

Abstract

A spectrophotometric technique has been applied for studying the reduction of chromium(VI) by poly(ethylene glycol) (PEG) as water-soluble and nontoxic synthetic polymer at a constant ionic strength of 4.0 mol dm–3 in the absence and presence of the ruthenium(III) catalyst. In the absence of the catalyst, the reaction orders in [Cr(VI)] and [PEG] were found to be unity and fractional first orders, respectively. The oxidation process was found to be acid-catalyzed with fractional second order in [H+]. The addition of Ru(III) was found to catalyze the oxidation rates with observation of zero-order reaction in [CrO42–] and fractional orders in both [PEG] and [Ru(III)], respectively. The PEG reduces the soluble toxic hexavalent Cr(VI) as a model pollutant to the insoluble nontoxic Cr(III) complex, which is known to be eco-friendly and more safer from the environmental points of view. The acid derivative of PEG was found to possess high affinity for the removal of poisonous heavy metal ions from contaminant matters by chelation. Formation of the 1:1 intermediate complex has been kinetically revealed. A consistent reaction mechanism of oxidation was postulated and discussed.
1. Introduction
Poly(ethylene glycol) (PEG) has commercial potentiality in a variety of fields such as biology, biomedical science, surface chemistry, and electrochemistry. This fact may be attributed to its unique properties such as chain flexibility, biocompatibility, non-immunogenicity, inert nature to most of biological molecules, and the basicity of the involved ether-oxygen into the main chain.1,2 Again, the PEG substrate and its derivatives have extensive applications in the synthesis of peptides of polymeric drugs,1,3 as carriers in polyamides4−7 and antistatic agents.8 It also used to improve the activity of the enzyme-metal ion,9−11 blood substitution,12 and blood circulation13 as well as a surface modifying agent of transition metal aluminides14 and for stabilization of water based on epoxy formulations.15 These advantages may be explained by the high hydrophilicity, no antigenicity, nontoxicity, and low cost of PEG along with the other mentioned properties.
Chemists and biochemists have paid much attention to the chemistry of chromium(VI) in the biological systems owing to its suspected carcinogen and mutagen symptoms caused by the high toxicity of this metal ion oxidant.16,17 The valences of chromium ions are ranging from +2 to +6 oxidation states. The toxic soluble chromium(VI) and nontoxic insoluble Cr(III) forms are the more stable oxidation states of chromium metal ion species. The latter being the more significantly safer and environmentally friendly.
Chromium(VI) is a labile oxyanion oxidant that existed in various forms such as hydro chromate (HCrO4–), chromate (CrO42–), or dichromate (Cr2O72–), depending on the pH of the solvent media. The flexibility to elucidate a variety of oxidation mechanisms in oxidation–reduction reactions containing Cr(VI) as an oxidant makes the respective redox system studies as a tool for the evidence of the chemical kinetics.
Although much attention has been focused by numerous investigators18−30 on the oxidation of organic alcohols by chromic acid, a few publications were reported on the oxidation of alcoholic macromolecules for either natural31−34 or synthetic polymers35 by this oxidant. This fact may be explained by the kinetic complexity arisen from the existence of either CrV or CrIV species as transient species formed during the reaction progression together with formation of Cr(III), which has a high affinity for chelation with the oxidation products to form a variety of insoluble complexes.29,30 However, the oxidation kinetics of PVA as a synthetic polymer by chromic acid has been studied in more details in acidic media;35 the natures of electron-transfer process and the transition states in the rate-determining steps still remain ambiguous and not complete.
In view of the above arguments and our interest on the oxidation kinetics of some alcoholic macromolecules using various oxidants in acidic media,32−35 the cited redox reaction seems to be of great significance to gain more knowledge on the role of the nature of both the reducing agent and the type of solvent on the kinetics and mechanistics of oxidation. Again, it is of interest to compensate the lack of information on the mechanism of the electron-transfer process whether of successive one-electron transfer in a sequence or of simultaneous two-electron changes in a single step as well as the nature of transition states in the rate-determining steps. Furthermore, this research work aims to present a simple method for the remediation of wastewater by eliminating the soluble carcinogenic Cr(VI) through the transformation of such species to the insoluble Cr(III) form together with synthesis of a low-cost coordination polymer oxidation product of the acid-PEG derivative as a new chelating agent. This method can be used for remediation of wastewater from simultaneous contaminated carcinogenic Cr(VI) and toxic heavy metal ions.
2. Results
2.1. Stoichiometry
The reaction stoichiometry was determined at constants of [H+] and ionic strength as described elsewhere.31−35 A stoichiometric ratio of 1.3 ± 0.1 mole for the ([Cr(VI)]consumed/[PEG]0) has been obtained after the reaction completion of the tested experimental runs. This result is corresponding to the following reaction stoichiometry
| 1 |
where H–(O–CH2–CH2)n–O–CH2–CH2OH corresponds to PEG, and H–(O–CH2–CH2)n–O–CH2COOH represents its acid derivative. The identification analyses were performed as described elsewhere36 The FTIR spectra (Figure 1) confirms the oxidation of the primary OH groups to the corresponding carboxylic acid of OCO at about 1640 (γas), an OCO of about 1420 (γs), and the observed broad peak at about 3504 cm–1 corresponds to the stretching vibration of O–H (γOH), respectively.
Figure 1.

