

Abstract
Background
In the EMBRACA phase III study (NCT01945775), talazoparib was associated with a significantly prolonged progression‐free survival (PFS) compared with physician's choice of chemotherapy (PCT) in germline BRCA1/2‐mutated HER2‐negative advanced breast cancer (ABC). Herein, the safety profile of talazoparib is explored in detail.
Materials and Methods
Overall, 412 patients received ≥1 dose of talazoparib (n = 286) or PCT (n = 126). Adverse events (AEs) were evaluated, including timing, duration, and potential overlap of selected AEs. The relationship between talazoparib plasma exposure and grade ≥3 anemia was analyzed. Time‐varying Cox proportional hazard models assessed the impact of dose reductions on PFS. Patient‐reported outcomes (PROs) in patients with common AEs and health resource utilization (HRU) were assessed in both treatment arms.
Results
The most common AEs with talazoparib were hematologic (195 [68.2%] patients) and typically occurred within the first 3–4 months of receiving talazoparib. Grade 3‐4 anemia lasted approximately 7 days for both arms. Overlapping grade 3‐4 hematologic AEs were infrequent with talazoparib. Higher talazoparib exposure was associated with grade ≥3 anemia. Permanent discontinuation of talazoparib due to hematologic AEs was low (<2%). A total of 150 (52.4%) patients receiving talazoparib had AEs associated with dose reduction. Hematologic toxicities were managed by supportive care medication (including transfusion) and dose modifications. Among patients with anemia or nausea and/or vomiting AEs, PROs favored talazoparib. After accounting for the treatment‐emergent period, talazoparib was generally associated with a lower rate of hospitalization and supportive care medication use compared with chemotherapy.
Conclusion
Talazoparib was associated with superior efficacy, favorable PROs, and lower HRU rate versus chemotherapy in gBRCA‐mutated ABC. Toxicities were manageable with talazoparib dose modification and supportive care.
Implications for Practice
Talazoparib was generally well tolerated in patients with germline BRCA‐mutated HER2‐negative advanced breast cancer in the EMBRACA trial. Common toxicities with talazoparib were primarily hematologic and infrequently resulted in permanent drug discontinuation (<2% of patients discontinued talazoparib due to hematologic toxicity). Hematologic toxicities typically occurred during the first 3–4 months of treatment and were managed by dose modifications and supportive care measures. A significant efficacy benefit, improved patient‐reported outcomes, lower rate of health resource utilization and a tolerable safety profile support incorporating talazoparib into routine management of germline BRCA‐mutated locally advanced/metastatic breast cancer.
Keywords: BRCA1, BRCA2, Breast cancer, Talazoparib, Chemotherapy
Short abstract
Talazoparib is a viable option for patients with germline BRCA‐mutated advanced breast cancer. This article presents detailed safety analyses for talazoparib, as a follow‐up to reported results from the EMBRACA trial, to highlight patterns of toxicity compared with chemotherapy and to outline guidelines for management of talazoparib toxicity in clinical practice via dose modifications and/or standard supportive care.
Introduction
Talazoparib is an inhibitor of poly (ADP‐ribose) polymerase (PARP) 1 and 2, which play important roles in DNA repair. Talazoparib exerts its cytotoxic effects by inhibition of PARP catalytic activity and by PARP trapping. This results in persistent single‐strand DNA breaks culminating in double‐strand DNA breaks that cannot be repaired accurately in tumors with defective DNA damage repair mechanisms, including tumors with mutations in the breast cancer susceptibility genes 1 or 2 (BRCA1/2) 1, 2, 3, 4. Although PARP inhibitors have emerged as effective cancer treatments, they also impact hematopoiesis, which explains some of the observed hematologic side effects of these drugs 5.
In clinical trials, talazoparib showed efficacy in patients with germline BRCA (gBRCA)‐mutated, locally advanced or metastatic human epidermal growth factor receptor 2 (HER2)‐negative breast cancer 6, 7. In the phase III EMBRACA study, progression‐free survival (PFS) was improved with talazoparib versus physician's choice of chemotherapy (PCT; hazard ratio [HR], 0.54; 95% confidence interval [CI], 0.41–0.71; p < .001), and talazoparib had a manageable safety profile 8. Significant overall improvement and delay in time to definitive clinically meaningful deterioration (TTD) in multiple patient‐reported, cancer‐related, and breast cancer‐specific symptoms, functioning, and global health status and quality of life (GHS/QoL) favored talazoparib over PCT 9.
