

Abstract
GTP hydrolysis is central to biology, being involved in regulating a wide range of cellular processes. However, the mechanisms by which GTPases hydrolyze this critical reaction remain controversial, with multiple mechanistic possibilities having been proposed based on analysis of experimental and computational data. In this mini-review, we discuss advances in our understanding of biological GTP hydrolysis based on recent computational studies and argue in favor of solvent-assisted hydrolysis as a conserved mechanism among GTPases. A concrete understanding of the fundamental mechanisms by which these enzymes facilitate GTP hydrolysis will have significant impact both for drug discovery efforts and for unraveling the role of oncogenic mutations.
Introduction
Guanosine triphosphate (GTP) hydrolysis is a biologically crucial reaction, involved in regulating almost all cellular processes. Therefore, GTPases are important drug targets and thus the topic of substantial experimental and computational research effort.1−6 However, there remain many open questions with regard to the preferred reaction pathway for GTP hydrolysis, as well as the nature of the corresponding transition states. This continued uncertainty hampers both our fundamental understanding of biological GTP hydrolysis and ongoing drug discovery efforts.
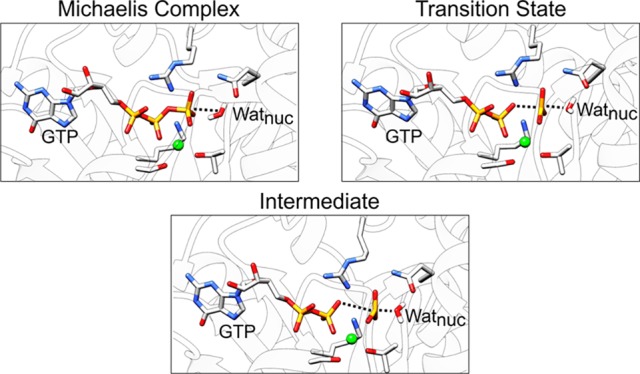
It should be noted that phosphoryl transfer is highly complex, and even in the seemingly simple case of nonenzymatic GTP hydrolysis, widely varying mechanistic proposals have been advanced, that can be grouped into two general categories: solvent-assisted pathways either with or without general-base catalysis (Figures 1A and B) or a substrate assisted-pathway in which the phosphate being attacked deprotonates the incoming nucleophile (Figure 1C, for detailed discussion see e.g. ref (7)). In the case of the corresponding GTPase-catalyzed reactions, similar mechanistic proposals have been put forward, focusing either on the involvement of an active site glutamine as the catalytic base that activates the nucleophilic water molecule or on substrate-assisted pathways in which a nonbridging phosphate oxygen of the GTP itself deprotonates the incoming nucleophile. Neither of these pathways, however, has been able to satisfactorily account for all key experimental observables, and no other candidate for the catalytic base exists in the active sites of these enzymes. It is highly curious how these enzymes are able to achieve efficient catalysis of GTP hydrolysis without the presence of an obvious candidate for the catalytic base, and it has therefore been questioned based on analysis of existing experimental data whether such a base is even necessary.2
Figure 1.

Potential mechanisms for both enzymatic and nonenzymatic GTP hydrolysis. Shown here are (A, B) solvent-assisted pathways that can proceed either with or without general-base catalysis, as well as (C) a substrate-assisted pathway in which the phosphate group receives a proton from the attacking water molecule. Reprinted with permission from ref (7) (direct link: https://pubs.acs.org/doi/10.1021/jacs.9b03193). Copyright 2019 American Chemical Society.
We recently provided significant advances in addressing this question by quantitatively reproducing experimental kinetic isotope effects for the nonenzymatic hydrolysis of a phosphate monoester dianion,8 allowing us to discriminate between different mechanisms. We then demonstrated that the mechanistic preference is highly leaving-group dependent, with phosphate monoesters with good leaving groups such as guanosine diphosphate (GDP) predominantly preferring to react via loose, dissociative solvent-assisted pathways, whereas those with poor leaving groups switch to a more compact substrate-assisted pathway.9 This provided further indications that maybe the answer to the mechanistic controversy with regard to enzyme-catalyzed GTP hydrolysis is simply that a solvent-assisted pathway does not require an explicit general base and that the dominant mechanism in solution is also the dominant mechanism in the GTPase active site.7,10 In this mini-review, we will provide a brief overview of the current state-of-the-art in modeling biological GTP hydrolysis, including discussing a number of recent QM/MM and metadynamics studies, in addition to our own recent work. We will highlight ongoing challenges in modeling these complex systems, as well as potential future directions for the field, as new mechanistic insights may make it finally possible to target these historically “undruggable” enzymes.