FTIR Spectra of PEG and its acid derivative of PEG.
2.2. Influence of [CrO42–] and [PEG] on the Reaction Rates
The good linearity obtained from ln(absorbance) versus time plots for more than 87% of reaction completion as well as the independence of the oxidation rates on the Cr(VI) concentration in the range of 3–9 × 10–4 mol dm–3 may confirm the unity order in [Cr(VI)]. The slopes obtained from such linear plots are corresponding to the observed pseudo first-order rate constants, (kobs). The calculated values of kobs using the method of least-squares are summarized in Table 1. Again, the fractional first-order kinetics in [PEG] was revealed from the nonconstancy obtained by dividing the (kobs/[PEG]0) at fixed [CrO42–], which confirmed by the equation (kobs = [PEG]n). Moreover, such observed linearity along the positive intercept obtained from the plots of the reciprocals of the observed rate constants kobs versus [PEG] (Figure 2) may indicate the obedience of the oxidation reaction to the Michaelis–Menten kinetics for the formation of 1:1 intermediate complexes.
Table 1. Pseudo-First-Order Rate Constants Values (104kobs, s–1)a.
| rates dependency | concentration (mol dm–3) | |
|---|---|---|
| 104kobs (s–1) | ||
| [PEG]b | 0.05 | 0.09 |
| 0.67 | 1.53 | |
| [H+]c | 2.0 | 3.0 |
| 0.78 | 1.53 | |
| 103 [MnSO4] | 0.7 | 1.0 |
| 1.44 | 1.35 | |
| 104 [Ru(III)] | 0.7 | 2.0 |
| 2.79 | 3.66 | |
[CrO2–4] = 7 × 10–4, [H+] = 3.0, and I = 4.0 mol dm–3 at 20 °C. Experimental errors (±3%).
[H+] = 3.0 mol dm–3.
[PEG] = 0.09 mol dm–3.
Figure 2.

Plot of 1/kobs vs 1/ [PEG]. [CrO42–] = 7.0 × 10–4, [H+] = 3.0, and I = 4.0 mol dm–3 at 20 °C.
Some experimental runs were performed using the dichromate ion (Cr2O72 +) oxidant to examine the influence of variation of the oxidant conformation on the reaction kinetics. The values of rate constant obtained from the two forms of chromium(VI) were found to be very nearly similar (1.53 × 10–4 s–1 in the case of CrO42– and 1.46 × 10–4 s–1 in the case of Cr2O72–, respectively), indicating that the Cr(VI) species is the sole reactive species in the oxidation processes in both two cases.
2.3. Influence of [H+] on the Reaction Rates
Different initial concentrations of HClO4 acid were used at constants of ionic strength (using NaClO4) and other reagents to examine the influence of the [H+] ion on the rates of oxidation. Increasing the acid concentration was found to be accompanied by an increase in the reaction rates, indicating that the oxidation process was of an acid-catalyzed nature. This result was based on the transformation of the oxyanion (CrO42–) of the tetrahedral structure to the hydrated cation (Cr(H2O)63+) of the octahedral configuration that needs some H+ ions to be consumed in this transformation.37 Both HCl and H2SO4 acids were used to examine the influence of nature of the anions of the acid on the oxidation rates. The oxidation rates obtained were found to increase in the order HCl < HClO4 < H2SO4 with the values of 1.53, 0.12, and 0.03 × 10–4 s–1, respectively, at 20 °C. This result means that the anion of the acid plays a remarkable role in the oxidation process. It is well known that both SO42– and Cl– anions have high tendency to form complexes,38 whereas the ClO4– anion is known to be extremely inert. Therefore, it may be possible that the use of SO42– and Cl– anion species tend to form some intermediate complexes prior to the rate-determining steps, and hence, these complexes will lead to retard the oxidation processes rather than that in the case of an inert ClO4– anion.
2.4. Influence of Ionic Strength on the Reaction Rates
The effect of variation of the ionic strength on the reaction rates was found to be of great significant to shed some light on the reactive species in the rate-determining step. Therefore, some kinetic runs were performed at constant [H+] = 3.0 mol dm–3 as NaClO4 concentration was increased to 5.0 mol dm–3. The values of kobs were found to increase with increasing the ionic strength. A plot of ln kobs against I0.5 gave a good straight line with a positive slope as shown in Figure 3. However, the ionic strength used was deviated from that needed by the above Debye–Hückel equation; the ionic strength dependence was qualitatively as expected when considering the charges involved.39
Figure 3.

Typical plot of the ionic strength dependence of the observed first-order rate constant. [CrO42–] = 7.0 × 10–4, [PEG] = 1.3 × 10–1, [H+] = 3.0 mol dm–3 at 20 °C.
2.5. Test of Free-Radical Intervention
The negative test of added 10% (v/v) acrylonitrile to the reaction mixture was found to be surprising and contrary to our expectations. Therefore, to confirm the absence or presence of such free radicals, both the ESR technique and mercuric chloride (HgCl2) reagent were also examined to ascertain such testing. These latter two tests indicated that no precipitate was formed confirming the absence of such free-radicals intervention.
