP2, DFT, and CASSCF levels.11,73−77 As far as we are aware, there have been no previous reports on detailed descriptions of vibrational frequencies of azofluorene compounds. In an effort to gain a better understanding of the vibrational frequencies of both cis and trans-isomers of our studied azo compounds, we have calculated IR and Raman scattering activities at the level of DFT-B3LYP/6-31+G(d,p). As fluorene (FL) moiety and the −N=N– are the major structural unit of our target AzoFL, at first we have calculated and discussed theoretically predicted IR and Raman scattering activity spectra of the parent DZ, DFDZ, and FL for comparison even though there is experimental49 as well as some theoretical work44 present in the literature.
The IR and the Raman activity spectra calculated by B3LYP/6-31+G(d,p) basis set at DFT level of theory of the trans- and cis-DZ, DFDZ, respectively, are shown in Figures 14 and 15 and their vibrational assignments of the fundamental modes along with their calculated IR and Raman activity intensities, frequencies, and normal mode of vibrations along with the respective force constants are given in Tables 9 and 10. Generally, force constants help us to know the strength of the bond and molecular stability.
Figure 14.

Calculated (a) IR; (b) Raman spectra of trans-DZ; (c) IR; (d) Raman spectra of cis-DZ at B3LYP/6-31+G (d,p). The calculated harmonic frequencies are represented with a Gaussian IR peak half-width at half height 4 cm–1.
Figure 15.

Calculated (a) IR; (b) Raman spectra of trans-DFDZ; (c) IR (d) Raman spectra of cis-DFDZ at DFT-B3LYP/6-31+G(d,p). The calculated harmonic frequencies are represented with a Gaussian IR peak half-width at half height 4 cm–1.
Table 9. Calculated IR and Raman Activity Frequencies for trans- and cis-DZ by Present Different Methods.
| AM1 |
HFb |
DFTc |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| modea | freqd | IIRe | kf | freqd | IIRe | IRamang | kf | freqd | IIRe | IRamang | kf | |
| trans-DZ | oop HNN | 1237.39 | 65.74 | 0.9696 | 1466.02 | 109.99 | 0.00 | 1.3354 | 1344.41 | 95.85 | 0.00 | 1.1446 |
| ip HNN | 1275.21 | 67.35 | 1.0298 | 1452.15 | 111.62 | 0.00 | 1.3610 | 1348.65 | 74.67 | 0.00 | 1.1518 | |
| HNNH defm | 1620.30 | 0.00 | 1.9728 | 1733.40 | 0.00 | 14.65 | 2.1834 | 1596.94 | 0.00 | 11.23 | 2.0720 | |
| str N=N | 2162.06 | 0.00 | 34.4968 | 1896.02 | 0.00 | 26.30 | 27.2443 | 1659.03 | 0.00 | 19.23 | 9.8621 | |
| sym str NH | 3280.27 | 0.00 | 6.7102 | 3592.80 | 0.00 | 239.56 | 8.1736 | 3251.63 | 0.00 | 277.76 | 6.6818 | |
| asym. str NH | 3312.97 | 6.68 | 6.9504 | 3626.00 | 2.54 | 0.00 | 8.3259 | 3280.65 | 21.98 | 0.00 | 6.8154 | |
| cis-DZ | oop HNN | 1289.82 | 58.36 | 1.0062 | 1399.11 | 0.00 | 0.58 | 1.3724 | 1269.07 | 0.00 | 1.74 | 1.1291 |
| HNNH sci | 1282.31 | 0.00 | 1.2137 | 1489.89 | 0.01 | 12.91 | 1.3311 | 1354.10 | 1.64 | 22.19 | 1.1166 | |
| HNNH roc | 1494.39 | 4.10 | 1.8079 | 1687.61 | 79.89 | 1.49 | 2.2188 | 1538.87 | 42.00 | 1.95 | 1.8468 | |
| str N=N | 2169.63 | 19.80 | 27.9352 | 1892.56 | 5.84 | 24.58 | 25.4603 | 1662.43 | 6.60 | 9.25 | 16.9573 | |
| asym str. NH | 3225.57 | 13.76 | 6.5184 | 3486.24 | 26.86 | 139.74 | 7.6871 | 3088.26 | 79.74 | 207.53 | 6.0310 | |
| sym. str. NH | 3261.52 | 13.08 | 6.7251 | 3555.23 | 13.23 | 129.71 | 8.0195 | 3185.08 | 51.12 | 163.88 | 6.4231 | |
Approximate description of mode; defm, deformation; tor, torsion; str, stretching; sym, symmetric; asym, asymmetric; oop, out-of-plane; ip, in-plane; sci, scissoring; roc, rocking.
HF/6-31+G(d,p).
B3LYP/6-31+G(d,p).
Vibrational frequencies in cm–1.
Infrared intensities in km/mol.
k, force constants in mDyne/A.
Raman intensities in Å4/AMU.
Table 10. Calculated IR and Raman Scattering Activities for trans- and cis-DFDZ by Present Different Methods.
| AM1 |
HFb |
DFTc |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| modea | freqd | IIRe | kg | freqd | IIRe | IRamanf | kg | freqd | IIRe | IRamanf | kg | |
| trans-DFDZ | ip FNN | 338.64 | 9.70 | 1.0649 | 484.39 | 14.22 | 0.00 | 1.7041 | 418.81 | 11.95 | 0.00 | 1.2131 |
| oop FNN | 345.83 | 0.51 | 1.1107 | 428.37 | 4.77 | 0.00 | 2.1789 | 361.41 | 2.82 | 0.00 | 1.6292 | |
| defm FNNF | 597.41 | 0.00 | 3.7344 | 707.74 | 0.00 | 12.06 | 5.4027 | 604.31 | 0.00 | 13.59 | 3.9914 | |
| asym. str NF + ip N=N. | 1304.92 | 121.86 | 15.8130 | 1205.13 | 309.84 | 0.00 | 13.4871 | 996.45 | 269.08 | 0.00 | 9.2159 | |
| sym. str NF + FNN defm. | 1328.96 | 0.00 | 15.7538 | 1261.13 | 0.00 | 23.59 | 13.8330 | 1034.89 | 0.00 | 15.11 | 9.2118 | |
| str N=N | 1934.25 | 0.00 | 30.8745 | 1969.90 | 0.00 | 17.06 | 32.0281 | 1628.78 | 0.00 | 7.72 | 21.8882 | |
| cis-DFDZ | sci FNNF | 240.22 | 1.40 | 0.6241 | 409.44 | 2.02 | 1.56 | 1.8490 | 330.82 | 0.33 | 2.71 | 1.2097 |
| oop FNN | 636.35 | 0.00 | 3.4565 | 631.98 | 0.00 | 0.97 | 3.4454 | 556.26 | 0.00 | 0.45 | 2.6647 | |
| defm FNN | 815.80 | 8.17 | 5.9643 | 901.05 | 62.69 | 6.48 | 7.7988 | 946.61 | 83.58 | 0.79 | 5.3934 | |
| sym. str NF + ip N=N | 1144.24 | 34.95 | 12.5026 | 1138.62 | 98.64 | 2.71 | 12.1812 | 910.57 | 96.49 | 13.88 | 7.7810 | |
| asym str NF + ip N=N | 1281.70 | 101.15 | 14.8780 | 1155.58 | 139.24 | 16.58 | 11.5335 | 740.60 | 111.95 | 8.91 | 7.5733 | |
| str N=N | 1967.24 | 26.35 | 31.9714 | 1963.07 | 25.99 | 8.14 | 31.8142 | 1643.26 | 27.42 | 1.65 | 22.2798 | |
Approximate description of mode; defm, deformation; tor, torsion; str, stretching; sym, symmetric; asym, asymmetric; oop, out-of-plane; ip, in-plane; sci, scissoring.
HF/6-31+G(d,p).
B3LYP/6-31+G(d,p).
Vibrational frequencies in cm–1.
Infrared intensities in km/mol.
Raman intensities in Å4/AMU. .
k, force constants in mDyne/Å.
2.4.1. N–H Vibration in DZ
Among six vibrational modes in trans-N2H2 (DZ), three modes were found as IR inactive, viz., 1596.94 (Ag, ip NH), 1659.03 (Ag, str N=N), and 3251.64 (Ag, sym str NH) cm–1 (Figure 14a) but found as Raman scattering active (Figure 14b). The asymmetric N–H stretching, in-plane and out-of-plane N–H vibrations observed at 3280.65 (Bu), 1348.64 (Bu), and 1344.41 (Au) cm–1 were found as IR active mode, but Raman inactive mode.
In cis-DZ, among the six vibration modes, five modes are found as IR active, for example, 1354.10 (A1), 1538.87 (B2), 1662.43 (A1), 3088.26 (B2), and 3185.08 (A1) cm–1. The out-of-plane twist mode of NH at 1269.07 (A2) cm–1 is Raman active but appears as very weak peak. In cis-isomer two peaks are observed for NH stretching vibration at 3088 for asymmetric and at 3185 cm–1 for symmetric stretching vibration in both the IR and Raman activity spectra (Figure 14c,d). The six vibrational modes of trans- and cis-DZ by DFT-B3LYP/6-31+G(d,p) calculation are shown in Figures S3 and S4.
On the other hand, in trans-DZ the asymmetric stretching of NH at 3280 cm–1 is IR active, but NH symmetric stretching vibration at 3251.63 cm–1 is IR inactive. Reversed trend is observed in Raman activity spectrum for trans-DZ (Table 9).
Jensen et al.78 mentioned the different vibrational mode as 1526 (ω1 N–N), 3154 (ω2 N–H sym), 3197 (ω3 N–H asym), 1663 (ω4 N–N–H sym), 1374 (ω5 N–N–H asym), and 1351 (ω6 tor) cm–1 for trans-DZ by CASSCF. Craig and Levin79 mentioned the experimental values as 1529 (N–N), 3128 (N–H sym), 3120 (N–H asym), 1582 (N–N–H sym), 1322 (N–N–H asym), and 1286 (tor) cm–1. On the other hand, Hwang and Mebel80 found the values at much higher frequencies at 1525 (N–N), 3382 (N–H sym), 3353 (N–H asym), 1628 (N–N–H sym), 1360 (N–N–H asym), and 1349 (tor) cm–1 by high-level G2M(MP2)//MP2/G-31G* calculation.
For cis-DZ, the vibrational modes are found by Jensen et al.78 at 1535 (ω1 N–N), 3144 (ω2 N–H sym), 3074 (ω3 N–H asym), 1416 (ω4 N–N–H sym), 1616 (ω5 N–N–H asym), and 1267 (ω6 tor) cm–1 by CASSCF.
The experimental values at 1558 (ω1 N–N), 2966 (ω2 N–H sym), 2884 (ω3 N–H asym), 1390 (ω4 N–N–H sym), 1439 (ω5 N–N–H asym), and 1259 (ω6 tor) cm–1 by Craig and Levin79 estimated from the approximate force field of trans-DZ. On the other hand Hwang and Mebel80 found the values at much higher frequencies at 1562 (ω1 N–N), 3306 (ω2 N–H sym), 3225 (ω3 N–H asym), 1373 (ω4 N–N–H sym), 1567 (ω5 N–N–H asym), and 1287 (ω6 tor) cm–1 by high level G2M(MP2)//MP2/G-31G* calculation. Biczysko et al.81 mentioned additional comparison for different parameters of both the trans-DZ and cis-DZ by different authors.
2.4.2. N–F Vibration in DFDZ
The different vibrational modes of trans-DFDZ at 361.43 (AU), 418.81 (BU), 604.31 (AG), 996.45 (BU), 1034.89 (AG), and 1628.78 (AG) cm–1 of our present calculation is very close to the experimental work82 viz. 364 (AU), 423 (BU), 603 (AG), 991 (BU), 1018 (AG), and 1523 (AG) cm–1. The six vibrational modes of trans- and cis-DFDZ by present DFT-B3LYP/6-31+G(d,p) calculation are shown in Figures S5 and S6.