Biological GTP Hydrolysis
GTPases are a large superclass of structurally and mechanistically diverse hydrolytic enzymes, which catalyze the conversion of guanosine triphosphate (GTP) to guanosine diphosphate (GDP) and inorganic phosphate (Pi). A key feature of these enzymes is their role as “molecular switches”, harnessing conformational transitions between active GTP-bound “ON” states and inactive GDP-bound “OFF” states to control various cellular processes. In doing so, they regulate all stages of cellular function, from signaling cascades to cell migration, polarity, adhesion, cytoskeletal organization, proliferation, and apoptosis.1 These transitions can involve fairly significant conformational changes and are facilitated by interaction with different external regulatory proteins, the so-called “GTPase activating proteins” (GAPs). These regulatory proteins also contribute to substantially increasing the rates of GTP hydrolysis by these enzymes by up to 105-fold, depending on the specific GTPase-regulatory protein combination.3
While a range of different GTPases exist with different structures and active site architectures based on biological context,3 the most studied include small GTPases such as Ras GTPase and Rho GTPase, as well as elongation factors such as elongation factor thermo-unstable (EF-Tu).5 Although all these GTPases catalyze the same chemical reaction, the mechanistic strategies by which this is achieved (and the corresponding catalytic rates) vary between these different enzymes through their use of active-site architectures with key differences in the identity and positioning of the main catalytic residues7 (Figure 2). However, two key residues are of interest in these enzymes: in small GTPases such as Ras, of particular interest is the role of an active site arginine, the so-called “arginine finger”,3 which is either inserted into the active site by the regulatory protein (as in Ras and Rab, Figures 2A and B) or intrinsic to the GTPase (as in Gαi, Figure 2C) and which provides additional electrostatic stabilization to the transition state. In addition, an active site glutamine (in the case of Ras, Rab, and Gαi, Figures 2A to C) or histidine (Figure 2D) acts to position the nucleophile in an optimal position for nucleophile attack on the γ-phosphate of the GTP.
Figure 2.
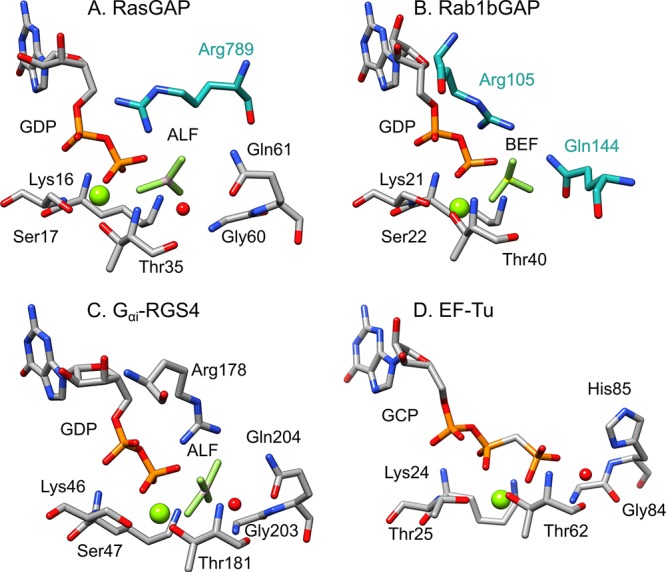
Active sites of three different signaling proteins: (A) RasGAP in complex with AlF3 (AF3) and a phosphonate GDP analogue (GNP) (PDB ID: 1WQ1(11,12)), (B) human Rab1 in complex with the GTPase activating protein (GAP) domain of TBC1D20 and BeF3– (BEF) (PDB ID: 4HLQ(12,13)), and (C) the α-subunit of a heterotrimeric G protein in complex with GDP and AlF4– (ALF) (PDB ID: 1AGR(12,14)). Shown here for comparison is also the active site of (D) Elongation Factor Thermo unstable (EF-Tu) in complex with a GTP analogue (GCP, E. coli numbering) (PDB ID: 4V5L(12,15)). Residues from each GTPase are highlighted in gray, and from the GAPs in cyan. The residues inserted by the regulatory proteins are specifically (A) the arginine finger in the RasGAP complex, (B) the arginine and glutamine fingers in the RabGAP complex. In case (C), the arginine (Arg178) and glutamine (Gln204) are intrinsic to the GTPase itself, and the catalytic stimulation by the regulatory protein is thus entirely allosteric through a so-called “dual-finger mechanism”. Adapted with permission from ref (7) (direct link: https://pubs.acs.org/doi/10.1021/jacs.9b03193). Copyright 2019 American Chemical Society.
