

Abstract
Intratumoral immunotherapies aim to trigger local and systemic immunologic responses via direct injection of immunostimulatory agents with the goal of tumor cell lysis, followed by release of tumor‐derived antigens and subsequent activation of tumor‐specific effector T cells. In 2019, a multitude of intratumoral immunotherapies with varied mechanisms of action, including nononcolytic viral therapies such as PV‐10 and toll‐like receptor 9 agonists and oncolytic viral therapies such as CAVATAK, Pexa‐Vec, and HF10, have been extensively evaluated in clinical trials and demonstrated promising antitumor activity with tolerable toxicities in melanoma and other solid tumor types. Talimogene laherparepvec (T‐VEC), a genetically modified herpes simplex virus type 1–based oncolytic immunotherapy, is the first oncolytic virus approved by the U.S. Food and Drug Administration for the treatment of unresectable melanoma recurrent after initial surgery. In patients with unresectable metastatic melanoma, T‐VEC demonstrated a superior durable response rate (continuous complete response or partial response lasting ≥6 months) over subcutaneous GM‐CSF (16.3% vs. 2.1%; p < .001). Responses were seen in both injected and uninjected lesions including visceral lesions, suggesting a systemic antitumor response. When combined with immune checkpoint inhibitors, T‐VEC significantly improved response rates compared with single agent; similar results were seen with combinations of checkpoint inhibitors and other intratumoral therapies such as CAVATAK, HF10, and TLR9 agonists. In this review, we highlight recent results from clinical trials of key intratumoral immunotherapies that are being evaluated in the clinic, with a focus on T‐VEC in the treatment of advanced melanoma as a model for future solid tumor indications.
Implications for Practice
This review provides oncologists with the latest information on the development of key intratumoral immunotherapies, particularly oncolytic viruses. Currently, T‐VEC is the only U.S. Food and Drug Administration (FDA)‐approved oncolytic immunotherapy. This article highlights the efficacy and safety data from clinical trials of T‐VEC both as monotherapy and in combination with immune checkpoint inhibitors. This review summarizes current knowledge on intratumoral therapies, a novel modality with increased utility in cancer treatment, and T‐VEC, the only U.S. FDA‐approved oncolytic viral therapy, for medical oncologists. This review evaluates approaches to incorporate T‐VEC into daily practice to offer the possibility of response in selected melanoma patients with manageable adverse events as compared with other available immunotherapies.
Keywords: Intratumoral immunotherapies, Talimogene laherparepvec, OPTiM, Melanoma, Immune checkpoint inhibitors
Short abstract
This review highlights recent results from clinical trials of key intratumoral immunotherapies that are being evaluated in the clinic, with a focus on talimogene laherparepvec in the treatment of advanced melanoma as a model for future solid tumor indications.
Intratumoral Immunotherapies
Cancer immunotherapy relies on the recognition of tumor‐associated antigens by immune cells in patients 1. Other cancer immune elimination mechanisms involve innate immunity components such as natural killer (NK) cells and NK cell–mediated antibody‐dependent cellular cytotoxicity, as well as release of proinflammatory cytokines such as tumor necrosis factor α and interferon‐γ 2, 3, 4. The first clinical success with cancer immunotherapy was reported over a century ago in patients with malignant tumors treated with intratumoral inoculation of live bacteria (Fig. 1) 5. Intratumoral immunotherapy offers enhanced locoregional efficacy and reduced systemic toxicity by enabling high bioavailability of the agent at the injected tumor sites while limiting systemic exposure 6. Using tumor as a source of antigens expressed across multiple tumor clones, intratumoral immunotherapies aim to initiate local recruitment of immune cells into the tumor microenvironment and subsequently prime T cells for a systemic polyclonal antitumor response, potentially addressing intra‐ and intertumoral heterogeneity 7, 8, 9. Several intratumoral therapies with differing mechanisms of action have successfully entered clinical trials and shown promising results (Table 1).
Using tumor as a source of antigens expressed across multiple tumor clones, intratumoral immunotherapies aim to initiate local recruitment of immune cells into the tumor microenvironment and subsequently prime T cells for a systemic polyclonal antitumor response, potentially addressing intra‐ and intertumoral heterogeneity.
Figure 1.
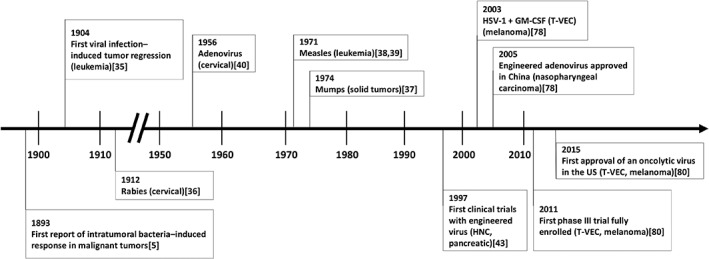
History of intratumoral therapies. Abbreviations: GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; HNC, head and neck cancer; T‐VEC, talimogene laherparepvec.
Table 1.
Summary of key intratumoral therapies
| Intratumoral therapy | Active agent; proposed MOA | Study phase, cancer type [reference], key outcomes | Key ongoing studies [http://clinicaltrials.gov identifier] |
|---|---|---|---|
| Nonviral oncolytics | |||
| PV‐10 | Rose bengal disodium; selective accumulation in lysosomes of tumor cells |
Phase II, melanoma 11 Efficacy ORR, 51% CR, 26% Safety Most AEs restricted to injection sites No treatment‐related grade 4 or 5 toxicities |
Phase III, melanoma [NCT02288897] Phase I, liver tumor [NCT00986661] |
| SD‐101 | Toll‐like receptor agonist; TLR9 activation |
Phase Ib (+ pembrolizumab), melanoma 23 Efficacy ORR, 78%, naive to prior anti–PD‐1/PD‐L1 ORR, 15%, received prior anti–PD‐1/PD‐L1 Safety Most AEs were grade 1–2 Mostly injection‐site reactions and flu‐like symptoms |
Phase III (+ pembrolizumab), prostate carcinoma [NCT03007732] Phase I (+ anti‐OX40 antibody + radiation), B‐cell non‐Hodgkin lymphoma [NCT03410901] |
| Tilsotolimod (IMO‐2125) | Toll‐like receptor agonist; TLR9 activation |
Phase I/II (+ ipilimumab), melanoma (refractory to anti–PD‐1) 25 Efficacy ORR, 38% Safety Mostly grade 1–2 injection‐related toxicities and flu‐like symptoms |
Phase III (+ ipilimumab), melanoma refractory to anti–PD‐1 [NCT03445533] |
| CMP‐001 | Toll‐like receptor agonist; TLR9 activation |
Phase Ib (+ pembrolizumab), melanoma (refractory to anti–PD‐1) 26 Efficacy ORR, 22.5% (weekly dosing) Safety Manageable acute toxicity profile |
Phase II (+ nivolumab), melanoma [NCT03618641] Phase Ib (+ pembrolizumab), melanoma [NCT02680184] |
| NKTR‐262 | Toll‐like receptor agonist; TLR7/8 activation |
Phase Ib (+ NKTR‐214, bempegaldesleukin), metastatic solid tumors 31 Efficacy ORR, 18.2% (2/11) Safety Well tolerated; no treatment‐related DLTs or SAEs |
Phase I/II (+ NKTR‐214, with or without nivolumab), metastatic solid tumors [NCT03435640] |
| Oncolytic viruses | |||
|
T‐VEC (approved by U.S. FDA) |
Genetically engineered HSV‐1 with GM‐CSF transgene; selected viral replication in tumor cells leading to tumor cell lysis |
Phase III (OPTiM), melanoma 46 Efficacy Durable response rate, 16.3% ORR, 26.4% CR, 10.8% Safety Well tolerated, with most common AEs being fatigue, chills, and pyrexia Phase II (+ ipilimumab), melanoma 95 Efficacy ORR, 39% CR, 13% Safety Well tolerated, with most common AEs being fatigue, chills, and pyrexia Phase Ib (+ pembrolizumab), melanoma 96 Efficacy Durable response rate, 16.3% ORR, 62% CR, 33% Safety Well tolerated, with most common AEs being fatigue, chills, and pyrexia |
Phase III (+ pembrolizumab), melanoma [NCT02263508] Phase II, melanoma [NCT02211131] Phase Ib/II (+ pembrolizumab), liver tumors [NCT02509507] Phase II (+ pembrolizumab), melanoma refractory to anti–PD‐1/PD‐L1 [NCT02965716] |
| CAVATAK | Genetically unaltered coxsackievirus A21; preferential infection of ICAM‐1‐expressing cells |
Phase II, melanoma 52 Efficacy ORR, 28.1% Durable response rate, 19.3% Safety No grade 3 or 4 AEs Phase I (+ ipilimumab), melanoma 50 Efficacy ORR, 50% Safety Minimal toxicity, with only 1 grade ≥ 3 treatment‐related fatigue attributable to ipilimumab |
Phase I (+ pembrolizumab), NSCLC [NCT02824965] and melanoma [NCT02565992] Phase Ib (+ ipilimumab), melanoma [NCT02307149] |
| Pexa‐Vec | Genetically engineered vaccinia virus; GM‐CSF transgene expression and thymidine kinase inactivation |
Phase II, hepatocellular carcinoma 63 Efficacy Intrahepatic response rate, 62% Safety Generally favorable safety profile Most common AEs were flu‐like symptoms |
Phase III (+ sorafenib), hepatocellular carcinoma [NCT02562755] Phase I/IIa (+ nivolumab), hepatocellular carcinoma [NCT03071094] |
| HF10 | Spontaneously mutated HSV‐1; no transgenes |
Phase II (+ ipilimumab), melanoma 73 Efficacy ORR, 41% CR, 16% Safety Grade ≥ 3 AEs, 37% Phase I (+ erlotinib, + gemcitabine), pancreatic cancer (unresectable) 72 Efficacy PFS, 6.3 mo OS, 15.5 mo Safety No AEs related to treatment |
Phase I (+ nivolumab), melanoma [NCT03259425] |
| PVS‐RIPO | Genetically engineered polio virus; selectively targeting GBM cells expressing Necl‐5; genetically modified to minimize neurovirulence |
Phase I, glioma grade IV 77 Efficacy OS, 21% at 24 mo, sustained to 36 mo Safety No neuropathogenicity No viral shedding |
Phase II (± lomustine), glioma |
| DNX‐2401 | Genetically engineered adenovirus; selective replication in Rb pathway‐deficient cells |
Phase I, recurrent malignant glioma 112 Efficacy 5 of 25 patients survived <3 y from treatment 3 patients had ≥3 y of PFS Safety No dose‐limiting toxicities 2 patients had treatment‐related AEs |
Phase II (+ pembrolizumab), recurrent glioblastoma or gliosarcoma [NCT02798406] |
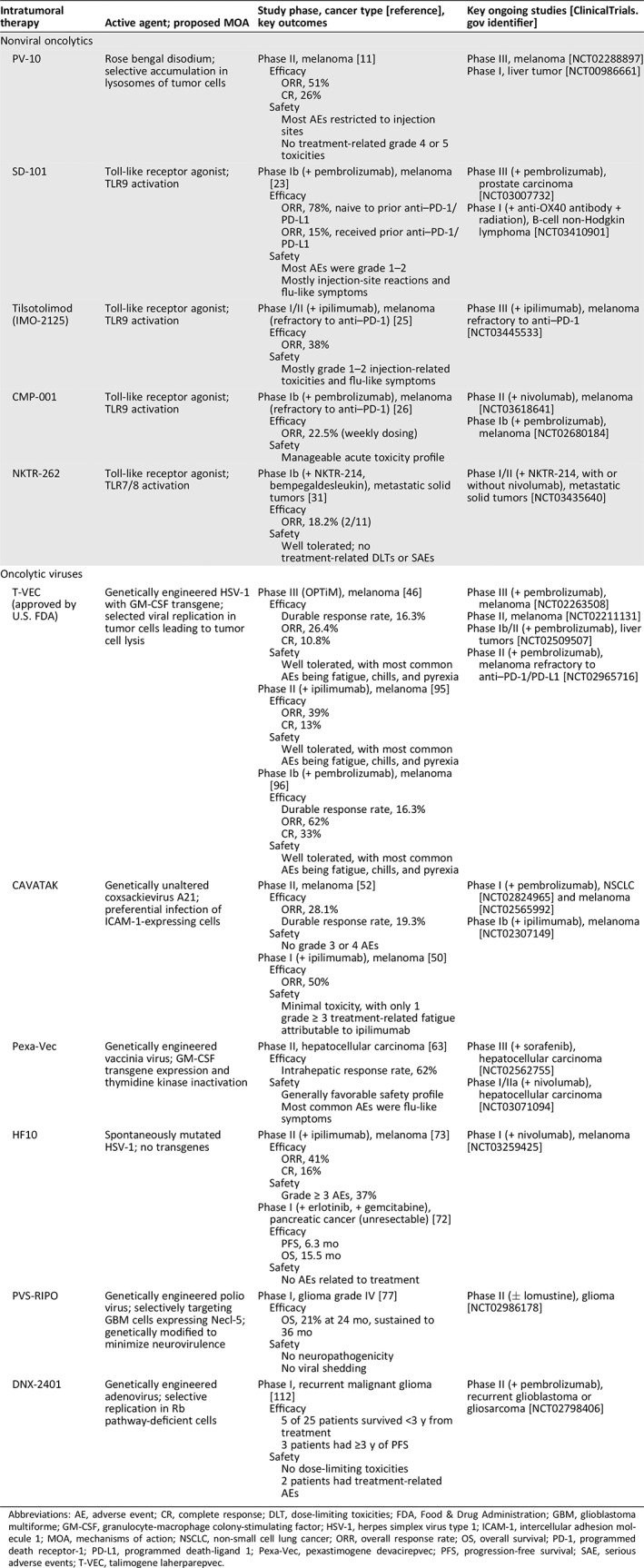
Abbreviations: AE, adverse event; CR, complete response; DLT, dose‐limiting toxicities; FDA, Food & Drug Administration; GBM, glioblastoma multiforme; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; HSV‐1, herpes simplex virus type 1; ICAM‐1, intercellular adhesion molecule 1; MOA, mechanisms of action; NSCLC, non‐small cell lung cancer; ORR, overall response rate; OS, overall survival; PD‐1, programmed death receptor‐1; PD‐L1, programmed death‐ligand 1; Pexa‐Vec, pexastimogene devacirepvec; PFS, progression‐free survival; SAE, serious adverse events; T‐VEC, talimogene laherparepvec.