2.6. Influence of [Mn2+] Ions on the Reaction Rates
Much debate has been recognized in the past18 with respect to the involvement of chromium(IV) as an intermediate in redox reactions involving chromium(VI) as an oxidant. It is well known that manganese(II) has high affinity for trapping the Cr(IV) intermediate. The estimated redox potentials of CrVI/CrIII (E° = 1.33 V) and MnIII/MnII (E° = 1. 51 V) couples indicate that the oxidation of MnII by CrVI is thermodynamically unfavorable.40,41 Hence, the influence of Mn2+ added to the reaction mixtures on the values of the oxidation rates may confirm the involvement of Cr(IV) as an intermediate during the reaction progression. This is attributed to the occurrence of the following reaction
| 2 |
The occurrence of such above reaction should lead to a decrease in the reaction rates as was experimentally observed.
Most reports of the oxidation of organic substrates by Cr(VI) were found to exhibit such behavior of decreasing the oxidation rates on the addition of the Mn(II) ion.42,43 The oxidation strength of the formed Mn(III) of the protonated substrates as a competitor oxidant is negligibly small compared to that of chromic acid owing to the rapid disproportionation of Mn(III) under our experimental conditions of [H+] used.44
Therefore, the involvement of Cr(IV) as an intermediate during the reaction progression has been examined by adding manganous ions19,45,46 to the reaction mixtures in the present work. The added salts showed a gradual decrease in the reaction rates with increasing the added Mn+2 ions as shown in Table 1.
2.7. Influence of [Ru(III)] Catalyst on the Reaction Rates
The oxidation rates were found to be accelerated by the addition of the Ru(III) catalyst to the reaction mixtures. The experimental results revealed zero order in [CrO42–] and fractional orders in both [PEG] and [Ru(III)], respectively, in the case of presence of the Ru(III) catalyst. The results are listed in Table 1.
3. Discussions
In general, two reaction mechanisms for the electron-transfer process in redox reactions involving chromium(VI) as an oxidant are usually suggested. The first mechanism corresponds to simultaneous transfer of two electrons in a single step as CrVI → CrIV → CrII. The second one represents to the transfer of successive one-electron transfer in the sequence as CrVI → CrV → CrIV → CrIII. Hence, those two mechanisms may be considered for the oxidation of PEG by Cr(VI) with formation of either CrV and/or CrIV intermediate species19,45,46 The decrease in the rates of oxidation on the addition of the Mn2+ ion to the reaction mixtures may be considered as an indirect evidence for involvement of chromium(IV) as an intermediate species rather than that of chromium(V) species during the progress of such present redox reaction.42,43
Under our experimental conditions of [H+] used, the observed non-integral second-order dependence in [H+] is reasonable when taking the protolytic equilibrium constant (K) values for Cr(VI) into consideration45,47,48 as well as that the quantity of formation of the chromium dimer is negligibly small. This means that chromic acid can be assumed to be the main reactive species of the chromium ion, which confirmed by the calculated quantity of the formed H2CrO4 using the values of K(45,47,48) (80–95% of [Cr(VI)]T).
In terms of the above arguments and the experimental observations of hydrogen ion concentration dependence of the oxidation rates, a plausible reaction mechanism consistent with the observed kinetic results may be postulated. In such mechanism, both the oxidant and substrate tend for protonation at the [H+] used.19 The protonation of the two reactant substrates can be expressed by the following protolytic equilibria
| 3 |
| 4 |
Here S and SH+ correspond to the PEG substrate and its protonated form. On the other hand, K1 and K are the protonation constants of chromic acid and PEG reactants, respectively. This protonation was followed by the fast attack of chromic acid on the protonated PEG reductant giving the intermediate complex (C1) depending on the nature of the formed intermediates as defined by eq 5
| 5 |
The intermediate (C1) is slowly decomposed in the rate-determining step giving the corresponding aldehyde substrate with formation of Cr(IV) and/or Cr(IV) (red) as initial oxidation products as follows
| 6 |
where the symbols S represents the nonfree-radical aldehyde substrate. Hence, the aldehyde substrate formed is rapidly oxidized by two reaction pathways. This means that the oxidation occurs by either picking up of a further new oxidant molecule or by one of the formed Cr(IV) and/or Cr(V) transient species to give rise to the final oxidation products as described by eqs 7 and 8, respectively,
| 7 |
| 8 |
The rate-law expression, which corresponds to the change of the rate constant with changing both the [H+] and PEG substrate concentrations, can be written as follows
| 9 |
When [PEG]0 > > [Ox]0 and rearrangement, one concludes that
| 10 |
where [PEG]T is the analytical total concentration of the PEG substrate.
The rate expression defined by eq 10 requires that plots of 1/kobs versus either 1/[PEG] at constant [H+] or 1/[H+]2 at constant [PEG] to be linear with positive intercepts on 1/kobs axes as was experimentally observed. In view of the small negligible intercept observed in (Figure 2) and rearrangement, eq 10 can be reduced to the following simple form
| 11 |
where the apparent rate constants ka′ and ka″ equal to kaKK1K2 and kaK1K2, respectively. Plots of [H+][PEG]T/kobs versus 1/[H+] of eq 11 were linear with positive intercepts on the [H+][PEG]T/kobs axis (Figure 4) from those slopes and intercepts, the values of the apparent rate constants, k’a and k″a, and the protonation constants (K) of the PEG substrate can be evaluated.
Figure 4.

Plots of [H+] [PEG]/kobs against 1/[H+] at different hydrogen concentrations and various temperatures. [CrO42–] = 7.0 × 10–4, [PEG] =1.3 × 10–1, and I = 4.0 mol dm–3.
