

Abstract
Background:
Bevacizumab (BEV), a monoclonal antibody against vascular endothelial growth factor-A (VEGF-A), is a standard component of medical therapy of metastatic colorectal cancer (mCRC). Activation of alternative angiogenesis pathways has been implicated in resistance to BEV. This phase II study examines the activity of combined vertical blockade of VEGF signaling with sorafenib and BEV as salvage therapy in patients with progressive disease (PD) on all standard therapy in mCRC.
Methods:
mCRC patients with documented PD on standard therapy, received sorafenib (200 mg orally twice daily, days 1–5 and 8–12) and BEV (5 mg/kg intravenously, day 1) every 2 weeks. Primary endpoint was 3-month progression-free survival (PFS) rate and secondary endpoints were overall survival (OS), response rate (RR), safety, and feasibility.
Results:
Of the 83 patients enrolled, 79 were evaluable. Of these, 42 (53%) were progression-free at 3 months. Median PFS was 3.5 months and median OS was 8.3 months. One patient had a partial response and 50 patients (63.3%) had at least one stable tumor assessment. Of 79 evaluable patients, 54 (68%) experienced grade 3/4 adverse events (AEs) at least possibly related to treatment. Most frequent grade 3/4 AEs were: fatigue (24.1%), hypertension (16.5%), elevated lipase (8.9%), hand-foot skin reaction (8.9%), diarrhea (7.6%), and proteinuria (7.6%). Reasons for treatment discontinuation were PD (72%), AEs (18%), patient refusal (8%), physician decision (1%), and death (1%).
Conclusions:
The combination of BEV and sorafenib as salvage therapy in heavily pretreated mCRC patients is tolerable and manageable, with evidence of promising activity.
ClinicalTrials.gov identifier:
NCT00826540, URL:http://clinicaltrials.gov/ct2/show/NCT00826540
Keywords: antiangiogenesis, bevacizumab, colorectal cancer, single nucleotide polymorphism, sorafenib
Introduction
Colorectal cancer (CRC) is one of the most common and deadliest malignancies in the United States, where it has been estimated that about 145,600 people were diagnosed with CRC and 51,020 patients died from this disease in 2019.1 Over the past decade, medical treatment options for metastatic colorectal cancer (mCRC) have greatly expanded with the introduction of novel chemotherapeutic agents (e.g. oxaliplatin and irinotecan), biologic inhibitors of the vascular endothelial growth factor (VEGF) system [e.g. bevacizumab (BEV)], and epidermal growth factor receptors in RAS wild-type cancers (panitumumab and cetuximab). Despite these improvements, the 5-year survival for mCRC patients is still only 11%.2 There is a great unmet need to develop novel therapeutic approaches that further improve outcome in mCRC, in particular in patients who have shown tumor progression after exhausting all standard treatment options.
It is well established that angiogenesis is essential for solid tumor growth, invasion, and metastases. Vascular endothelial growth factor-A (VEGF-A), a pro-angiogenic factor, is the most potent mediator of angiogenesis, and has been shown to be overexpressed in a variety of human cancers, including mCRC. Thus, VEGF-A is an appropriate and attractive target for biologic therapy. BEV, a recombinant humanized version of a murine anti-human VEGF-A monoclonal antibody, inhibits VEGF-A interaction with its receptors, VEGFR-1 and VEGFR-2, thereby neutralizing VEGF-A activity.3 Although single-agent treatment with BEV has shown little activity in mCRC, BEV treatment exhibits synergistic therapeutic effects when combined with standard cytotoxic drugs, resulting in statistically significant increased progression-free survival (PFS) and overall survival (OS) in mCRC patients in the first- and second-line setting,4–6 independent of KRAS (Kirsten rat sarcoma viral oncogene homolog) status.7,8
Unfortunately, the integration of BEV into treatment algorithms has led to only incremental improvements of a few months in PFS and OS, and for patients on ongoing BEV-containing therapy in a palliative setting, tumor progression will invariably occur. Resistance to anti-VEGF therapy can be mediated via overexpression of VEGF receptors, increase in VEGF levels, and upregulation of alternate angiogenesis signaling pathways, such as platelet derived growth factor receptor (PDGFR) signaling.9 Therefore, a complete blockage of the VEGF-signaling pathway by combining a ligand inhibitor, such as BEV, with a multi-targeted kinase inhibitor blocking the VEGF system on a receptor level, at the same time also targeting potentially compensatory pro-angiogenic mechanisms, could result in synergistic inhibition of tumor angiogenesis. Sorafenib is a multi-kinase inhibitor that targets several serine-threonine and tyrosine kinases involved in tumor progression and angiogenesis, including all VEGFRs, PDGFR-β, RET, Flt3, and c-KIT. Sorafenib has demonstrated proof-of-efficacy in the treatment of advanced renal cell carcinoma, unresectable hepatocellular carcinoma, and thyroid cancer.10–12 Sorafenib inhibition of angiogenesis receptors has the potential to complement BEV activity by completely vertically blocking VEGF signaling and inhibiting other angiogenic pathways potentially involved in the mediation of resistance to BEV. Based on these considerations, we evaluated the therapeutic effect of dual angiogenesis inhibition with sorafenib and BEV as salvage therapy in mCRC patients in North Central Cancer Treatment Group (NCCTG) trial N054C. NCCTG is now a part of the Alliance for Clinical Trials in Oncology. Given the fact that, in a salvage therapy setting, most patients without active therapy develop RECIST-documented tumor progression within the first 2 months,13,14 a 50% rate of patients progression-free at 3 months was chosen as primary endpoint of the study. In addition, previous studies have shown that genetic single nucleotide polymorphisms (SNPs) involved in the VEGF/VEGFR2 pathway are associated with anti-angiogenesis treatment efficacy and toxicity.15 We genotyped DNA for VEGF, VEGFR2, and HIF-1a SNPs from enrolled patients, and evaluated their associations with treatment outcome and safety in correlative studies.