Among six vibrational modes in trans-N2F2 (DFDZ), three modes were found as IR active (Figure 15a) by our B3LYP/6-31+G(d,p) calculation. The out-of-plane FNN, in-plane FNN, and asymmetric N–F stretching vibrations observed at 361.43, 418.86, and 996.20 cm–1 were found as IR active mode, but Raman inactive. On the other hand the IR inactive modes at 604.30, 1034.48, and 1628.71 cm–1 for FNNF torsion, symmetric stretching of NF and stretching vibration of N=N were found as Raman active mode in trans-DFDZ (Figure 15b).
The different vibrational modes of cis-DFDZ at 330.82 (A1), 556.26 (A2), 740.60 (B2), 910.57 (A1), 946.61 (B2), and 1643.26 (A1) cm–1 are also close to the experimental work,82 for example, 332 (A1), 546 (A2), 731 (B2), 897 (A1), 957 (B2), and 1492 (A1) cm–1. In cis-DFDZ, all of the vibrations were found as IR active except out-of-plane of FNN at 556.26 cm–1, which is Raman active however appears as very weak peak (Figure 15c).
The other five peaks at 1643.27 (str N=N), 946.61 (oop N=N), 910.57(sym str NF), 740.60 (asym str NF), and 330.82 (oop FNN) cm–1, respectively, are both the IR and Raman active (Figure 15d).
The resulting vibrational frequencies for the optimized geometries and predicted vibrational assignments of the fundamental modes of both the trans- and cis-AzoFL along with the theoretically calculated harmonic vibrational frequencies, IR intensities, Raman scattering activities, and normal mode of vibrations are given in Tables 11 and 12, respectively, using B3LYP/6-31+G(d,p) basis set at DFT level of theory. Some of the vibrational modes of both the trans- and cis-AzoFL are shown in Figures S7 and S8. In aromatic cyclic compounds, almost all of the modes are delocalized over the whole molecule;83 hence, assignments of several vibrational modes are very difficult. However, the assignment of the calculated frequencies is aided by the animation option of Gauss View 6 graphical interface for Gaussian program, which gives a visual presentation of the shape of the vibrational modes.
Table 11. Calculated IR and Raman Activity Frequencies of trans-AzoFL with B3LYP/6-31+G(d,p) in the Ground State.
| mode no. | syma | freqb | IIRc | IRamand | ke | approximate description of modef |
|---|---|---|---|---|---|---|
| 1 | A | 15.40 | 0.1306 | 0.0000 | 0.0006 | twist (FL1 wrt FL2) |
| 2 | A | 20.19 | 0.1109 | 0.0000 | 0.0013 | wag (FL1 wrt FL2) + wag (N=N) |
| 3 | B | 35.05 | 0.3784 | 0.0000 | 0.0044 | tor FL ring |
| 4 | B | 47.18 | 0.0000 | 2.9250 | 0.1080 | defm FL ring + oop (CH) |
| 5 | A | 101.53 | 0.0041 | 0.0000 | 0.3050 | defm FL ring + oop (CH) + oop (N=N) |
| 6 | A | 122.77 | 0.0000 | 9.8997 | 0.0539 | tor FL ring |
| 7 | B | 125.22 | 0.0000 | 2.0646 | 0.0568 | defm ring + oop (CH) + oop (N=N) |
| 8 | B | 134.25 | 0.0000 | 0.0527 | 0.0408 | defm ring + oop (CH) |
| 9 | A | 137.57 | 0.3727 | 0.0000 | 0.0422 | defm ring + oop (CH) |
| 10 | A | 165.20 | 0.0000 | 2.8101 | 0.1067 | tor ring |
| 11 | B | 206.31 | 7.5155 | 0.0000 | 0.1305 | tor ring (A, C) + (A′, C′) |
| 12 | B | 240.13 | 0.0000 | 1.5216 | 0.1119 | ring defm + twist (N=N) + oop (CH) |
| 13 | A | 243.89 | 16.1469 | 0.0000 | 0.0805 | defm ring + oop (C9H) |
| 14 | B | 250.70 | 0.0000 | 1.1733 | 0.1230 | defm ring + twist (N=N) + oop (CH) |
| 15 | A | 256.87 | 0.0646 | 0.0001 | 0.1974 | defm ring + oop (N=N) + oop (CH) |
| 16 | A | 287.18 | 0.0000 | 11.3418 | 0.2415 | sci ring (A, C) + (A′, C′) + tor (CNNC) |
| 17 | B | 346.18 | 0.0000 | 8.1400 | 0.3737 | twist ring + twist (N=N) |
| 18 | B | 378.25 | 2.3707 | 0.0000 | 0.7009 | ip (ring + N=N) |
| 19 | A | 384.53 | 1.5695 | 0.0000 | 0.4152 | wag (ring A, C) + wag (N=N) + oop (CH) |
| 20 | B | 430.51 | 0.0000 | 0.3769 | 0.3059 | twist (FL1, FL2) |
| 21 | A | 433.15 | 11.5592 | 0.0000 | 0.3052 | wag (FL1, FL 2) |
| 22 | A | 445.17 | 0.0997 | 0.0000 | 0.3415 | ring defm + rot (C9Hs) |
| 23 | B | 448.40 | 0.0000 | 2.1609 | 0.3527 | twist ring |
| 24 | B | 466.63 | 26.6329 | 0.0000 | 0.6957 | tor ring |
| 25 | A | 480.19 | 0.0000 | 7.5415 | 0.8080 | defm angle |
| 26 | B | 506.58 | 0.0000 | 0.7934 | 0.4932 | twist FL1 + twist FL2 + twist (N=N) |
| 27 | A | 513.21 | 0.0000 | 5.0313 | 0.6578 | sci FL1 + sci FL2 + ip (N=N) |
| 28 | A | 524.41 | 0.0087 | 0.0000 | 0.5675 | twist FL1 + twist FL2+ wag (N=N) |
| 29 | B | 547.75 | 1.2942 | 0.0000 | 0.9779 | tors ring + ip (N=N) |
| 30 | A | 557.17 | 0.0000 | 31.4107 | 1.0816 | tor ring |
| 31 | B | 571.35 | 9.1429 | 0.0000 | 1.0384 | sci (FL1 wrt FL2) + ip (N=N) |
| 32 | B | 582.70 | 0.0000 | 0.3227 | 0.6431 | twist (FL1 wrt FL2) |
| 33 | B | 595.31 | 0.0467 | 0.0000 | 1.4851 | CCC defm + ip (CNNC) |
| 34 | A | 623.01 | 4.8431 | 0.0001 | 0.7032 | wag (ring A + ring A′) + twist (ring C, C′) |
| 35 | A | 648.30 | 0.0000 | 91.0341 | 1.6913 | defm CCC + defm CCN |
| 36 | B | 660.49 | 26.9341 | 0.0000 | 1.6296 | defm CCC + sci (ring A, A′) + ip (N=N) |
| 37 | A | 675.62 | 0.0000 | 22.4051 | 1.6754 | defm CCC + defm CCN |
| 38 | B | 708.48 | 0.0000 | 2.3301 | 1.0291 | twist (FL1 wrt FL2) |
| 39 | A | 716.97 | 0.1941 | 0.0000 | 0.9115 | wag (ring A, ring A′) + wag (ring C, C′) + twist (ring A, C) + twist (ring A, C′) |
| 40 | B | 733.72 | 14.7894 | 0.0000 | 1.7404 | defm CCC + ip (CNN) |
| 41 | B | 743.38 | 0.0000 | 0.1909 | 0.5322 | wag (ring CH of ring C, C′) + twist (ring C, ring C′) |
| 42 | A | 747.36 | 110.7069 | 0.0000 | 0.5334 | wag (CH) |
| 43 | A | 758.63 | 0.0000 | 271.7288 | 1.8586 | breathing (FL1 + FL2) |
| 44 | B | 773.10 | 0.9828 | 0.0000 | 1.9565 | defm CCC |
| 45 | B | 781.15 | 0.0000 | 18.5025 | 0.7666 | twist (FL1 wrt FL2) |
| 46 | A | 784.35 | 50.9480 | 0.0000 | 0.8703 | wag (FL1 wrt FL2) + rot (C9H) |
| 47 | A | 830.40 | 0.0000 | 150.7548 | 1.6810 | defm CCC + ip (CNN) |
| 48 | B | 837.65 | 1.1534 | 0.0000 | 1.9166 | Defm (CCC) |
| 49 | B | 850.84 | 0.0000 | 0.7253 | 0.6349 | twist (ring A, ring A) + wag (CH of ring A, ring A′) |
| 50 | A | 854.15 | 30.9248 | 0.0007 | 0.6399 | wag (CH) + wag (ring A, A′) |
| 51 | A | 864.12 | 0.0000 | 211.1922 | 2.7803 | defm [(CNN) + (CCC)] |
| 52 | B | 876.04 | 0.0000 | 0.9303 | 0.6194 | wag (CH of ring C, C′) |
| 53 | A | 876.38 | 0.4976 | 0.0000 | 0.6233 | twist (CH of ring C, C′) |
| 54 | B | 901.99 | 0.0000 | 0.2642 | 0.6911 | twist (CH ring A, CH ring A′) |
| 55 | A | 905.86 | 18.6493 | 0.0000 | 0.7332 | Wag (CH ring A, A′) |
| 56 | B | 945.51 | 0.0000 | 0.9680 | 0.7815 | twist CH ring C + twist CH ring C′+ twist (FL1, FL2) |
| 57 | A | 946.12 | 2.4263 | 0.0000 | 0.7810 | CH Ring |
| 58 | B | 957.95 | 4.9996 | 0.0000 | 1.6768 | roc (CH ring A, CH ring A′) |
| 59 | A | 962.52 | 0.0000 | 25.2250 | 1.9971 | str (CC) + defm (CNN, ring A, ring A′) |
| 60 | B | 972.04 | 0.0000 | 0.6574 | 0.9856 | twist (ring A, C) + wag (FL1 + FL2) |
| 61 | A | 972.60 | 11.8089 | 0.0000 | 1.0041 | twist (ring A, C) + twist (A′, C′) + twist (FL1, FL2) |
| 62 | B | 984.72 | 0.0000 | 1.7196 | 0.7933 | twist (ring A + A′) |
| 63 | A | 985.26 | 0.2813 | 0.0001 | 0.7891 | twist A + twist A′ |
| 64 | B | 992.07 | 0.0000 | 2.3310 | 0.7479 | twist (ring C + ring C) + twist (FL1, FL2) |
| 65 | A | 992.08 | 0.1066 | 0.0000 | 0.7479 | twist (ring C) + twist (ring C′) |
| 66 | A | 1020.42 | 0.0000 | 279.0661 | 4.3569 | ip (CCC) + ip (CC) |
| 67 | B | 1020.57 | 17.0765 | 0.0000 | 4.3326 | Ip (CCC) |
| 68 | A | 1049.26 | 0.0000 | 661.2481 | 1.3802 | ip (CHs) + ip (CCC) |
| 69 | B | 1049.38 | 5.8000 | 0.0000 | 1.3780 | Ip (CCC) |
| 70 | A | 1107.31 | 0.0000 | 10612.1266 | 1.5274 | ip (CH) + ip (CCC) + sym str C–N |
| 71 | B | 1115.72 | 23.0272 | 0.0000 | 1.4555 | ip (CHs) |
| 72 | A | 1128.81 | 0.0000 | 1479.4138 | 1.3370 | ip (CHs) |
| 73 | B | 1130.87 | 8.1503 | 0.0000 | 1.2852 | sci (CHs) |
| 74 | B | 1156.71 | 0.0000 | 9.7794 | 0.8964 | ip (C9Hs + C9′Hs) |
| 75 | A | 1156.84 | 0.0250 | 0.0007 | 0.8968 | ip (C9Hs + C9′Hs) |
| 76 | A | 1159.19 | 0.0000 | 435.9443 | 1.0448 | sci (CHs) + roc (CHs) |
