

Abstract
Background
With the accelerated development of next‐generation sequencing (NGS), identified variants, and targeted therapies, clinicians who confront the complicated and multifarious genetic information may not effectively incorporate NGS‐based circulating tumor DNA (ctDNA) analysis into routine patient care. Consequently, standardized ctDNA testing reports are of vital importance. In an effort to guarantee high‐quality reporting performance, we conducted an investigation of the current detection and reporting practices for NGS‐based ctDNA analysis.
Materials and Methods
A set of simulated ctDNA samples with known variants at known allelic frequencies and a corresponding case scenario were distributed to 66 genetic testing laboratories for ctDNA analysis. Written reports were collected to evaluate the detection accuracy, reporting integrity, and information sufficiency using 21 predefined criteria.
Results
Current reporting practices for NGS‐based ctDNA analysis were found to be far from satisfactory, especially regarding testing interpretation and methodological details. Only 42.4% of laboratories reported the results in complete concordance with the expected results. Moreover, 74.2% of reports only listed aberrations with direct and well‐known treatment consequences for the tumor type in question. Genetic aberrations for which experimental agents and/or drug access programs are available may thus be overlooked. Furthermore, methodological details for the interpretation of results were missing from the majority of reports (87.9%).
Conclusion
This proof‐of‐principle study suggests that the capacity for accurate identification of variants, rational interpretation of genotypes, comprehensive recommendation of potential medications, and detailed description of methodologies need to be further improved before ctDNA analysis can be formally implemented in the clinic.
Implications for Practice
Accurate, comprehensive, and standardized clinical sequencing reports can help to translate complex genetic information into patient‐centered clinical decisions, thereby shepherding precision oncology into daily practice. However, standards, guidelines, and quality requirements for clinical reports of next‐generation sequencing (NGS)‐based circulating tumor DNA (ctDNA) analysis are currently absent. By using a set of simulated clinical ctDNA samples and a corresponding case scenario, current practices were evaluated to identify deficiencies in clinical sequencing reports of ctDNA analysis. The recommendations provided here may serve as a roadmap for the improved implementation of NGS‐based ctDNA analysis in the clinic.
Keywords: Circulating tumor DNA, Next‐generation sequencing, Clinical reports, Quality control, Standardization
Short abstract
Understanding the whole picture of tumor variations will allow for customized treatment decisions. Circulating tumor DNA (ctDNA) is emerging as a valuable tool to guide targeted therapy. This study analyzed clinical reports of next‐generation sequencing‐based ctDNA analysis for detection accuracy and reporting capacity.
Introduction
Precision medicine is an inevitable trend in the development of cancer treatment. By getting the whole picture of tumor variations, it can help patients make their way into optimum custom treatment decisions 1, 2, 3. Because circulating tumor DNA (ctDNA) can provide a comprehensive profiling of tumor aberrations, ctDNA analysis is emerging as a valuable tool for the guidance of targeted therapy 3, 4, 5, the observation of tumor dynamics 6, 7, and the assessment of resistance to treatment 8, 9. So far, many different approaches, including the amplification refractory mutation system (ARMS), digital polymerase chain reaction (dPCR), and next‐generation sequencing (NGS), have been applied for the detection of tumor aberrations in ctDNA 10, 11, 12. Because of the increased affordability, accessibility, high throughput, and unrestricted detection range, NGS‐based ctDNA analysis is now replacing ARMS, dPCR, and other ctDNA analysis approaches stepwise and is being extensively used in the clinical setting 12.
Currently, an increasing number of clinical and commercial laboratories have been performing NGS‐based ctDNA analysis and issuing corresponding clinical reports to provide information to oncologists for the care of their patients 12. However, because of the use of different NGS panels (commercial NGS panels or laboratory‐developed test panels), target enrichment strategies (targeted capture or multiplex PCR), sequencing platforms, improvement countermeasures, bioinformatics analysis pipelines, and databases (public databases or self‐built databases) by different laboratories, the detected variants and relevant information regarding medication in each report also differ 13, 14. Although these laboratories usually self‐proclaim that they have accurately detected tumor‐specific alterations with high clinical sensitivity and specificity and have comprehensively reported the latest medication information, the authenticity and reliability of the clinical ctDNA analysis report is still doubtful 15, 16. As accurate, comprehensive, and standardized test reports serve as the cornerstone for clinicians to identify the best treatment strategies for patients with cancer, the existing inconsistent reports have made this a challenging time for clinicians to exactly fathom out the reliable reports and effectively incorporate these reports into routine patient care 17, 18, 19.
To address this situation, a multidisciplinary molecular tumor board (MTB) has been proposed 17, 18 that brings together clinicians, pathologists, geneticists, molecular biologists, and bioinformaticians to assess appropriate tumor profiling and to discuss acceptable therapeutic options for patients. However, owing to the low penetration rate, the low level of patient coverage, and the long lead time for gathering experts, MTB is not currently a viable option 20. Therefore, guaranteeing the accuracy, comprehensiveness, and standardization of clinical reports is of great importance to help integrate the genomic information into appropriate clinical care decisions. In 2017, the Association for Molecular Pathology (AMP), American Society of Clinical Oncology (ASCO), and College of American Pathologists (CAP) jointly published a consensus recommendation for the interpretation and reporting of sequence variants in cancer 21, 22. However, this consensus recommendation is typically used for tumor tissue. Because of the unique biological characteristics of ctDNA and the particular clinical significance of ctDNA analysis (such as readily available for observation of tumor dynamics and assessment of resistance to treatment), some additional information, including description of detailed clinical information and previous medication, may be needed for the reporting of variants detected in ctDNA. Currently, ctDNA report experiences are scarcely shared in the literature and standardized clinical reports for NGS‐based ctDNA analysis have not yet been published 20. Moreover, little is known about whether accurate and sufficient information about detected variants, relevant medications, and methodological details is being reported to clinicians.
Hence, to determine the root causes of this inconsistent reporting and to help standardize the reporting process, the National Center for Clinical Laboratories of China and the Royal College of Pathologists Australasia Quality Assurance Programs of Australia jointly conducted an investigation of the detecting and reporting practices for NGS‐based ctDNA analysis. Using a set of simulated clinical ctDNA samples and a corresponding case scenario, official clinical reports of NGS‐based ctDNA analysis from 66 genetic testing laboratories were assessed for their detection accuracy and reporting capacity.