PV‐10
PV‐10 (Provectus Biopharmaceuticals, Inc., Knoxville, TN) is an injectable formulation of rose bengal disodium, a water‐soluble xanthene dye, designed to be injected into solid tumors. PV‐10 has received orphan drug designation from the U.S. Food & Drug Administration (FDA) for its metastatic melanoma and hepatocellular carcinoma (HCC) indications 10, 11, 12. After intratumoral injection, PV‐10 accumulates in lysosomes and causes rapid tumor cell lysis, which can potentially induce secondary T cell–mediated systemic antitumor immunity 13, 14. In vitro and in vivo experiments demonstrated that PV‐10 selectively accumulates in lysosomes of transformed cells but not normal cells 13, 15. In a preclinical study that used the mouse B16 melanoma model, a single injection of intratumoral PV‐10 led to regression of both injected lesions and distant lesions in the lung. Splenocytes isolated from mice treated with PV‐10 showed enhanced tumor‐specific interferon (IFN)‐γ production compared with splenocytes obtained from control‐treated mice. A significant increase in B16 cell lysis was observed after PV‐10 treatment 14. In addition, combining PV‐10 with anti–programmed death receptor‐1(PD‐1) and programmed death‐ligand 1 (PD‐L1) agents resulted in an enhanced antitumor effect in melanoma‐bearing mice 16.
In a phase I study, 11 patients with locoregionally recurrent melanoma received one intratumoral injection of PV‐10. The treatment was well tolerated with no serious adverse events (AEs) observed in the study cohort. The most common AEs were transient mild‐to‐moderate pain, local inflammation, and pruritus at injection sites. The overall response rate (ORR; complete response [CR] or partial response [PR]) was 55% at 12 weeks 17. In a phase II study of intratumoral PV‐10 in refractory metastatic melanoma, 41 of 80 patients (51%) had a response, and 21 patients (26%) achieved CR. The median duration of response was 4 months, with 8% of patients being disease‐free after 52 weeks. AEs were predominantly restricted to the injection sites without any treatment‐related grade 4 or 5 toxicities 11.
A phase 3 study of PV‐10 is ongoing to assess PV‐10 monotherapy versus systemic chemotherapy with dacarbazine or temozolomide or T‐VEC for the treatment of locally advanced cutaneous melanoma in patients who are BRAF V600 wild type and have failed or are not candidates for at least one immune checkpoint inhibitor (http://clinicaltrials.gov identifier: NCT02288897). A phase Ib–II study of intratumoral PV‐10 in combination with pembrolizumab, a PD‐1–blocking antibody, for the treatment of metastatic melanoma is currently enrolling participants (http://clinicaltrials.gov identifier: NCT02557321). Safety and efficacy of PV‐10 in liver tumors from either primary HCC or liver metastases from distant tumors are currently being investigated in a phase I study (http://clinicaltrials.gov identifier: NCT00986661).
Toll‐Like Receptor Agonists
Toll‐like receptors (TLRs) are a family of pattern recognition receptors that are essential components of the innate immunity. Recognition of pathogens derived from bacteria, viruses, and fungi, or specific agonists by TLRs initiates a cascade of downstream proinflammatory events, resulting in both innate and adaptive immune responses 18. TLRs also play an important role in the development of cancer, and agonists of TLRs have demonstrated potential for cancer treatment 19. Results from preclinical studies and early‐phase clinical trials support the use of TLR9 agonists for the treatment of solid tumors and hematologic malignancies 20, 21, 22. Using a mouse model of cervical carcinoma, Baines and Celis reported that repeated administration of synthetic oligodeoxynucleotides bearing CpG motifs, an adjuvant to trigger T‐cell response via TLR9, caused significant antitumor effects and that the tumor regression correlated with increased infiltration of CD8+ effector T cells into the tumor 21.
A phase I trial was conducted to evaluate the safety profile of CpG‐28, a TLR agonist administered intratumorally, in 24 patients with recurrent glioblastoma. Overall, CpG‐28 was well tolerated, with major treatment‐related AEs being transient worsening of neurological condition, fever, and reversible lymphopenia. Response was observed in two patients, and the median overall survival was 7.2 months 20. In another phase Ib multicenter study, patients with unresectable or metastatic malignant melanoma were treated with the combination of intratumoral SD‐101 (Dynavax Technologies, Berkeley, CA), a synthetic TLR9 agonist, and intravenous pembrolizumab 23. The combination resulted in an ORR of 78% among nine patients who were naive to prior anti–PD‐1 and PD‐L1 therapy and an ORR of 15% among 13 patients who received prior anti–PD‐1 and PD‐L1 therapy. In patients naive to prior anti–PD‐1/PD‐L1 therapy, the estimated 12‐month progression‐free survival (PFS) rate was 88%, and the overall survival rate was 89%. The most common AEs were injection‐site reactions and transient flu‐like symptoms. In a phase I/II dose escalation study of intratumoral SD‐101 in combination with low‐dose radiation (http://clinicaltrials.gov identifier: NCT02266147), 29 patients with low‐grade, treatment‐naive B‐cell lymphoma received 4 Gy of radiation followed by five weekly injections of SD‐101. No treatment‐related grade 4 or 5 serious AEs occurred. Five patients had PR, and one had CR. Tumor reduction at untreated sites was seen in 24 patients, and nearly all patients had tumor reduction at their treated sites. The treatment led to an increase in CD4+/CD8+ effector T cells and a decrease in T follicular helper cells and regulatory T cells in the tumor microenvironment 24.
Combinations of other TLR9 agonists and checkpoint inhibitors, such as tilsotolimod (IMO‐2125; Idera Pharmaceuticals, Cambridge, MA) plus ipilimumab (a cytotoxic T‐lymphocyte antigen‐4 [CTLA‐4]–blocking antibody), as well as CMP‐001 (Checkmate Pharmaceuticals, Cambridge, MA) plus pembrolizumab, have been shown to trigger response in patients with melanoma refractory to PD‐1 inhibition. In the phase I/II ILLUMINATE‐204 study, the combination of tilsotolimod plus ipilimumab led to an ORR of 38% (8/21) in patients with metastatic melanoma refractory to anti–PD‐1 therapy 25. In a phase Ib trial evaluating CMP‐001 in combination with pembrolizumab in a similar patient population, the ORR was 22.5% (9/40 patients) 26.
Intratumoral injection of another TLR9 agonist, PF‐3512676, resulted in complete regression of tumors established by the A20 B‐cell lymphoma cell line in the preclinical xenograft model. When mice were inoculated with two tumors followed by intratumoral injection of PF‐3512676 in just one tumor, both tumors regressed, indicating that the immune response triggered by PF‐3512676 was systemic 27. In a phase I study (http://clinicaltrials.gov identifier: NCT00185965), 15 patients with low‐grade B‐cell lymphoma received PF‐3512676 in combination with low‐dose radiotherapy at the same single tumor site. The regimen was well tolerated, with only grade 1 or 2 systemic AEs and no treatment‐limiting AEs. One patient had CR, three had PR, and two had stable disease (SD). Memory CD8+ T cells were induced by treatment in blood and pleural fluid of responding patients, as evidenced by increased cell surface expression of both CD45RO, memory marker, and CD137, activation marker, measured by flow cytometry; however, it is not known if these induced memory CD8+ T cells were tumor specific. One patient who responded to the first treatment course relapsed and subsequently received a second course of vaccination, which led to a second and more rapid clinical response 28. Currently, TLR9 agonists are being evaluated in combination with checkpoint inhibitors or radiotherapy in a variety of tumor types, including prostate carcinoma (http://clinicaltrials.gov identifier: NCT03007732), lymphoma (http://clinicaltrials.gov identifier: NCT03410901), metastatic colorectal cancer (http://clinicaltrials.gov identifier: NCT03507699), and melanoma (http://clinicaltrials.gov identifiers: NCT02644967, NCT03445533, NCT02680184, and NCT03618641).