There has been significant debate about the roles of both the glutamine and histidine in catalysis by these enzymes, focusing in particular on whether they act as an active site base to activate catalytic water molecule or merely act to position it in the active site through hydrogen bonding interactions. In the case of EF-Tu, structural information suggested that the histidine acts as a general base,15 but computational studies have indicated that this histidine is in fact most likely protonated due to a greatly elevated pKa.16,17 It has similarly been suggested that the active site glutamine can take on a similar role in Ras and related enzymes, largely due to the absence of other obvious general bases in the active site (see discussion in e.g. refs (2 and 7)); however, once again due to the extremely low pKa of the carbonyl group (estimated to be ∼ –2), this scenario is unlikely, raising the question of what the mechanism of catalysis actually is, and, here, computational work can play a significant role in unraveling putative mechanistic pathways as well as the role of the regulatory proteins in facilitating catalysis. We note that we have discussed the potential biological role of the Mg2+ in detail in earlier work7 (see also references cited therein) and, therefore, do not repeat it here.
Recent Computational Studies
Molecular simulations have historically played an important role in expanding our understanding of biological GTP hydrolysis.5 Our focus here will be on contributions made by recent computational work on Ras and related enzymes; note that in the interest of space conservation, we do not discuss here computational work on elongation factors, as this has been discussed in great detail elsewhere.18
There has been extensive research efforts in understanding the mechanism of GTPases, including the protonation state of the substrate. In particular, a neutron diffraction study of Ras in complex with the GTP analogue GppNHp suggested potential protonation of the analogue,19 arguing that the Ras active site significantly increases the pKa of the bound nucleotide and questioned the conventional view that the nucleotide is fully deprotonated. However, this was challenged by Mann and co-workers,20 who computed infrared and NMR spectra from QM/MM simulations, which were compared to prior work. They also provided novel spectra of GTP and several GTP analogues bound to lipidated Ras, on a membrane system under near-native conditions, confirming that the nucleotide is indeed bound to Ras in its fully deprotonated form at the start of the hydrolysis reaction and is not protonated. In detailed crystallographic and quantum chemical analysis of an analogous system, a quaternary complex of Rho·GAP·GDP·Pi,21 the authors argue for a complex network of hydrogen bonds and sequential proton transfers, including two low-barrier hydrogen bonds (LBHB) introduced by the GAP, that facilitate reaction progress and product release.
We note that the proton shuttling proposed in ref (21) is based on the assumption of a substrate-assisted pathway (Figure 1C), in which the attacking water molecule in the Rho active site is initially deprotonated by the bound nucleotide. Similar assumptions of mobile protons have been put forward for Ras22 and hGBP1,23,24 in the former case involving the active site glutamine shown in Figure 2, and in the latter case a more complex proton transfer chain involving multiple active site side chains and an intermediary water molecule. However, both ref (22) and refs (23 and 24) have suggested that the mechanisms involved for Ras and hGBP1, respectively, are complex and that the preferred mechanism can be modulated by mutations.