Materials and Methods
Study Design
To investigate the current reporting practices for NGS‐based ctDNA analysis and to assess the reporting consistency between different laboratories, a set of simulated ctDNA samples and a corresponding case scenario were distributed to genetic testing laboratories for ctDNA analysis. The sample design and corresponding clinical information originated from recently published case reports 23. Four commonly detected and clinically relevant variants in the EGFR and TP53 genes, with variant allelic frequencies (VAF) ranging from 0.1% to 10%, were incorporated into our panel. Official clinical reports of NGS‐based ctDNA analysis on particular simulated clinical samples were requested to submit for critical appraisal. Table 1 summarizes the composition of the panel, and the clinical details for the samples are described in the Case Scenario Design section.
Table 1.
The intended results and validated results of the scheme panel
| Sample No. | Intended results | Validated results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ARMS | dPCR | Targeted NGS | ||||||||
| Gene | Transcript ID | Variant | Allele Frequency, % | Variant | Allele Frequency, % | Variant | Allele Frequency, % | Variant | Allele Frequency, % | |
| A | TP53 | NM_000546.5 | c.742C>G (p.Arg248Gly) | 10 | ND | ND | ND | ND | c.742C>G (p.Arg248Gly) | 15.32 |
| EGFR | NM_005228.3 | c.2235_2249del (p.Glu746_Ala750del) | 5 | c.2235_2249del (p.Glu746_Ala750del) | ND | c.2235_2249del (p.Glu746_Ala750del) | 7.68 | c.2235_2249del (p.Glu746_Ala750del) | 6.66 | |
| B | TP53 | NM_000546.5 | c.742C>G (p.Arg248Gly) | 5 | ND | ND | ND | ND | c.742C>G (p.Arg248Gly) | 6.31 |
| EGFR | NM_005228.3 | c.2235_2249del (p.Glu746_Ala750del) | 1 | c.2235_2249del (p.Glu746_Ala750del) | ND | c.2235_2249del (p.Glu746_Ala750del) | 0.70 | c.2235_2249del (p.Glu746_Ala750del) | 0.25 | |
| EGFR | NM_005228.3 | c.2369C>T (p.Thr790Met) | 0.5 | c.2369C>T (p.Thr790Met) | ND | c.2369C>T (p.Thr790Met) | 0.44 | c.2369C>T (p.Thr790Met) | 0.89 | |
| C | TP53 | NM_000546.5 | c.742C>G (p.Arg248Gly) | 1 | ND | ND | ND | ND | c.742C>G (p.Arg248Gly) | 0.94 |
| EGFR | NM_005228.3 | c.2235_2249del (p.Glu746_Ala750del) | 0.1 | c.2235_2249del (p.Glu746_Ala750del) | ND | c.2235_2249del (p.Glu746_Ala750del) | 0.13 | c.2235_2249del (p.Glu746_Ala750del) | 0.10 | |
| EGFR | NM_005228.3 | c.2369C>T (p.Thr790Met) | 0.1 | c.2369C>T (p.Thr790Met) | ND | c.2369C>T (p.Thr790Met) | 0.11 | c.2369C>T (p.Thr790Met) | 0.16 | |
| EGFR | NM_005228.3 | c.2390G>C (p.Cys797Ser) | 0.25 | ND | ND | ND | ND | c.2390G>C (p.Cys797Ser) | 0.13 | |
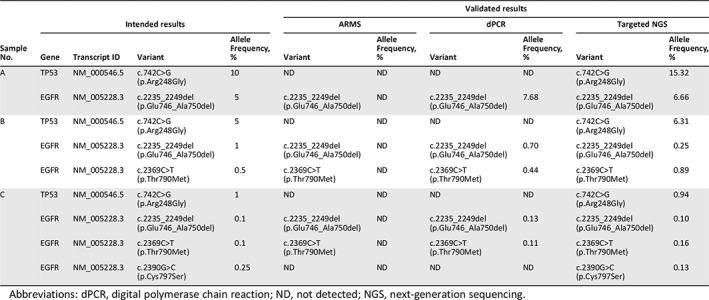
Abbreviations: dPCR, digital polymerase chain reaction; ND, not detected; NGS, next‐generation sequencing.
Sixty‐six participating laboratories were chosen from a pool of 89 potential candidates that completed a selection survey. The selection criteria included NGS panel content (to ensure most of the mutations in our panel could be detected), clinical diagnostic workload (to ensure laboratories delivering a clinical ctDNA testing service were included), and global location (to ensure sample stability during transportation). Subsequently, three vials of simulated ctDNA samples (sample A–C) were shipped on dry ice to each laboratory. Detailed specifications of storage conditions and assay procedures were provided. In addition to the three test samples, sample normal control (NC) was also delivered as normal genomic DNA extracted from blood cells to filter out irrelevant mutations. These participating laboratories were requested to provide feedback for the official written reports within 3 weeks. Additional information was also collected, including detectable gene range, enrichment strategy, sequencing platform, bioinformatics pipeline, adopted database, limit of detection (LOD), and relevant quality metrics.
Preparation and Validation of Simulated ctDNA Samples
The simulated ctDNA samples for somatic mutation detection were generated by mixing sheared site‐directed mutagenesis DNA fragments with digested genomic DNA fragments. The digested genomic DNA fragments were prepared from the lymphoblastoid cell line, GM12878 (Coriell Cell Repositories, Camden, NJ), by micrococcal nuclease (MNase) digestion. The sheared DNA fragments, containing different variants, were prepared by PCR‐based site‐directed mutagenesis and ultrasonication. Further details of the preparation of synthetic ctDNA samples are described in our previous report 24. The primers used to construct each variant are listed in supplemental online Table 1. In the present study, between two and four different mutated fragments were pooled with the digested genomic DNA in controlled proportions in each sample. The total quantity of nucleic acid in each sample was at least 125 ng. A QuantStudio 3D dPCR system (ThermoFisher Scientific, Waltham, MA) was used to ascertain the VAF. Prepared simulated ctDNA samples with known mutations at known VAFs were validated by ARMS, dPCR, and NGS. The protocols for ARMS, dPCR, and NGS are described in full in supplemental online Appendix 1.
Case Scenario Design
To simulate a real clinical setting, a case scenario was designed and subsequently delivered along with the simulated ctDNA samples. Details of the designed case scenario are as follows.
In March 2016, a 45‐year‐old man with no history of smoking was subjected to medical examination at X Hospital due to a persistent cough. Computed tomography (CT) revealed a 3.8 cm right middle lobe mass and bilateral nodules, with the largest measuring 8 mm. In addition, a single 13 mm hepatic metastatic lesion and several bone lesions were reported. Bronchoscopic biopsy was performed and histological examination of the biopsy identified the presence of adenocarcinoma (T4N2M1b). To determine whether targeted therapy was necessary, mutation detection of the tumor biopsy sample and plasma sample (Sample A) was performed. According to the results of detection, the patient began treatment with targeted agents in April 2016. CT scans performed in June 2016 were indicative of a significant decrease in the size of the right middle lobe mass. Plasma of the patient was then collected every 2 months for monitoring (Sample B was collected in December 2016). By March 2017, disease progression was observed. Therefore, paclitaxel and carboplatin treatment was initiated in April 2017. After 4 cycles, however, the condition of the patient was still no clear improvement. Subsequently, ctDNA detection was performed, and based on the results, another targeted agent was used in June 2017. After 1 week, the patient's condition was improved, and plasma of the patient was subsequently collected every 2 months to monitoring the patient's progress (Sample C was collected in February 2018). Samples A–C are ctDNA samples, extracted from the plasma of this patient. Sample NC is genomic DNA extracted from the white blood cells of this patient.