Clinical evaluation of TLR7/8 agonists is also underway. NKTR‐262 (NEKTAR, San Francisco, CA) is a small molecule agonist of TLR7/8 29. The phase I/II trial named REVEAL assesses safety and antitumor activity of the combination of intratumoral NKTR‐262 and NKTR‐214, a CD122‐biased agonist administered via IV infusion, with or without nivolumab. Eligible patients had advanced melanoma, Merkel cell carcinoma, colorectal cancer, urothelial carcinoma, or sarcoma. The phase I dose escalation part will identify the recommended phase II dose for the NKTR‐262 + NKTR‐214 doublet and for NKTR‐262 + NKTR‐214 + nivolumab triplet. Additional patients will be enrolled into the phase II part to further evaluate the antitumor activity of these combinations (http://clinicaltrials.gov identifier: NCT03435640) 30. Preliminary data with the doublet has been presented. Of 11 evaluable patients from the dose escalation cohort, 2 had PR, and 3 had SD, and 6 had progressive disease, resulting in a disease control rate of 45.5%. The combination of NKTR‐262 and NTKR‐214 has been well tolerated with no treatment‐related serious adverse events or dose‐limiting toxicities 31.
Intratumoral Oncolytic Viruses
Several intratumoral therapies that are either approved or under development are oncolytic viruses (OVs), defined as genetically modified or naturally occurring viruses that can preferentially infect tumor cells and, as a result, cause tumor cell lysis without affecting normal cells. OVs are recognized as a novel class of antitumor agents that offer promising therapeutic potential 7. The first evidence of viral infection–induced tumor regression was reported over a century ago, followed by an increasing number of case reports describing remission of malignancies after naturally acquired, systemic viral infection or vaccination (Fig. 1) 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43. Advances made in the techniques of genetic engineering and characterization of the viral genome have led to the current generation of genetically engineered OVs, some of which express foreign genes encoding immune modulators, such as cytokines and chemokines 32. These immune modulators can synergize with the immunostimulatory nature of OVs and potentiate an enhanced antitumor immune response 44. In 2005, an engineered H101 variant of adenovirus (Oncorine; Shanghai Sunway Biotech Co., Ltd, Shanghai, China) was approved in China for the treatment of nasopharyngeal carcinoma in combination with chemotherapy, making it the first approved OV worldwide 45. In 2015, talimogene laherparepvec (T‐VEC; IMLYGIC; Amgen Inc., Thousand Oaks, CA), a genetically modified oncolytic viral therapy, was approved by the U.S. FDA for the local treatment of unresectable cutaneous, subcutaneous, and nodal lesions in patients with melanoma recurrent after initial surgery 46. T‐VEC is the only approved OV in the U.S. to date.
In this section, we highlight the mechanisms of action and key clinical trial results for several intratumoral OVs that are still under clinical development but likely to become treatment options in the future (Table 1; DNX‐2401 [DNAtrix Therapeutics, Houston, TX], an engineered adenovirus not described in the article, is included in Table 1).
CAVATAK
Coxsackievirus A21 (CAVATAK, Merck & Co., Inc. Kenilworth, NJ) is a naturally occurring, genetically unaltered OV (without foreign gene expression), which preferentially infects tumor cells that express increased level of intercellular adhesion molecule 1 on the cell surface, leading to tumor cell lysis 47. CAVATAK has demonstrated activity against in vitro and in vivo melanoma cell lines and xenografts 48, 49.
A phase Ib study entitled Melanoma Intra‐Tumoral CAVATAK and Ipilimumab (MITCI) was conducted to evaluate the safety and efficacy of the combination of CAVATAK and ipilimumab, in up to 50 patients with treated or untreated unresectable stage IIIC–IVM1c melanoma. Interim results showed that there were no dose‐limiting toxicities reported in the 23 enrolled patients. The combination had minimal toxicity, with only one grade ≥ 3 treatment‐related fatigue that was attributable to ipilimumab. Of the 18 patients evaluable for response assessment, the confirmed ORR was 50% (9/18 patients). ORR was 60% (6/10) in patients who were naive to checkpoint inhibitors and 38% (3/8) in those who had experienced checkpoint inhibitors. Notably, in patients with stage IVM1c disease, ORR was 57.1% (4/7 patients). Responses were seen in both injected and uninjected lesions, as well as in patients with progressive disease following checkpoint inhibitor therapy 50. The phase I/II CANON study evaluated tolerability of intravesical administration of CAVATAK in patients with nonmuscle invasive bladder cancer. Interim analysis showed clinical activity of CAVATAK, evidenced by complete tumor response, viral replication within tumor, and viral‐induced apoptosis 51. In a phase II, multicenter, open‐label study entitled CAVATAK in Late‐stage Melanoma (CALM), intratumoral injection of CAVATAK was administered in 57 patients with unresectable stage IIIC–IVM1c melanoma 52. At 6 months, 21 of 57 (36.8%) evaluable patients achieved immune‐related PFS, which met the study's primary endpoint. ORR and durable response rate (defined as rate of continuous CR or PR lasting ≥6 months) per immune‐related Response Evaluation Criteria In Solid Tumors (RECIST) were 28.1% and 19.3%, respectively. No grade 3 or 4 AEs were reported.
Combination of CAVATAK with another checkpoint inhibitor, pembrolizumab, is being evaluated in a phase 1 clinical trial in patients with advanced melanoma (CAPRA trial, http://clinicaltrials.gov identifier: NCT02565992).
Pexastimogene Devacirepvec
Pexastimogene devacirepvec (Pexa‐Vec; SillaJen, Inc., Busan, Republic of Korea) is a vaccinia virus–based oncolytic immunotherapy that has been genetically modified to inactivate the viral gene encoding thymidine kinase and express human granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) and β‐galactosidase. Under normal conditions, physiological level of GM‐CSF in the serum ranges from undetectable to 20–100 pg/mL in humans 53; it can rise to high levels in serum and local tissues upon stimulation by proinflammatory signals such as cytokines, antigens, and microbial products 54. There are no data on the intratumoral or systemic level of GM‐CSF after treatment with Pexa‐Vec or T‐VEC. The replication and spread of Pexa‐Vec is dependent on the activation of epidermal growth factor receptor signaling pathway and high intercellular level of thymidine kinase, both of which are hallmarks of proliferating cancer cells 55. Deactivation of the viral gene for thymidine kinase interferes with its ability to efficiently replicate in normal cells. The insertion of genes encoding GM‐CSF allows for activation of antigen‐presenting cells 56. Both intravenous and intratumoral administration of Pexa‐Vec have been clinically proven to be well tolerated in adult patients with various types of solid tumors and in pediatric cancer patients 57, 58, 59, 60, 61. Pexa‐Vec has been tested in over 300 patients; the most commonly reported AEs were transient flu‐like symptoms, including fever and chills 62.
Currently, intratumoral administration of Pexa‐Vec, alone or in combination with sorafenib or immune checkpoint inhibitors, is mainly being assessed for the treatment of HCC in later phase trials. In a randomized phase II study of intratumoral Pexa‐Vec in 30 patients with unresectable primary HCC, Pexa‐Vec was given at doses of 108 and 109 plaque‐forming units per mL. Both doses had a favorable safety profile, with the most common AEs being flu‐like symptoms. A high dose of Pexa‐Vec resulted in an intrahepatic response rate of 62% and was associated with improved overall survival when compared with the low‐dose group (median, 14.1 months vs. 6.7 months; hazard ratio [HR] 0.39; p = .020) 63. However, the TRAVERSE trial, a randomized phase IIb study of Pexa‐Vec plus best supportive care versus best supportive care alone in patients with advanced HCC refractory to sorafenib, did not meet its primary endpoint of prolonging survival 59.
A randomized, open‐label, phase III study of intrahepatic injection of Pexa‐Vec followed by sorafenib versus sorafenib alone in patients with advanced HCC is currently enrolling participants (PHOCUS trial; http://clinicaltrials.gov identifier: NCT02562755) 64. In addition, the combination of Pexa‐Vec and nivolumab is being evaluated as first‐line treatment for advanced HCC in a phase I/IIa trial (http://clinicaltrials.gov identifier: NCT03071094).
HF10
HF10 (Takara Bio Inc., Kusatsu, Shiga, Japan) is a spontaneously mutated OV derived from herpes simplex virus type 1 (HSV‐1) without insertion of any foreign genes 65. HF10 was proved effective in treating disseminated peritoneal colon cancer and breast cancer in preclinical mouse models 66, 67 and was subsequently studied in phase I trials involving patients with recurrent breast cancer 68, head and neck cancer 69, unresectable pancreatic cancer 70, and melanoma 71. In a single‐arm, open‐label, phase I trial, 12 patients with unresectable, locally advanced pancreatic cancer received up to four intratumoral injections of HF10 (under the guidance of endoscopic ultrasound once every 2 weeks) in combination with erlotinib and gemcitabine. Of the nine patients who completed treatment, three had PR, four had SD, and two had progressive disease. AEs recorded for this study were deemed unrelated to HF10 treatment 72. Similarly, tolerable toxicities and preliminary antitumor activity were seen in other early phase trials, warranting subsequent clinical investigation of HF10.
In a phase II trial in patients with unresectable metastatic melanoma, the combination of intratumoral HF10 and systemic ipilimumab was shown to be well tolerated and demonstrated an ORR of 41% (18/44 patients) and a CR rate of 16% (7/44 patients) at 24 weeks. The duration of response ranged from 12 weeks to longer than 48 weeks, approximately, with 42.9% of responders having response ongoing after 24 weeks 73. Currently, HF10 is being tested in combination with nivolumab as a neoadjuvant therapy in resectable stage IIIB, IIIC, or IVM1a melanoma (http://clinicaltrials.gov identifier: NCT03259425).
PVS‐RIPO
Poliovirus is a single‐strand, positive‐sense RNA enterovirus that causes poliomyelitis in humans. The poliovirus receptor, nectin‐like molecule 5 (Necl‐5; also known as CD155), is widely upregulated in high‐grade glioblastoma multiforme (GBM) and several other solid tumors. Poliovirus shows natural target tropism for GBM cells expressing Necl‐5, allowing for selective infection and subsequent tumor cell lysis 74. However, Necl‐5 is also expressed in the central nervous system, and tropism for Necl‐5 is a major cause of the neuropathogenicity of poliovirus. PVS‐RIPO (Istari Oncology Inc., Research Triangle Park, NC) is a recombinant nonpathogenic poliovirus derived from the live attenuated Sabin poliovirus vaccine. To minimize the neurovirulence of wild‐type poliovirus, PVS‐RIPO is genetically modified with a heterologous human rhinovirus type 2 internal ribosomal entry site, which preferentially blocks viral translation in neurons while maintaining affinity for tumor cells 75.
PVS‐RIPO was shown to cause oncolysis of malignant glioma in vivo without adapting to a pathogenic phenotype, supporting its further evaluation in clinical trials 76. In a phase I study, 61 adult patients with recurrent malignant glioma received intratumoral infusion of PVS‐RIPO. No evidence of neuropathogenicity or viral shedding was observed. The dose level of 5.0 × 107 median tissue culture infectious dose was identified as the safety dose for phase II. The overall survival rate reached a plateau of 21% (13/61 patients; 95% confidence interval [Cl], 11%–33%) at 24 months and sustained at 36 months, which was higher than that in historical controls 77. In May 2016, PVS‐RIPO received breakthrough therapy and orphan drug designations from the U.S. FDA for glioblastoma. A phase II, randomized study of PVS‐RIPO alone or in combination with single‐cycle lomustine in patients with recurrent World Health Organization grade IV malignant glioblastoma is currently ongoing (http://clinicaltrials.gov identifier: NCT02986178).