In a recent study, we proposed a much simpler model for GTP hydrolysis by Ras and related enzymes, building on our early work on the nonenzymatic reaction.7,8,10 Specifically, we performed detailed empirical valence bond simulations to compare solvent- and substrate-assisted pathways in three distinct GTPases: Ras, Rab, and the Gαi subunit of a heterotrimeric G-protein, both in the presence and in the absence of the corresponding GTPase activating (GAP/RGS) proteins.7 Our results, which are in excellent agreement with experimental observables, demonstrate that, in all cases, the preferred mechanism for GTP hydrolysis is a conserved solvent-assisted pathway by at least 7 kcal mol–1, eliminating the need for a general base in the active site (Figure 3). This pathway involves the rate-limiting nucleophilic attack of a water molecule, leading to a short-lived intermediate that tautomerizes to form H2PO4– and GDP as the final products (Figures 1A and 3). This solvent assisted model is in excellent agreement with experimental data, that have suggested a loose dissociative transition state for GTP hydrolysis in the Ras and RasGAP active sites, as well as analysis of kinetic data, which suggests that there is little kinetic benefit to deprotonation of the nucleophile (for details, see e.g. ref (2) and references cited therein). We note that a similar transition state has also been hinted at for RhoA-catalyzed GTP hydrolysis, based on 19F NMR experiments and quantum chemical calculations,25 further strengthening our hypothesis that this mechanism is conserved among Ras and related enzymes.7
Figure 3.

Calculated free energy profiles for the (A) solvent- and (B) substrate-assisted hydrolysis of GTP in aqueous solution, and as catalyzed by Ras and the RasGAP complex. (C) Activation free energies for each of the two mechanisms, as catalyzed by a range of GTPases (Ras, Rab and Gαι, both with and without their corresponding regulatory proteins, as well as the Ras Q61H mutant). Note that as shown in panel A, the initial phosphoryl transfer reaction in the solvent-assisted hydrolysis of GTP leads to a short-lived intermediate (INT), which will then tautomerize to yield the final product, which is chemically identical to the final product state for the substrate-assisted pathway. A similar pathway was observed in quantum chemical calculations of phosphate esters in aqueous solution.8−10 For further details, see ref (7). Reprinted with permission from ref (7) (direct link: https://pubs.acs.org/doi/10.1021/jacs.9b03193). Copyright 2019 American Chemical Society.
In our recent work,7 we also made two key observations with regard to the role of the GAP protein (or RGS protein in the case of Gαι): (1) that the GAP/RGS protein sequesters the active site from solvent, thus affecting the local dielectric constant of the active site, contributing to the enhancement of the GTP hydrolysis rate, and (2) that the GAP/RGS protein rigidifies the active site, both reducing its volume and reducing the conformational space of the catalytically critical arginine and glutamine residues (Figure 2), thus creating a better preorganized active site and facilitating efficient catalysis. Our first observation is further supported by molecular dynamics and QM/MM studies performed by Tichauer et al.,26,27 who have explored water distributions and electron density within wild-type NRas and Q61 mutants. This work has shown that the precise placement of water molecules in the active site is important and that substitution of Q61 (ideally GDP-like) not only leads to displacement of water molecules from the active site but also disrupts the electron density of the bound nucleotide, which can explain the reduced hydrolysis rate when this residue is mutated. Interestingly, however, and as also pointed out by the authors, significant differences in the water distribution at the active site are observed when comparing results from pure classical molecular dynamics to those obtained from hybrid QM/MM molecular dynamics simulations.27 Here, the QM/MM molecular dynamics results provide better agreement with a putative catalytically favorable conformation of the enzyme. Our second observation is further supported by a μs time scale molecular dynamics simulation study by Sayyed-Ahmed et al.,28 who explored the dynamics of both wild-type H- and K-Ras as well as variants carrying the oncogenic G12D, G12V, and G13D variants. They demonstrated that the oncogenic variants led to altered conformational ensembles of the protein, in particular involving differences in both flexibility of and coupling between the switch regions of the protein, as well as in intraprotein and intermolecular interactions and fluctuations. This in turn has implications for Ras GTPase regulation and activity, including effector or exchange factor binding, as well as Ras dimerization and Ras–membrane interactions.