| 77 | B | 1165.34 | 1.7000 | 0.0000 | 1.0925 | sci (CHs) + roc (CHs) |
| 78 | A | 1181.07 | 0.0000 | 142.5714 | 0.9725 | sci (CHs) + asym sci (FL1, FL2 CHs) |
| 79 | B | 1181.39 | 7.2494 | 0.0000 | 0.9680 | asym sci (CHs FL1, FL2) |
| 80 | A | 1200.55 | 0.0000 | 104.9316 | 1.6617 | ip (CCC) + sci (CHs C9Hs + C9′Hs) |
| 81 | B | 1202.04 | 3.5008 | 0.0000 | 1.7514 | sci (CHs C9Hs + C9′Hs) + ip (CCC) |
| 82 | A | 1209.63 | 0.0000 | 12583.3187 | 3.1893 | ip (CCC) + sci CHs + roc CHs + sym str (C–N) |
| 83 | B | 1222.40 | 56.0718 | 0.0000 | 1.8815 | ip CHs |
| 84 | A | 1225.19 | 0.0000 | 745.0504 | 1.5251 | ip CHs |
| 85 | B | 1231.20 | 54.5108 | 0.0000 | 1.7872 | sci CHs + roc CHs + breathing (FL1, FL2) + asym str C–N |
| 86 | A | 1264.60 | 0.0000 | 12036.5886 | 2.5479 | sci CHs + breathing (FL1, FL2) + sym str C–N |
| 87 | B | 1289.01 | 75.3458 | 0.0000 | 3.1057 | roc (CH) + breathing (A, A′ ring) + asym str (C–N). |
| 88 | B | 1308.45 | 1.8658 | 0.0000 | 1.8966 | roc CHs |
| 89 | A | 1313.87 | 0.0000 | 7.1549 | 1.8008 | roc CHs |
| 90 | A | 1330.98 | 0.0000 | 4202.1486 | 1.8113 | roc CHs |
| 91 | B | 1332.51 | 4.3273 | 0.0000 | 1.9298 | roc CHs |
| 92 | B | 1356.62 | 20.1559 | 0.0000 | 7.4855 | roc CHs |
| 93 | A | 1360.72 | 0.0000 | 1265.4991 | 7.6120 | str Ar (C=C) + ip CC |
| 94 | B | 1381.32 | 28.0791 | 0.0000 | 4.9607 | breathing B, B′ ring, roc CHs + sci CHs |
| 95 | A | 1383.50 | 0.0000 | 6199.7816 | 5.3489 | ip CCC |
| 96 | A | 1452.84 | 0.0000 | 1222.2265 | 1.4355 | sci C9Hs + asym CHs (FL1, FL2) |
| 97 | B | 1453.11 | 15.7754 | 0.0000 | 1.4073 | sci (C9Hs + C9′Hs) |
| 98 | A | 1463.43 | 0.0000 | 328.5309 | 4.9904 | sci CHs + ip CC |
| 99 | B | 1465.09 | 23.0809 | 0.0000 | 5.0421 | sci CHs + str Ar (C=C) |
| 100 | A | 1484.09 | 0.0000 | 11663.8821 | 3.1207 | sci (CHs FL1 wrt CHs FL2) + roc CHs |
| 101 | B | 1490.76 | 28.1557 | 0.0000 | 3.1617 | roc all CHs |
| 102 | A | 1497.01 | 0.0000 | 13011.3686 | 4.1202 | roc CHs + str N=N + sci CHs |
| 103 | B | 1502.90 | 4.2781 | 0.0000 | 3.7202 | str Ar (C=C) + roc CHs |
| 104 | A | 1513.48 | 0.0000 | 521.0812 | 4.3967 | str Ar (C=C) + str N=N + roc CHs |
| 105 | B | 1520.07 | 20.6099 | 0.0000 | 4.3109 | str Ar (C=C) + sci C9Hs + roc CHs |
| 106 | A | 1529.83 | 0.0000 | 28230.3369 | 7.2216 | str Ar (C=C) + str N=N + roc CHs |
| 107 | B | 1604.96 | 17.9432 | 0.0000 | 10.0612 | str Ar (C=C) + roc CHs |
| 108 | A | 1613.87 | 0.0000 | 141.7826 | 11.2227 | str Ar (C=C) + str N=N + roc CHs |
| 109 | B | 1625.55 | 3.3940 | 0.0000 | 9.5645 | str Ar (C=C) |
| 110 | A | 1625.72 | 0.0000 | 210.1051 | 9.7473 | str Ar (C=C) |
| 111 | B | 1652.38 | 32.7979 | 0.0000 | 10.3799 | str Ar (C=C) |
| 112 | A | 1653.34 | 0.0000 | 9012.2045 | 10.4304 | str Ar (C=C) + str (N=N) |
| 113 | B | 1655.07 | 77.6056 | 0.0000 | 11.0443 | str Ar (C=C) |
| 114 | A | 1658.61 | 0.0000 | 7430.5054 | 11.7092 | str Ar (C=C) + str (N=N) |
| 115 | B | 3033.36 | 25.8446 | 0.0001 | 5.7475 | sym str (C9Hs + C9′Hs) + asym str (C9Hs wrt C9′Hs) |
| 116 | A | 3033.37 | 0.0000 | 387.4819 | 5.7475 | sym str (C9Hs + C9′Hs) + sym str (C9Hs wrt C9′Hs) |
| 117 | B | 3062.81 | 0.0000 | 176.3528 | 6.0918 | asym str (C9Hs + C9′Hs) + sym str (C9Hs wrt C9′Hs) |
| 118 | A | 3062.82 | 12.3280 | 0.0004 | 6.0918 | asym str (C9H + C9′H) + asym str (C9Hs wrt C9′Hs) |
| 119 | B | 3174.85 | 15.1999 | 0.0000 | 6.4524 | asym str CHs |
| 120 | A | 3174.87 | 0.0000 | 84.2479 | 6.4525 | (sym + asym) str CHs |
| 121 | B | 3181.31 | 13.2558 | 0.0000 | 6.4913 | asym str (CH) |
| 122 | A | 3181.32 | 0.0000 | 335.5393 | 6.4913 | asym str CHs |
| 123 | B | 3184.62 | 17.9921 | 0.0000 | 6.5110 | asym str CHs |
| 124 | A | 3184.72 | 0.0000 | 126.8227 | 6.5114 | asym str (C4H, C4′H) |
| 125 | A | 3192.56 | 0.0000 | 370.0879 | 6.5627 | sym str (FL1, FL2 CHs) |
| 126 | B | 3192.56 | 52.0569 | 0.0000 | 6.5627 | asym str (FL1, FL2 CHs) |
| 127 | B | 3194.96 | 5.4030 | 0.0000 | 6.5625 | asym str (C1Hs, C1H) |
| 128 | A | 3195.04 | 0.0000 | 138.1649 | 6.5632 | sym str (C1H, C1H) |
| 129 | B | 3204.19 | 82.7763 | 0.0000 | 6.6368 | asym str (CH ring C, CH ring C′) |
| 130 | A | 3204.26 | 0.0000 | 925.6424 | 6.6370 | sym str (CH FL1 + CH FL2) |
| 131 | A | 3226.84 | 0.0000 | 79.2692 | 6.6985 | sym str (C3H, C3′H) |
| 132 | B | 3226.98 | 4.7576 | 0.0000 | 6.6996 | asym str (C3H, C3′H) + sym str (C4H, C4′H) + asym str (C3H, C3′H) |
sym, symmetry.
Vibrational frequencies in cm–1.
Infrared intensities in km/mol.
Raman scattering activities A4/AMU.
k, force constants in mDyne/A.
defm, deformation; tor, torsion; str, stretching; sym, symmetric; asym, asymmetric; oop, out-of-plane bending; ip, in-plane bending; sci, scissoring; roc. rocking; wrt, with respect to.
Table 12. Calculated IR and Raman Activity Frequencies of cis-AzoFL with B3LYP/6-31+G(d,p) in the Ground State.
| mode no. | syma | freqb | IIRc | IRamand | ke | approximate description of modef |
|---|---|---|---|---|---|---|
| 1 | A | 16.10 | 0.0354 | 21.1321 | 0.0010 | sci (FL1 wrt FL2) + wag (N=N) |
| 2 | B | 23.20 | 0.9214 | 2.5838 | 0.0013 | twist (FL 1 wrt FL2) |
| 3 | A | 33.03 | 0.0118 | 15.8790 | 0.0026 | twist (FL 1 wrt FL2) |
| 4 | B | 57.74 | 1.1551 | 1.0155 | 0.0115 | defm FL ring + oop (CH) |
| 5 | A | 81.21 | 0.0049 | 5.9617 | 0.0219 | twist FL1 + twist FL2 + wag (N=N) |
| 6 | A | 114.28 | 0.3861 | 56.0888 | 0.0453 | roc FL ring + oop defm + twist (N=N) |
| 7 | B | 128.35 | 0.2046 | 4.2641 | 0.0424 | twist ring + oop (CHs) |
| 8 | A | 149.78 | 0.4114 | 12.4845 | 0.0496 | twist ring + oop (CHs) |
| 9 | B | 150.11 | 2.7730 | 0.1719 | 0.0652 | twist ring + oop (CHs) |
| 10 | B | 196.72 | 13.3724 | 0.6190 | 0.1326 | wag (ring A, C) + wag (N=N) + wag (ring A′, C′) |
| 11 | A | 204.52 | 0.1416 | 1.1113 | 0.1282 | tor ring (A, C) + (A′,C′) |
| 12 | A | 236.47 | 0.0025 | 28.4148 | 0.1555 | sci (A, C) + wag (CNNC) + sci (A′, C′) |
| 13 | B | 243.51 | 5.7391 | 0.9453 | 0.0752 | defm ring + oop (C9H) + oop (CHs) |
| 14 | A | 247.94 | 6.4319 | 26.3393 | 0.0851 | defm ring + oop (CHs) |
| 15 | B | 285.45 | 3.3517 | 10.0966 | 0.2542 | defm ring + defm (CNNC) |
| 16 | A | 310.85 | 0.2273 | 251.3607 | 0.3552 | defm ring + defm (CNNC) |
| 17 | B | 322.27 | 13.0652 | 0.7972 | 0.3900 | twist ring + ip (CNNC) |
| 18 | B | 366.62 | 0.6868 | 0.9933 | 0.5795 | ip (ring + CNNC) |
| 19 | A | 400.35 | 1.3971 | 202.2271 | 0.4316 | wag (ring A, C)+ wag (N=N) + oop (CHs) |
| 20 | B | 425.73 | 13.7947 | 4.7201 | 0.3012 | wag (FL 1 + FL2) |
| 21 | A | 438.84 | 1.0108 | 0.2072 | 0.3462 | wag (A, C) + wag (FL1, FL2) |
| 22 | B | 440.49 | 1.2049 | 0.8478 | 0.3341 | defm ring + rot (C9H) |
| 23 | A | 441.40 | 0.4384 | 6.0995 | 0.3370 | twist ring + ip C9H |
| 24 | A | 481.22 | 0.0118 | 190.0508 | 0.6275 | tor ring + twist (N=N) + defm C9H |
| 25 | B | 493.78 | 0.9959 | 4.4919 | 0.5385 | defm CCC + oop (CNNC) |
| 26 | B | 507.46 | 5.8230 | 0.0114 | 0.5664 | Oop (CCC) |
| 27 | A | 514.04 | 1.2477 | 6.5145 | 0.6680 | twist FL1 + twist FL2 + oop (N=N) + defm C9Hs |
| 28 | A | 535.70 | 0.8259 | 158.0208 | 0.7761 | twist FL1 + twist FL2 + oop (N=N) |
| 29 | B | 537.59 | 0.3535 | 1.7202 | 1.0500 | ring tors + ip (N=N) |
| 30 | A | 560.82 | 0.3996 | 31.3759 | 1.0440 | ring tor + oop (CCC) + ip (CCC) |
| 31 | B | 570.01 | 0.3014 | 2.2451 | 1.0294 | defm (CCC) + ip (CNNC) + ip (C9Hs) |
| 32 | B | 585.51 | 1.5405 | 13.0525 | 0.7889 | twist (FL1 wrt FL2) + defm (CNNC) |
| 33 | A | 597.31 | 5.4282 | 57.6224 | 1.1750 | CCC defm + oop (CNNC) |
| 34 | A | 634.99 | 5.3238 | 285.0074 | 0.9178 | wag (CHs ring A + CHs ring A′) + twist (ring C + C′) |
| 35 | B | 648.25 | 7.4542 | 0.0203 | 1.6524 | defm CCC + defm CNN |
| 36 | A | 664.35 | 0.2260 | 69.2317 | 1.5363 | defm CCC + sci (ring A, A′) + twist (N=N) |
| 37 | B | 699.46 | 4.7357 | 10.6299 | 1.2211 | defm CCC + defm CNN + defm (H–C9–H) |
| 38 | A | 703.54 | 0.0008 | 3.5729 | 1.7322 | defm CCC + wag (CNNC) |
| 39 | B | 711.51 | 2.9284 | 0.2414 | 1.3482 | ip (CNNC) + mixing of ip + oop CHs |