Introduction of T‐VEC and Data from Clinical Trials of T‐VEC Monotherapy
T‐VEC is an HSV‐1–derived OV designed to selectively replicate in tumor cells and produce GM‐CSF to trigger release of tumor‐derived antigens via lysis of tumor cells, followed by antigen presentation and systemic antitumor immune responses 78. Several genetic modifications have been made to T‐VEC to enhance tumor cell selectivity, restore antigen presentation, and enhance immune recognition of HSV‐infected tumor cells while minimizing toxicity. Two copies of human transgene encoding GM‐CSF, an immune modulator playing a critical role in the maturation of monocytes and dendritic cells, are inserted into the viral genome, priming for antigen presentation to T cells and subsequent antitumor immune responses (Fig. 2)78.
Figure 2.
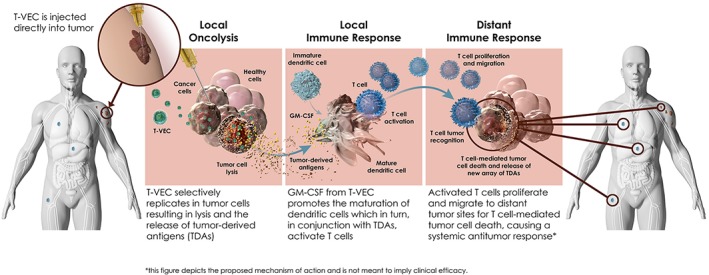
Proposed mechanisms of action for T‐VEC and effect of T‐VEC on immune cell populations. Abbreviations: GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; T‐VEC, talimogene laherparepvec. Image courtesy of Amgen Inc.
T‐VEC is classified as a biosafety level 1 agent in the U.S., a level that is not known to consistently cause disease in healthy adults 79. T‐VEC is administered via intratumoral injection into cutaneous, subcutaneous, and/or nodal lesions that are visible, palpable, or detectable by ultrasound guidance (Fig. 3) 80. The injection volume depends on lesion size and ranges from up to 0.1 mL for lesions ≤0.5 cm to up to 4 mL for lesions >5 cm. For the initial treatment with T‐VEC, the largest lesions should be injected first. For subsequent injections, new lesions that have developed since the initial injection should be prioritized for injection. The recommended maximum injection volume per treatment is 4 mL. In a phase II study evaluating the biodistribution and shedding of T‐VEC in patients with advanced melanoma, transient and low levels of T‐VEC were detected in blood and urine during treatment (no blood or urine samples were tested positive 30 days after the final dose), and occlusive dressings provided an effective barrier to viral transmission as the live virus was never detected on the exterior surface. The transmission of T‐VEC from patients to close contacts was not detected with proper administration and handling process 81.
Figure 3.
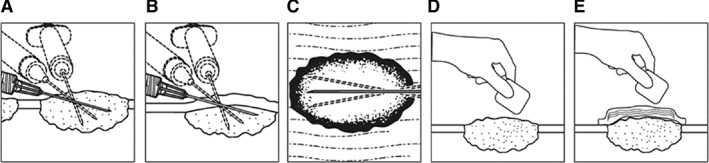
Talimogene laherparepvec (T‐VEC) injection procedures and recommended dosing schedule. Illustration of T‐VEC administration for (A) cutaneous lesions, (B) subcutaneous lesions, and (C) nodal lesions. A new needle is used for each injected lesion. (D): After injection, the injection site and surrounding area should be swabbed with alcohol and an absorbent pad and dry occlusive dressing should be applied. (E): The exterior of the occlusive dressing should also be swabbed with alcohol.
The antitumor activity of T‐VEC has been demonstrated in a variety of in vitro human cancer cell lines and in vivo mouse models. In xenograft mouse models receiving injection of T‐VEC, shrinkage or clearance was observed in both injected tumors and tumors that were not directly injected; treated mice were protected against further tumor cell rechallenge 78. In mouse tumor models treated with T‐VEC in combination with ipilimumab, the combination resulted in a significant increase in median survival and complete regressions compared with either agent alone. All injected tumors were cured, and 80% of uninjected contralateral tumors showed regression (with 6/10 cures) 82.
T‐VEC was initially tested in a phase I study in patients with refractory cutaneous, subcutaneous, or nodal metastases from a variety of tumor types, including breast, head and neck, and gastrointestinal cancers, as well as malignant melanoma 83. After demonstration of tolerability and biological activity, T‐VEC was further evaluated in a phase II study in 50 patients with unresectable, stage IIIC–IV malignant melanoma. ORR was 26% per RECIST, with responses seen in both injected and uninjected lesions. AEs were limited to transient flu‐like symptoms 84.
Results from phase I and II studies supported the initiation of an open‐label, randomized, phase III pivotal trial that investigated T‐VEC in patients with unresectable stage IIIB/C or IV melanoma (OPTiM; http://clinicaltrials.gov identifier: NCT00769704, EudraCT number: 2008‐006140‐20) 46, 83, 84. A total of 436 patients were randomized 2:1 to receive intratumoral T‐VEC (295 patients) or subcutaneous recombinant GM‐CSF (141 patients). Overall, 57% had stage IIIB, IIIC, or IVM1a disease, and 47% had not received prior systemic therapy in a metastatic setting.
At the time of primary analysis, median treatment duration was 23.0 weeks (0.1–78.9) for the T‐VEC arm and 10.0 weeks (0.6–72.0) for the GM‐CSF arm; median follow‐up time was 44.4 months (32.4–58.7). Both treatments were well tolerated, with no treatment‐related deaths and few cases of treatment discontinuation caused by AEs (4% for T‐VEC and 2% for GM‐CSF). Flu‐like symptoms such as fatigue, chills, and pyrexia were the most common AEs with T‐VEC treatment, as seen in previous studies 83, 84. Incidence of treatment‐related grade 3 or 4 AEs was 11% with T‐VEC and 5% with GM‐CSF.
The primary endpoint was durable response rate, defined as the rate of CR or PR lasting ≥6 months continuously and beginning within the first 12 months. Treatment with T‐VEC resulted in a significantly improved durable response rate compared with GM‐CSF treatment (16.3% vs. 2.1%; unadjusted odds ratio, 8.9; 95% Cl, 2.7–29.2; p < .001). ORR with T‐VEC was 26.4% (95% Cl, 21.4%–31.5%), higher than that with GM‐CSF (5.7%; 95% Cl, 1.9%–9.5%; p < .001). Thirty‐two patients treated with T‐VEC achieved CR, resulting in a CR rate of 10.8%, which was higher than that reported historically for other single‐agent immunotherapies 85, 86. The median time to response among T‐VEC responders was 4.1 months (1.2–16.7). The median duration of treatment was 23.0 weeks (0.1–78.9) in the T‐VEC arm and 10.0 weeks (0.6–72.0) in the GM‐CSF arm (Table 2). Patients responding to T‐VEC had a prolonged duration of response; in a follow‐up analysis of OPTiM, 40 of 48 patients (83%) who had a durable response to T‐VEC had responses ongoing after a median follow‐up duration of 18.4 months (10.8–19.2; Fig. 4A) 87.
Table 2.
Efficacy of T‐VEC alone or in combination with checkpoint inhibitors in advanced melanoma
| Endpoints | OPTiM, phase III | T‐VEC + ipilimumab, phase II | T‐VEC + pembrolizumab (MASTERKEY‐265), phase Ib | ||
|---|---|---|---|---|---|
| T‐VEC (n = 295) | GM‐CSF (n = 141) | T‐VEC + ipilimumab (n = 98) | Ipilimumab (n = 100) | T‐VEC + pembrolizumab (n = 21) | |
| Allow prior systemic therapy | Yes | Yes | No | ||
| Median follow‐up time, months | 44.4 | 15.6 | 13.3 | 36.8 | |
| Durable response rate, % (95% Cl) | 16.3(12.1–20.5) | 2.1(0.0–4.5) | N/A | N/A | N/A |
| p value | <.001 | ||||
| ORR, % | 26.4 | 5.7 | 39 | 18 | 67 |
| p value | <.001 | .002 | N/A | ||
| CR, % | 10.8 | < 1 | 13 | 7 | 43 |
| PR, % | 15.6 | 5.0 | 26 | 11 | 24 |
| SD, % | 45.4 | 50.4 | 19 | 24 | 10 |
| DCR, % | 76.3 | 56.7 | 58 | 42 | 76 |
| Median PFS, mo | N/A | N/A | 8.2 | 6.4 | NE |
| HR (95% Cl), p value | 0.83 (0.56–1.23), .35 | N/A | |||
| Median OS, mo | 23.3 | 18.9 | NE | NE | NE |
| OS rates, % | |||||
| At 12 mo | 74 | 69 | N/A | N/A | 95.2 |
| At 24 mo | 50 | 40 | N/A | N/A | 76.2 |
| At 36 mo | 39 | 30 | N/A | N/A | 71.4 |
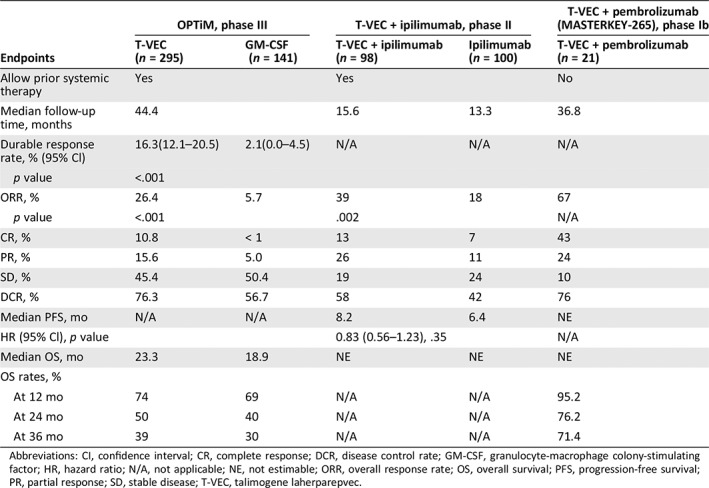
Abbreviations: CI, confidence interval; CR, complete response; DCR, disease control rate; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; HR, hazard ratio; N/A, not applicable; NE, not estimable; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival; PR, partial response; SD, stable disease; T‐VEC, talimogene laherparepvec.
Figure 4.