Assessment of the Reporting Performance
To better evaluate the reporting performance of the participating laboratories, a series of predefined scoring criteria were used to evaluate the written reports 21, 22, 25 (Table 2). These predefined evaluation criteria involved five main components: patient identification, sample identification, testing interpretation, methodological details, and general demand, as well as evaluation on the detection accuracy, reporting integrity, and information sufficiency. The overall score for all evaluation criteria was 100 points. Full marks were awarded if the element was presented correctly. Partial marks were allocated if the element was present but did not fully comply with the evaluation criteria. No marks were assigned if the element was incorrect or missing. Special attention was paid to the detected variants, the interpretation of somatic mutations, and the recommendations for potential targeted drugs and trials, because these elements determine the therapeutic strategy of patients.
Table 2.
Evaluation criteria for NGS‐based ctDNA analysis reports
| Evaluation criteria | Marks |
|---|---|
| Patient Identification | |
| Name, gender, age, etc. | 2.0 |
| Pathological diagnosis | 3.0 |
| Sample identification | |
| Sample ID | 3.0 |
| Sample type | 3.0 |
| Sample quality | 3.0 |
| Collected and received dates | 3.0 |
| Testing interpretation | |
| Detected variantsa | 20.0 |
| Clinical interpretationb | 15.0 |
| Drug recommendationc | 15.0 |
| Further recommendation | 10.0 |
| Methodologic details | |
| Brief description of method | 3.0 |
| Genes or genomic regions included in the panel | 3.0 |
| Assay performance characteristics | 3.0 |
| Critical quality metrics | 3.0 |
| Version of reference sequence | 3.0 |
| Testing limitations | 3.0 |
| General demand | |
| No clerical error | 1.0 |
| Reporting date | 1.0 |
| Pages of report | 1.0 |
| Signature of operator | 1.0 |
| Reference | 1.0 |
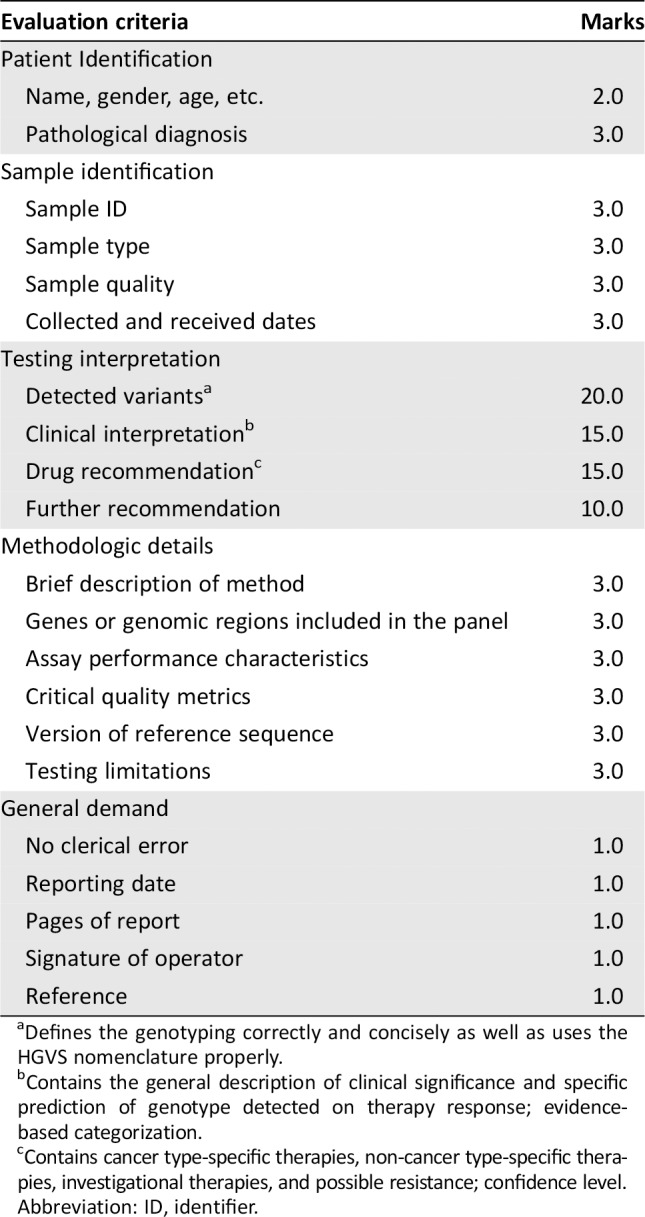
Defines the genotyping correctly and concisely as well as uses the HGVS nomenclature properly.
Contains the general description of clinical significance and specific prediction of genotype detected on therapy response; evidence‐based categorization.
Contains cancer type‐specific therapies, non‐cancer type‐specific therapies, investigational therapies, and possible resistance; confidence level.
Abbreviation: ID, identifier.
The assessment of detected variants was analyzed based on the respective panel content, LOD, and intended results. Variants out of the specific detectable range were not considered in the scoring process. False‐negative (FN) and false‐positive (FP) results, for which the reported genotype differed from the expected results, were considered as critical errors because the treatment outcome would be affected. In addition, results for which a genotype was reported in which it was below the stated LOD (without additional verification or explanation) were also taken as errors, because laboratories offering diagnostic mutational analysis of ctDNA should test for the selected clinically relevant variants. The interpretation of somatic mutations and recommendations of potential targeted drugs and trials were assessed according to the joint recommendations of the AMP, ASCO, and CAP 21. Reports of somatic variants should indicate their category based on their clinical impact. For example, tier I variants have a strong clinical significance (level A and B evidence), tier II variants have potential clinical significance (level C or D evidence), tier III variants are of unknown clinical significance, and tier IV variants are benign or likely benign 21. Potential targeted drugs or trials should cite references and indicate their classifications (level A–level D) 21. In addition, another 18 essential elements of reports were evaluated, as listed in Table 2. Accurately documenting the general information of patients was a basic requirement of any report. Sample type and sample quality were also included, as this may help to decrease the sample's negative effects and ensure the testing accuracy. A statement outlining the testing methodology and its limitations plays a vital role in determining the causes of FN or FP results and test failures. Extensive analyses were performed on these items to stress the importance of accurate patient and sample identification and clear statements about testing results and comprehensive clinical interpretations.