Patients and methods
Patient selection
Patients aged 18 years or older with a diagnosis of stage IV colorectal cancer (histologic proof of metastatic disease was not required) were eligible for enrollment. Pertinent inclusion criteria included: measurable disease; progressive disease during or within 6 months after standard therapy including BEV, fluoropyrimidine, oxaliplatin, irinotecan, and epidermal growth factor receptor (EGFR) antibodies (for KRAS wild-type cancers) or ineligibility for one or more of these agents; KRAS status documentation in medical record; life expectancy ⩾6 months, an Eastern Cooperative Oncology Group (ECOG) performance score (PS) of 0 or 1, and adequate bone marrow, renal function, and hepatic function. Patients had to be willing to provide mandatory blood samples for correlative research studies.
Patients with known brain metastases were excluded from the study. Patients with uncontrolled hypertension (i.e. systolic BP > 150 mm Hg or diastolic BP > 100 mm Hg on antihypertensive medications), and patients with uncontrolled clinically relevant medical conditions were not eligible.
Concurrent therapy restrictions included: no prior sorafenib therapy; no prior discontinuation of BEV due to adverse events (AEs); no concurrent anticoagulant, except low-dose warfarin or heparin for deep venous thrombosis prophylaxis (not treatment); and no other concurrent investigational agent for this cancer. Full inclusion and exclusion criteria are provided in the study protocol, available online.
This study was approved by the Mayo Clinic Institutional Review Board (IRB 08-004165) and the North Central Cancer Treatment Group (NCCTG-Alliance N054C). Patients provided IRB-approved, protocol-specific written informed consent prior to initiating therapy for study-specific treatment and inclusion in the present study.
Treatment
Patients received sorafenib (200 mg orally twice a day on days 1–5 and 8–12) and BEV (5 mg/kg intravenously on day 1) every 2 weeks. This starting dose was chosen in view of the results of a prior phase I trial that demonstrated that the combination of BEV and sorafenib could not be administered at full doses.16,17 If any sorafenib doses were missed, they were not made up, and cycle length remained 14 days. Patients continued treatment until the occurrence of progressive disease or treatment-limiting toxicities. Dose adjustments were made depending on the type and severity of treatment-related toxicities. Dose reduction steps for sorafenib were as follows: starting dose: 200 mg twice daily, on days 1–5 and 8–12, dose reduction 1: 200 mg once daily continuously, dose reduction 2: 200 mg once daily on days 1–5 and 8–12 of each cycle. Sorafenib was discontinued if not tolerated after two dose reductions. BEV was discontinued if unmanageable BEV-related AEs occurred; there were no dose adjustments for BEV.
Patient evaluation
Hypertension was monitored through routine evaluation of blood pressure, weekly during the first 6 weeks of treatment, and prior to each BEV treatment thereafter. AEs were collected using the National Cancer Institute Common Toxicity Criteria for Adverse Events Version 3.0 (NCI CTCAE V3.0, http://ctep.cancer.gov/reporting/ctc.html).
Disease assessment
Imaging for tumor measurements was conducted within 4 days before planned treatment and every 6 weeks thereafter for assessment of response. Tumor response evaluation and response definitions were according to Response Evaluation Criteria in Solid Tumors (RECIST 1.0). Patients were also considered to have progressed in cases of significant clinical deterioration that could not be attributed to study treatment or other medical conditions. These conditions included worsening of tumor-related symptoms, ⩾10% weight loss, or a decline in PS of >1 level on ECOG scale.