The calculated values of ka’ and ka″ by the method of least-squares are listed in Table 2. The values of protonation constants (K) were found to be in good agreement and with the same order of magnitude to that reported previously for oxidation of poly(vinyl alcohol) as synthetic alcoholic polymer by this oxidant.31 This result may be considered as indicative evidence to support the suggested mechanism.
Table 2. Apparent Rate Constants (k’a and k″a) and Protonation Constants (K) Valuesa.
| temperature
(°C) |
|||
|---|---|---|---|
| constants | 27 | 35 | 40 |
| 104k’a, dm9 mol–3 s–1 | 3.10 | 3.32 | 3.54 |
| 103 k″a, dm6 mol–2 s–1 | 2.85 | 3.80 | 4.39 |
| 10 K, dm3 mol–1 | 1.11 | 0.95 | 0.81 |
[CrO42–] = 7 × 10–4 and I = 4.0 mol dm–3 at different temperatures. Experimental errors (±3%).
The non-availability of the formation constants (K2) at different temperatures makes it difficult to evaluate the values of the rate constants of the elementary reaction ka. Therefore, the listed apparent rate constants are composite quantities of the rate, protonation, and the formation constants. Arrhenius and Eyring equations were applied to evaluate the activation parameters of the (ka’ and ka″) and second-order rate constant (kn). The calculated values of those rate constants using the least-square method along with those thermodynamic values for protonation of PEG are summarized in Table 3.
Table 3. Activation Parameters of Apparent Rate Constants and the Second-Order Rate Constants and Thermodynamic Parameters of Protonation Constant (K)a.
| parameter |
|||||
|---|---|---|---|---|---|
| constant | ΔH≠ (kJ mol–1) | ΔS≠(J mol–1 K–1) | ΔG293≠ (kJ mol–1) | Ea≠ (kJ mol–1) | A (mol–1 s–1) |
| ka’ | 5.57 | –293.60 | 93.07 | 8.17 | 1.19 × 102 |
| ka″ | 22.35 | –219.50 | 87.76 | 24.92 | 0.6 × 102 |
| kn | 14.37 | –249.25 | 73.05 | 16.87 | 1.62 × 102 |
| K | ΔH° (kJ mol–1) | ΔS°300 (J mol–1 K–1) | ΔG°300 (kJ mol–1) | ||
| +18.59 | +43.70 | +5.48 | |||
[CrO42–] = 7.0 × 10–4, [PEG] = 1.3 × 10–1, and I = 4.0 mol dm–3. Experimental error ±3%. Second-order rate constant measured at [H+] = 4.0 mol dm–3.
The observed negative value of ΔH° indicates that the protonation of the PEG substrate is of an exothermic nature. On the other hand, the large positive values for ΔG≠ may confirm the nonspontaneity of the complex formation prior to the rate-determining steps, that is, the activated complexes could be more ordered and more compactness than that of the reactants. This suggestion was supported by the observed large negative values of entropies of activations (ΔS≠).
The observed small activation energy value, Ea≠ (Table 3), means that the reactants to overcome the transition state or form the intermediate complexes do not need much energy for such purposes.
Generally, the values of activation entropies, ΔS≠, for redox reactions of innersphere nature were reported to be of negative signs, whereas that of positive ΔS≠ values were usually refer to redox reactions of outersphere mechanisms.45−50 Consequently, the observed negatively ΔS≠ values in Table 3 may indicate the predomination of the innersphere mechanism than that of the outersphere type for reduction of chromic acid by the PEG substrate.
Moreover, the small differences observed between the two rates of oxidation on using CrO42– and Cr2O72– species may be interpreted by the large steric hindrance faced the attack of Cr2O72– species of a large size on the protonated substrate of PEG to form the complex (C1) compared to that of small size CrO42– species. Moreover, the great similarity in the magnitude of the values of the rate constant using those two oxidants (Cr2O72– and CrO42–) may indicate that the reactive species in both cases are the Cr(VI) ion.
In general, the reaction mechanism of the present work may suggest the existence of three pathway routes for reduction of the chromium(VI) ion by PEG in the case of the absence of the Ru(III) catalyst as shown in Scheme 1A–C. The experimental results and data interpretations may suggest to neglect the pathway (B) since it involves the intervention of the free-radical mechanism, and the reaction was of unity order in [H+]. Again, reaction pathway (B) can be also excluded since it was based on the intervention of the free-radical mechanism.
Scheme 1. Speculated Mechanism of Reduction of Potassium Chromate by Poly(ethylene glycol) in Aqueous Perchlorate Solutions.