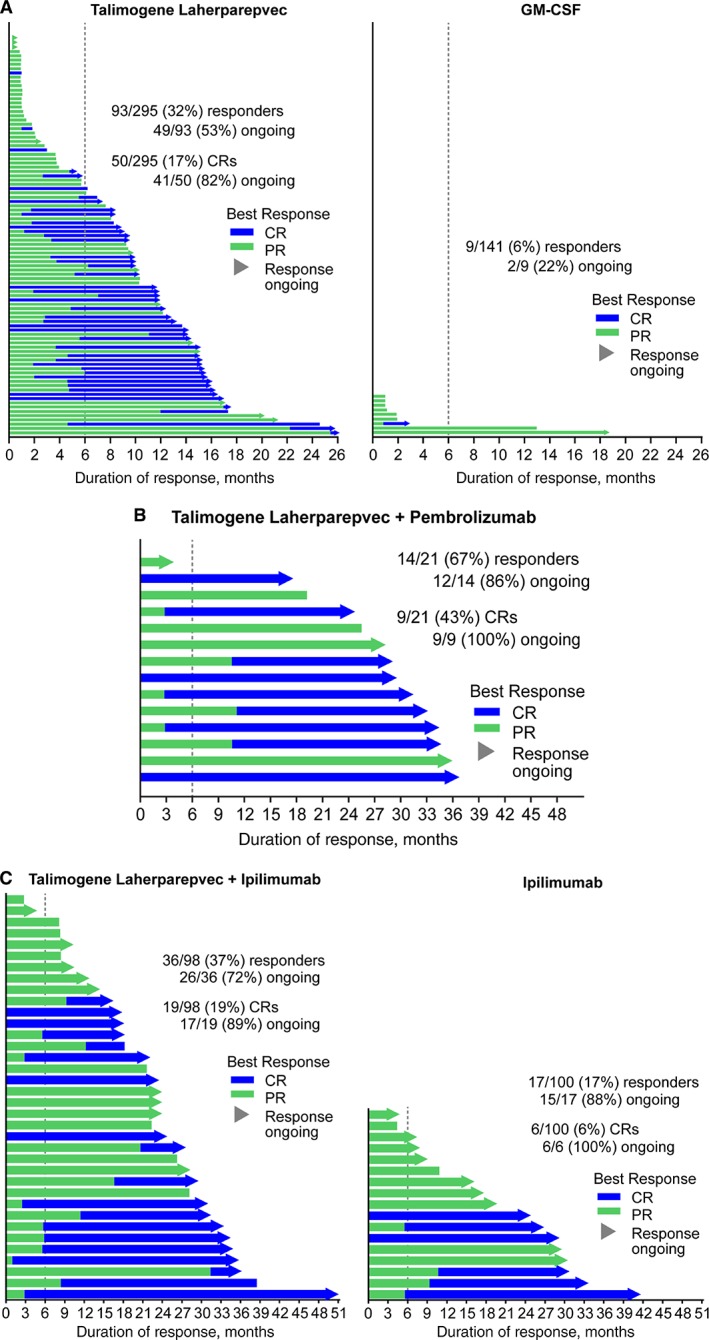
Duration of response in responders from OPTiM and T‐VEC combination trials. Duration of response for responders in the OPTiM trial (A) and combination trials of T‐VEC and pembrolizumab (B) (MASTERKEY‐265 phase 1b) or ipilimumab (C). Duration of response was defined as the longest period of response from entering response to first documented evidence of patient no longer meeting criteria for response. Response was evaluated by central Endpoint Assessment Committee in OPTiM and by investigators in MASTERKEY‐265 phase 1b and the study of T‐VEC plus ipilimumab. Abbreviations: CR, complete response; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; PR, partial response; T‐VEC, talimogene laherparepvec. Sources: Figure 4A was published in Kaufman et al., 2017 [88], which is open access and distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/) that permits unrestricted use, distribution, and reproduction in any medium. Figure 4B and 4C courtesy of Amgen Inc. (data on file).
To date, single‐agent use of T‐VEC in OPTiM has not demonstrated significant superiority over GM‐CSF on overall survival. Median overall survival was 23.3 months (95% Cl, 19.5–29.6) with T‐VEC and 18.9 months (95% Cl, 16.0–23.7) with GM‐CSF (HR, 0.79; 95% Cl, 0.62–1.00; p = .051) at the time of primary analysis (Fig. 5A; Table 2). In a subgroup analysis, patients with earlier stage IIIB–IVM1a disease derived more OS benefit from T‐VEC than patients with late‐stage IVM1b–c disease (median OS, 41.1 months [30.6–not estimable] with stage IIIB–IVM1a vs. 13.4 months [11.4–16.2] with stage IVM1b–c). In a landmark analysis of OPTiM assessing association between durable response and overall survival, achieving durable response was found to be significantly associated with improvement in OS at the landmarks of 9 months (HR, 0.07; p = .0003), 12 months (HR, 0.05; p < .0001), and 18 months (HR, 0.11; p = .0002; Fig. 5B–D) 88. In the final survival analysis at 3 years after the last randomization with a median follow‐up of 49 months, T‐VEC continued to yield a persistent effect on overall survival, with only one additional event occurring since the primary analysis 89.
Figure 5.

Overall survival and association between durable response and overall survival in OPTiM. Kaplan‐Meier plots of overall survival in the OPTiM ITT population (A), and in patients who achieved a durable response vs. patients who did not achieve durable response prior to landmark times of 9 months (B), 12 months (C), and 18 months (D) from randomization. Abbreviations: CI, confidence interval; DR, durable responder; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; HR, hazard ratio; ITT, intention to treat; NE, not estimable; OS, overall survival; T‐VEC, talimogene laherparepvec. Sources: Figure 5A has been adapted from: Kaufman HL, Andtbacka RHI, Collichio FA et al. Primary overall survival (OS) from OPTiM, a randomized phase 3 trial of talimogene laherparepvec (T‐VEC) versus subcutaneous (SC) granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) for the treatment of unresected stage IIIB/C and IV melanoma. Oral presentation from the 2014 Annual Meeting of the American Society of Clinical Oncology; May 30–June 4, 2014; Chicago, IL. Figure 5B–D were published in Kaufman et al., 2017 [88], which is open access and distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/) that permits unrestricted use, distribution, and reproduction in any medium.
After its approval by the U.S. FDA, T‐VEC has subsequently been approved by the European Medicines Agency, Australia's Therapeutic Goods Administration, the Swiss Agency for Therapeutic Products, and Israel's Ministry of Health.
Clinical Studies of Combination of T‐VEC and Checkpoint Inhibitors in Advanced Melanoma
Combining immunotherapies with complementary mechanisms of action may enhance the efficacy of either therapy alone. Because of the favorable safety profile and oncolytic properties, T‐VEC is an ideal candidate for combination regimen with immunotherapies such as checkpoint‐blocking antibodies, ipilimumab, pembrolizumab, and nivolumab. These immunotherapies have resulted in improvements in response and survival and are approved for the treatment of melanoma and other tumor types 90, 91, 92. The efficacy of T‐VEC in combination with immune checkpoint inhibitors or as monotherapy is summarized in Table 2.
Combining immunotherapies with complementary mechanisms of action may enhance the efficacy of either therapy alone. Due to the favorable safety profile and oncolytic properties, T‐VEC is an ideal candidate for combination regimen with immunotherapies such as checkpoint‐blocking antibodies, ipilimumab, pembrolizumab, and nivolumab. These immunotherapies have resulted in improvements in response and survival and are approved for the treatment of melanoma and other tumor types.
T‐VEC, in combination with pembrolizumab, is currently being evaluated in a phase Ib/III clinical trial of advanced melanoma (MASTERKEY‐265, http://clinicaltrials.gov identifier: NCT02263508). The phase Ib part of the trial was a multicenter, open‐label, single‐arm study of intratumoral T‐VEC combined with intravenous pembrolizumab in patients with previously untreated, unresectable stage IIIB–IV metastatic melanoma. The T‐VEC dosing schedule was consistent with that in OPTiM. Pembrolizumab 200 mg was given intravenously every 2 weeks after the initial dose of T‐VEC. At the time of primary analysis, patients had been followed up for a median of 18.6 months (17.7–20.8). No dose‐limiting toxicities were observed among 21 patients enrolled in the study. The most common treatment‐related AEs were fatigue, chills, and fever, which have been previously reported in T‐VEC and pembrolizumab monotherapy studies 46, 93. Additional pembrolizumab‐related AEs that frequently occurred were rash and arthralgia. Overall, the combination regimen was generally well tolerated and did not increase the toxicities from single‐agent therapies. The combination of T‐VEC plus pembrolizumab resulted in an impressively high confirmed ORR of 62% with a CR rate of 33%, evaluated per immune‐related response criteria by investigators.
In a follow‐up efficacy analysis of phase Ib MASTERKEY‐265 after a median follow‐up time of 36.8 months, two patients’ tumor response converted from previous partial response and stable disease to CR, resulting in an improved confirmed ORR of 67%, with the CR rate increased to 43% 94. The median duration of response was not reached; 85.7% of responders remained in response at the time of follow‐up analysis (Fig. 4B). The median overall survival was not estimable. The 12‐month, 24‐month, and 36‐month overall survival rates were 95.2%, 76.2%, and 71.4%, respectively. No additional safety signals were detected.
A randomized, open‐label, phase II study was conducted to evaluate the efficacy and safety of T‐VEC in combination with ipilimumab versus ipilimumab alone in patients with advanced unresectable melanoma 95. Patients with histologically confirmed stage IIIB–IVM1c melanoma were enrolled. Ninety‐eight patients received the combination of T‐VEC plus ipilimumab and 100 patients received ipilimumab alone. At the time of primary analysis, the median follow‐up time was 68 weeks (0–156) for the combination arm and 58 weeks (0–152) for the ipilimumab arm.
This study met its primary endpoint: the investigator‐assessed ORR per immune‐related response criteria was significantly higher with the combination of T‐VEC plus ipilimumab than with ipilimumab alone (39% vs. 18%; odds ratio, 2.9; 95% Cl, 1.5–5.5; p = .002). The response rate favored the combination arm regardless of disease stage, baseline tumor burden, and prior line of therapy. CR rate was 13% (13/98) in the combination arm and 7% (7/100) in the ipilimumab arm. PR rate was 26% (25/98) in the combination arm and 11% (11/100) in the ipilimumab arm.
At the time of primary analysis, duration of response was not reached in either arm; 89% of responders in the combination arm and 83% in the ipilimumab arm were still in response. The median time to response was 5.8 months (5.4–10.9) in the combination arm and not estimable in the ipilimumab arm. The median PFS was 8.2 months (4.2–21.5) in the combination arm and 6.4 months (3.2–16.5) in the ipilimumab arm (HR, 0.83; 95% Cl, 0.56–1.23; p = .35). The analysis of overall survival was immature, and patients are still being followed up for overall survival. In a follow‐up analysis that occurred 2 years after the last patient was randomized, CR rate with the combination increased to 19% (19/98), and 72% of responders (26/36) remained in response (Amgen data on file, Fig. 4C).
The combination of T‐VEC and ipilimumab was tolerable for melanoma patients, with no unexpected AEs and no meaningful increase in grade ≥ 3 AEs with either single agent. The incidence of grade ≥ 3 ipilimumab‐related AEs was 19% in the combination arm and 18% in the ipilimumab arm, and the incidence of grade ≥ 3 T‐VEC–related AEs was 15%. In addition to ipilimumab‐related AEs, additional AEs observed with the combination therapy were mostly grade 1 or 2 flu‐like events that were attributable to T‐VEC treatment. Overall, ORR and CR with the combination of T‐VEC and ipilimumab were approximately twice as high as those with single‐agent ipilimumab across all stages of melanoma (IIIB–IVM1c). Importantly, there were no additional safety concerns raised by the combination.
In summary, according to findings from previous studies described above, the combination of T‐VEC and checkpoint inhibitors synergistically improves the depth and durability of response compared with single‐agent checkpoint inhibitors, without additional safety signals (Fig. 4).
Systemic Antitumor Immunity Induced by T‐VEC
The proposed dual mechanisms of action of T‐VEC involve a direct lysis of tumor cells stimulating a local response in the tumor microenvironment, followed by a systemic immune response via release of GM‐CSF and a plethora of tumor‐derived antigens and subsequent activation of effector T cells in distant metastases (Fig. 2) 78.