Genetic variant association study
Genomic DNA was extracted from peripheral blood of evaluable patients in this study. Genotyping was performed in the Genotyping Shared Resource, Mayo Clinic, Rochester, MN, using TaqMan Drug Metabolism Genotyping Assays (Applied Biosystems, Foster City, CA) or direct sequencing for genotyping VEGF (rs25648, rs3025039, rs2010963, rs1005230, rs833061, rs699947, rs1570360, rs10434), VEGFR2 (rs2305948, rs2219471, rs2071559, rs1870377), and HIF-1a (rs11549465, rs11549467) SNPs. For each SNP analyzed, frequencies for each SNP subgroup of patients (i.e. homozygous wild type versus heterozygous versus homozygous for mutant allele) were analyzed using contingency tables and either Chi-square or Fisher’s exact test to determine genotype associations with the study’s primary endpoint, progression-free rate at 3 months of follow up, as well as genotype associations with the most common grade ⩾3 AEs (i.e. fatigue, hypertension, skin reaction) at least possibly related to treatment.
Statistical considerations
A two-stage Simon design used 40 or 72 patients to test the null hypothesis that the true success proportion in a given patient population is, at most, 50%.18 Definition of success: the primary endpoint of this trial was the PFS rate at 3-months from registration in patients with mCRC treated with sorafenib and BEV. All patients meeting the eligibility criteria who have signed a consent form and have begun treatment will be considered evaluable. Patients lost to follow up before 3 months (e.g. progression, refusing further treatment, etc.) will be considered treatment failures. All eligible patients will be followed until death, or a minimum of 3 years. Thus, defining success as ‘no progression by 3 months’, we assume that less than 50% success is justification to recommend against further study, and that greater than 65% success is justification to recommend for further study. A Simon design based on the minimizing the maximum sample size calls for a sample size of 72 patients to provide a significance of 0.1 and a power of 90%. In stage 1, if ⩽19 successes were observed in the first 40 evaluable patients, the regimen was considered ineffective and the study terminated; if >19 successes were observed, the study proceeded to stage 2. In stage 2, an additional 32 patients were enrolled in the study; if ⩽41 successes were observed the regimen was considered ineffective in this patient population and if ⩾42 successes were observed this regimen was to be recommended for further testing of this regimen in subsequent studies in this patient population.
Secondary endpoints included response rate (RR), OS, safety, and feasibility. Confirmed responses were responses maintained for two consecutive cycles. Patients demonstrating tumor progression within the first six cycles were classified as nonresponders. PFS was defined as the time to first occurrence of progression or death, with nonprogression living patients being censored at the time of their last disease assessment. OS was defined as the time from registration to death. Patients lost to follow up for this endpoint were censored at the date of last contact (i.e. date last known alive). The distribution of OS was estimated using Kaplan–Meier methodology. Simple frequency analysis was conducted to see if RR was related to prior treatment. Descriptive statistics were used to investigate how prior treatments affected various other measures as well.
Data collection and statistical analyses were conducted by the Alliance Statistics and Data Center. Data quality was ensured by review of data by the Alliance Statistics and Data Center and by the study chairperson following Alliance policies.
Results
Patient characteristics
Data for analyses were frozen as of 15 February 2011. A total of 83 patients were enrolled between 3 June 2009 and 20 October 2009; 1 patient canceled participation before receiving treatment, and 3 were later deemed ineligible, leaving 79 evaluable patients for primary endpoint analysis. Median age was 62 years (range: 36–88 years). The patient baseline demographics, disease characteristics, and previous treatment are summarized in Table 1.
Table 1.
Patient baseline characteristics.
| Patient characteristics | Median (range) | Patients (n = 79) | |
|---|---|---|---|
| Number | Percent | ||
| Age (years) | 62 (36–88) | ||
| Gender | |||
| Male | 43 | 54% | |
| Female | 36 | 46% | |
| KRAS status | |||
| Wild-type | 39 | 49% | |
| Mutated | 40 | 51% | |
| ECOG performance status | |||
| 0 | 44 | 56% | |
| 1 | 35 | 44% | |
| Prior radiation therapy | 25 | 32% | |
| Number of metastatic sites | |||
| 1 | 12 | 15% | |
| 2 | 26 | 33% | |
| 3 | 25 | 32% | |
| 4 | 14 | 18% | |
| 5 | 2 | 3% | |
| Most common metastatic site | |||
| Liver | 63 | 80% | |
| Lung | 52 | 66% | |
| Nodal | 46 | 58% | |
ECOG, Eastern Cooperative Oncology Group; KRAS, Kirsten rat sarcoma viral oncogene homolog.