Consequently, the reaction pathway (A) of the nonfree radical intervention may be suggested as the more probable one for reduction of chromium(VI) by poly(ethylene glycol). This suggestion can be confirmed by the observed results of the second-order reaction in [H+] and the formation of Cr(IV) but not Cr(V) as an initial oxidation product (based on the results of the added Mn2+ ions). Furthermore, the kinetic results obtained from the ionic strength dependence of the reaction rates may support a reaction mechanism of two-electron changes of innersphere nature based on the signs of the reactive species in the rate-slow step together with the fact that it does not appear any report of the two-electron-transfer process of the outersphere nature up until now.49,50
Furthermore, the kinetics of the present redox reaction has been study in presence of Ru(III) to examine the effect of such a catalyst on the kinetics and mechanistic of the present oxidation–reduction reaction. Ruthenium(III) has been widely used as a catalyst51,52 in many other redox reactions that involving one or two equivalent oxidants in acidic or in alkaline solutions; It found that the addition of Ru(III) to the present reaction mixture was accompanied by an increase in the oxidation rates with first-order kinetics in both the catalyst and hydrogen ion concentrations, whereas zero order with respect to the [oxidant] was observed. This result indicates that the Ru(III) catalyst is first reacted with the chromic acid oxidant to form the intermediate complex (C2). Then, the formed complex (C2) reacts with the PEG to give a further intermediate complex (C3) with subtraction of a proton by a water molecule prior to the rate-determining step, followed by the transfer of electrons from the PEG substrate to the chromic acid in the rate-determining step to give the substrate (C̀3) and the reduced form of the oxidant as initial oxidation products as follows
| 12 |
| 13 |
Then, the substrate radical is rapidly decomposed giving the final oxidation product
| 14 |
In a similar manner to that applied for derivation of the rate-law expression of the former mechanism the rate-law for the changing of the reactants concentration can be expressed by
| 15 |
where [S]T is the analytical total concentration of the PEG and equals ([S]T = [S] + [SH+] + [C3]). Consequently, eq 15 on rearrangement yields
| 16 |
where 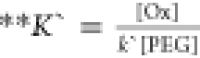
According to eq 16, a plot of the reciprocals of the observed rate constants (1/k′obs) against 1/ [Ru(III)] should be straight line with positive intercepts on the 1/k′obs axis as was experimentally observed. A typical plot is shown in Figure 5.
Figure 5.

Plot of 1/k′obs versus 1/[Ru (III)]. [CrO42–] = 7.0 × 10–4, [PEG] =1.3 × 10–1, [H+] = 3.0, and I = 4.0 mol dm–3 at 25 °C.
4. Conclusions
A conventional spectrophotometric technique has been applied for studying the reduction of Cr(VI) by PEG as a synthetic polymer at a constant ionic strength of 4.0 mol dm–3 in the absence and presence of the Ru(III) catalyst. In the absence of the Ru(III) catalyst, a unity order in [Cr(VI)] and fractional first-order kinetics in [PEG] were observed. The [H+] dependence of the rate-constants indicated that the oxidation process was of acid-catalyzed nature with fractional second–order kinetics in [H+]. Adding the Ru(III) catalyst to the reaction mixtures was found to catalyze the oxidation process with results of zero-order reaction in [CrO42–] and fractional first-order reactions in both [PEG] and [Ru (II)], respectively. The PEG reduces the soluble carcinogenic Cr(VI) to nontoxic insoluble Cr(III), whereas the oxidation product of PEG tends to remove the toxic polyvalent metal ions may present in the solvent media by chelation. The kinetic parameters were evaluated, and a suitable reaction mechanism for the oxidation process in terms of two-electron transfer of an inner-sphere nature was suggested and discussed.
5. Experimental Section
5.1. Materials and Preparations
Poly(ethylene glycol) 6000 (Merck KGA, 64271 Darmstadt, Germany) was used without further purification. Solution of PEG was prepared by adding the requisite amount of reagent powder to doubly distilled water. All other reagents were prepared and analyzed as described earlier.31−35 Sodium perchlorate as an inert electrolyte was used to adjust the ionic strength.
5.2. Kinetic Measurements
The kinetic measurements were conducted under pseudo-first-order conditions where polyethylene glycol (PEG) was present in a large excess over that of the oxidant’s concentration [CrO42–]. The course of reaction was followed by recording the decrease in absorbance of chromic acid at its absorption maximum, 350 nm, as a function of time as described elsewhere.31,53,54 It found that no interference from the reagents or the products occurred at this wavelength during the experimental measurements.
A Shimadzu UV-2101/3101 PC automatic scanning double-beam spectrophotometer fitted with a wavelength program controller using cells with a path length of 1 cm. The temperature was controlled within ±0.05 °C. The scanning spectral changes during the oxidation reaction are shown in Figure 6.
Figure 6.

Spectral changes (250–800 nm) in the reduction of potassium chromate by poly(ethylene glycol) in aqueous perchloric acid. [CrO42–] = 7.0 × 10–4, [PEG] =1.3 × 10–1, [H+] = 3.0, and I = 4.0 mol dm–3 at 20 °C. (a) In absence of Ru(III), (b) after reaction completion, and (c) in presence of Ru(III).
Acknowledgments
This work supported by Chemistry Department, Faculty of Science, Assiut University, Assiut 71516, Egypt.
Appendix (I)
Under the cited experimental conditions of the [H+] used, both reactants have high tendency for protonation in aqueous acidic media forming the corresponding reactive protonated forms as follows
| 3 |
| 4 |
Then, the redox reaction takes place by attacking the chromic acid oxidant on the protonated PEG (SH+) forming an intermediate complex (C1) prior to the rate-determining step with releasing a water molecule as follows
| 5 |
Then, a slow decomposition of the formed complex (C1) occurs in slow step giving (C̀) and (red) as initial oxidation products.