| 40 | A | 717.21 | 0.0938 | 52.0916 | 0.9457 | tor CNNC + twist (CHs ring A, CHs ring C) + twist (CHs ring A′, CHs ring C′) |
| 41 | B | 740.14 | 32.3067 | 4.5337 | 0.5761 | wag (CHs of ring C+ CHs of ring C′) + twist (ring C wrt C′) + twist (C9Hs) |
| 42 | A | 746.57 | 33.9975 | 48.0180 | 0.5371 | wag (CHs of ring C, CHs of ring C′) + twist (C9Hs) + twist (CHs of ring A, ring A′) |
| 43 | B | 759.94 | 14.0169 | 33.7320 | 1.3485 | breathing (FL1 + FL2) |
| 44 | A | 768.83 | 0.5835 | 104.4676 | 1.9254 | defm CCC |
| 45 | B | 775.17 | 97.4902 | 0.6544 | 0.7040 | wag (CHs of A, CHs of C) + wag (CHs of A′, CHs of C′) + twist (FL 1 wrt FL2) |
| 46 | A | 782.63 | 12.2314 | 19.2813 | 0.8340 | wag (FL1 wrt FL2) + ip (C9Hs) |
| 47 | B | 817.47 | 19.7106 | 55.4728 | 0.9403 | twist (CHs of A, CHs of C) + twist (CHs of A′, CHs of C′) + oop (CNN) |
| 48 | A | 834.03 | 0.0093 | 64.5449 | 1.7862 | defm (CCC) + ip C9Hs |
| 49 | B | 836.07 | 0.4532 | 16.7616 | 1.8038 | defm CCC (FL1 + FL2) + ip (HC9H) |
| 50 | A | 844.32 | 3.5224 | 147.8764 | 0.6390 | wag (C3H, C4H) + wag (C3′H, C4′H) |
| 51 | B | 858.96 | 37.5160 | 69.5819 | 0.8108 | twist (C1H, C3H), twist (C1′H, C3′H), wag (C3H, C4H) + wag (C3′H, C4′H) + defm (CNN) |
| 52 | A | 876.08 | 0.0805 | 1.1247 | 0.6205 | oop (CH of ring C + CH of ring C′) |
| 53 | B | 876.82 | 2.0257 | 4.6846 | 0.6344 | oop (CH of ring C) + oop (CH of ring C′) |
| 54 | A | 896.50 | 3.9054 | 44.8419 | 0.7112 | wag (C1H wrt C1′H) |
| 55 | B | 905.64 | 28.6929 | 11.4053 | 0.8903 | twist (C1H, C1′H), defm (CNNC) |
| 56 | A | 915.04 | 0.2439 | 159.0103 | 1.9629 | defm CCC + sci (C1H, C9H) + sci (C1′H, C9′H) + wag (N=N) |
| 57 | B | 935.55 | 1.2191 | 60.3666 | 1.9948 | ip (C9H + C9′H) + ip (CCC + CCN + CNN) |
| 58 | A | 943.50 | 0.4601 | 0.6865 | 0.7927 | twist (CHs FL1 + CHs FL2) |
| 59 | B | 944.14 | 3.5314 | 0.1416 | 0.7962 | twist (CHs FL1) + twist (CHs FL2) |
| 60 | B | 963.21 | 10.4801 | 2.5244 | 0.7617 | twist (C3H, C4H) + twist (C3′H,C4′H) |
| 61 | A | 963.32 | 0.2611 | 12.4265 | 0.7626 | twist (C3H, C4H) + twist (C3′H, C4′H) |
| 62 | B | 974.85 | 3.0234 | 1.2928 | 1.0137 | twist (CHs FL1 + CHs FL2) |
| 63 | A | 974.89 | 2.5418 | 8.6102 | 1.0040 | twist (CHs FL1 + twist CHs) |
| 64 | B | 991.63 | 0.1159 | 0.3457 | 0.7476 | twist (CHs ring C + CHs ring C′) |
| 65 | A | 991.65 | 0.0290 | 1.2768 | 0.7476 | twist (CHs ring A) + twist (CHs ring A′) |
| 66 | A | 1020.15 | 0.8340 | 28.3434 | 4.2962 | ip (CCC) + ip (CC) |
| 67 | B | 1020.18 | 8.3474 | 0.2482 | 4.3072 | ip (CCC) + ip (CC) ip CH) |
| 68 | A | 1049.36 | 5.1414 | 207.8673 | 1.3839 | ip (CHs) + ip (CCCC) |
| 69 | B | 1049.46 | 2.5014 | 26.8516 | 1.3826 | ip (CCCC) + CHs ip |
| 70 | A | 1103.13 | 0.0056 | 1454.4185 | 1.4552 | sci (C1H, C3H) + sci (C1′H, C3′H) + ip (CCC) |
| 71 | B | 1113.52 | 4.1562 | 176.5421 | 1.5513 | sci (C1H, C3H) + sci (C1′H, C3′H) |
| 72 | A | 1126.01 | 0.9559 | 45.0660 | 1.3721 | ip (CHs FL1 + CHs FL2) |
| 73 | B | 1128.52 | 2.6721 | 22.3835 | 1.3585 | ip (CHs) |
| 74 | B | 1155.73 | 4.1544 | 0.8568 | 1.1050 | ip (C9Hs + C9′Hs) |
| 75 | A | 1156.95 | 0.2232 | 7.4892 | 0.9379 | ip (C9Hs + C9′Hs) + sci (C3H, C4H) + sci (C3′H, C4′H) |
| 76 | B | 1158.02 | 0.8281 | 1.5203 | 0.9553 | sci (CHs) + roc (CHs) |
| 77 | A | 1159.84 | 0.7999 | 2.2940 | 1.1079 | sci (C3H, C4H) + sci (C3H, C4H) + rot (C9Hs, C9′Hs) |
| 78 | A | 1180.51 | 0.1856 | 60.0919 | 0.9888 | sci (CHs ring C + C′+ C9H+ C9′H) |
| 79 | B | 1180.70 | 1.1577 | 9.0697 | 0.9804 | ip (CHs ring C + C′) |
| 80 | A | 1197.11 | 0.9682 | 166.3222 | 1.5546 | ip (CHs ring C + C′) + ip (C9Hs + C9′Hs) |
| 81 | B | 1198.66 | 10.8312 | 0.0735 | 1.6174 | ip (CHs ring C + C′) + ip (C9Hs + C9′Hs) |
| 82 | A | 1209.59 | 2.2357 | 1118.7989 | 2.4233 | defm (CCC) + ip CHs + sym str CN |
| 83 | B | 1215.31 | 8.1421 | 185.1219 | 2.3385 | ip CCC + ip CHs + asym str CN |
| 84 | B | 1224.42 | 8.8441 | 11.8070 | 1.5404 | ip CHs |
| 85 | A | 1224.43 | 1.3426 | 35.1919 | 1.5615 | ip CHs |
| 86 | A | 1260.81 | 0.5616 | 1171.3536 | 2.3165 | ip CHs + breathing (FL1, FL2) + sym str CN |
| 87 | B | 1261.34 | 6.3533 | 368.2336 | 2.4705 | ip (CHs) + breathing (FL1, FL2 ring) + asym str CN |
| 88 | B | 1311.72 | 8.2546 | 94.3526 | 1.8547 | ip CHs |
| 89 | A | 1313.53 | 0.0144 | 270.3249 | 1.7991 | ip CHs |
| 90 | B | 1330.04 | 1.1844 | 153.2975 | 1.7712 | roc (CHs ring C + C′) + ip (CHs + CCC) |
| 91 | A | 1330.52 | 0.0032 | 536.8398 | 1.7683 | ip CHs |
| 92 | B | 1345.36 | 33.9201 | 100.7305 | 6.3844 | str Ar (C=C) |
| 93 | A | 1351.73 | 5.0802 | 163.4050 | 7.3540 | str Ar (C=C) + ip (C9H + C9′H) |
| 94 | B | 1378.91 | 4.1174 | 94.8485 | 4.7393 | str Ar (C=C), (breathing B, B′) + ip CHs |
| 95 | A | 1380.86 | 1.1511 | 530.3869 | 4.8915 | str Ar (C=C), (breathing B, B′) + ip CHs |
| 96 | B | 1452.45 | 9.4167 | 12.3591 | 1.5043 | sci (C9Hs + C9′Hs) |
| 97 | A | 1452.55 | 0.9079 | 42.8585 | 1.5021 | sci (C9Hs + C9′Hs) |
| 98 | B | 1459.31 | 12.2895 | 23.2562 | 3.9503 | sci CHs + ip CC |
| 99 | A | 1459.79 | 10.1687 | 345.8181 | 3.7889 | sci CHs + str C=C |
| 100 | A | 1486.39 | 3.2886 | 101.6084 | 3.0486 | sci (CHs FL1 wrt CHs FL2) +roc CHs |
| 101 | B | 1487.37 | 31.3337 | 2.0133 | 3.0645 | roc all CHs |
| 102 | B | 1501.64 | 7.8461 | 125.2425 | 3.5549 | roc CHs + sci CHs |
| 103 | A | 1502.81 | 6.7233 | 490.6604 | 3.6339 | str Ar (C=C) + roc CHs |
| 104 | A | 1514.83 | 0.9425 | 170.6483 | 4.0351 | str Ar (C=C) + roc CHs |
| 105 | B | 1515.05 | 5.6894 | 283.2436 | 4.0391 | str Ar (C=C) + sci C9Hs + roc CHs |
| 106 | A | 1581.21 | 77.5397 | 6875.2755 | 4.6318 | str Ar (C=C) + str N=N + roc CHs |
| 107 | B | 1601.91 | 0.4325 | 130.5009 | 9.6223 | str Ar (C=C) + roc CHs |
| 108 | A | 1614.82 | 9.2285 | 598.8006 | 10.9474 | str Ar (C=C) + str N=N + roc CHs |
| 109 | B | 1625.92 | 2.1664 | 26.2751 | 9.5054 | str Ar (C=C) |
| 110 | A | 1626.08 | 0.3527 | 79.5832 | 9.6688 | str Ar (C=C) |
| 111 | B | 1651.10 | 1.4909 | 2076.4003 | 10.6608 | str Ar (C=C) |
| 112 | A | 1653.75 | 1.6761 | 2460.3547 | 10.5096 | str Ar (C=C), str N=N, ip CH, defm CCC |
| 113 | B | 1654.53 | 8.4589 | 98.6487 | 10.7875 | Ar (C=C), ip CH, defm CCC |
| 114 | A | 1657.49 | 0.6588 | 981.3998 | 11.8993 | str Ar (C=C), str N=N, ip CH, defm CCC |
| 115 | B | 3033.87 | 21.7959 | 64.7313 | 5.7495 | sym str (C9Hs, + C9′H) + asym str (C9H wrt C9′Hs) |
| 116 | A | 3033.89 | 7.3265 | 388.4236 | 5.7496 | sym str (C9Hs, + C9′H) + sym (C9Hs wrt C9′H) |
| 117 | A | 3063.31 | 3.2802 | 136.3858 | 6.0937 | asym str (C9Hs, + C9′Hs) + asym str (C9Hs wrt C9′Hs) |
| 118 | B | 3063.31 | 7.8622 | 56.9552 | 6.0937 | asym str (C9Hs, + C9′Hs) + sym str (C9Hs wrt C9′Hs) |
| 119 | B | 3174.81 | 14.7400 | 40.7724 | 6.4521 | str CHs ring C + str CHs ring C′ |
| 120 | A | 3174.83 | 0.2286 | 51.5632 | 6.4522 | str CHs ring C + str CHs ring C′ |
| 121 | B | 3181.12 | 2.5596 | 68.7996 | 6.4898 | str CHs ring C + str CHs ring C′ |
| 122 | A | 3181.12 | 4.9886 | 184.4987 | 6.4898 | str CHs ring (C + C′) |
| 123 | B | 3186.67 | 6.7277 | 12.6357 | 6.5170 | asym str (C4H, C4′H) |
| 124 | A | 3186.82 | 4.5728 | 22.0377 | 6.5177 | str (CHs) |
| 125 | B | 3190.95 | 5.9328 | 78.6016 | 6.5459 | asym str (C1H, C1′H) |
| 126 | A | 3190.97 | 6.0211 | 138.6614 | 6.5461 | sym str (C1H, C1′H) |
| 127 | B | 3192.37 | 42.6610 | 108.7342 | 6.5613 | asym str (CHs ring C, C′) |
| 128 | A | 3192.40 | 2.5487 | 184.9013 | 6.5612 | asym str (CH ring C + CH ring C′) |
| 129 | B | 3204.14 | 32.2569 | 178.3448 | 6.6365 | sym str (CHs ring C + CHs ring C′) + asym str (CHs ring C, CHs ring C′) |
| 130 | A | 3204.20 | 30.1293 | 552.0913 | 6.6367 | sym str (CH ring C + CH ring C′) |
| 131 | A | 3215.99 | 0.0045 | 116.4154 | 6.6591 | sym str (C3H, C4H) + sym (C3′H, C4′H) + sym str (C3H, wrt C3′H) |
| 132 | B | 3216.10 | 8.9671 | 1.6723 | 6.6601 | sym str (C3H, C4H), sym str (C3′H, C4′H) + asym str (C3H, wrt C3′H) |
sym, symmetry.