All statistical analyses were performed using SPSS 23.0 (IBM, Armonk, NY). Performances were compared by using Fisher's exact test with a two‐tailed statistical significance at p < .05.
Results
Study Design and Preparation of Simulated ctDNA Samples
In our study, a set of simulated ctDNA samples and a corresponding case scenario were developed to evaluate the reporting performance of NGS‐based ctDNA analysis across different laboratories. Commonly found mutations (EGFR 19del), resistant mutations (EGFR T790M and EGFR C797S), and undefined mutations (TP53 R248G) were finally incorporated in our scheme with the VAF ranging from 0.1% to 10% (Table 1). By design, 66.7% (6/9) of these mutations had VAFs within the 0.1%–1% range.
By the detection of capillary electrophoresis, the distribution of DNA fragments of our synthetic ctDNA materials were confirmed to be similar to plasma cell‐free DNA species 26, 27 (Fig. 1A). Subsequently, the relationship between the VAF obtained by theoretical calculation and the VAF measured by dPCR was determined. Sheared DNA fragments with EGFR 19del were serially diluted and blended with MNase‐digested DNA fragments at precise ratios of 20%, 10%, 5%, 2.5%, 1.25%, 0.625%, 1%, 0.1%, and 0.01%, whereas EGFR T790M were mixed at precise ratios of 2%, 1%, 0.5%, 0.25%, 0.125%, and 0.0625%. For both EGFR T790M and EGFR 19del, the theoretical VAFs of serially diluted samples were linearly related to the VAFs detected by dPCR (EGFR T790M: slope, 0.25; R2 = 0.964; EGFR 19del: slope, 0.27; R2 = 0.982; Fig. 1B). Using theoretical formulae, the VAF in our samples could be estimated in a predictable pattern. The simulated ctDNA samples were further confirmed to contain the designed variants, with the intended VAFs, using ARMS, dPCR, and NGS. No FN or FP results were reported. The results of our panel validation are summarized in Table 1.
Figure 1.
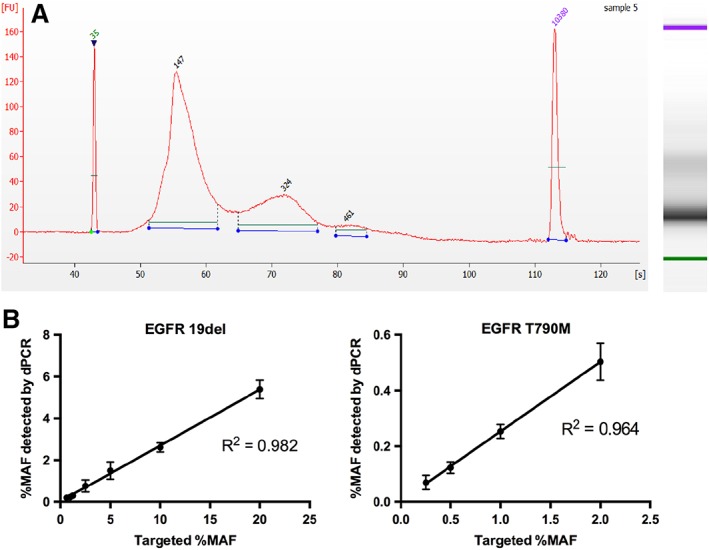
Preparation of the simulated ctDNA samples. (A): The validation results of our simulated ctDNA samples by using capillary electrophoresis. (B): The validation results of our simulated ctDNA samples by using digital PCR.
Abbreviation: dPCR, digital polymerase chain reaction; MAF, mutant allele frequency.
Characteristics of Participants
Overall, 84 results were received from 89 clinical laboratories (84 from China and 5 from Australia) before the deadline. Among these responses, 66 laboratories (63 from China and 3 from Australia), including 60 commercial laboratories and 6 clinical laboratories, were evaluated as they submitted complete datasets. Of these 66 laboratories, 65.2% employed hybrid capture enrichment (43/66), whereas 34.8% employed multiplex PCR enrichment (23/66, Fig. 2A). A variety of platforms and panels were used by participating laboratories (Fig. 2B). The most commonly used platforms were the NextSeq CN500 (Illumina, San Diego, CA; 25/66, 37.9%) and HiSeq X Ten (Illumina, 10/66, 15.2%), followed by the NextSeq 500 (Illumina) (9/66, 13.6%). Notably, 56 of the 66 laboratories (84.8 %) used Illumina platforms, while only 9 (13.6%) used ThermoFisher Scientific platforms and one used a fully automated GeneReader NGS system (QIAGEN, Hilden, Germany). The LOD of the assays among participating laboratories ranged from 0.03% to 0.5%, with 48.5% (32/66) reporting an LOD below 0.1% (Fig. 2C). All laboratories declared that the quantity and quality of our simulated ctDNA samples met their method‐specific requirements for ctDNA testing.
Figure 2.
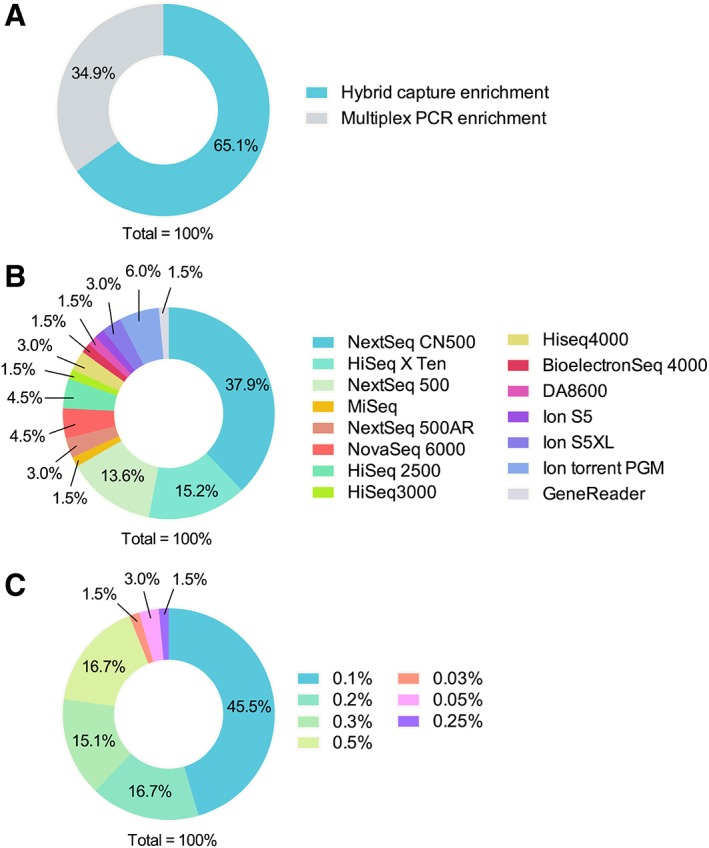
The distribution of laboratories in different characteristics. (A): The distribution of laboratories using different targeted enrichment strategies. (B): The distribution of laboratories using specific sequencing platforms. (C): The distribution of laboratories with different limit of detection.