Outcome measures
Of the 79 evaluable patients at the time of this analysis, 70 (89%) had progressed and 64 (81%) had died. Of the 15 patients still alive, median follow-up time was 12.4 months (range: 3.8–15.6 months). All patients had sufficient follow up to be evaluated for the primary endpoint, PFS rate at 3 months after study entry. Of the 79 evaluable patients, 42 (53%) were progression-free at 3 months, 1 patient (1%) had a partial response, and 50 (63%) patients had at least one stable tumor measurement, for a total disease control rate (DCR) of 64% (Figure 1). At their first tumor assessment, 22 patients (28%) were found to have progressed, and 6 (8%) went off treatment before having any tumor assessments (5 for AEs and 1 refusal to continue treatment). Median PFS was 3.5 months [95% confidence interval (CI) 2.7–4.2 months].
Figure 1.
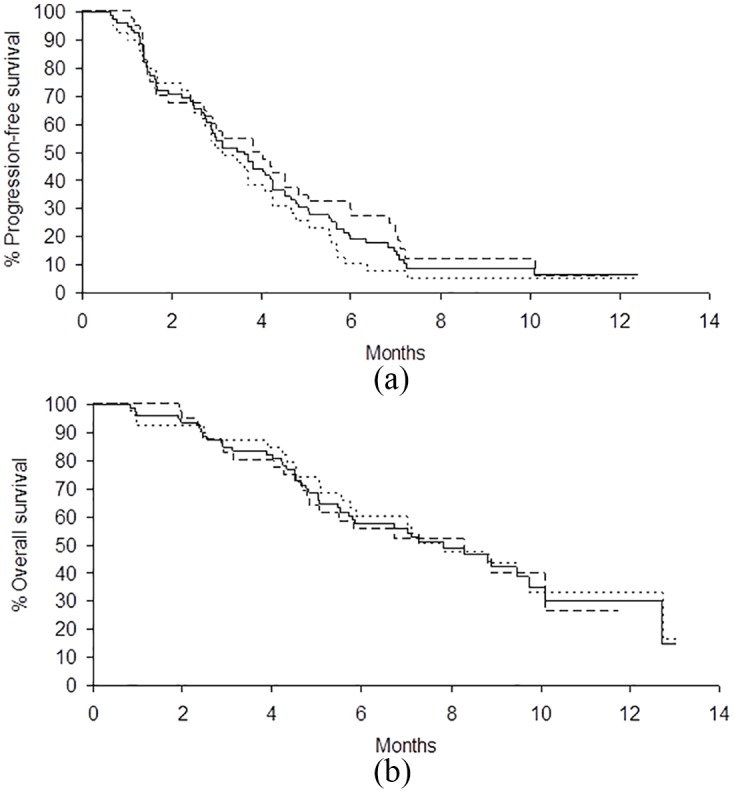
Kaplan–Meier curves for (a) PFS and (b) OS for wild-type KRAS (n = 39, dotted line), mutated KRAS (n = 40, dashed line), and all patients combined (n = 79, solid line) being treated with sorafenib plus BEV as salvage therapy.
BEV, bevacizumab; KRAS, Kirsten rat sarcoma viral oncogene homolog; OS, overall survival; PFS, progression-free survival.
Outcome by KRAS status
Median PFS for KRAS mutated patients was slightly longer than that for KRAS wild type, but was not statistically significant [3.8 versus 3.1 months; hazard ratio (HR) = 0.81, p = 0.36]. Median OS for all patients was 8.3 months (95% CI 5.5–11.3 months). There was no difference in OS by KRAS status (mutated 8.3 months versus wild-type 7.8 months; HR = 0.93, p = 0.79).
Treatment summary
A total of 565 treatment cycles were administered. The median percent dose administered was 100% for BEV and 85% for sorafenib (Figure 2). Thirty-one patients (39%) delayed treatment at least once during their time on study, 13 patients (16%) omitted a dose of BEV at least once during treatment, 26 patients (33%) omitted a dose of sorafenib at least once during treatment, and 49 patients (62%) reduced their sorafenib at least once during treatment. Reasons for treatment discontinuation were: disease progression (57/76; 72%), AEs (14/79, 18%), refused further treatment (6/79, 8%), physician decision (1/79, 1%), and death on study (1/79, 1%). Best percent change in sum of target lesions from baseline in patients with available post-baseline tumor measurements (Figure 3).
Figure 2.

Average sorafenib dose and number of patients treated per 2-week cycle (target dose: 4000 mg per 2-week cycle).
Figure 3.