Vibrational frequencies in cm–1.
Infrared intensities in km/mol.
Raman scattering activities in A4/AMU.
k, force constants in mDyne/A.
defm, deformation; tor, torsion; str, stretching; sym, symmetric; asym, asymmetric; oop, out-of-plane bending; ip, in-plane bending; sci, scissoring; roc, rocking; wag, waging; wrt, with respect to.
The model compound AzoFL has 46 atoms; hence, there are 138 motions, 3 of which are translational, 3 of which are rotational, and 132 (τ3N-6′) of which are vibrational modes. The azo compound AzoFL belongs to C2 point group symmetry. Sixty-six vibrational modes are IR active and 66 modes are IR inactive. All of the IR inactive modes are found as Raman active modes.
The theoretically predicted IR and Raman scattering activity spectra by using B3LYP/6-31+G(d,p) basis set at DFT level of theory for both the trans- and cis-AzoFL with the FL are shown in Figures 16 and 17 by using B3LYP/6-31+G(d,p) basis set at DFT level of theory.
Figure 16.

Calculated (a) IR (b) Raman spectra of trans-AzoFL (c) IR (d) Raman spectra of cis-AzoFL at B3LYP/6-31+G (d,p). The calculated harmonic frequencies are represented with a Gaussian IR peak half-width at half height 4 cm–1.
Figure 17.

Calculated (a) IR and (b) Raman scattering activity spectra of FL at DFT-B3LYP/6-31+G(d,p). The calculated harmonic frequencies are represented with a Gaussian IR peak half-width at half height 4 cm–1.
2.4.3. N=N Vibration
The stretching vibrations of azo N=N unit is usually observed11,73 around at 1556–1420 cm–1. The nature of the compound is very important in analyzing spectra of azo compounds. The stretching vibration of N=N is found to vary for the different-nitrogen containing compounds. The N=N stretching vibration of a symmetrical trans-azo compound is forbidden in the IR due to no change in the dipole moment. Thus, the identification of this vibration and to distinguish between the cis- and trans-isomers is somewhat problematic due to its weakness or absence in the IR. Hence, the IR spectrum alone is not straightforward to analyze for such type of compounds. The trans-DZ at 1659 cm–1 for N=N stretching vibration shows zero intensity in IR but is Raman scattering active. However, the cis-azo (N=N) compounds due to nonzero dipole moment is expected to show active IR bands. The calculated N=N stretching vibrations at 1658.61, 1653.34, 1613.87, 1529.83, 1513.48, and 1497.01 cm–1 is found with zero intensity for the trans-AzoFL in the present work but are Raman scattering active. Conjugation with FL ring lowers the frequency of the N=N double bond in AzoFL. At the present study, the same N=N stretching vibration was found at 1657.49, 1653.75, 1614.82, and 1581.21 cm–1, respectively, for the cis-AzoFL. The parent cis-DZ due to its isolated and stronger N=N double bond character shows IR band at higher frequency at 1662 cm–1, which reflects the 0.01 Å shorter bond length of cis-DZ compared to cis-AzoFL. Minisini et al.84 found N=N stretching vibration at 1591 and 1544 cm–1 for cis-4-hydoxyazobenzene and trans-4-hydoxyazobenzene, respectively, by DFT calculation.
The N=N stretching frequency of cis-DFDZ at 1643.26 cm–1 shifted at 1628.78 cm–1 in trans-DFDZ, a 14.48 cm–1 shift to lower frequency is observed by Raman activity spectrum. The N=N stretching frequency of cis-DZ at 1662.43 cm–1 shifted at 1659.03 cm–1 in trans-DZ, a 3.40 cm–1 shift to lower frequency is observed by Raman activity spectrum. It should be noted that though the cis-DZ has higher N=N stretching vibration (1662.43 cm–1) compared to that of cis-DFDZ (1643.26 cm–1), the force constant is considerably lower (16.96 vs 22.28 mDyne/Å) in cis-DZ (Tables 9 and 10). Similarly, even though the trans-DZ has higher N=N stretching frequency (1659.03 cm–1) compared to that of cis-and trans-DFDZ, its force constant was found as lower value (9.86 mDyne/Å) by our B3LYP/6-31+G(d,p) calculation (Tables 9 and 10). Normally, bonds with stronger force constants have higher vibrational frequencies; however, in this case, we have observed the anomalies.
The in-plane vibration of N=N was observed at 322.27, 366.62, and 537.59 cm–1 as rocking mode with weak intensity band in cis-AzoFL. The CNNC angle deformation was found at 905.64 cm–1 as moderate weak band. The out-of-plane vibration of N=N appeared at low frequency at 16.10, 196.72, and 915.04 cm–1 as wagging vibration mode, whereas the 114.28, 481.22, 535.70, and 664.35 cm–1 bands appeared as twisting mode in cis-AzoFL.
The in-plane vibration of N=N appeared at 571.35 and 660.49 with moderate strong band but zero intensity in Raman activity scattering spectrum for trans-AzoFL.
2.4.4. C–N Vibration
The C–N stretching bands generally appear around at 1000–1300 cm–1.65,85 The identification of this vibration is somewhat difficult due to the mixing of vibrations in this region. In trans-AzoFL, the asymmetric C–N vibrations were found at 1231.20 and 1289.01 cm–1 as strong band in IR which is Raman inactive, whereas the symmetric stretching of C–N at 1264.60 cm–1 with zero intensity is Raman scattering active.
Our calculated C–N vibration mode in cis-AzoFL appeared at 1209.59, 1215.31, 1260.81, and 1261.34 cm–1 as mixing mode with in-plane CH vibration and CCC deformation. All of the modes are IR and Raman active. The in-plane and out-of-plane bending vibrations assigned for AzoFL are also presented in Tables 11 and 12.
2.4.5. Aromatic C=C Vibrations
Ar(C=C) stretching vibrations usually found at 1625–1430 cm–1.65,85 For the model trans-AzoFL, the calculated Ar(C=C) stretching vibration appears at 1502.90, 1513.48, 1520.07, 1604.96, 1613.87, and 1529.83 cm–1 together with other modes. The vibrations at 1652.38 and 1655.07 cm–1 appear as a strong peak for Ar(C=C) stretching vibration. The stretching vibration at 1625.72, 1653.34, and 1658.61 cm–1 for Ar(C=C) appears as zero intensity in IR spectra but as strong peak in Raman activity spectrum. The in-plane vibration of Ar(C=C) was observed at 513.35 and 547.75 cm–1 with zero intensity in IR spectrum.
For the model cis-AzoFL, the calculated Ar(C=C) stretching vibration appears at 1502.81, 1514.83, 1515.05, 1601.91, and 1614.82, cm–1 together with the other mode. The vibrations at 1625.92 and 1626.08 and 1651.10 cm–1 appear as a strong peak for only Ar(C=C) stretching vibration.
For the parent fluorene (FL), the calculated Ar(C=C) stretching vibration appears at 1623.76, 1628.97, 16.54.80, and 1654.87 cm–1. The calculated IR spectra of FL at B3LYP/6-31+G(d,p) are shown in Figure 17. The frequencies of different bonds, their IR intensities, Raman scattering activities, and force constants are listed in Table S4.
2.4.6. C–H Vibrations
The aromatic C–H stretching typically exhibits65,85 several weak-to-moderate bands above 3000 cm–1. In trans-AzoFL, the four C–H bonds from C9Hs and C9′Hs are stretches at 3033.36, 3033.37, 3062.81, and 3062.82 cm–1 as moderate strong band. The two C–H bonds at C9 position stretches both symmetrically and asymmetrically among themselves and with respect to other fluorene ring C9′Hs as well. Among the two symmetric modes for the two C–H bonds at C9 position, one is asymmetric at 3033.36 cm–1 with respect to other ring found as IR active. However, the other one at 3033.37 cm–1 that is symmetric with respect to other ring is found as IR inactive but Raman active. The same trend is observed for the asymmetric stretching vibration of the two C9–H bonds. The stretching vibration of rest aromatic 14 C–H bonds from two fluorenyl ring appeared together at 3174.85, 3174.87, 3181.31, 3181.32, 3184.62, 3184.72, 3192.56, 3194.96, 3195.04, 3204.19, 3204.26, 3226.84, and 3226.98 cm–1. Among those seven modes are IR inactive but Raman active, while seven IR active modes are Raman inactive. A similar spectral pattern was observed for the aromatic C–H absorption band region. The entire vibration modes in this region are found as both IR and Raman active with low intensity. The two C–H bonds in C9 of fluorene appears at 3032.08 and 3060.75 cm–1 as doublet, one symmetric and the other for asymmetric stretching, respectively. The calculated harmonic frequencies for the AzoFL molecule are related to the gaseous phase, but the reported values from experimental works are done in the solid phase. Hence, a slight disagreement between the present calculated and reported experimental frequencies can be expected. Aromatic C–H in-plane bending vibrations usually occur in the region of 1390–990 cm–1 and are very useful for characterization and identification of aromatic compounds, whereas C–H out-of-plane deformations generally appears at 1000–700 cm–1.65,85 Both the in-plane and out-of-plane bending vibrations within the fluorene ring and between the two fluorene ring in different pattern for 18 C–H groups as scissoring, rocking, twisting, and wagging mode were observed. The out–of plane wagging vibration for aromatic ring C–H appeared at 754.23 cm–1 as a strong band together with fluorene ring breathing at 754.67 cm–1 in parent FL. The same wagging mode in trans-AzoFL shifted to 747.35 and at 775.17 cm–1 in cis-AzoFL as strong band. Though the C–H bonds in both the FL ring of cis-AzoFL vibrate in wagging mode, they twist as a net result with respect to one another ring.