Abbreviation: PCR, polymerase chain reaction.
Performance of the Written Reports
According to our predefined evaluation criteria, the average score for the reports was 52.0 ± 16.1 points. No laboratory gained full marks and only three laboratories received scores higher than 70 (3/66, 4.55%). The scoring for most reports (37/66, 56.0%) ranged from 50 to 70 points, with 24 labs receiving marks lower than 60 (24/37, 64.9%; Fig. 3A).
Figure 3.
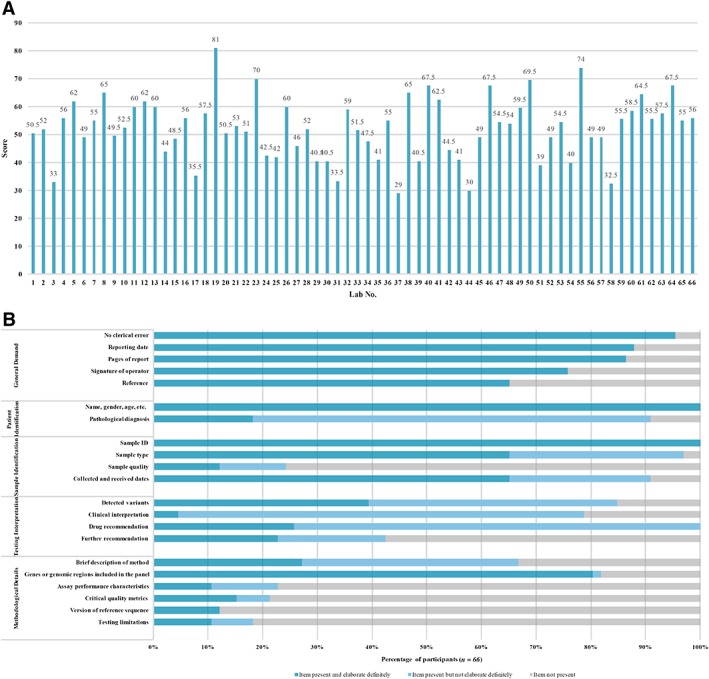
Overview of the whole reporting performance of 66 participants. (A): The scoring record of the written reports from 66 participating laboratories. (B): The detail reporting performance of 66 participating laboratories.
Abbreviation: ID, identifier.
Figure 3 shows the details of the reporting performance of all the participating laboratories. The content of the reports was found to vary between different laboratories. General items, such as general demand, patient identification, and sample identification, were well documented in most reports (Fig. 3B). However, items specific to testing interpretation and methodological details not satisfactorily reported (Fig. 3B).
The most important observation was that the accuracy of ctDNA detection was poor. Only 42.4% of laboratories (28/66) reported the results completely in concordance with the mutations in our scheme. By further analyzing the sources of discordance, 60 errors were identified, including 49 FNs (49/60, 81.67%), 6 FPs (6/60, 10.0%), and 5 results below the LOD, without annotation (5/60, 8.33%, Fig. 4). It was noteworthy that most of the FNs were observed for EGFR 19del and EGFR T790M testing in sample C (39/49, 79.6%), indicating that the detection failure may be caused by the low VAF of 0.1%. The majority of the FP results (83.3%, 5/6) were rare mutations with low VAFs (<0.1%), which raises questions about the accuracy of ctDNA analysis for variants with VAFs below 0.1%. Consistent with these concerns, the concordance with expected results improved significantly (up to 56 of 66, 84.8%) when comparisons were limited to VAFs ≥0.2%. To further analyze other sources of FN results, different combinations of target enrichment strategies and sequencing platforms were also assessed. For laboratories using Illumina platforms, 27 (27/297, 9.1%) FN results occurred in laboratories adopting hybrid capture enrichment strategies, and 11 (11/131, 8.4%) FN results were reported by those that used multiplex PCR strategies. By contrast, among labs employing semiconductor sequencing systems, all 11 of the (11/94, 11.7%) FN results were reported from labs adopting multiplex PCR enrichment. There were no significant differences between target enrichment strategies and sequencing platforms in the number of FN results (p = .738 and p = .301, respectively). Interestingly, when participants separated into those using commercially available panels (19/66, 28.8%) and those using laboratory developed testings (LDTs) (47/66, 71.2%), the commercial NGS methods showed excellent scores, whereas the latter presented higher error rates (p < .05).
Figure 4.
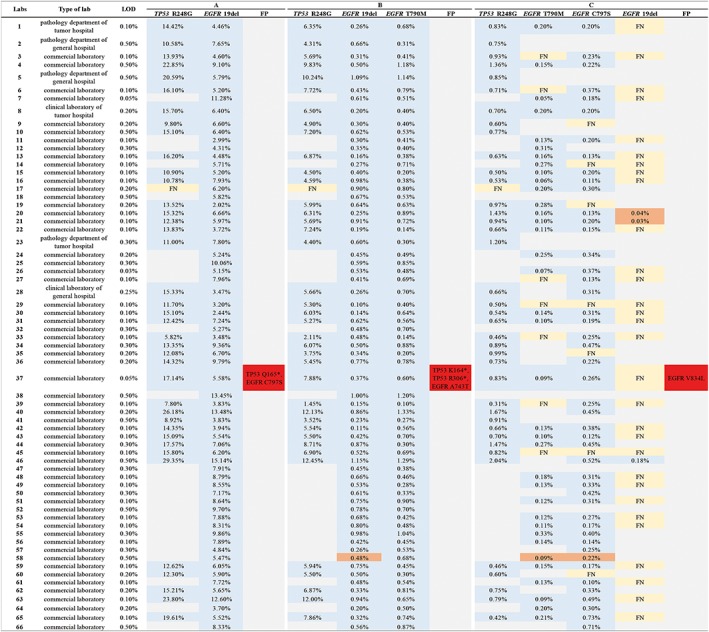
Overview of the performance for qualitative detection. The testing results of each laboratory are indicated by columns. A blue box indicates that the intended variants were correctly reported; a gray box indicates that the intended variants were not reported as the expected variant beyond the specific detectable range or lower than the validated limit of detection (LOD); a yellow box stand for a false‐negative result; a red box stand for a false‐positive result; an orange box stand for the result lower than LOD. The allele frequencies reported are shown inside the boxes.