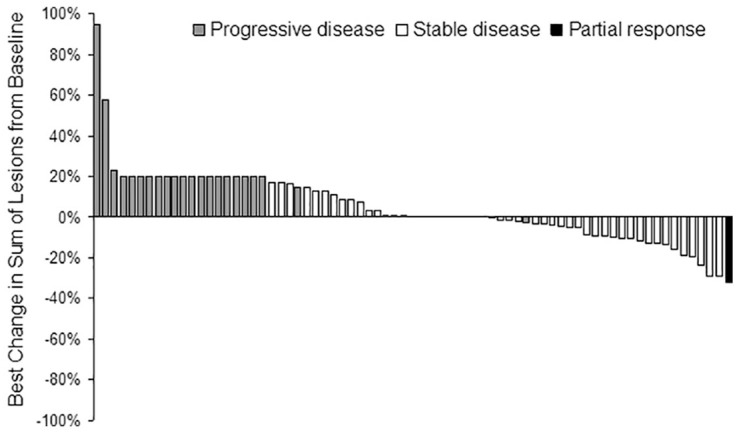
Best percent change in sum of target lesions from baseline in patients with available post-baseline tumor measurements (n = 73). Patients who went off treatment before any scans and had PD at any point during follow up, or patients who had PD without any scans during treatment were assigned 20% change (RECIST PD threshold). PD patients with a percent change <20% were due to clinical progression.
PD, progressive disease.
AE summary
All 79 evaluable patients had at least one toxicity assessment. A total of 60 patients (76%) experienced a grade 3 or 4 AE, with 54 (68%) patients having a grade 3 or 4 AE that was at least possibly related to treatment. Of the 11 patients (14%) experiencing a grade 4 event, 10 (13%) had a grade 4 event at least possibly related to treatment. No grade 5 AEs occurred. Table 2 lists the grade 3 and higher toxicities reported as at least possibly related to study treatment.
Table 2.
All grade 3–4 AEs with >2% incidence at least possibly related to study treatment.
| Toxicity | Frequency (%) | ||
|---|---|---|---|
| Grade 3 | Grade 4 | Total grade 3–4 | |
| Fatigue | 18 (22.8) | 1 (1.3) | 19 (24.1) |
| Hypertension | 13 (16.5) | 0 | 13 (16.5) |
| Lipase increased | 6 (7.6) | 1 (1.3) | 7 (8.9) |
| Hand-foot skin reaction | 7 (8.9) | 0 | 7 (8.9) |
| Diarrhea | 5 (6.3) | 1 (1.3) | 6 (7.6) |
| Proteinuria | 6 (7.6) | 0 | 6 (7.6) |
| Anorexia | 4 (5.1) | 0 | 4 (5.1) |
| Abdominal pain | 2 (2.6) | 1 (1.3) | 3 (3.9) |
| Alkaline phosphatase increased | 3 (3.9) | 0 | 3 (3.9) |
| Bilirubin | 3 (3.9) | 0 | 3 (3.9) |
| Creatinine increased | 3 (3.9) | 0 | 3 (3.9) |
| Hyponatremia | 3 (3.9) | 0 | 3 (3.9) |
| Nausea | 3 (3.9) | 0 | 3 (3.9) |
| Weight loss | 2 (2.6) | 0 | 2 (2.6) |
| Amylase | 1 (1.3) | 1 (1.3) | 2 (2.6) |
| Anemia | 1 (1.3) | 1 (1.3) | 2 (2.6) |
| Hypokalemia | 2 (2.6) | 0 | 2 (2.6) |
AEs, adverse events.
Genetic variant association results
Of 79 evaluable patients enrolled in this study, 78 provided peripheral blood for germline DNA extraction. The SNPs analyzed and genotype frequencies are summarized in Supplemental Table S1, and the genotype associations with the 3-month PFS rate and the most common AEs are provided in Table 3. In univariate analysis, the presence of a mutant T allele in the 5’ UTR of VEGF (rs25648) was associated with fewer successes in progression-free rate at 3-month follow up (p = 0.026, Supplemental Table S2). The presence of a mutant C allele in the promoter region of VEGFR2 (rs2071559) was associated with fewer grade ⩾3 hypertension AEs related to treatment (p = 0.010, Supplemental Table S3). However, after correcting for multiple comparisons using the Benjamini–Hochberg method, none of the p values were significant at a 0.05 level. No differences were observed between the genotype subgroups and either progression-free rate at 3 months or the most common grade ⩾3 AE related to treatment for the remaining SNPs that were analyzed, before or after correction.
Table 3.