In cis-AzoFL, the four C–H bonds from C9Hs and C9′Hs are stretches at 3033.87, 3033.89, 3063.31, and 3063.31 cm–1 as moderate strong band. The stretching vibration of rest aromatic 14 C–H bonds from two fluorenyl ring appeared together at 3174.81, 3174.83, 3181.12, 3181.12, 3186.67, 3186.82, 3190.95, 3190.97, 3192.37, 3192.40, 3204.14, 3204.20, 3215.99, and 3216.10 cm–1. The C–H bonds at different positions stretch both symmetrically and asymmetrically among themselves within the ring and with respect to other fluorene ring as well. Unlike the trans-AzoFl, the vibrational frequencies of C–H bonds of cis-AzoFl are found as both the IR and Raman scattering active. All of the vibrational modes of cis-AzoFL for the different C–H bonds, their IR intensities, Raman scattering activities, and force constants are listed in Table 12. All of the frequencies were found to be well matched within the characteristics region and the details are presented in Tables 11 and 12 for both the isomers.
In FL, the two C–H bonds from C9Hs stretch symmetrically and asymmetrically at 3032.08 and 3060.75 cm–1 as moderate strong band, respectively. The stretching vibrations from other C–H bonds are observed at 3173.37, 3173.71, 3179.55, 3181.13, 3190.85, 3192.06, 3203.15, and 3203.80 cm–1, respectively.
2.4.7. Ring Vibration
The fluorenyl ring breathing vibration at 759.94 cm–1 in cis-AzoFL matches nicely with the literature value.86 The breathing mode at 758.63 cm–1 in trans-AzoFL is IR inactive but Raman scattering active mode. The breathing mode in parent fluorene ring appears at 754.67 cm–1 (Figure 17) as a very weak peak in our present work.
Overall, the present computations show that both the trans- and cis-isomers possess different vibrational frequencies for the same structural −N=N– unit; hence, both the isomers were characterized and distinguished. The isolated N=N stretching vibration of trans-diazene appears at 1659.03 cm–1 in Raman scattering spectra whereas the same vibration mode appears at 1662.43 cm–1 for cis-diazene in both the IR and Raman scattering spectra. The N=N group in both the trans and cis-DFDZ vibrates at ∼30 and ∼19 cm–1 lower frequency at 1628.71 and 1643.27 cm–1, respectively, compared to that of respective DZ. We can safely conclude that the isolated N=N stretching vibration in the presence of substituents shifts toward the shorter wavelength in symmetrically disubstituted azo compounds. For cis-diazene, both the asymmetric and symmetric stretching vibration bands at 3088.26 and 3185.08 cm–1 were observed for the two N–H groups in IR and Raman scattering spectra, whereas for trans-isomer only one, the asymmetric stretching vibration band at 3313 cm–1 was found as IR active and the other one, symmetric vibration at 3280 cm–1 was Raman scattering active. The same trend was observed for difluorodiazene, for example, two absorption bands of the two N–F groups, asymmetric and symmetric absorption bands at 740.60 and 910.57 cm–1, were observed both in the IR and Raman scattering spectra for cis-DFDZ. The asymmetric stretching vibration band of N–F bonds at 996.45 cm–1 was found as IR active and on the other hand, symmetric vibration at 1034.89 cm–1 was found as Raman scattering active for trans-DFDZ. Similar patterns were observed for the model compound trans- and cis-AzoFL. Among different bands of stretching vibration, the two asymmetric stretching vibrations at 1289.01 and 1231.20 cm–1 for C–N bond are IR active, whereas the band is found as inactive mode in Raman scattering spectra. The IR inactive mode of symmetric stretching vibration at 1264 and 1209.63 cm–1 for the same C–N bond are found as Raman active mode.
3. Conclusions
In order to gain insight into the azo −N=N– backbone, studies on the molecular geometry, vibrational frequencies, infrared intensities, force constants, and Raman scattering activities were carried out at the DFT with B3LYP functional using 6-31+G(d,p) basis set for trans- and cis-bis(9H-fluoren-2-yl)diazene (AzoFL). The work has been extended with the π-conjugation spacer fluorene, the parent trans-/cis-diazene and difluorodiazene by the same method (DFT) and same basis set 6-31+G(d,p). The influences of substituents on the azo −N=N– backbone of parent trans- and cis-diazene by using (i) electron rich π-bonded aromatic fluorene rings and (ii) electron-rich lone pairs of F atoms having electron withdrawing nature, for example, in model AzoFL and difluorodiazene were observed. Introducing fluorene ring into the −N=N– backbone causes an increase of the N=N bond length due to the extensive π-bond conjugation in AzoFL. The longer bond length reflects on the stretching vibration of AzoFl. Both the trans- and cis-AzoFL vibrates at a much lower frequency compared to that of parent trans- and cis-diazene. A reverse trend (shorter −N=N– bond length) is observed by introducing F atoms into the −N=N– backbone. Though it is expected that compounds having shorter bond length should vibrate at higher frequency but unexpectedly both the trans- and cis-difluorodiazene vibrates at lower frequency compared to that of parent diazene. It should be noted that though the trans-AzoFL is stable by 16.33 kcal/mol than the cis-AzoFL, the trans-DFDZ is less stable than its cis-counterpart.
To study the electronic properties, viz. UV–vis spectra, excitation energies and the maximum absorption wavelength, oscillator strength, energies of HOMO, LUMO, and energy difference between them, Eg (HOMO–LUMO), electronic transitions, and transition probabilities for the model trans- and cis-AzoFL by TD-DFT calculation using B3LYP/6-31+G(d,p) starting from the initial optimized geometry by DFT-B3LYP/6-31+G(d,p) in gas phase were performed. Both the UV–vis spectral and vibrational analyses show a substantial influence on the −N=N– backbone and a significant variation were observed. Critical comparisons were carried out with the above-mentioned compounds using TD-DFT and ZIndo method.
Compared to parent trans-diazene (λmax 178.97 nm), a significant variation to longer wavelength (∼245 nm) is observed due to incorporation of the fluorene (FL) ring into the −N=N– backbone. The co-planarity of the two FL ring with the longer N=N bond length compared to the unsubstituted parent diazene indicates the effective red shift due to the extended π-conjugation in trans-AzoFL. The nonplanarity of cis-AzoFL (48.1° tilted about the C–N bond relative to the planar N=N–C bond) reflects its ∼64 nm blue shift compared to that of trans-counterpart. However, two F atoms into the backbone of −N=N– causes only ∼10 nm red shift in trans-DFDZ but ∼15 nm opposite blue shift in cis-DFDZ respectively for π–π* transition band compared to that of trans- and cis-diazene.
The same trend is observed for n−π* transition as well, that is, the n−π* band shifts to longer wavelength (λmax 517.82 nm) in cis-AzoFL, on the other hand the same band shifts to shorter wavelength (λmax ≈ 190 nm) in cis-DFDZ compared to that of parent cis-diazene (λmax 371.78 nm). Present calculation shows that the ZIndo method is reasonably good to explain the absorption behavior of the cis-/trans-isomers of studied azo compounds.
These findings can provide the basic understanding on the electronic properties of geometric cis–trans azo isomers. The opposite absorption behavior between AzoFL and DFDZ clearly imply that the aromatic fluorene (FL) ring and fluorine atoms (F) as substituents on the azo −N=N– backbone play a vital role among the different pair of cis–trans azo compounds under study. Because all of the calculations were performed in the same platform, it allowed us to compare and investigate the behaviors of the azo compounds more accurately. Isac and co-workers87 observed charge-transfer transitions in azobenzene when substituted with maleimide functional group. Compared to azobenzene, our model azofluorene compounds have extended π-conjugation framework and thus might have the possibility to play a potential role in such type of charge transfer transitions. We believe that the findings of the present work are of substantial interest in the field of optoelectronic properties of π-conjugated azo polymers.
4. Computational Methods
The ground-state geometries of six azo compounds, viz., trans- and cis-isomers of diazene (DZ), difluorodiazene (DFDZ), our model compound bis(9H-fluoren-2-yl)diazene (AzoFL) respectively, and the π-conjugation spacer, fluorene (FL) were calculated at the DFT level of theory. The B3LYP hybrid functional88,89 using 6-31+G(d,p) basis set was employed to perform the calculations in gas phase for all of the above-mentioned compounds in neutral state. The geometries for all of the DFT calculations were initially optimized at the semi-empirical AM190 level. The resulting geometry is then employed as starting geometry for optimization at the DFT/B3LYP level of theory for better description. Geometry optimization by ab initio Hartree–Fock calculations were also performed using HF/6-31+G(d,p), HF/6-31++G(d,p), and HF/6-311+G(d,p) basis set for DZ and DFDZ. Bernys optimization algorithm91 was used to run the complete geometry optimization for both the trans- and cis-AzoFL and all other above-mentioned compounds. The optimized structural parameters of DFT calculations and all other calculations at the same level of theory and basis set were used in the vibrational frequency calculations. Vibrational frequency calculations were performed with high degree of accuracy, and no imaginary frequencies were found. Hence, true minimum on the potential energy surface were obtained in each case. The unscaled calculated harmonic frequencies, force constants, infrared intensities, and Raman scattering activities were obtained from the output result of the frequency calculations.
The GaussView program92 which is a graphical user interface designed to be used with Gaussian,93 has been used to predict the vibrational modes, intensities, and spectra by visual animation for the verification of the normal mode assignments. The defined coordinates form a complete set and match quite well with the motions observed using the gauss view 6.0.16 program. Density functional time-dependent, TD/DFT94−97 excited-state calculations with the B3LYP/6-31+G(d,p) basis set based on the optimized geometries of B3LYP/6-31+G(d,p) were carried out on the three lowest spin allowed singlet–singlet transitions for the model compound AzoFL, other mentioned azo compounds and FL in the gas phase to get the excitation energies, UV–vis absorption maximum wavelengths (λmax), oscillator strength (f) and UV–vis absorption spectra, HOMO, LUMO energies, and the FMO orbitals. Based on the optimized geometry from AM1, ZIndo98−100 calculations were performed in similar fashion. All of the calculations mentioned above were performed by Gaussian 1693 and Gauss View 6.0.1692 program package by intel core i3-6006U CPU@2.00 GHz, 1.99 GHz on note book computer by windows version 10.
Acknowledgments
Both the authors are grateful to Shofiqule Azam for giving the Gaussian 16 Revision-A.03 and Gauss View 6.0.16 program package software to continue the research work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03839.
Additional material with computational results which includes (Figures and Tables) ZIndo calculation, TD-DFT//B3LYP/6-31+G(d,p) calculation with different initial geometry HF/6-31+G(d,p) methods, excitation energies, electronic transitions, transition probabilities, and different modes of vibrational frequencies of different compounds (PDF)
Author Contributions
The manuscript was written through contributions of both authors. Both the authors have given approval to the final version of the manuscript.