In addition, another important observation was that the genetic and medication information regarding the detected variants in these clinical sequencing reports were also insufficient. In common with the reporting recommendations for formalin‐fixed paraffin‐embedded (FFPE) tissue samples 22, 28, evidence‐based categorization of cancer‐related variants is of critical importance. However, only six laboratories (9.1%, 6/66) classified and reported the detected variants according to the level of evidence. In addition, variant‐related clinical interpretations, drug recommendations, and comprehensive further recommendations were absent, insufficient, or not relevant to the clinical case scenario in most reports (Fig. 3). In particular, for the drug recommendations, 74.2% (49/66) of reports only listed drugs approved by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), or the National Medical Products Administration (NMPA) for the actionable variants (EGFR 19del and EGFR T790M), but without clinical trials of potential benefits and comprehensive clinical interpretations referring to the provided clinical information. Likewise, for the variants with potential clinical significance (TP53 R248G and EGFR C797S), several clinical trials were casual and unordered cited but without any interpretation based on the utility of these clinical trials.
Furthermore, many reports have also lost the information of methodological details, particularly assay performance characteristics (such as LOD [52/66, 78.8%] and minimal depth of sequencing coverage [58/66, 87.9%]), and critical quality control (QC) metrics (52/66, 78.8%). It is significant to state that the assay performance characteristics and QC metrics were critical to reliably interpret the results as negative or positive and thus have a direct impact on treatment decisions 12. However, several reports overinterpreted the absence of a relevant mutation, without providing such information. Furthermore, of those reports that stated LOD and QC metrics, three reported variants with VAFs under the LOD but without any explanation. Because only mutations reported within the detectable range are regarded as clinically useful by clinicians, attention should be paid to the information sufficiency of these results to avoid compromising therapy decision making.
Discussion
Currently, the quality of molecular targeted treatments largely depends on the decision of the clinicians 18. However, because the number of new NGS panels, approved targeted therapies, and recently published clinical trials are ever increasing, and because clinicians are confronting large amounts of complicated and multifarious genetic information, a wide gap is rapidly growing between the potential of genetic tests and clinical practice in cancer care 18. To address this disparity, collaboration between clinicians and laboratories is greatly needed for assuring patients get appropriate treatments. For that to occur, clinicians need to be assured that the report they are getting is calling mutations accurately and reporting relative information comprehensively, and then clinicians can work with necessary teams (such as tumor board) to identify best treatment strategies. However, by evaluating the reporting performance of 66 genetic testing laboratories using our simulated ctDNA materials, only three commercial laboratories who performing commercially available panels got a score higher than 70. It is demonstrated that the current reporting practice of NGS‐based ctDNA analysis was far from satisfactory, especially with regard to testing interpretation and methodological details.
For testing interpretation, the most important observation was the low accuracy of detected variants. By distributing identical ctDNA samples, only 28 laboratories (42.4%) reported the results entirely in concordance with the expected results. As these samples were synthetically generated in the form of extracted ctDNA, any confounding influences of tumor heterogeneity and ctDNA extraction bias were eliminated. By further analyzing the source and nature of the errors, it was revealed that most of the detection errors were due to the lower VAFs (especially ≤0.1%), even though many laboratories claimed that they can reliably detect mutations with VAFs of 0.03%–0.1%, with a specificity higher than 95%. No significant differences in error frequencies were found between different target enrichment strategies or sequencing platforms (p = .738 and p = .301, respectively). In contrast, when participants were separated into those using commercially available panels and those using LDTs, the commercial NGS methods showed excellent scores while the latter presented higher error rates (p < .05). These findings highlight a potential source of errors for these laboratory tests, raising concerns of whether analytical validation procedures are as rigorous as claimed 10. Similar results were also found in several recently published orthogonal comparison studies and the recent European pilot schemes, which also underscores the importance of analytically validated assays with sensitivity below 1% VAF 16, 29, 30. Thus, in the future, stricter experiments for the confirmation of performance parameters (especially LOD) need to be further developed in all laboratories by using materials with natural ctDNA characteristics and precise VAF rather than clinical samples or plasmids 16.
Apart from the low accuracy of variant detection, appropriate clinical interpretations of ctDNA genotypes and comprehensive medication recommendations were also found to be problematic (Fig. 3). The majority (95.4%) of reports did not classify the detected variants according to the level of evidence. Furthermore, 74.2% of the reports only listed the FDA‐, EMA‐, or NMPA‐approved drugs (level A) or well‐powered studies with consensus from experts in the field (level B) for the actionable variants (EGFR 19del and EGFR T790M), but without clinical or preclinical trials of potential benefits (level C–D) and comprehensive clinical interpretations referring to the provided clinical information. Likewise, for the variants with potential clinical significance (TP53 R248G and EGFR C797S), different clinical trials were unordered listed but without any interpretation based on the utility of these clinical trials in most of reports. Because comprehensive clinical reports is the cornerstone for identifying best treatment strategies, clinical reports without appropriate clinical interpretations of ctDNA genotypes and with insufficient medication information may make it difficult for clinicians to decipher the data and take appropriate actions 21, 31. Hence, in common with the reporting recommendations for FFPE tissue samples 22, 28, evidence‐based categorization of cancer‐related variants and sufficient medication information is recommended to be well documented in the ctDNA analysis reports, although they are not mandatory requirement. Meanwhile, as the most commonly performed manual interpretation is prone to miss less well‐known treatment options or recent therapies, laboratories should place a great deal of importance should be placed on updating informative databases in a timely manner.
Moreover, as a new technology, clinical reports of plasma ctDNA testing are also suggested to include some specific contents (such as the information on sample quality, QC metrics, and a description of the methodology), in addition to some required elements (such as LOD) 21, 22, 32. However, many laboratories in our study did not include a description of the methodology (especially LOD and minimal depth of sequencing coverage), sample quality (input DNA), and QC metrics. Because the properties of the testing method (sensitivity, specificity, LOD, and minimal depth of sequencing coverage) and the amount of input DNA are the basis of establishing FN or failed results and, thus, have a direct impact on treatment decisions 2, 33, 34, these parameters are proposed to clearly indicated in the written reports.