Conclusions and Outlook
It is clear that due to their biological importance and their role in regulating cellular signaling pathways, GTPases are important drug targets, in particular for cancer therapeutics. However, targeting these enzymes is nontrivial, leading GTPases such as Ras to be described as “undruggable” (although recent years have seen growing reason for optimism29−31). Part of the challenge has come from the fact that, despite a tremendous wealth of kinetic, biochemical, structural, and computational data, the catalytic mechanisms and mode of regulation of these enzymes remain controversial,2,4,5 in no small part due to the tremendous complexity of this seemingly simple reaction. Here, we show that molecular simulation can be a valuable tool to rationalize experimental data and predict mechanisms, provided the simulation model can predict all key experimental observables, and a number of key recent studies have been described in this mini-review. We believe that the strongest evidence from the computational indicates that a solvent-assisted pathway is the most likely operative pathway in Ras and related enzymes, as expected from experimental work.2 The insight these studies provide is an important step forward both to be able to finally target the undruggable as well as for rationalizing the role of oncogenic mutants.
Acknowledgments
This work was funded by Stiftelsen Olle Engkvist Byggmästare (grant 190-0335) and the Knut and Alice Wallenberg Foundation (Wallenberg Academy Fellowship followed by Wallenberg Scholar fellowship to S.C.L.K., KAW 2018.0140 and 2019.0431, respectively).
Biographies

Ana Rita Calixto received her Bachelor (2011) and Master (2013) degrees in Biochemistry from the University of Porto, Portugal. In 2018, she completed her Ph.D. in Sustainable Chemistry, in the Theoretical and Computational Biochemistry Research group at the same university, under the supervision of Professor Pedro Alexandrino Fernandes and Professor Maria João Ramos. Currently, she is a postdoctoral fellow at Uppsala University with Professor Lynn Kamerlin. Her scientific research focuses on applying different computational methods to understand biological processes, such as enzyme catalysis and protein dynamics. Her recent work is focused on phosphate hydrolysis by biological molecules. She is studying a large range of GTPases to understand their catalytic mechanism and regulation.

Cátia Moreira received her Bachelor (2009) and Master (2011) degrees in Biochemistry from the University of Porto, Portugal. During her first years as a researcher, she worked in microbiology at University of Porto (Portugal, 2008–2012), Plymouth Marine Laboratory (UK, 2010–2011), and Fundación Medina (Spain, 2012). Since then she switched fields, moving into computational biochemistry, in 2012. She holds a Ph.D. in Sustainable Chemistry (2017; double degree by University of Porto and NOVA University Lisbon) with research developed under the supervision of Professors Maria João Ramos and Pedro Fernandes. She is currently a postdoctoral fellow at Uppsala University with Professor Lynn Kamerlin, where her research is focused on unraveling the chemistry behind the regulation, mechanism, and evolution of GTPases.

Lynn Kamerlin is a Professor of Structural Biology at Uppsala University, where she has been a member of the faculty since 2011. She received her doctoral training with Dr. John Wilkie at the University of Birmingham, followed by postdoctoral training with Profs. Stefan Boresch (University of Vienna) and Arieh Warshel (University of Southern California). Her research interests focus on computational physical organic chemistry, as well as on using molecular simulation to unravel the evolution of new enzyme functions. She has served as Chair of the Young Academy of Europe and is currently a Fellow of the Royal Society of Chemistry and a Wallenberg Scholar. She is a member of the editorial boards of Electronic Structure and Chemistry Methods and a member of the editorial advisory boards of ACS Catalysis, The Journal of Physical Chemistry, Journal of Chemical Informatics, and ACS Omega. She is also the director of the board of the UPPMAX supercomputing center at Uppsala University. Her nonacademic interests include science policy, playing the piano, amateur photography, and languages.
The authors declare no competing financial interest.