Genotype associations with primary endpoint and most common AEs.
| SNP (n = 78) | 3-month PFS p value |
Fatigue p value |
Hypertension p value |
Skin reaction p value |
|---|---|---|---|---|
| rs699947 | 0.839a | 0.804a | 0.432b | 0.318b |
| rs1005230 | 0.839a | 0.804a | 0.432b | 0.318b |
| rs833061 | 0.659a | 0.553a | 0.392b | 0.471b |
| rs1570360 | 0.450a | 0.618a | 0.847b | 0.304b |
| rs2010963 | 0.705b | 0.801a | 0.260a | 1.000b |
| rs25648 | 0.026b | 0.797b | 0.289b | 0.737b |
| rs3025039 | 0.851b | 0.130b | 0.228b | 1.000b |
| rs10434 | 0.646a | 0.152a | 0.665b | 0.741b |
| rs2305948 | 0.972a | 0.331b | 0.277b | 0.114b |
| rs2071559 | 0.133a | 0.399a | 0.010b | 0.089b |
| rs1870377 | 0.289b | 1.000b | 0.819a | 0.513b |
| rs2219471 | 0.385b | 0.741a | 0.641a | 0.848b |
| rs11549465 | 0.972a | 0.748b | 0.464b | 0.330b |
| rs11549467 | 1.000b | 1.000b | 1.000b | 0.149b |
AEs, adverse events; PFS, progression-free survival; SNP, single nucleotide polymorphism.
Chi-square p values.
Fisher’s exact test p values.
Discussion
Our study demonstrates that the combination of a small molecule multi-kinase inhibitor, sorafenib, and BEV is tolerable, with a manageable toxicity profile when the kinase inhibitor is used at a reduced dose compared with its approved dose when used as single agent in hepatocellular, renal cell carcinoma, and thyroid cancer. The 68% grade 3 or higher AE at least possibly related to treatment is comparable with 65% grade 3/4 AE rate from cetuximab plus irinotecan, one of the standards of care for mCRC.19 The main side-effects observed were fatigue, hypertension, and skin reactions, which were less pronounced compared with studies that combined BEV with sorafenib at higher doses and at different schedules.16 The initial phase I study of BEV and sorafenib identified a high rate of hypertension, affecting 23 of 39 patients (67%), with 13 patients (33%) experiencing grade 3/4 hypertension, which emerged as the most frequent dose-limiting toxicity. In that early study, however, the addition of only one antihypertensive agent was allowed. This is in contrast to our study, where the use of antihypertensive agents was less regulated, and, whereas we saw a similar rate of grade 3 hypertension (16%), it only rarely led to treatment discontinuation. Importantly, only a minority of patients in our study discontinued therapy for reasons other than progressive disease, which again underscores the tolerability of the combination regimen at the investigated doses and with the toxicity management strategy outlined per protocol. In addition, it has to be kept in mind that the rate of grade 3/4 AEs for other agents tested in a similar ‘last line’ setting in colorectal cancer were found to be in the same range as in our study (79% for cetuximab, 54% for regorafenib).13,20 The dose and schedule used mirror the approach of Lee and colleagues, which confirmed the tolerability and activity of this combination in various tumor types.17
The demonstration of combinability of sorafenib and BEV can have considerable implications for the future development of kinase inhibitors in malignancies like advanced colorectal cancer, where BEV is a standard component of medical therapy. In colorectal cancer, the role of BEV was recently further emphasized with the positive findings of a phase III trial that investigated the efficacy of BEV administered beyond progression when added to chemotherapy; that trial confirmed that protracted VEGF-A inhibition is associated with improved outcomes in this malignancy.8 Our study results tie into these findings since all patients had demonstrated progressive disease on, or intolerability to, all available chemotherapeutic or biologic agents, including BEV, before entering the study.
Although preclinical models suggested some anticancer activity of sorafenib in colorectal cancer,21,22 clinical studies of this agent in combination with chemotherapy have generated disappointing results.23,24 Most prominently, a large randomized placebo-controlled phase IIb study comparing modified FOLFOX6 with or without sorafenib (at full dose of 400 mg twice daily continuously) as first-line therapy did not demonstrate improvement in PFS but significantly increased toxicity.24
The combination of the two biologic agents, sorafenib and BEV, however, appears to have clinically relevant activity in a salvage therapy setting in heavily pretreated patients with mCRC, independent of KRAS mutation status. It is important to note that the main eligibility criteria for our single-arm phase II trial were identical to those of a recently presented phase III trial, which investigated single-agent regorafenib, a multi-kinase inhibitor similar in structure and kinase inhibitory profile to sorafenib, as salvage therapy in mCRC.20 With all caveats of cross-trial comparisons, the outcomes data of the sorafenib/BEV combination in this treatment-refractory patient population compare favorably with the results reported for regorafenib in terms of DCR (sorafenib/BEV 64%, regorafenib 41%), PFS (3.5 months and1.9 months, respectively), and OS (8.3 months and 6.4 months, respectively). At the same time, the toxicity profile was very similar to regorafenib, with fatigue, hand-foot skin reaction, and hypertension being most prominent. PFS and OS in our study were also similar to the results reported for EGFR monoclonal antibodies as salvage therapy in patients with KRAS wild-type colorectal cancer.25,26 Of note is that about 30% of patients demonstrated progressive disease at the very first tumor assessment at 6 weeks.