In comparison with the external quality assurance schemes that were organized by the IQN Path for ctDNA analysis 16, the overall performance of participants was poorer in our scheme. Several required laboratory performances (such as sample identification, detecting accuracy, and LOD reporting) and recommended laboratory performance (such as categorization of variants and/or medications and specific reporting preferences) for NGS‐based ctDNA analysis were found to be problematic. However, we designed a clinical case scenario and included variants at a VAF of 0.1%–10% (rather than at 1% and 5% as previously reported) in our study to resemble patient material as closely as possible. By using these variants with low VAF and the corresponding case scenario, our study demonstrates that there is still room for improvement with regard to the clinical reports in the Asia‐Pacific region. Although laboratories are only required to report accurate mutations with correct interpretations, evidence‐based testing interpretation and specific methodological details also play a significant role in building accurate, comprehensive, and standardized clinical reports. To better improve the reporting capacity of laboratories, Figure 5 summarizes the future directions for further development of a comprehensive NGS‐based ctDNA analysis report.
Figure 5.
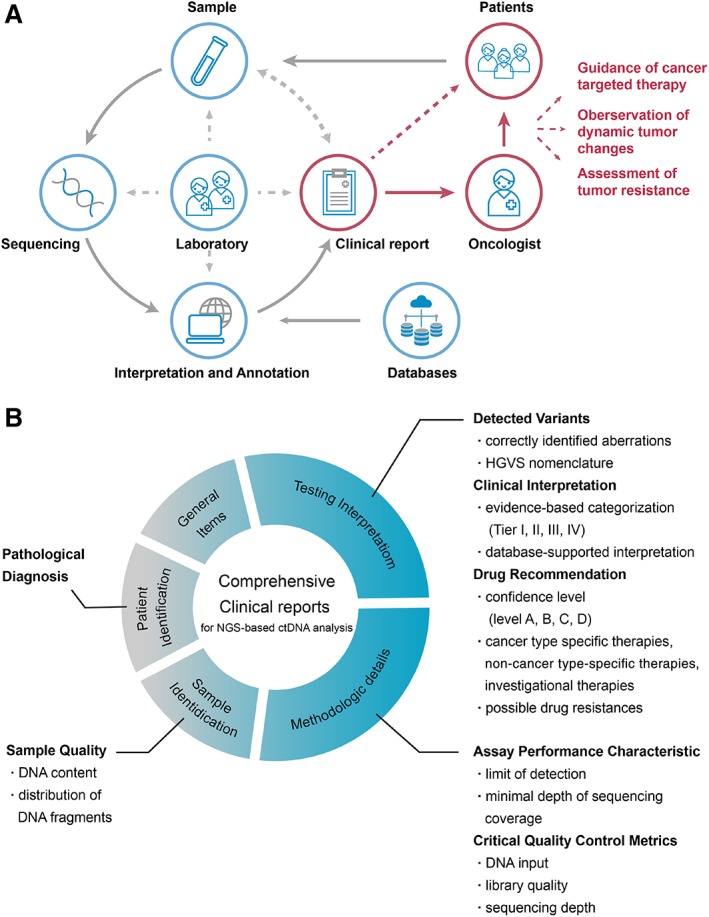
An overview of the critical elements next‐generation sequencing (NGS)‐based circulating tumor (ctDNA) analysis. (A): The importance of clinical reports in NGS‐based ctDNA analysis. (B): The critical elements for a comprehensive ctDNA analysis report.
Abbreviation: HGVS, Human Genome Variation Society.
Despite some advantages of our study, there are still several limitations that bear additional consideration. First, with a time lag, the case scenario in our scheme is a bit dated because osimertinib is now considered as the first‐line therapy for patients with EGFR mutant non‐small cell lung cancer. Nevertheless, this patient case did not impact our analysis, as it does capture clinically relevant timepoints of baseline and progression. Second, detailed characteristics of participants such as working seniority, volume of business, and certified status are insufficient in our study. This could potentially result in a risk of diminishing findings. Therefore, further study collecting additional metrics of participating laboratories is needed to strengthen our findings. Third, although no associations are identified between detection of our manufactured variants and assay design, this finding may need further confirmation for other classes of variants, such as copy number variation and fusions. Lastly, many other technologies (such as dPCR and ARMS) have also been applied for the detection of tumor aberrations in ctDNA; however, only laboratories performing NGS‐based ctDNA analysis were enrolled and analyzed in our scheme. Therefore, we cannot determine whether other ctDNA analysis technologies are more accurate than NGS for detecting mutations. In the future, more definitive studies are needed to investigate the overall performance of NGS platforms compared with non‐NGS methods.
Conclusion
Our study offers insights into a transformative period for the provision of precision medicine. Although NGS‐based ctDNA analyses have been implemented in the clinic for 5 years, our findings confirmed that many laboratories’ ability of ctDNA testing for variants with low VAF (≤0.1%) and capacity of rational interpretation and medication recommendations during reporting still need to be further improved. Our study evaluated the current practices, assessed the deficiencies of clinical sequencing reports for ctDNA analysis, and provides further recommendations that may serve as a roadmap for the improved implementation of NGS‐based ctDNA analysis. As numerous genetic testing laboratories have been performing ctDNA analysis for thousands of patients with cancer each year, these findings are clinically relevant and cautionary.
Author Contributions
Conception/design: Rongxue Peng, Rui Zhang, Martin P. Horan, Jinming Li
Provision of study material or patients: Rongxue Peng, Rui Zhang, Martin P. Horan, Li Zhou, Sze Yee Chai, Nalishia Pillay, Kwang Hong Tay, Tony Badrick, Jinming Li
Collection and/or assembly of data: Rongxue Peng, Rui Zhang, Martin P. Horan, Li Zhou, Nalishia Pillay, Kwang Hong Tay, Tony Badrick, Jinming Li
Data analysis and interpretation: Rongxue Peng, Rui Zhang, Martin P. Horan, Sze Yee Chai, Jinming Li
Manuscript writing: Rongxue Peng, Rui Zhang, Martin P. Horan, Li Zhou, Sze Yee Chai, Nalishia Pillay, Kwang Hong Tay, Tony Badrick, Jinming Li
Final approval of manuscript: Rongxue Peng, Rui Zhang, Martin P. Horan, Li Zhou, Sze Yee Chai, Nalishia Pillay, Kwang Hong Tay, Tony Badrick, Jinming Li
Disclosures
The authors indicated no financial relationships.
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supplementary Informtion
Supplemental Table S1
Acknowledgments
We thank Geneseeq Technology Inc. and AmoyDx Technology Inc. for their technical supports. This work was supported by the National Nature Science Foundation of China (81601848 to R.Z.), the Beijing Natural Science Foundation (7174345 to R.Z.), and Beijing Hospital Nova Project (BJ‐2018‐136 to R.Z.). The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Disclosures of potential conflicts of interest may be found at the end of this article.