Responses were seen in both injected and uninjected lesions, including visceral lesions, across previous studies of T‐VEC as monotherapy or in combination with checkpoint inhibitors 46, 95. In a lesion‐level analysis of OPTiM, T‐VEC monotherapy resulted in a ≥ 50% decrease in size in 64% of injected lesions, 34% of uninjected nonvisceral lesions, and 15% of uninjected visceral lesions; complete resolution of lesions occurred in 47% of injected lesions, 22% of uninjected lesions, and 9% of visceral lesions 87. In the combination arm of the phase II study of T‐VEC plus ipilimumab, 57% of patients had a decrease in uninjected nonvisceral lesions, and 52% had a decrease in uninjected visceral lesions 95. Visceral lesions were not injected in either study. Similar results were seen in the phase Ib study of T‐VEC plus pembrolizumab in advanced melanoma 96. Although the treatment effect of T‐VEC was seen in uninjected lesions in OPTiM, the possibility that similar effects seen in the combination trials could be attributable to checkpoint inhibitors cannot be excluded; the ongoing randomized, double‐blind, phase III study of T‐VEC plus pembrolizumab will provide information to elucidate this additive effect of T‐VEC to checkpoint inhibitors.
At the cellular level, it was shown that treatment with T‐VEC was associated with increases in both local and systemic melanoma‐associated antigen recognized by T cells–specific T cells and decreases in immune suppressor cell populations such as CD4+ and forkhead box P3 (FOXP3) + regulatory T cells, CD8+ and FOXP3+ suppressor T cells, and myeloid‐derived suppressive cells in the tumor microenvironment 96, 97.
In a phase II, single‐arm, biomarker study of T‐VEC monotherapy (EudraCT number: 2013‐005552‐15), analysis using biopsy samples from uninjected lesions showed that T‐VEC treatment resulted in increases in CD8+ tumor‐infiltrating lymphocytes, granzyme B+ effector CD8+ T cells, memory CD8+ T cells, and CD8+ T cells expressing checkpoint markers PD‐1 and CTLA‐4 but not macrophages 98. Additionally, in patients with melanoma treated with T‐VEC and pembrolizumab combination, CD8+ T cell increases after T‐VEC treatment correlated with clinical response, and the level of PD‐L1 expression was also shown to be increased after T‐VEC treatment. The response to the combination did not appear to be associated with baseline PD‐L1 status or CD8+ T‐cell infiltration 96. These immunological changes observed in uninjected lesions from patients treated with T‐VEC support a systemic effect induced by T‐VEC. However, this topic remains an active area of investigation, and relevant available data are currently limited.
It is worth noting that progression before response was commonly observed in patients treated with T‐VEC alone or in combination with checkpoint inhibitors, consistent with that seen with other immunotherapies. This pattern of pseudoprogression reinforces the importance of continuing treatment with T‐VEC in the event of appearance of new lesions or increase in existing lesions 87, 95, 99, 100, 101.
Real‐World Experiences with T‐VEC in the Treatment of Melanoma
Franke et al. from The Netherlands reported a single‐institution experience with T‐VEC monotherapy 102. In this study, 26 patients with stage IIIB–IVM1a melanoma treated with T‐VEC were included, with a follow‐up time ≥ 6 months. The T‐VEC treatment schedule was consistent with the OPTiM study protocol. Three patients (11.5%) underwent prior systemic treatment, including BRAF and/or MEK inhibitors (3.8%), ipilimumab plus nivolumab (3.8%), and treatment in a phase I trial (3.8%). The median follow‐up time was 12.5 months. Of the 26 patients, 16 (61.5%) had CR and 7 (26.9%) had PR, resulting in an ORR of 88.5%. The disease control rate was 92.3%. No new AEs were observed compared with those reported in OPTiM.
Another single‐institution experience with T‐VEC by Perez et al. included 27 patients with stage IIIB–IVM1c melanoma, 14 (52%) of whom had no prior treatment and 5 (19%) had prior treatment with immunotherapies. Five patients (19%) had stage IV disease. The median follow‐up time was 8.6 months. Of 23 patients who met the criteria for response analysis, 10 patients (43.5%) achieved CR and 3 (13.1%) had PR, leading to an ORR of 56.5%. Disease was controlled in 78.3% of patients. AEs were consistent with previous reports 103.
In a multi‐institutional experience with T‐VEC monotherapy published by Louie et al., 80 patients who had stage IIIB–IV melanoma and had been followed up for at least 3 months were evaluated 104. The median follow‐up time was 9 months (3–28). When evaluating locoregional response, thirty‐one (39%) patients had CR and 14 (18%) had PR, resulting in an ORR of 57%. AEs were generally mild, with the most common flu‐like symptoms seen in 22 (28%) patients.
These real‐world data showed a significantly higher ORR than that observed in OPTiM, possibly due to a greater number of patients (70%) with stage IV disease enrolled in OPTiM 46. As demonstrated in OPTiM subgroup analyses, patients with earlier‐stage melanoma are more likely to have a response to T‐VEC compared with those having late‐stage disease 105. In addition, tumor assessments were different in real‐world studies as compared with OPTiM. Therefore, these data should be interpreted with caution because of the small number of patients and limited follow‐up time, as well as the retrospective nature. There was also evidence supporting that T‐VEC is a viable option for use with a checkpoint inhibitor or as a monotherapy in patients who had previously received a checkpoint inhibitor 104, 106, 107.
Ongoing Clinical Trials of T‐VEC
Currently, T‐VEC is being evaluated as a neoadjuvant therapy in patients with resectable melanoma (http://clinicaltrials.gov identifier: NCT02211131). Results from an interim analysis of this trial have been previously presented 108.
The clinical evaluation of the combination of T‐VEC plus checkpoint inhibitors is currently ongoing in a variety of injectable tumor types, including those with cutaneous, subcutaneous, and nodal lesions, as well as noncutaneous lesions. The phase III MASTERKEY‐265 study is a randomized, multicenter trial of T‐VEC in combination with pembrolizumab for the treatment of patients with unresectable stage IIIB–IVM1c melanoma (http://clinicaltrials.gov identifier: NCT02263508). Results from the phase Ib part of MASTERKEY‐265 have been previously presented and are described above 96. Phase III has completed enrollment and is currently ongoing. A phase Ib/II, multicenter, open‐label trial is currently underway to evaluate the safety of T‐VEC injected into liver tumors alone and in combination with systemic pembrolizumab in patients with primary HCC or liver metastases from non‐HCC tumors, including breast cancer, colorectal cancer, gastroesophageal cancer, melanoma, non‐small cell lung cancer, and clear cell renal cell carcinoma (MASTERKEY‐318, http://clinicaltrials.gov identifier: NCT02509507). Early safety data of MASTERKEY‐318 have been previously presented 109. Intrahepatic injection of T‐VEC in combination with intravenous atezolizumab is also being evaluated in a phase Ib study involving patients with metastatic triple‐negative breast cancer or colorectal cancer with liver metastases (http://clinicaltrials.gov identifier: NCT03256344).
A significant number of patients with melanoma do not respond to anti–PD‐1 and PD‐L1 agents because of lack of preexisting tumor antigen–specific T cells in the tumor 110. With the hypothesis that intratumoral T‐VEC can address this lack of immune activation through its unique mechanisms of action, a phase II clinical trial of intratumoral T‐VEC in combination with intravenous pembrolizumab is currently recruiting patients with advanced melanoma who have disease progression on prior treatment with anti–PD‐1 and PD‐L1 agents (SWOG‐S1607, http://clinicaltrials.gov identifier: NCT02965716) 111.
Conclusions and Future Directions
According to evidence from preclinical and clinical studies, intratumoral immunotherapies have the capability to trigger a systemic antitumor immune response by increasing tumor‐specific T‐cell populations and potentially turning “cold” tumors into “hot” ones. Most of the intratumoral therapies described in this review have demonstrated promising efficacy in melanoma, with a few of them actively being evaluated in other solid tumor types. Combination with immune checkpoint inhibitors is the predominant focus in the clinical development of these therapies. Intratumoral immunotherapy is an optimal option for patients with advanced melanoma who have injectable disease. Being the first and only U.S. FDA‐approved oncolytic immunotherapy, T‐VEC provides durable clinical benefits with tolerable toxicities in advanced melanoma. When combined with systemic checkpoint inhibitors, T‐VEC significantly improves the depth and durability of the systemic antitumor response without additional safety signals, supporting the rationale that combining immunotherapies that leverage two separate areas of the cancer immunity cycle may further enhance the response. As the data of combination trials are emerging, which may better define the safety and efficacy signals, the combination of intratumoral T‐VEC and checkpoint inhibitors may become the a more widely applied treatment option for selected melanoma patients with injectable tumors in the near future.
Data Sharing
Qualified researchers may request data from Amgen clinical studies. Complete details are available at the following: http://www.amgen.com/datasharing.