References
- Sprang S. R. G Protein Mechanisms: Insights From Structural Analysis. Annu. Rev. Biochem. 1997, 66, 639–678. 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- Lassila J. K.; Zalatan J. G.; Herschlag D. Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Annu. Rev. Biochem. 2011, 80, 669–702. 10.1146/annurev-biochem-060409-092741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kötting C.; Gerwert K. The Dynamics of the Catalytic Site in Small GTPases, Variations on a Common Motif. FEBS Lett. 2013, 587, 2025–2027. 10.1016/j.febslet.2013.05.021. [DOI] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Sharma P. K.; Prasad R. B.; Warshel A. Why Nature Really Chose Phosphate. Q. Rev. Biophys. 2013, 46, 1–132. 10.1017/S0033583512000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho A. T.; Szeler K.; Vavitsas K.; Åqvist J.; Kamerlin S. C. Modeling the Mechanisms of Biological GTP Hydrolysis. Arch. Biochem. Biophys. 2015, 582, 80–90. 10.1016/j.abb.2015.02.027. [DOI] [PubMed] [Google Scholar]
- Gasper R.; Wittinghofer F. The Ras Switch in Structural and Historical Perspective. Biol. Chem. 2019, 401, 143–163. 10.1515/hsz-2019-0330. [DOI] [PubMed] [Google Scholar]
- Calixto A. R.; Moreira C.; Pabis A.; Kötting C.; Gerwert K.; Rudack T.; Kamerlin S. C. L. GTP Hydrolysis Without an Active Site Base: A Unifying Mechanism for Ras and Related GTPases. J. Am. Chem. Soc. 2019, 141, 10684–10701. 10.1021/jacs.9b03193. [DOI] [PubMed] [Google Scholar]
- Duarte F.; Åqvist J.; Williams N. H.; Kamerlin S. C. L. Resolving Apparent Conflicts Between Theoretical and Experimental Models of Phosphate Monoester Hydrolysis. J. Am. Chem. Soc. 2015, 137, 1081–1093. 10.1021/ja5082712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte F.; Barrozo A.; Åqvist J.; Williams N. H.; Kamerlin S. C. L. The Competing Mechanisms of Phosphate Monoester Dianion Hydrolysis. J. Am. Chem. Soc. 2016, 138, 10664–10673. 10.1021/jacs.6b06277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrozo A.; Blaha-Nelson D.; Williams N. H.; Kamerlin S. C. L. The Effect of Magnesium Ions on Triphosphate Hydrolysis. Pure Appl. Chem. 2017, 89, 715–727. 10.1515/pac-2016-1125. [DOI] [Google Scholar]
- Scheffzek K.; Ahmadian M. R.; Kabsch W.; Wiesmuller L.; Lautwein A.; Schmitz F.; Wittinghofer A. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and its Loss in Oncogenic Ras Mutants. Science 1997, 277, 333–338. 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucelic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavriljuk K.; Gazdag E.-M.; Itzen Y.; Kötting C.; Goody R. S.; Gerwert K. Catalytic Mechanism of Mammalian Rab*RabGAP Complex in Atomic Detail. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 21348–21353. 10.1073/pnas.1214431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer J. J.; Berman D. M.; Gilman A. G.; Sprang S. R. Structure of RGS4 Bound to ALF4– - Activated Giα1: Stabilization of the Transition State for GTP Hydrolysis. Cell 1997, 89, 251–261. 10.1016/S0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Voorhees R. M.; Schmeing T. M.; Kelley A. C.; Ramakrishnan V. The Mechanism for Activation of GTP Hydrolysis on the Ribosome. Science 2010, 330, 835–838. 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin G.; Kamerlin S. C. L.; Åqvist J. Energetics of Activation of GTP Hydrolysis on the Ribosome. Nat. Commun. 2013, 4, 1733. 10.1038/ncomms2741. [DOI] [PubMed] [Google Scholar]
- Mondal D.; Warshel A. EF-Tu and EF-G are Activated by Allosteric Effects. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3386–3391. 10.1073/pnas.1800054115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock L. V.; Kolar M. H.; Grubmuller H. Molecular Simulations of the Ribosome and Associated Translation Factors. Curr. Opin. Struct. Biol. 2018, 49, 27–35. 10.1016/j.sbi.2017.11.003. [DOI] [PubMed] [Google Scholar]