Reference
- 1. Oellerich M, Schütz E, Beck J et al. Using circulating cell‐free DNA to monitor personalized cancer therapy. Crit Rev Clin Lab Sci 2017;54:205–218. [DOI] [PubMed] [Google Scholar]
- 2. Diaz LA, Jr , Bardelli A. Liquid biopsies: Genotyping circulating tumor DNA. J Clin Oncol 2014;32:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zugazagoitia J, Ramos I, Trigo JM et al. Clinical utility of plasma‐based digital next‐generation sequencing in patients with advance‐stage lung adenocarcinomas with insufficient tumor samples for tissue genotyping. Ann Oncol 2019;30:290–296. [DOI] [PubMed] [Google Scholar]
- 4. Tomasetti M, Amati M, Neuzil J et al. Circulating epigenetic biomarkers in lung malignancies: From early diagnosis to therapy. Lung Cancer 2017;107:65–72. [DOI] [PubMed] [Google Scholar]
- 5. Vendrell JA, Mau‐Them FT, Béganton B et al. Circulating cell free tumor DNA detection as a routine tool forlung cancer patient management. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fan G, Zhang K, Ding J et al. Prognostic value of EGFR and KRAS in circulating tumor DNA in patients with advanced non‐small cell lung cancer: A systematic review and meta‐analysis. Oncotarget 2017;8:33922–33932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fan G, Zhang K, Yang X et al. Prognostic value of circulating tumor DNA in patients with colon cancer: Systematic review. PLoS One 2017;12:e0171991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thierry AR, Pastor B, Jiang ZQ et al. Circulating DNA demonstrates convergent evolution and common resistance mechanisms during treatment of colorectal cancer. Clin Cancer Res 2017;23:4578–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Normanno N, Maiello MR, Chicchinelli N et al. Targeting the EGFR T790M mutation in non‐small‐cell lung cancer. Expert Opin Ther Targets 2017;21:159–165. [DOI] [PubMed] [Google Scholar]
- 10. Veldore VH, Choughule A, Routhu T et al. Validation of liquid biopsy: Plasma cell‐free DNA testing in clinical management of advanced non‐small cell lung cancer. Lung Cancer (Auckl) 2018;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Las Casas CM, Gonzalez‐Cao M, Ramirez SV et al. Usefulness of circulating free DNA for monitoring epidermal growth factor receptor mutations in advanced non‐small cell lung cancer patients: A case report. Transl Lung Cancer Res 2016;5:532–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richman SD, Fairley J, Hall JA et al. Results of the uk neqas for molecular genetics reference sample analysis. J Clin Pathol 2018;71:989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roy S, LaFramboise WA, Nikiforov YE et al. Next‐generation sequencing informatics: Challenges and strategies for implementation in a clinical environment. Arch Pathol Lab Med 2016;140:958–975. [DOI] [PubMed] [Google Scholar]
- 14. Goodwin S, McPherson JD, McCombie WR. Coming of age: Ten years of next‐generation sequencing technologies. Nat Rev Genet 2016;17:333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Torga G, Pienta KJ. Patient‐paired sample congruence between 2 commercial liquid biopsy tests. JAMA Oncol 2018;4:868–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keppens C, Dequeker EMC, Patton SJ et al. International pilot external quality assessment scheme for analysis and reporting of circulating tumour DNA. BMC Cancer 2018;18:804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rolfo C, Manca P, Salgado R et al. Multidisciplinary molecular tumour board: A tool to improve clinical practice and selection accrual for clinical trials in patients with cancer. ESMO Open 2018;3:e000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van der Velden DL, van Herpen CML, van Laarhoven HWM et al. Molecular tumor boards: Current practice and future needs. Ann Oncol 2017;28:3070–3075. [DOI] [PubMed] [Google Scholar]
- 19. Harada S, Arend R, Dai Q et al. Implementation and utilization of the molecular tumor board to guide precision medicine. Oncotarget 2017;8:57845‐57854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singer F, Irmisch A, Toussaint NC et al. SwissMTB: Establishing comprehensive molecular cancer diagnostics in swiss clinics. BMC Med Inform Decis Mak 2018;18:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li MM, Datto M, Duncavage EJ et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017;19:4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Merker JD, Oxnard GR, Compton C et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol 2018;36:1631–1341. [DOI] [PubMed] [Google Scholar]
- 23. Arulananda S, Do H, Musafer A et al. Combination osimertinib and gefitinib in C797S and T790M EGFR‐mutated non‐small cell lung cancer. J Thorac Oncol 2017;12:1728–1732. [DOI] [PubMed] [Google Scholar]
- 24. Zhang R, Peng R, Li Z et al. Synthetic circulating cell‐free DNA as quality control materials for somatic mutation detection in liquid biopsy for cancer. Clin Chem 2017;63:1465–1475. [DOI] [PubMed] [Google Scholar]
- 25. Plon SE, Eccles DM, Easton D et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008;29:1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cole HA, Cui F, Ocampo J, et al. Novel nucleosomal particles containing core histones and linker DNA but no histone H1. Nucleic Acids Res 2016;44:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henikoff JG, Belsky JA, Krassovsky K et al. Epigenome characterization at single base‐pair resolution. Paper presented at: The National Academy of Sciences; November 2011;108:18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ivanov M, Laktionov K, Breder V et al. Towards standardization of next‐generation sequencing of FFPE samples for clinical oncology: Intrinsic obstacles and possible solutions. J Transl Med 2017;15:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haselmann V, Ahmad‐Nejad P, Geilenkeuser WJ et al. Results of the first external quality assessment scheme (EQA) for isolation and analysis of circulating tumour DNA (ctDNA). Clin Chem Lab Med 2018;56:220–228. [DOI] [PubMed] [Google Scholar]
- 30. Stetson D, Ahmed A, Xu X et al. Orthogonal comparison of four plasma NGS tests with tumor suggests technical factors are a major source of assay discordance. JCO Precis Oncol 2019. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 31. Rolfo C, Mack PC, Scagliotti GV et al. Liquid biopsy for advanced non‐small cell lung cancer (NSCLC): A statement from the IASLC. J Thorac Oncol 2018;13:1248–1268. [DOI] [PubMed] [Google Scholar]
- 32. Han Y, Zhang R, Lin G et al. Quality assessment of reporting performance for EGFR molecular diagnosis in non‐small cell lung cancer. The Oncologist 2017;22:1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nikolaev S, Lemmens L, Koessler T et al. Circulating tumoral DNA: Preanalytical validation and quality control in a diagnostic laboratory. Anal Biochem 2018;542:34–39. [DOI] [PubMed] [Google Scholar]
- 34. Normanno N, Denis MG, Thress KS et al. Guide to detecting epidermal growth factor receptor (EGFR) mutations in ctdna of patients with advanced non‐small‐cell lung cancer. Oncotarget 2017;8:12501–12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supplementary Informtion
Supplemental Table S1