The present work is carried out by personal expenses of the authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Zollinger H.Color Chemistry: Syntheses, Properties and Applications of Organic Dyes and Pigments, 3rd ed.; Verlag Helvetica Chimica Acta: Zurich, 2003. [Google Scholar]
- Croot P. L.; Johansson M. Determination of iron speciation by cathodic stripping voltammetry in seawater using the competing ligand 2-(2-thiazolylazo)-p-cresol (tac). Electroanalysis 2000, 12, 565–576. . [DOI] [Google Scholar]
- Choi D.; Lee S. K.; Chung T. D.; Kim H. Electrochemical determination of adsorption isotherm of mordant red 19 on mercury and its analytical application for the indirect determination of uranium. Electroanalysis 2000, 12, 477–482. . [DOI] [Google Scholar]
- Ahmad T.; Kandil F.; Moustapha C. Preparation and characterization of some new azo dyes, azomethine dyes and heterocyclic -schiff bases Derivatives. AASCIT J. Chem. 2015, 2, 24–31. [Google Scholar]
- Xu H.; Zeng X. Synthesis of diaryl-azo derivatives as potential antifungal agents. Bioorg. Med. Chem. Lett. 2010, 20, 4193–4195. 10.1016/j.bmcl.2010.05.048. [DOI] [PubMed] [Google Scholar]
- Tonelli M.; Vazzana I.; Tasso B.; Boido V.; Sparatore F.; Fermeglia M.; Paneni M. S.; Posocco P.; Pricl S.; Colla P. L.; Ibba C.; Secci B.; Collu G.; Loddo R. Antiviral and cytotoxic activities of aminoarylazo compounds and aryltriazene derivatives. Bioorg. Med. Chem. 2009, 17, 4425–4440. 10.1016/j.bmc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrahpour A.; Zarei M. Efficient one-pot synthesis of 2-azetidinones from acetic acid derivatives and imines using methoxymethylene-n,n-dimethyliminium salt. Tetrahedron 2010, 66, 5017–5023. 10.1016/j.tet.2010.05.009. [DOI] [Google Scholar]
- Jarrahpour A.; Zarei M. DMF-dimethyl sulfate as a new reagent for the synthesis of β-lactams. Tetrahedron Lett. 2009, 50, 1568–1570. 10.1016/j.tetlet.2009.01.092. [DOI] [Google Scholar]
- Zarei M.; Mohamadzadeh M. 3-Thiolated 2-azetidinones: synthesis and in vitro antibacterial and antifungal activities. Tetrahedron 2011, 67, 5832–5840. 10.1016/j.tet.2011.05.043. [DOI] [Google Scholar]
- Zhao X.; Li C.; Zeng S.; Hu W. Discovery of highly potent agents against influenza A virus. Eur. J. Med. Chem. 2011, 46, 52–57. 10.1016/j.ejmech.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Biswas N.; Umapathy S. Structures, vibrational frequencies, and normal modes of substituted azo dyes: infrared, raman, and density functional calculations. J. Phys. Chem. A 2000, 104, 2734–2745. 10.1021/jp9929263. [DOI] [Google Scholar]
- Bach H.; Anderle K.; Fuhrmann T.; Wendorff J. H. Biphoton-induced refractive index change in 4-amino-4′-nitroazobenzene/polycarbonate. J. Phys. Chem. 1996, 100, 4135–4140. 10.1021/jp952094i. [DOI] [Google Scholar]
- Ramanujam P. S.; Hvilsted S.; Andruzzi F. Novel biphotonic holographic storage in a side-chain liquid crystalline polyester. Appl. Phys. Lett. 1993, 62, 1041–1043. 10.1063/1.108788. [DOI] [Google Scholar]
- Lee G. J.; Kim D.; Lee M. Photophysical properties and photoisomerization processes of Methyl Red embedded in rigid polymer. Appl. Opt. 1995, 34, 138–143. 10.1364/ao.34.000138. [DOI] [PubMed] [Google Scholar]
- Ramanujam P. S.; Hvilsted S.; Zebger I.; Siesler H. W. On the explanation of the biphotonic processes in polyesters containing azobenzene moieties in the side chain. Macromol. Rapid Commun. 1995, 16, 455–461. 10.1002/marc.1995.030160701. [DOI] [Google Scholar]
- Hvilsted S.; Andruzzi F.; Ramanujam P. S. Side-chain liquid-crystalline polyesters for optical information storage. Opt. Lett. 1992, 17, 1234–1236. 10.1364/ol.17.001234. [DOI] [PubMed] [Google Scholar]
- Willner I.; Rubin S. Control of the structure and functions of biomaterials by light. Angew. Chem., Int. Ed. Engl. 1996, 35, 367–385. 10.1002/anie.199603671. [DOI] [Google Scholar]
- Wagner-Wysiecka E.; Łukasik N.; Biernat J. F.; Luboch E. Azo group(s) in selected macrocyclic compounds. J. Inclusion Phenom. Macrocyclic Chem. 2018, 90, 189–257. 10.1007/s10847-017-0779-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino E.; Ribagorda M. Control over molecular motion using the cis–trans photoisomerization of the azo group. Beilstein J. Org. Chem. 2012, 8, 1071–1090. 10.3762/bjoc.8.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.-C.; Chang Y.-C.; Tsai B.-K.; Lin T.-C.; Lin J.-H.; Hsiao V. K. S. trans/cis-Isomerization of fluorene-bridged azo chromophore with significant two-photon absorbability at near-infrared wavelength. Chem.—Asian J. 2014, 9, 3390–3396. 10.1002/asia.201402896. [DOI] [PubMed] [Google Scholar]
- Smart Light-Responsive Materials: Azobenzene-Containing Polymers and Liquid Crystals; Zhao Y.; Ikeda T., Eds.; John Wiley & Sons: New Jersey, 2009. [Google Scholar]
- Deloncle R.; Caminade A.-M. Stimuli-responsive dendritic structures: The case of light-driven azobenzene-containing dendrimers and dendrons. J. Photochem. Photobiol., C 2010, 11, 25–45. 10.1016/j.jphotochemrev.2010.02.003. [DOI] [Google Scholar]
- Han M.; Honda T.; Ishikawa D.; Ito E.; Hara M.; Norikane Y. Realization of highly photoresponsive azobenzene-functionalized monolayers. J. Mater. Chem. 2011, 21, 4696–4702. 10.1039/c0jm03697h. [DOI] [Google Scholar]
- Samanta S.; Beharry A. A.; Sadovski O.; McCormick T. M.; Babalhavaeji A.; Tropepe V.; Woolley G. A. Photoswitching azo compounds in vivo with red light. J. Am. Chem. Soc. 2013, 135, 9777–9784. 10.1021/ja402220t. [DOI] [PubMed] [Google Scholar]
- Chen S.-L.; Chu C.-C.; Hsiao V. K. S. Reversible light-modulated photoluminescence from azobenzene-impregnated porous silicon. J. Mater. Chem. C 2013, 1, 3529–3531. 10.1039/c3tc30329b. [DOI] [Google Scholar]
- Liu M.; Yin L.; Wang L.; Miao T.; Cheng X.; Wang Y.; Zhang W.; Zhu X. Synthesis of monodisperse aromatic azo oligomers toward gaining new insight into the isomerization of π-conjugated azo systems. Polym. Chem. 2019, 10, 1806–1811. 10.1039/c9py00001a. [DOI] [Google Scholar]
- Liu Y.; Wu W.; Wei J.; Yu Y. Visible light responsive liquid crystal polymers containing reactive moieties with good processability. ACS Appl. Mater. Interfaces 2017, 9, 782–789. 10.1021/acsami.6b11550. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Yoshida K.; Fujiki M.; Zhu X. Unpolarized-light-driven amplified chiroptical modulation between chiral aggregation and achiral disaggregation of an azobenzene-alt-fluorene copolymer in limonene. Macromolecules 2011, 44, 5105–5111. 10.1021/ma2012128. [DOI] [Google Scholar]
- Anwar N.; Willms T.; Grimme B.; Kuehne A. J. C. Light-switchable and monodisperse conjugated polymer particles. ACS Macro Lett. 2013, 2, 766–769. 10.1021/mz400362g. [DOI] [PubMed] [Google Scholar]
- Wang K.; Yin L.; Miu T.; Liu M.; Zhao Y.; Chen Y.; Zhou N.; Zhang W.; Zhu X. Design and synthesis of a novel azobenzene-containing polymer both in the main- and side-chain toward unique photocontrolled isomerization properties. Mater. Chem. Front. 2018, 2, 1112–1118. 10.1039/c8qm00035b. [DOI] [Google Scholar]
- Kraft A.; Grimsdale A. C.; Holmes A. B. Electroluminescent conjugated polymers—seeing polymers in a new light. Angew. Chem., Int. Ed. 1998, 37, 402–428. . [DOI] [PubMed] [Google Scholar]
- Bernius M.; Inbasekaran M.; Woo E.; Wu W.; Wujkowski L. Fluorene-based polymers-preparation and applications. J. Mater. Sci.: Mater. Electron. 2000, 11, 111–116. 10.1023/a:1008917128880. [DOI] [Google Scholar]
- Karim M. A.; Cho Y.-R.; Park J. S.; Kim S. C.; Kim H. J.; Lee J. W.; Gal Y.-S.; Jin S.-H. Novel fluorene-based functional ‘click polymers’ for quasi-solid-state dye-sensitized solar cells. Chem. Commun. 2008, 1929. 10.1039/b800032h. [DOI] [PubMed] [Google Scholar]
- Bagheri S. N.; Hashemianzadeh S. M. Density functional theory study of new azo dyes with different π-spacers for dye-sensitized solar cells. Spectrochim. Acta, Part A 2015, 143, 20–34. 10.1016/j.saa.2015.02.026. [DOI] [PubMed] [Google Scholar]
- Heeney M.; Bailey C.; Giles M.; Shkunov M.; Sparrowe D.; Tierney S.; Zhang W.; McCulloch I. Alkylidene fluorene liquid crystalline semiconducting polymers for organic field effect transistor devices. Macromolecules 2004, 37, 5250–5256. 10.1021/ma049798n. [DOI] [Google Scholar]
- Romain M.; Chevrier M.; Bebiche S.; Mohammed-Brahim T.; Rault-Berthelot J.; Jacques E.; Poriel C. The structure–property relationship study of electron-deficient dihydroindeno[2,1-b]fluorene derivatives for n-type organic field effect transistors. J. Mater. Chem. C 2015, 3, 5742–5753. 10.1039/c5tc00355e. [DOI] [Google Scholar]
- Becke A. D. Perspective: fifty years of density–functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. 10.1063/1.4869598. [DOI] [PubMed] [Google Scholar]
- Devlin F. J.; Finley J. W.; Stephens P. J.; Frisch M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields: a comparison of local, nonlocal, and hybrid density functionals. J. Phys. Chem. 1995, 99, 16883–16902. 10.1021/j100046a014. [DOI] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Handy N. C.; Murray C. W.; Amos R. D. Study of methane, acetylene, ethene, and benzene using Kohn-Sham theory. J. Phys. Chem. 1993, 97, 4392–4396. 10.1021/j100119a023. [DOI] [Google Scholar]
- Johnson B. G.; Gill P. M. W.; Pople J. A. The performance of a family of density functional methods. J. Chem. Phys. 1993, 98, 5612–5626. 10.1063/1.464906. [DOI] [Google Scholar]
- Wheelless C. J. M.; Zhou X.; Liu R. Density functional theory study of vibrational spectra. 2. Assignment of fundamental vibrational frequencies of fulvene. J. Phys. Chem. 1995, 99, 12488–12492. 10.1021/j100033a019. [DOI] [Google Scholar]
- Handy N. C.; Maslen P. E.; Amos R. D.; Andrews J. S.; Murray C. W.; Laming G. J. The harmonic frequencies of benzene. Chem. Phys. Lett. 1992, 197, 506–515. 10.1016/0009-2614(92)85808-n. [DOI] [Google Scholar]
- Lee S. Y.; Boo B. H. Density functional theory study of vibrational spectra of fluorene. J. Phys. Chem. 1996, 100, 8782–8785. 10.1021/jp960020g. [DOI] [Google Scholar]
- Demaison J.; Hegelund F.; Bürger H. Experimental and ab initio equilibrium structure of trans-diazene HNNH. J. Mol. Struct. 1997, 413-414, 447–456. 10.1016/s0022-2860(97)00019-7. [DOI] [Google Scholar]
- Pu X.; Wong N. B.; Zhou G.; Gu J.; Tian A. Substituent effects on the trans/cis isomerization and stability of diazenes. Chem. Phys. Lett. 2005, 408, 101–106. 10.1016/j.cplett.2005.04.011. [DOI] [Google Scholar]