Considering the analytical total concentration of PEG [S]T defined by eq i
| i |
The concentration of PEG can be evaluated by the following equation,
| ii |
Since the general rate-law equation of the present redox reaction is expressed by eq iii
| iii |
Then, the change of the rate constant with changing the [H+] and [PEG] can be expressed by the following equation
Under the pseudo-first-order conditions of [PEG]0 ≥ [CrVI], the rate-law expression is usually expressed by the formula,
| iv |
Comparing both eqs iv and 9, one concludes that
| v |
| 10 |
The small intercept observed
in Figure 2 leads to
neglecting the term  in eq 10 to give
the following simple relationship
in eq 10 to give
the following simple relationship
| 11 |
where ka` = kaKK1K2 and ka″ = kaK1K2.
The authors declare no competing financial interest.
References
- Harris J. M.Poly(ethylene Glycol) chemistry, introduction to chemistry and biological applications of poly (ethylene glycol); Plenum Press: New York, 1993. [Google Scholar]
- Kacar G. Characterizing the structure and properties of dry and wet polyethylene glycol using multi-scale simulations. Phys. Chem. Chem. Phys. 2018, 20, 12303–12311. 10.1039/C8CP01802B. [DOI] [PubMed] [Google Scholar]
- Royer G. P.; Anantharmaiah G. M. Peptide synthesis in water and the use of immobilized carboxypeptidase Y for deprotection. J. Am. Chem. Soc. 1979, 101, 3394–3396. 10.1021/ja00506a051. [DOI] [Google Scholar]
- Ulbrich K.; Strohalm J.; Kopec̆ek J. Poly(ethylene glycol)s containing enzymatically degradable bonds. Makromol. Chem. 1986, 187, 1131–1144. 10.1002/macp.1986.021870510. [DOI] [Google Scholar]
- Ouchi T.; Hagihara Y.; Takahashi K.; Takano Y.; Igarashi I. Synthesis and antitumor activity of poly(ethylene glycol)s linked to 5-fluorouracil via a urethane or urea bond. Drug Des. Discovery 1992, 9, 93–105. [PubMed] [Google Scholar]
- Greenwald R. B.; Gilbert C. W.; Pendri A.; Conover C. D.; Xia J.; Martinez A. Drug delivery systems: Water soluble taxol 2′-poly(ethylene glycol) ester prodrugs design and in vivo effectiveness. J. Med. Chem. 1996, 39, 424–431. 10.1021/jm950475e. [DOI] [PubMed] [Google Scholar]
- Zalipsky S.; Gilon C.; Zilkha A. Attachment of drugs to polyethylene glycols. Eur. Polym. J. 1983, 19, 1177–1183. 10.1016/0014-3057(83)90016-2. [DOI] [Google Scholar]
- Pechar M.; Strohalm J.; Ulbrich K.; Schacht E. Biodegradable drug carriers based on poly(ethylene glycol) block copolymers. Macromol. Chem. Phys. 1997, 198, 1009–1020. 10.1002/macp.1997.021980408. [DOI] [Google Scholar]
- Jan I.; Frantisek C. Inst. Entomol. – genetic control of pest lepidoptera, genotoxicity testing in drosophila. Czech Pat. 1987, 234, 832–835. [Google Scholar]
- Veyarma N.; Nakata M.; Nakamura A. Conjugates with enzyme-metal ion for improved stability and activity of enzymes. Poly J. (Tokkyo) 1985, 17, 721–727. [Google Scholar]
- Tukayuki Y.; Takeshi M.; Katsunobu T.; Yuji S.; Yukata T.; Yuji I. Conjugates with enzyme-metal ion for improved stability and activity of enzymes. Biochem. Biophys. Res. Commun. 1987, 145, 908–914.3593378 [Google Scholar]
- Katsunobu T.; Kimiko O.; Takayuki Y.; Yuji S.; Yoh K.; Ayako M.; Yuji I. Conjugates with enzyme-metal ion for improved stability and activity of enzymes. J. Biotechnol. 1988, 8, 135–140. [Google Scholar]
- Yabuki A.; Yamaji K.; Ohki H.; Iwashita Y. Characterization of a pyridoxalated hemoglobin-polyoxyethylene conjugate as a physiologic oxygen carrier. Transfusion 1990, 30, 516–520. 10.1046/j.1537-2995.1990.30690333482.x. [DOI] [PubMed] [Google Scholar]
- Blume G.; Cevc G. Molecular mechanism of the lipid vesicle longevity in vivo. Biochim. Biophys. Acta 1993, 1146, 157–168. 10.1016/0005-2736(93)90351-Y. [DOI] [PubMed] [Google Scholar]
- Takerkart G.; Segard E.; Monsigny M. Partition of trypsin in two-phase systems containing a diamidino-α, ω-diphenylcarbamyl poly (ethylene glycol) as competitive inhibitor of trypsin. FEBS Lett. 1974, 42, 218–220. 10.1016/0014-5793(74)80789-1. [DOI] [PubMed] [Google Scholar]
- Losi M. E.; Amrhein C.; Frankenberger W. T. Jr. Environmental biochemistry of chromium. Rev. Environ. Contam. Toxicol. 1994, 136, 91–121. 10.1007/978-1-4612-2656-7_3. [DOI] [PubMed] [Google Scholar]