A previous study demonstrated that the T-604C SNP (rs2071559) structurally alters the VEGFR2 promoter binding site for the E2F transcription factor, resulting in a 68% reduction in transcriptional activity and an increased risk of coronary heart disease.15 The relationship between reduced VEGFR2 transcription and fewer grade ⩾3 hypertension AEs related to treatment in mCRC patients with the mutant C allele (rs2071559) of VEGFR2 needs to be further elucidated. VEGF rs25648 and VEGFR2 rs2071559 were not prognostic in multivariable analysis, which was independently confirmed in hepatocellular carcinoma patients receiving sorafenib.27
Limitations of this single-arm phase II study included that there is no clear evidence that sorafenib adds to BEV and vice versa due to lack of randomization. Neither BEV nor sorafenib have documented relevant single-agent activity in mCRC in this setting. Inevitably, at the time this trial was done, no patient in this study had extended molecular testing, and no adjustment for sidedness was done according to 2019 standards. Acknowledging the limitations of this single-arm phase II study, the tolerability and antitumor activity observed for sorafenib combined with BEV warrants further evaluation of this regimen in a randomized setting and in combination with conventional chemotherapy in earlier lines of therapy, in particular in patients with KRAS mutated colorectal cancer who have more limited treatment options.
Supplemental Material
Supplemental material, Supplemental_Tables for Dual VEGF inhibition with sorafenib and bevacizumab as salvage therapy in metastatic colorectal cancer: results of the phase II North Central Cancer Treatment Group study N054C (Alliance) by Hao Xie, Jacqueline M. Lafky, Bruce W. Morlan, Philip J. Stella, Shaker R. Dakhil, Gerald G. Gross, William S. Loui, Joleen M. Hubbard, Steven R. Alberts and Axel Grothey in Therapeutic Advances in Medical Oncology
Supplemental material, Supplemental_Text_R1 for Dual VEGF inhibition with sorafenib and bevacizumab as salvage therapy in metastatic colorectal cancer: results of the phase II North Central Cancer Treatment Group study N054C (Alliance) by Hao Xie, Jacqueline M. Lafky, Bruce W. Morlan, Philip J. Stella, Shaker R. Dakhil, Gerald G. Gross, William S. Loui, Joleen M. Hubbard, Steven R. Alberts and Axel Grothey in Therapeutic Advances in Medical Oncology
Acknowledgments
This work was previously presented in part in 2010 J Clin Oncol 28:15s (suppl; abstr 3549) and 2011 Cancer Res 71:8s (abstr 4131).
Footnotes
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U24CA196171, U10CA180821 and U10CA180882 (to the Alliance for Clinical Trials in Oncology), UG1CA232760 and UG1CA189808; also supported in part by funds from Bayer and Genentech. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest statement: The Mayo Clinic Foundation received funding for research activities carried out by A. Grothey from Genentech/Roche and Bayer Oncology.
All remaining authors have declared no conflicts of interest.
ORCID iD: Hao Xie  https://orcid.org/0000-0002-9640-4055
https://orcid.org/0000-0002-9640-4055
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Hao Xie, Division of Medical Oncology, Mayo Clinic, Rochester, MN, USA.
Jacqueline M. Lafky, Alliance Statistics and Data Center, Mayo Clinic, Rochester, MN, USA Department of Biostatistics, Mayo Clinic, Rochester, MN, USA
Bruce W. Morlan, Department of Biostatistics, Mayo Clinic, Rochester, MN, USA
Philip J. Stella, St Joseph Mercy Health System, Ann Arbor, MI, USA
Shaker R. Dakhil, Wichita Community Clinical Oncology Program, Wichita, KS, USA
Gerald G. Gross, MeritCare Health System, Fargo, ND, USA
William S. Loui, University of Hawaii Cancer Center, Honolulu, HI, USA
Joleen M. Hubbard, Division of Medical Oncology, Mayo Clinic, Rochester, MN, USA
Steven R. Alberts, Division of Medical Oncology, Mayo Clinic, Rochester, MN, USA
Axel Grothey, Medical Oncology, West Cancer Center, 9745 Wolf River Blvd, Germantown, TN 38138-1762, USA.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019; 69: 7–34. [DOI] [PubMed] [Google Scholar]
- 2. Center MM, Jemal A, Smith RA, et al. Worldwide variations in colorectal cancer. CA Cancer J Clin 2009; 59: 366–378. [DOI] [PubMed] [Google Scholar]
- 3. Grothey A, Allegra C. Antiangiogenesis therapy in the treatment of metastatic colorectal cancer. Ther Adv Med Oncol 2012; 4: 301–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giantonio BJ, Catalano PJ, Meropol NJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern cooperative oncology group study E3200. J Clin Oncol 2007; 25: 1539–1544. [DOI] [PubMed] [Google Scholar]