- Spada R. F. K.; Ferraro L. F. A.; Cardoso D. V. V.; Roberto-Neto O.; Machado F. B. C. Thermochemistry and kinetics of the trans-N2H2 + N reaction. Chem. Phys. Lett. 2013, 557, 37–42. 10.1016/j.cplett.2012.12.011. [DOI] [Google Scholar]
- Back R. A.; Willis C.; Ramsay D. A. The near-ultraviolet absorption spectrum of diimide vapor. Can. J. Chem. 1974, 52, 1006–1012. 10.1139/v74-160. [DOI] [Google Scholar]
- Nordhoff K.; Anders E. MO theoretical investigation of the cis effect observed in mono- and dihalodiazenes. J. Org. Chem. 1999, 64, 7485–7491. 10.1021/jo990779z. [DOI] [Google Scholar]
- Bree A.; Zwarich R. Vibrational assignment of fluorene from the infrared and Raman spectra. J. Chem. Phys. 1969, 51, 912–920. 10.1063/1.1672155. [DOI] [Google Scholar]
- Burns D. M.; Iball J. The crystal and molecular structure of fluorene. Proc. R. Soc. London, Ser. A 1955, 227, 200. 10.1098/rspa.1955.0004. [DOI] [Google Scholar]
- Pitzer K. S.; Hollenberg J. L. cis- and trans-Dichloroethylenes. The infrared spectra from 130–400 cm.-1 and the thermodynamic properties. J. Am. Chem. Soc. 1954, 76, 1493–1496. 10.1021/ja01635a010. [DOI] [Google Scholar]
- Bohn R. K.; Bauer S. H. An electron diffraction study of the structures of cis- and trans- N2F2. Inorg. Chem. 1967, 6, 309–312. 10.1021/ic50048a025. [DOI] [Google Scholar]
- Kuczkowski R. L.; Wilson E. B. Jr. Microwave spectrum, structure, and dipole moment of ‘Cis’-N2F2. J. Chem. Phys. 1963, 39, 1030. 10.1063/1.1734353. [DOI] [Google Scholar]
- Banerjee D.; Ghosh A.; Chattopadhyay S.; Ghosh P.; Chaudhuri R. K. Revisiting the ‘cis-effect’ in 1,2-difluoro derivatives of ethylene and diazene using ab initio multireference methods. Mol. Phys. 2014, 112, 3206–3224. 10.1080/00268976.2014.938710. [DOI] [Google Scholar]
- Yamamoto T.; Kaneno D.; Tomoda S. The importance of lone pair delocalizations: theoretical investigations on the stability of cis and trans isomers in 1,2-halodiazenes. J. Org. Chem. 2008, 73, 5429–5435. 10.1021/jo8006896. [DOI] [PubMed] [Google Scholar]
- Wolfe S. Gauche effect. Stereochemical consequences of adjacent electron pairs and polar bonds. Acc. Chem. Res. 1972, 5, 102–111. 10.1021/ar50051a003. [DOI] [Google Scholar]
- Epiotis N. D.; Yates R. L.; Larson J. R.; Kirmaier C. R.; Bernardi F. Directional effects of .sigma. conjugation on geometrical isomerism. J. Am. Chem. Soc. 1977, 99, 8379–8388. 10.1021/ja00468a001. [DOI] [Google Scholar]
- Jacquemin D.; Perpète E. A.; Scuseria G. E.; Ciofini I.; Adamo C. Extensive TD-DFT investigation of the first electronic transition in substituted azobenzenes. Chem. Phys. Lett. 2008, 465, 226–229. 10.1016/j.cplett.2008.09.071. [DOI] [Google Scholar]
- Jacquemin D.; Wathelet V.; Perpète E. A.; Adamo C. Extensive TD-DFT benchmark: Singlet-excited states of organic molecules. J. Chem. Theory Comput. 2009, 5, 2420–2435. 10.1021/ct900298e. [DOI] [PubMed] [Google Scholar]
- Jacquemin D.; Preat J.; Perpète E. A.; Vercauteren D. P.; André J.-M.; Ciofini I.; Adamo C. Absorption spectra of azobenzenes simulated with time-dependent density functional theory. Int. J. Quantum Chem. 2011, 111, 4224–4240. 10.1002/qua.22910. [DOI] [Google Scholar]
- Laurent A. D.; Jacquemin D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. 10.1002/qua.24438. [DOI] [Google Scholar]
- Mohammed I. A.; Mustapha A. Synthesis of new azo compounds based on N-(4-hydroxypheneyl)maleimide and N-(4-methylpheneyl)maleimide. Molecules 2010, 15, 7498–7508. 10.3390/molecules15107498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lednev I. K.; Ye T.-Q.; Matousek P.; Towrie M.; Foggi P.; Neuwahl F. V. R.; Umapathy S.; Hester R. E.; Moore J. N. Femtosecond time-resolved UV-visible absorption spectroscopy of trans-azobenzene: dependence on excitation wavelength. Chem. Phys. Lett. 1998, 290, 68–74. 10.1016/s0009-2614(98)00490-4. [DOI] [Google Scholar]
- Pavia D. L.; Lampman G. M.; KriZ G. S.; Vyvyan J. R.. Introduction to spectroscopy, 5th ed.; Cengage Learning: Stamford CT, 2015. [Google Scholar]
- Microencapsulation: Innovative Applications; Giamberini M., Prieto S. F., Tylkowsky B., Eds.; Walter de Gruyter GmbH & Co KG, 2015. [Google Scholar]
- Foresman J. B.; Frisch A.. Exploring Chemistry with Electronic Structure Methods, 3rd ed.; Gaussian, Inc.: Wallingford, CT, 2015. [Google Scholar]
- Fleming I.Frontier Orbitals and Organic Chemical Reactions; John Wiley: London, 1976. [Google Scholar]
- Kosar B.; Albayrak C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta, Part A 2011, 78, 160–167. 10.1016/j.saa.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Pearson R. G. Chemical hardness and density functional theory. J. Chem. Sci. 2005, 117, 369–377. 10.1007/bf02708340. [DOI] [Google Scholar]
- Parr R. G.; Pearson R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. 10.1021/ja00364a005. [DOI] [Google Scholar]
- Charles A. M. DFT study on structure, electronic properties, and reactivity of cis-isomers of [(NC5H4-S)2Fe(CO)2]. J. Chem. Sci. 2011, 123, 727–731. 10.1007/s12039-011-0131-2. [DOI] [Google Scholar]
- Armstrong D. R.; Clarkson J.; Smith W. E. Vibrational analysis of trans-azobenzene. J. Phys. Chem. 1995, 99, 17825–17831. 10.1021/j100051a005. [DOI] [Google Scholar]
- Kurita N.; Tanaka S.; Itoh S. Ab initio molecular orbital and density functional studies on the stable structures and vibrational properties of trans- and cis- azobenzenes. J. Phys. Chem. A 2000, 104, 8114–8120. 10.1021/jp001307n. [DOI] [Google Scholar]
- Fliegl H.; Köhn A.; Hättig C.; Ahlrichs R. Ab initio calculation of the vibrational and electronic spectra of trans- and cis-azobenzene. J. Am. Chem. Soc. 2003, 125, 9821–9827. 10.1021/ja034433o. [DOI] [PubMed] [Google Scholar]
- Gagliardi L.; Orlandi G.; Bernardi F.; Cembran A.; Garavelli M. A theoretical study of the lowest electronic states of azobenzene: the role of torsion coordinate in the cis–trans photoisomerization. Theor. Chem. Acc. 2004, 111, 363–372. 10.1007/s00214-003-0528-1. [DOI] [Google Scholar]
- Harabuchi Y.; Ishii M.; Nakayama A.; Noro T.; Taketsugu T. A multireference perturbation study of the NN stretching frequency of trans-azobenzene in nπ* excitation and an implication for the photoisomerization mechanism. J. Chem. Phys. 2013, 138, 064305. 10.1063/1.4790611. [DOI] [PubMed] [Google Scholar]
- Jensen H. J. A.; Joergensen P.; Helgaker T. Ground-state potential energy surface of diazene. J. Am. Chem. Soc. 1987, 109, 2895–2901. 10.1021/ja00244a007. [DOI] [Google Scholar]
- Craig N. C.; Levin I. W. Vibrational assignment and potential function for trans-diazene (diimide): predictions for cis-diazene. J. Chem. Phys. 1979, 71, 400–407. 10.1063/1.438084. [DOI] [Google Scholar]
- Hwang D.-Y.; Mebel A. M. Reaction mechanism of N2/H2 conversion to NH3: a theoretical study. J. Phys. Chem. A 2003, 107, 2865–2874. 10.1021/jp0270349. [DOI] [Google Scholar]
- Biczysko M.; Poveda L. A.; Varandas A. J. C. Accurate MRCI study of ground-state N2H2 potential energy surface. Chem. Phys. Lett. 2006, 424, 46–53. 10.1016/j.cplett.2006.04.073. [DOI] [PubMed] [Google Scholar]
- Huber K. P.; Herzberg G.. Molecular Spectra and Molecular Structure: IV. Constants of Diatomic Molecules; Van Nostrand Reinhold Co.: New York, 1979. [Google Scholar]
- Lee S. Y. Density functional theory calculation of molecular structure and vibrational spectra of dibenzothiophene in the ground and the lowest triplet state. J. Phys. Chem. A 2001, 105, 8093–8097. 10.1021/jp010991i. [DOI] [Google Scholar]
- Minisini B.; Fayet G.; Tsobnang F.; Bardeau J. F. Density functional theory characterisation of 4-hydroxyazobenzene. J. Mol. Model. 2007, 13, 1227–1235. 10.1007/s00894-007-0244-1. [DOI] [PubMed] [Google Scholar]
- Silverstein R. M.; Bassler G. C.; Morrill T. C.. Spectrometric Identification of Organic Compounds; Wiley: New York, 1981. [Google Scholar]
- Teimouri A.; Chermahini A. N.; Emami M. Synthesis, characterization, and DFT studies of a novel azo dye derived from racemic or optically active binaphthol. Tetrahedron 2008, 64, 11776–11782. 10.1016/j.tet.2008.09.104. [DOI] [Google Scholar]
- Isac D. L.; Airinei A.; Maftei D.; Humelnicu I.; Mocci F.; Laaksonen A.; Pinteală M. On the charge-transfer excitations in azobenzene maleimide compounds. A theoretical study. J. Phys. Chem. A 2019, 123, 5525–5536. 10.1021/acs.jpca.9b02082. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Dewar M. J. S.; Zoebisch E. G.; Healy E. F.; Stewart J. J. P. Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. 10.1021/ja00299a024. [DOI] [Google Scholar]
- Schlegel H. B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. 10.1002/jcc.540030212. [DOI] [Google Scholar]
- Dennington R.; Keith T. A.; Milam J. M.. GaussView; Semichem Inc.: Shawnee Mission, KS, 2016.
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M. A.; Marenich V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr., Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers N.; et al. Gaussian 16, Revision A. 03; Gaussian, Inc.: Wallingford CT, 2016.
- Runge E.; Gross E. K. U. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997–1000. 10.1103/physrevlett.52.997. [DOI] [Google Scholar]
- Petersilka M.; Gossmann U. J.; Gross E. K. U. Excitation energies from time-dependent density-functional theory. Phys. Rev. Lett. 1996, 76, 1212–1215. 10.1103/physrevlett.76.1212. [DOI] [PubMed] [Google Scholar]
- Vignale G. Real-time resolution of the causality paradox of time-dependent density-functional theory. Phys. Rev. A 2008, 77, 062511. 10.1103/physreva.77.062511. [DOI] [Google Scholar]
- Hohenberg P.; Kohn W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. 10.1103/physrev.136.b864. [DOI] [Google Scholar]
- Ridley J.; Zerner M. An intermediate neglect of differential overlap technique for spectroscopy: pyrrole and the azines. Theor. Chim. Acta 1973, 32, 111–134. 10.1007/bf00528484. [DOI] [Google Scholar]
- Zerner M. C.Semiempirical molecular orbital methods. In Reviews in Computational Chemistry II; Lipkowitz K. B., Boyd D. B., Eds.; Wiley-VCH: New York, 1991; pp. 313–365. [Google Scholar]
- Neto J. D. D. M.; Zerner M. C. New parametrization scheme for the resonance integrals (Hμν) within the INDO/1 approximation. Main group elements. Int. J. Quantum Chem. 2001, 81, 187–201. . [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.