- a Saha R.; Nandi R.; Saha B. Sources and toxicity of hexavalent chromium. J. Coord. Chem. 2011, 64, 1782–1806. 10.1080/00958972.2011.583646. [DOI] [Google Scholar]; b Saha R.; Saha I.; Nandi R.; Ghosh A.; Basu A.; Ghosh S. K.; Saha B. Application of Chattim tree (devil tree, Alstonia scholaris) saw dust as a biosorbent for removal of hexavalent chromium from contaminated water. Can. J. Chem. Eng. 2013, 91, 814–821. 10.1002/cjce.21703. [DOI] [Google Scholar]
- Das A. K. Micellar effect on the kinetics and mechanism of chromium (VI) oxidation of organic substrates. Coord. Chem. Rev. 2004, 248, 81–99. 10.1016/j.cct.2003.10.012. [DOI] [Google Scholar]
- Khan Z.; Din K. U. Effect of manganese (II) ions on the oxidation of malic and oxaloethanoic acids by aqueous HCrO–4. Transition Met. Chem. 2001, 26, 672–678. 10.1023/A:1012056310950. [DOI] [Google Scholar]
- Lee D. G.The oxidation of organic compounds by permanganate ion and hexavalent chromium; Open Court: La Salle IL,1980. [Google Scholar]
- Wiberg K. B.; Schafer H. Direct observation of intermediates in the chromic acid oxidation of secondary alcohols. J. Am. Chem. Soc. 1967, 89, 455–457. 10.1021/ja00978a054. [DOI] [Google Scholar]
- Acharjee A.; Rakshit A.; Chowdhury S.; Malik S.; Barman M. K.; Ali M. A.; Saha B. Micellar catalysed and heteroaromatic base promoted rate enhancement of oxidation of an alicyclic alcohol in aqueous medium. J. Mol. Liq. 2019, 277, 360–371. [Google Scholar]
- Sar P.; Ghosh A.; Malik S.; Ray D.; Das B.; Saha B. Selective heteroaromatic nitrogen base promoted chromium(VI) oxidation of isomeric pentanols in aqueous micellar media at room temperature. J. Ind. Eng. Chem. 2016, 42, 53–62. 10.1016/j.jiec.2016.07.028. [DOI] [Google Scholar]
- Mukherjee K.; Saha R.; Ghosh A.; Ghosh S. K.; Saha B. Efficient combination of promoter and catalyst for chromic acid oxidation of propan-2-ol to acetone in aqueous acid media at room temperature. Spectrochim. Acta, Part A 2013, 101, 294–305. 10.1016/j.saa.2012.09.095. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Saha R.; Saha B. Suitable combination of promoter and micellar catalyst for kilo fold rate acceleration on propanol to propionaldehyde conversion in aqueous media. J. Ind. Eng. Chem. 2014, 20, 345–355. 10.1016/j.jiec.2013.03.028. [DOI] [Google Scholar]
- Saha R.; Ghosh A.; Saha B. Combination of best promoter and micellar catalyst for chromic acid oxidation of 1-butanol to 1-butanal in aqueous media at room temperature. Spectrochim. Acta, Part A 2014, 124, 130–137. 10.1016/j.saa.2013.12.101. [DOI] [PubMed] [Google Scholar]
- Chowdhury K. M.; Mandal J.; Saha B. Micellar catalysis of chromium (VI) oxidation of ethane-1, 2-diol in the presence and absence of 2, 2′-bipyridine in aqueous acid media. J. Coord. Chem. 2009, 62, 1871–1878. 10.1080/00958970802687547. [DOI] [Google Scholar]
- Ghosh S. K.; Basu A.; Saha R.; Ghosh A.; Mukherjee K.; Saha B. Micellar catalysis on picolinic acid promoted hexavalent chromium oxidation of glycerol. J. Coord. Chem. 2012, 65, 1158–1177. 10.1080/00958972.2012.669035. [DOI] [Google Scholar]
- Beattie J. K.; Haight G. P. Jr. Chromium (VI) oxidations of inorganic substrates. Inorg. React. Mech. 1972, 17, 93–145. [Google Scholar]
- Mitewa M.; Bontchev P. R. Chromium(V) coordination chemistry. Coord. Chem. Rev. 1985, 61, 241–272. 10.1016/0010-8545(85)80006-0. [DOI] [Google Scholar]
- Abdel-Hamid M. I.; Khairou K. S.; Hassan R. M. Kinetics and mechanism of permanganate oxidation of pectin polysaccharide in acid perchlorate media. Eur. Polym. J. 2003, 39, 381–387. 10.1016/S0014-3057(02)00217-3. [DOI] [Google Scholar]
- Ahmed G. A.-W.; Khairou K. S.; Hassan R. M. Kinetics and mechanism of oxidation of chitosan polysaccharide by permanganate ion in aqueous perchlorate solutions. J. Chem. Res. 2003, 2003, 182–183. 10.3184/030823403103173598. [DOI] [Google Scholar]
- Hassan R. M. New coordination polymers. III: Oxidation of poly (vinyl alcohol) by permanganate ion in alkaline solutions. Kinetics and mechanism of formation of intermediate complex with a spectrophotometric detection of manganate(VI) transient species. J. Poly. Sci. Poly. Inter. 1993, 30, 5–9. [Google Scholar]