- Knihtila R.; Holzapfel G.; Weiss K.; Meilleur F.; Mattos C. Neutron Crystal Structure of RAS GTPase Puts in Question the Protonation State of the GTP gamma-Phosphate. J. Biol. Chem. 2015, 290, 31025–31036. 10.1074/jbc.M115.679860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann D.; Guldenhaupt J.; Schartner J.; Gerwert K.; Kötting C. The Protonation States of GTP and GppNHp in Ras Proteins. J. Biol. Chem. 2018, 293, 3871–3879. 10.1074/jbc.RA117.001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molt R. W. Jr.; Pellegrini E.; Jin Y. A GAP-GTPase-GDP-Pi Intermediate Crystal Structure Analyzed by DFT Shows GTP Hydrolysis Involves Serial Proton Transfers. Chem. - Eur. J. 2019, 25, 8484–8488. 10.1002/chem.201901627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorenko B. L.; Kots E. D.; Nemukhin A. V. Diversity of Mechanisms in Ras-GAP Catalysis of Guanosine Triphosphate Hydrolysis Revealed by Molecular Modeling. Org. Biomol. Chem. 2019, 17, 4879–4891. 10.1039/C9OB00463G. [DOI] [PubMed] [Google Scholar]
- Tripathi R.; Glaves R.; Marx D. The GTPase hGBP1 Converts GTP to GMP in Two Steps via Proton Shuttle Mechanisms. Chem. Sci. 2017, 8, 371–380. 10.1039/C6SC02045C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi R.; Noetzel J.; Marx D. Exposing Catalytic Versatility of GTPases: Taking Reaction Detours in Mutants of hGBP1 Enzyme Without Additional Energetic Cost. Phys. Chem. Chem. Phys. 2019, 21, 859–867. 10.1039/C8CP06343E. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Molt R. W. Jr.; Waltho J. P.; Richards N. G.; Blackburn G. M. 19F NMR and DFT Analysis Reveal Structural and Electronic Transition State Features for RhoA-Catalyzed GTP Hydrolysis. Angew. Chem., Int. Ed. 2016, 55, 3318–3322. 10.1002/anie.201509477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichauer R. H.; Favre G.; Cabantous S.; Landa G.; Hemeryck A.; Brut M. Water Distribution within Wild-Type NRas Protein and Q61 Mutants during Unrestrained QM/MM Dynamics. Biophys. J. 2018, 115, 1417–1430. 10.1016/j.bpj.2018.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichauer R. H.; Favre G.; Cabantous S.; Brut M. Hybrid QM/MM vs Pure MM Molecular Dynamics for Evaluating Water Distribution within p21N-ras and the Resulting GTP Electronic Density. J. Phys. Chem. B 2019, 123, 3935–3944. 10.1021/acs.jpcb.9b02660. [DOI] [PubMed] [Google Scholar]
- Sayyed-Ahmad A.; Prakash P.; Gorfe A. A. Distinct Dynamics and Interaction Patterns in H- and K-Ras Oncogenic P-loop Mutants. Proteins: Struct., Funct., Genet. 2017, 85, 1618–1632. 10.1002/prot.25317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I.; Rhett J. M.; O’Bryan J. P. Therapeutic Targeting of RAS: New Hope for Drugging the “Undruggable. Biochim. Biophys. Acta, Mol. Cell Res. 2020, 1867, 118570. 10.1016/j.bbamcr.2019.118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J.; Rex K.; Saiki A. Y.; Mohr C.; Cooke K.; Bagal D.; Gaida K.; Holt T.; Knutson C. G.; Koppada N.; Lanman B. A.; Werner J.; Rapaport A. S.; San Miguel T.; Ortiz R.; Osgood T.; Sun J. R.; Zhu X.; McCarter J. D.; Volak L. P.; Houk B. E.; Fakih M. G.; O’Neil B. H.; Price T. J.; Falchook G. S.; Desai J.; Kuo J.; Govindan R.; Hong D. S.; Ouyang W.; Henary H.; Arvedson T.; Cee V. J.; Lipford J. R. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Hallin J.; Engstrom L. D.; Hargis L.; Calinisan A.; Aranda R.; Briere D. M.; Sudhakar N.; Bowcut V.; Baer B. R.; Ballard J. A.; Burkard M. R.; Fell J. B.; Fischer J. P.; Vigers G. P.; Xue Y.; Gatto S.; Fernandez-Banet J.; Pavlicek A.; Velastagui K.; Chao R. C.; Barton J.; Pierobon M.; Baldelli E.; Patricoin E. F. 3rd; Cassidy D. P.; Marx M. A.; Rybkin I. I.; Johnson M. L.; Ou S. I.; Lito P.; Papadopoulos K. P.; Janne P. A.; Olson P.; Christensen J. G.. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery 2020, 10, 54. 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]