Disclosures
Omid Hamid: Amgen, Bristol‐Myers Squibb, Merck, Novartis, Roche (C/A), Amgen, Array, Bristol‐Myers Squibb, Genentech, Nvoartis, Sanofi, Regeneron (SAB), Amgen, Arcus, Astellas, AstraZeneca, Bristol‐Myers Squibb, Celldex, Cytomx, Genentech, GlaxoSmithKline, Immunocore, Incyte, Iovance, Merck, Merck Serono, Medimmune, Nextcure, Novartis, Parker, Pfizer, Polynoma, Regeneron, Roche (RF); Rubina Ismail: Amgen (E/OI); Igor Puzanov: C/A for Roche, AbbVie, Inovio, 4SC, Amgen (C/A), Amgen (H, other—travel and accommodation). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Acknowledgments
We thank Yang Li, Ph.D. (Amgen Inc.) for medical writing assistance in the preparation of this article. This work was supported by Amgen Inc.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Schuster M, Nechansky A, Kircheis R. Cancer immunotherapy. Biotechnol J 2006;1:138–147. [DOI] [PubMed] [Google Scholar]
- 2. Vesely MD, Kershaw MH, Schreiber RD et al. Natural innate and adaptive immunity to cancer. Ann Rev Immunol 2011;29:235–271. [DOI] [PubMed] [Google Scholar]
- 3. Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene 2008;27:5932–5943. [DOI] [PubMed] [Google Scholar]
- 4. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565–1570. [DOI] [PubMed] [Google Scholar]
- 5. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Am J Med Sci 1893;105:487–510. [PubMed] [Google Scholar]
- 6. Sloot S, Rashid OM, Zager JS. Intralesional therapy for metastatic melanoma. Expert Opin Pharmacother 2014;15:2629–2639. [DOI] [PubMed] [Google Scholar]
- 7. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci 2016;107:1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Todo T, Rabkin SD, Sundaresan P et al. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication‐competent herpes simplex virus. Hum Gene Ther 1999;10:2741–2755. [DOI] [PubMed] [Google Scholar]
- 9. Marabelle A, Tselikas L, de Baere T et al. Intratumoral immunotherapy: Using the tumor as the remedy. Ann Oncol 2017;28(suppl 12):xii33–xii43. [DOI] [PubMed] [Google Scholar]
- 10. Foote M, Read T, Thomas J et al. Results of a phase II, open‐label, non‐comparative study of intralesional PV‐10 followed by radiotherapy for the treatment of in‐transit or metastatic melanoma. J Surg Oncol 2017;115:891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thompson JF, Agarwala SS, Smithers BM et al. Phase 2 study of intralesional PV‐10 in refractory metastatic melanoma. Ann Surg Oncol 2015;22:2135‐2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tan CY, Neuhaus SJ. Novel use of rose bengal (PV‐10) in two cases of refractory scalp sarcoma. ANZ J Surg 2013;83:93. [DOI] [PubMed] [Google Scholar]
- 13. Wachter EA, Dees C, Harkins J et al. Imaging photosensitizer distribution and pharmacology using multiphoton microscopy. International Symposium on Biomedical Optics, Optical Diagnostics of Living Cells, San Jose, CA; January 19–25, 2002;4622:112–118. [Google Scholar]
- 14. Toomey P, Kodumudi K, Weber A et al. Intralesional injection of rose bengal induces a systemic tumor‐specific immune response in murine models of melanoma and breast cancer. PloS One 2013;8:e68561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maker AV, Prabhakar B, Pardiwala K. The potential of intralesional rose bengal to stimulate t‐cell mediated anti‐tumor responses. J Clin Cell Immunol 2015;6:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu H, Weber A, Morse J et al. T cell mediated immunity after combination therapy with intralesional PV‐10 and blockade of the PD‐1/PD‐L1 pathway in a murine melanoma model. PloS One 2018;13:e0196033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thompson JF, Hersey P, Wachter E. Chemoablation of metastatic melanoma using intralesional rose bengal. Melanoma Res 2008;18:405–411. [DOI] [PubMed] [Google Scholar]
- 18. Takeda K, Kaisho T, Akira S. Toll‐like receptors. Ann Rev Immunol 2003;21:335–376. [DOI] [PubMed] [Google Scholar]
- 19. Shi M, Chen X, Ye K et al. Application potential of toll‐like receptors in cancer immunotherapy: Systematic review. Medicine 2016;95:e3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carpentier A, Laigle‐Donadey F, Zohar S et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol 2006;8:60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baines J, Celis E. Immune‐mediated tumor regression induced by CpG‐containing oligodeoxynucleotides. Clin Cancer Res 2003;9:2693–2700. [PubMed] [Google Scholar]
- 22. Link BK, Ballas ZK, Weisdorf D et al. Oligodeoxynucleotide CpG 7909 delivered as intravenous infusion demonstrates immunologic modulation in patients with previously treated non‐hodgkin lymphoma. J Immunol 2006;29:558–568. [DOI] [PubMed] [Google Scholar]
- 23. Ribas A, Medina T, Kummar S et al. SD‐101 in combination with pembrolizumab in advanced melanoma: Results of a phase Ib, multicenter study. Cancer Discov 2018;8:1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frank MJ, Reagan PM, Bartlett NL et al. In situ vaccination with a TLR9 agonist and local low‐dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov 2018;8:1258–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Diab A, Haymaker C, Benatchez C et al. Intratumoral (IT) injection of the tlr9 agonist tilsotolimod (IMO‐2125) in combination with ipilimumab (ipi) triggers durable responses in PD‐1 inhibitor refractory metastatic melanoma (rMM): Results from a multicenter, phase I/II study. Ann Oncol 2018;29:viii442–viii466. [Google Scholar]
- 26. Milhem M, Gonzales R, Medina T et al. Intratumoral toll‐like receptor 9 (TLR9) agonist, CMP‐001, in combination with pembrolizumab can reverse resistance to PD‐1 inhibition in a phase Ib trial in subjects with advanced melanoma. Presented at: AACR Annual Meeting 2018; April 14–18, 2018; Chicago, IL:CT144.
- 27. Varghese B, Widman A, Do J et al. Generation of CD8+ T cell‐mediated immunity against idiotype‐negative lymphoma escapees. Blood 2009;114:4477–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brody JD, Ai WZ, Czerwinski DK et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: A phase I/II study. J Clin Oncol 2010;28:4324–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kivimae S, Hennessy M, Pena R et al. Comprehensive antitumor immune activation by a novel TLR7/8 targeting agent NKTR‐262 combined with CD122‐biased immunostimulatory cytokine NKTR‐214. Presented at: AACR Annual Meeting 2018; April 14–18, 2018; Chicago, IL:3755.
- 30. Diab A, Marcondes M, Tagliaferri MA et al. Reveal: A phase I/II, open‐label, multicenter, dose escalation and dose expansion study of NKTR‐262 [TLR 7/8 agonist] plus NKTR‐214 [CD122‐biased agonist] with or without nivolumab (nivo) in patients (pts) with locally advanced or metastatic solid tumor malignancies. Ann Oncol 2018;29(suppl 8):mdy279.433. [Google Scholar]
- 31. Diab A, Marcondes M, Cattaruzza F et al. Oral presentation: Phase 1b: Preliminary clinical activity and immune activation for NKTR‐262 [TLR 7/8 agonist] plus bempegaldesleukin (NKTR‐214) [CD122‐biased agonist] in patients (pts) with locally advanced or metastatic solid tumors (reveal phase 1b/2 trial). Presented at: 2019 ASCO‐SITC Clinical Immuno‐Oncology Symposium; February 28–March 2, 2019; San Francisco, CA.
- 32. Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: A century of promise, a decade of progress. Nat Clin Pract Oncol 2007;4:101–117. [DOI] [PubMed] [Google Scholar]
- 33. Bierman HR, Crile DM, Dod KS et al. Remissions in leukemia of childhood following acute infectious disease: Staphylococcus and streptococcus, varicella, and feline panleukopenia. Cancer 1953;6:591–605. [DOI] [PubMed] [Google Scholar]
- 34. Hansen RM, Libnoch JA. Remission of chronic lymphocytic leukemia after smallpox vaccination. Arch Intern Med 1978;138:1137–1138. [PubMed] [Google Scholar]
- 35. Dock G. The influence of complicating diseases upon leukaemia. Am J Med Sci 1904;127:563–592. [Google Scholar]
- 36. De Pace N. Sulla scomparsa di un enorme cancro vegetante del collo dell'utero senza cura chirurgica. Ginecologia 1912;9:82–89. [Google Scholar]
- 37. Asada T. Treatment of human cancer with mumps virus. Cancer 1974;34:1907–1928. [DOI] [PubMed] [Google Scholar]
- 38. Pasquinucci G. Possible effect of measles on leukaemia. Lancet 1971;1:136. [DOI] [PubMed] [Google Scholar]
- 39. Bluming AZ, Ziegler JL. Regression of Burkitt's lymphoma in association with measles infection. Lancet 1971;2:105–106. [DOI] [PubMed] [Google Scholar]
- 40. Huebner R, Rowe W, Schatten W et al. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer 1956;9:1211–1218. [DOI] [PubMed] [Google Scholar]
- 41. Toolan H, Saunders E, Southam C et al. H‐1 virus viremia in the human. Proc Soc Exp Biol Med 1965;119:711–715. [DOI] [PubMed] [Google Scholar]
- 42. Wheelock EF, Dingle JH. Observations on the repeated administration of viruses to a patient with acute leukemia. A preliminary report. N Engl J Med 1964;271:645–651. [DOI] [PubMed] [Google Scholar]
- 43. Melcher A, Parato K, Rooney CM et al. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol Ther 2011;19:1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Graaf JF, de Vor L, Fouchier RAM et al. Armed oncolytic viruses: A kick‐start for anti‐tumor immunity. Cytokine Growth Factor Rev 2018;41:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liang M. Oncorine, the world first oncolytic virus medicine and its update in China. Current Cancer Drug Targets 2018;18:171–176. [DOI] [PubMed] [Google Scholar]
- 46. Andtbacka RH, Kaufman HL, Collichio F et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015;33:2780–2788. [DOI] [PubMed] [Google Scholar]
- 47. Xiao C, Bator‐Kelly CM, Rieder E et al. The crystal structure of coxsackievirus A21 and its interaction with ICAM‐1. Structure 2005;13:1019–1033. [DOI] [PubMed] [Google Scholar]
- 48. Shafren DR, Au GG, Nguyen T et al. Systemic therapy of malignant human melanoma tumors by a common cold‐producing enterovirus, coxsackievirus a21. Clini Cancer Res 2004;10:53–60. [DOI] [PubMed] [Google Scholar]
- 49. Au GG, Lindberg AM, Barry RD et al. Oncolysis of vascular malignant human melanoma tumors by coxsackievirus a21. Int J Oncol 2005;26:1471–1476. [DOI] [PubMed] [Google Scholar]
- 50. Curti B, Richards J, Hallmeyer S et al. The MITCI (phase 1b) study: A novel immunotherapy combination of intralesional coxsackievirus A21 and systemic ipilimumab in advanced melanoma patients with or without previous immune checkpoint therapy treatment. Presented at: American Association for Cancer Research Annual Meeting 2017; Apr 1–5, 2017; Washington, DC;CT114.