- 5. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–2342. [DOI] [PubMed] [Google Scholar]
- 6. Tebbutt NC, Wilson K, Gebski VJ, et al. Capecitabine, bevacizumab, and mitomycin in first-line treatment of metastatic colorectal cancer: results of the Australasian gastrointestinal trials group randomized phase III MAX study. J Clin Oncol 2010; 28: 3191–3198. [DOI] [PubMed] [Google Scholar]
- 7. Ince WL, Jubb AM, Holden SN, et al. Association of k-ras, b-raf, and p53 status with the treatment effect of bevacizumab. J Natl Cancer Inst 2005; 97: 981–989. [DOI] [PubMed] [Google Scholar]
- 8. Bennouna J, Sastre J, Arnold D, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol 2013; 14: 29–37. [DOI] [PubMed] [Google Scholar]
- 9. Kopetz S, Hoff PM, Morris JS, et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol 2010; 28: 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brose MS, Nutting CM, Jarzab B, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 2014; 384: 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007; 356: 125–134. [DOI] [PubMed] [Google Scholar]
- 12. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359: 378–390. [DOI] [PubMed] [Google Scholar]
- 13. Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357: 2040–2048. [DOI] [PubMed] [Google Scholar]
- 14. van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 2007; 25: 1658–1664. [DOI] [PubMed] [Google Scholar]
- 15. Wang Y, Zheng Y, Zhang W, et al. Polymorphisms of KDR gene are associated with coronary heart disease. J Am Coll Cardiol 2007; 50: 760–767. [DOI] [PubMed] [Google Scholar]
- 16. Azad NS, Posadas EM, Kwitkowski VE, et al. Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. J Clin Oncol 2008; 26: 3709–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee JM, Sarosy GA, Annunziata CM, et al. Combination therapy: intermittent sorafenib with bevacizumab yields activity and decreased toxicity. Br J Cancer 2010; 102: 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials 1989; 10: 1–10. [DOI] [PubMed] [Google Scholar]
- 19. Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351: 337–345. [DOI] [PubMed] [Google Scholar]
- 20. Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013; 381: 303–312. [DOI] [PubMed] [Google Scholar]
- 21. Gulhati P, Zaytseva YY, Valentino JD, et al. Sorafenib enhances the therapeutic efficacy of rapamycin in colorectal cancers harboring oncogenic KRAS and PIK3CA. Carcinogenesis 2012; 33: 1782–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wehler TC, Hamdi S, Maderer A, et al. Single-agent therapy with sorafenib or 5-FU is equally effective in human colorectal cancer xenograft–no benefit of combination therapy. Int J Colorectal Dis 2013; 28: 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Azad N, Dasari A, Arcaroli J, et al. Phase I pharmacokinetic and pharmacodynamic study of cetuximab, irinotecan and sorafenib in advanced colorectal cancer. Invest New Drugs 2013; 31: 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tabernero J, Garcia-Carbonero R, Cassidy J, et al. Sorafenib in combination with oxaliplatin, leucovorin, and fluorouracil (modified FOLFOX6) as first-line treatment of metastatic colorectal cancer: the RESPECT trial. Clin Cancer Res 2013; 19: 2541–2550. [DOI] [PubMed] [Google Scholar]
- 25. Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26: 1626–1634. [DOI] [PubMed] [Google Scholar]
- 26. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-RAS mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359: 1757–1765. [DOI] [PubMed] [Google Scholar]
- 27. Scartozzi M, Faloppi L, Svegliati Baroni G, et al. VEGF and VEGFR genotyping in the prediction of clinical outcome for HCC patients receiving sorafenib: the ALICE-1 study. Int J Cancer 2014; 135: 1247–1256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplemental_Tables for Dual VEGF inhibition with sorafenib and bevacizumab as salvage therapy in metastatic colorectal cancer: results of the phase II North Central Cancer Treatment Group study N054C (Alliance) by Hao Xie, Jacqueline M. Lafky, Bruce W. Morlan, Philip J. Stella, Shaker R. Dakhil, Gerald G. Gross, William S. Loui, Joleen M. Hubbard, Steven R. Alberts and Axel Grothey in Therapeutic Advances in Medical Oncology
Supplemental material, Supplemental_Text_R1 for Dual VEGF inhibition with sorafenib and bevacizumab as salvage therapy in metastatic colorectal cancer: results of the phase II North Central Cancer Treatment Group study N054C (Alliance) by Hao Xie, Jacqueline M. Lafky, Bruce W. Morlan, Philip J. Stella, Shaker R. Dakhil, Gerald G. Gross, William S. Loui, Joleen M. Hubbard, Steven R. Alberts and Axel Grothey in Therapeutic Advances in Medical Oncology