- Hassan R. M.; Abdel-Kader D. A.; Ahmed S. M.; Fawzy A.; Zaafarany I. A.; Asghar B. H.; Takagi H. D. Acid-catalyzed oxidation of carboxymethyl cellulose. Kinetics and mechanism of permanganate oxidation of carboxymethyl cellulose in acid perchlorate solutions. Catal. Commun. 2009, 11, 184–190. 10.1016/j.catcom.2009.09.023. [DOI] [Google Scholar]
- Abdel-Hamid M. I.; Ahmed G. A.-W.; Hassan R. M. Kinetics and mechanism of oxidation of poly(vinyl alcohol) macromolecule by chromic acid in aqueous perchloric acid. Eur. Polym. J. 2001, 37, 2201–2206. 10.1016/S0014-3057(01)00128-8. [DOI] [Google Scholar]
- Pretch E.; Clerc T.; Seibl J.; Simon W.. Tables of spectral data for structure determination of organic compounds; Springer-Verlag: Berlin, Heidelberg, 1983. [Google Scholar]
- Hicks K. W. Kinetics of the permanganate ion-potassium octacyanotungstate (IV) reaction. J. Inorg. Nucl. Chem. 1976, 38, 1381–1383. 10.1016/0022-1902(76)80159-5. [DOI] [Google Scholar]
- Cotton F. A.; Wilkinson W. G.; Murillo C. A.; Bochmann M.. Advanced Inorganic Chemistry; Wiley-Eastern: New York, 1996. [Google Scholar]
- Laidler K. J.Chemical Kinetics; McGraw-Hill: New York, 1965. [Google Scholar]
- Manhas M. S.; Kumar P.; Mohammed F.; Khan Z. Oxidative degradation of non-ionic surfactant (TritonX-100) by chromium (VI). Colloids Surf., A 2008, 320, 240–246. 10.1016/j.colsurfa.2008.02.003. [DOI] [Google Scholar]
- Milazzo G.; Caroli S.; Sharma V. K.. Tables of standard electrode metal potentials; Wiley & Sons: New York, 1978. [Google Scholar]
- Gupta K. K. S.; Chakladar J. K. Kinetics of the chromic acid oxidation of arsenic (III). J. Chem. Soc., Dalton Trans. 1974, 222–225. 10.1039/dt9740000222. [DOI] [Google Scholar]
- Sengupta K. K.; Chakladar J. K.; Chatterjee A. K. Kinetics of the oxidation of hypophosphorous and phosphorous acids by chromium (VI). J. Inorg. Nucl. Chem. 1973, 35, 901–908. 10.1016/0022-1902(73)80460-9. [DOI] [Google Scholar]
- Adler S. J.; Noyes R. M. The mechanism of the permanganate-oxalate reaction. J. Am. Chem. Soc. 1955, 77, 2036–2042. 10.1021/ja01613a002. [DOI] [Google Scholar]
- Espenson J. H. Oxidation of transition metal complexes by chromium (VI). Acc. Chem. Res. 1970, 3, 347–353. 10.1021/ar50034a004. [DOI] [Google Scholar]
- Stewart R.Oxidation in Organic Chemistry; Wiberg K. B., Ed.. Academic Press: New York,1965. [Google Scholar]
- Banaś B. A stopped-flow study of the oxidation reaction of ascorbic acid with chromic acid. Inorg. Chim. Acta 1981, 53, L13–L15. 10.1016/S0020-1693(00)84725-8. [DOI] [Google Scholar]
- Tong J. Y.-P.; King E. L. A spectrophotometric investigation of the equilibria existing in acidic solutions of chromium (VI)1-3. J. Am. Chem. Soc. 1953, 75, 6180–6186. 10.1021/ja01120a022. [DOI] [Google Scholar]
- Hassan R. M.; Mousa M. A.; Wahdan M. H. Kinetics and mechanism of oxidation of β-phenylalanine by permanganate ion in aqueous perchloric acid. J. Chem. Soc., Dalton Trans. 1988, 605–609. 10.1039/DT9880000605. [DOI] [Google Scholar]
- Hassan R. M. A mechanistic approach to the kinetics of oxidation of uranium (IV) by hexachloroplatinate (IV) in aqueous perchlorate solutions. Evidence of the formation of a binuclear intermediate complex. J. Phys. Chem. A 2011, 115, 13338–13345. 10.1021/jp204991w. [DOI] [PubMed] [Google Scholar]
- Vijayasri K.; Rajaram J.; Kuriacose J. C. RuCl2(PPh3)3 Catalyzed oxidation of secondary alcohols with N-methylmorpholine N-oxide. J. Chem. Sci. 1986, 97, 125–132. 10.1007/BF03040768. [DOI] [Google Scholar]
- Manikyamba P. Ru(III)-Catalyzed oxidation of pyruvic acid by iodate-A kinetic study. React. Kinet. Catal. Lett. 2003, 78, 169–173. 10.1023/A:1022534521041. [DOI] [Google Scholar]
- Hassan R. M.; Ahmed S. M.; Fawzy A.; Abdel-Kader D. A.; Ikeda Y.; Takagi H. D. Acid-catalyzed oxidation of carboxymethyl cellulose polysaccharide by chromic acid in aqueous perchlorate solutions. A kinetics study. Catal. Commun. 2010, 11, 611–615. 10.1016/j.catcom.2010.01.006. [DOI] [Google Scholar]
- Zaafarany I. A.; Khairou K. S.; Hassan R. M. Acid-catalysis of chromic acid oxidation of kappa-carrageenan polysaccharide in aqueous perchlorate solutions. J. Mol. Catal. A: Chem. 2009, 302, 112–118. 10.1016/j.molcata.2008.12.005. [DOI] [Google Scholar]