- 51. Pandha HS, Annels N, Arif M et al. Phase I/II canon study: Oncolytic immunotherapy for the treatment of non‐muscle invasive bladder (NMIBC) cancer using intravesical coxsackievirus A21. Ann Oncol 2016;27:e16016a. [Google Scholar]
- 52. Andtbacka R, Curti, BD , Kaufman H et al. Final data from CALM: A phase II study of coxsackievirus A21 (CVA21) oncolytic virus immunotherapy in patients with advanced melanoma. J Clin Oncol 2015;33:9030a. [Google Scholar]
- 53. Shi Y, Liu CH, Roberts AI et al. Granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) and T‐cell responses: What we do and don't know. Cell Res 2006;16:126–133. [DOI] [PubMed] [Google Scholar]
- 54. Conti L, Gessani S. GM‐CSF in the generation of dendritic cells from human blood monocyte precursors: Recent advances. Immunobiology 2008;213:859–870. [DOI] [PubMed] [Google Scholar]
- 55. Breitbach CJ, Bell JC, Hwang TH et al. The emerging therapeutic potential of the oncolytic immunotherapeutic Pexa‐Vec (JX‐594). Oncolytic Virother 2015;4:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Breitbach CJ, Parato K, Burke J et al. Pexa‐Vec double agent engineered vaccinia: Oncolytic and active immunotherapeutic. Curr Opin Virol 2015;13:49–54. [DOI] [PubMed] [Google Scholar]
- 57. Park SH, Breitbach CJ, Lee J et al. Phase 1b trial of biweekly intravenous Pexa‐Vec (JX‐594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol Ther 2015;23:1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cripe TP, Ngo MC, Geller JI et al. Phase 1 study of intratumoral Pexa‐Vec (JX‐594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol Ther 2015;23:602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Downs‐Canner S, Guo ZS, Ravindranathan R et al. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Mol Ther 2016;24:1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Heo J, Breitbach CJ, Moon A et al. Sequential therapy with JX‐594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: Preclinical and clinical demonstration of combination efficacy. Mol Ther 2011;19:1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hwang TH, Moon A, Burke J et al. A mechanistic proof‐of‐concept clinical trial with JX‐594, a targeted multi‐mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol Ther 2011;19:1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lawler SE, Speranza MC, Cho CF et al. Oncolytic viruses in cancer treatment: A review. JAMA Oncol 2017;3:841–849. [DOI] [PubMed] [Google Scholar]
- 63. Breitbach CJ, Moon A, Burke J et al. A phase 2, open‐label, randomized study of Pexa‐Vec (JX‐594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Methods Mol Biol 2015;1317:343–357. [DOI] [PubMed] [Google Scholar]
- 64. Abou‐Alfa GK, Galle PR, Chao Y et al. PHOCUS: A phase 3 randomized, open‐label study comparing the oncolytic immunotherapy Pexa‐Vec followed by sorafenib (SOR) vs SOR in patients with advanced hepatocellular carcinoma (HCC) without prior systemic therapy. J Clin Oncol 2017;34(suppl)TPS4146a. [Google Scholar]
- 65. Eissa IR, Naoe Y, Bustos‐Villalobos I et al. Genomic signature of the natural oncolytic herpes simplex virus HF10 and its therapeutic role in preclinical and clinical trials. Front Oncol 2017;7:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kimata H, Takakuwa H, Goshima F et al. Effective treatment of disseminated peritoneal colon cancer with new replication‐competent herpes simplex viruses. Hepatogastroenterology 2003;50:961–966. [PubMed] [Google Scholar]
- 67. Teshigahara O, Goshima F, Takao K et al. Oncolytic viral therapy for breast cancer with herpes simplex virus type 1 mutant HF 10. J Surg Oncol 2004;85:42–47. [DOI] [PubMed] [Google Scholar]
- 68. Nakao A, Kimata H, Imai T et al. Intratumoral injection of herpes simplex virus HF10 in recurrent breast cancer. Ann Oncol 2004;15:988–989. [DOI] [PubMed] [Google Scholar]
- 69. Fujimoto Y, Mizuno T, Sugiura S et al. Intratumoral injection of herpes simplex virus hf10 in recurrent head and neck squamous cell carcinoma. Acta Otolaryngol 2006;126:1115–1117. [DOI] [PubMed] [Google Scholar]
- 70. Kasuya H, Kodera Y, Nakao A et al. Phase iIdose‐escalation clinical trial of HF10 oncolytic herpes virus in 17 Japanese patients with advanced cancer. Hepatogastroenterology 2014;61:599–605. [PubMed] [Google Scholar]
- 71. Gildener‐Leapman N, Ferris RL, Ohr J et al. A phase I trial of intratumoral administration of hf10 in patients with refractory superficial cancer: Immune correlates of virus injection. J Clin Oncol 2013;31(suppl):3099a. [Google Scholar]
- 72. Hirooka Y, Kasuya H, Ishikawa T et al. A phase I clinical trial of EUS‐guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer 2018;18:596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Andtbacka RHI, Ross MI, Agarwala SS et al. Final results of a phase II multicenter trial of HF10, a replication‐competent HSV‐1 oncolytic virus, and ipilimumab combination treatment in patients with stage IIIB‐IV unresectable or metastatic melanoma. J Clin Oncol 2017;35(suppl):9510a. [Google Scholar]
- 74. Goetz C, Gromeier M. Preparing an oncolytic poliovirus recombinant for clinical application against glioblastoma multiforme. Cytokine Growth Factor Rev 2010;21:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Foreman PM, Friedman GK, Cassady KA et al. Oncolytic virotherapy for the treatment of malignant glioma. Neurotherapeutics 2017;14:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dobrikova EY, Broadt T, Poiley‐Nelson J et al. Recombinant oncolytic poliovirus eliminates glioma in vivo without genetic adaptation to a pathogenic phenotype. Mol Ther 2008;16:1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Desjardins A, Gromeier M, Herndon JE II et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med 2018;379:150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liu BL, Robinson M, Han ZQ et al. Icp34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti‐tumour properties. Gene Ther 2003;10:292–303. [DOI] [PubMed] [Google Scholar]
- 79. Amgen inc . Imlygic material safety data sheet. Available at http://msds.Amgen.Com/∼/media/amgen/repositorysites/msds‐amgen‐com/imlygicsds.Ashx. Accessed May 1, 2019.
- 80. Imlygic (talimogene laherparepvec) . Full prescribing information. Thousand Oaks, CA: Amgen inc.; 2017. [Google Scholar]
- 81. Andtbacka RHI, Mehnert JM, Nemunaitis JJ et al. Phase 2 trial evaluating biodistribution and shedding of talimogene laherparepvec in patients with unresectable stage IIIB–IV melanoma. Presented at: Society for Melanoma Research Congress Annual Meeting; October 18–21, 2017; Brisbane, Australia.
- 82. Moesta AK, Cooke K, Piasecki J et al. Local delivery of oncovex(mgm‐csf) generates systemic antitumor immune responses enhanced by cytotoxic T‐lymphocyte‐associated protein blockade. Clin Cancer Res 2017;23:6190–6202. [DOI] [PubMed] [Google Scholar]
- 83. Hu JC, Coffin RS, Davis CJ et al. A phase I study of oncoVEXGM‐CSF, a second‐generation oncolytic herpes simplex virus expressing granulocyte macrophage colony‐stimulating factor. Clin Cancer Res 2006;12:6737–6747. [DOI] [PubMed] [Google Scholar]
- 84. Senzer NN, Kaufman HL, Amatruda T et al. Phase II clinical trial of a granulocyte‐macrophage colony‐stimulating factor‐encoding, second‐generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 2009;27:5763–5771. [DOI] [PubMed] [Google Scholar]
- 85. Hodi FS, O'Day SJ, McDermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Robert C, Schachter J, Long GV et al; KEYNOTE‐006 investigators. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–2532. [DOI] [PubMed] [Google Scholar]
- 87. Andtbacka RH, Ross M, Puzanov I et al. Patterns of clinical response with talimogene laherparepvec (T‐VEC) in patients with melanoma treated in the optim phase III clinical trial. Ann Surg Oncol 2016;23:4169–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kaufman HL, Andtbacka RHI, Collichio FA et al. Durable response rate as an endpoint in cancer immunotherapy: Insights from oncolytic virus clinical trials. J Immunother Cancer 2017;5:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Andtbacka RHI, Collichio FA, Amatruda T et al. Final planned overall survival (OS) from OPTiM, a randomized phase III trial of talimogene laherparepvec (T‐VEC) versus GM‐CSF for the treatment of unresected stage IIIB/C/IV melanoma (NCT00769704). J Immunother Cancer 2014;2(suppl 3):P263a. [Google Scholar]
- 90. Keytruda (pembrolizumab) . Full prescribing information. Kenilworth, NJ: Merck & Co., Inc.; 2019. [Google Scholar]
- 91. Opdivo (nivolumab) . Full prescribing information. New York, NY: Bristol‐Myers Squibb; 2019. [Google Scholar]
- 92. Yervoy (ipilimumab) . Full prescribing information. New York, NY: Bristol‐Myers Squibb; 2018. [Google Scholar]
- 93. Ribas A, Hamid O, Daud A et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016;315:1600–1609. [DOI] [PubMed] [Google Scholar]
- 94. Long GV, Dummer R, Andtbacka RHI et al. Follow‐up analysis of MASTERKEY‐265 phase 1b (ph1b) study of talimogene laherparepvec (T‐VEC) in combination (combo) with pembrolizumab (pembro) in patients (pts) with unresectable stage IIIB–IVM1c melanoma (MEL). Presented at: Society for Melanoma Research Fifteenth International Congress; October 24–27, 2018; Manchester, England.
- 95. Chesney J, Puzanov I, Collichio F et al. Randomized, open‐label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol 2018;36:1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ribas A, Dummer R, Puzanov I et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti‐PD‐1 immunotherapy. Cell 2017;170:1109–1119.e1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kaufman HL, Kim DW, DeRaffele G et al. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM‐CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol 2010;17:718–730. [DOI] [PubMed] [Google Scholar]
- 98. Gogas H, Samoylenko I, Schadendorf D et al. Talimogene laherparepvec (T‐VEC) treatment increases intratumoral effector t‐cell and natural killer (NK) cell density in noninjected tumors in patients (pts) with stage IIIB–IVM1C melanoma: Evidence for systemic effects in a phase 2, single‐arm study. Ann Oncol 2018;29:viii442–viii466. [Google Scholar]
- 99. Wolchok JD, Hoos A, O'Day S et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune‐related response criteria. Clin Cancer Res 2009;15:7412–7420. [DOI] [PubMed] [Google Scholar]
- 100. Brahmer JR, Tykodi SS, Chow LQ et al. Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Weber JS, O'Day S, Urba W et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–5956. [DOI] [PubMed] [Google Scholar]
- 102. Franke V, Berger DMS, Klop WMC et al. High response rates for T‐VEC in early metastatic melanoma (stage IIIB/C‐IVM1a). Int J Cancer 2019;145:974–978. [DOI] [PubMed] [Google Scholar]
- 103. Perez MC, Miura JT, Naqvi SMH et al. Talimogene laherparepvec (TVEC) for the treatment of advanced melanoma: A single‐institution experience. Ann Surg Oncol 2018;25:3960–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Louie RJ, Perez MC, Jajja MR et al. Real‐world outcomes of talimogene laherparepvec therapy: A multi‐institutional experience. J Am Coll Surg 2019;228:644–649. [DOI] [PubMed] [Google Scholar]
- 105. Harrington KJ, Andtbacka RH, Collichio F et al. Efficacy and safety of talimogene laherparepvec versus granulocyte‐macrophage colony‐stimulating factor in patients with stage IIIB/C and IVM1a melanoma: Subanalysis of the phase III OPTIM trial. Onco Targets Ther 2016;9:7081–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Perez M, Amatruda T, Conry R et al. Checkpoint‐inhibitor (CPI)‐experienced patients (pts) from COSMUS‐1, a clinical observational study of talimogene laherparepvec (T‐VEC) in United States practice. J Clin Oncol 2019;37(suppl):142a. [Google Scholar]
- 107. Gogas H, Gutzmer R, Malvehy J et al. Response rates with talimogene laherparepvec monotherapy in patients with stage IIIB–IVM1C melanoma previously treated with a checkpoint inhibitor. Presented at: 15th EADO Congress; April 24–27, 2019; Paris, France.
- 108. Andtbacka RHI, Dummer R, Gyorki DE et al. Interim analysis of a randomized, open‐label phase 2 study of talimogene laherparepvec (T‐VEC) neoadjuvant treatment (neotx) plus surgery (surgx) vs surgx for resectable stage IIIB‐IVM1a melanoma (MEL). J Clin Oncol 2018;36(suppl):9508a. [Google Scholar]
- 109. Hecht JR, Pless M, Cubillo A et al. Early safety from a phase 1, multicenter, open‐label clinical trial of talimogene laherparepvec (T‐VEC) injected into liver tumors. J Clin Oncol 2018;36(suppl):2438a. [Google Scholar]
- 110. Daud AI, Loo K, Pauli ML et al. Tumor immune profiling predicts response to anti‐PD‐1 therapy in human melanoma. J Clin Invest 2016;126:3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hu‐Lieskovan S, Moon J, Campos D et al. Reversing resistance to PD‐1 blockade by combination of talimogene laherparepvec (T‐VEC) with pembrolizumab (pembro) in advanced melanoma patients following progression on a prior PD‐1 inhibitor: SWOG S1607 (NCT#02965716). J Clin Oncol 2018;36:TPS9603a. [Google Scholar]
- 112. Lang FF, Conrad C, Gomez‐Manzano C et al. Phase I study of DNX‐2401 (delta‐24‐RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol 2018;36:1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]