Many patients with breast cancer associated with a BRCA mutation are treated with chemotherapy, which is associated with a high degree of toxicity and significant deterioration of patient‐reported outcomes (PROs) 9, 10. Talazoparib, with its favorable efficacy, safety, and PRO profile versus chemotherapy, represents a viable option for patients with gBRCA‐mutated advanced breast cancer. These results supported the U.S. Food and Drug Administration (FDA) and European Medicines Agency's approval of talazoparib in this setting 11, 12, 13. We present more detailed safety analyses for talazoparib from EMBRACA to highlight its patterns of toxicity compared with chemotherapy, and to outline guidelines for management of talazoparib toxicity in clinical practice via dose modifications and/or standard supportive care.
Materials and Methods
Study Design and Treatments
EMBRACA is an ongoing open‐label, randomized, international, phase III study comparing the efficacy, PROs, and safety of oral talazoparib to PCT (capecitabine, eribulin, gemcitabine, or vinorelbine) in patients with HER2‐negative, gBRCA‐mutated locally advanced or metastatic breast cancer (http://clinicaltrials.gov identifier: NCT01945775). All patients randomized to talazoparib started on 1 mg once daily. Detailed study information was published 8. CONSORT diagram is shown (Fig. 1) 8.
Figure 1.

CONSORT diagram.
Hematologic inclusion criteria were hemoglobin ≥9.0 g/dL with last transfusion ≥14 days before randomization, neutrophils ≥1500 × 106/L, and platelets ≥100 × 109/L. During the study, talazoparib dosing guidance for managing adverse events (AEs) was amended (details below and supplemental online Table 1), which primarily affected management of grade ≥3 hematologic toxicities. For patients randomized to PCT, dosing guidance for each chemotherapy was given to investigators; however, flexibility was allowed for adjustment of PCT dosing following local institutional practice and the country prescribing information. Study treatment continued until disease progression, unacceptable toxicity, withdrawal of consent, or per discretion of investigator.
As detailed in supplemental online Tables 1 and 2, initial protocol requirements after a grade ≥3 anemia AE (hemoglobin <8 g/dL) required that talazoparib dose be interrupted until hemoglobin levels recovered to grade 1 (≥10 g/dL) or baseline before resuming talazoparib at a lower dose level, whereas the inclusion criteria permitted a hemoglobin value ≥9 g/dL. The protocol was amended as follows: in the case of a grade ≥3 anemia (<8 g/dL), hemoglobin levels must return to grade 1 or meet study eligibility criteria (≥9 g/dL) before talazoparib could resume at a lower dose level. Supportive medications (antiemetics, antidiarrheals, bisphosphonates and denosumab, and gonadotropin‐releasing hormones) could be provided prophylactically or therapeutically at the discretion of investigator. Growth factors and transfusions were administered as supportive care (see supplemental online data section 1.0 and supplemental online Table 1).
Table 1.
Hematologic toxicity based on laboratory values during treatment‐emergent period
| Laboratory values | Talazoparib (n = 286) | Overall PCT (n = 126) |
|---|---|---|
| Hemoglobin values,a n (%) | ||
| Hemoglobin ≥9.0 g/dL throughout treatment‐emergent periodb | 150 (52.4) | 107 (84.9) |
| 8.0 g/dL ≤ hemoglobin <9.0 g/dL at least once during treatment‐emergent period | 24 (8.4) | 8 (6.3) |
| Hemoglobin <8.0 g/dL at least once during treatment‐emergent period | 111 (38.8) | 8 (6.3) |
| Neutrophil values,a , c n (%) | ||
| Neutrophils ≥1500 × 106/L throughout treatment‐emergent period | 132 (46.2) | 49 (38.9) |
| 1000 × 106/L ≤ neutrophils <1500 × 106/L at least once during treatment‐emergent period | 93 (32.5) | 26 (20.6) |
| Neutrophils <1000 × 106/L at least once during treatment‐emergent period | 60 (21.0) | 48 (38.1) |
| Platelet values,a n (%) | ||
| Platelets ≥75 × 109/L throughout treatment‐emergent period | 209 (73.1) | 117 (92.9) |
| 50 × 109/L ≤ platelets <75 × 109/L at least once during treatment‐emergent period | 34 (11.9) | 4 (3.2) |
| Platelets <50 × 109/L at least once during treatment‐emergent period | 42 (14.7) | 2 (1.6) |
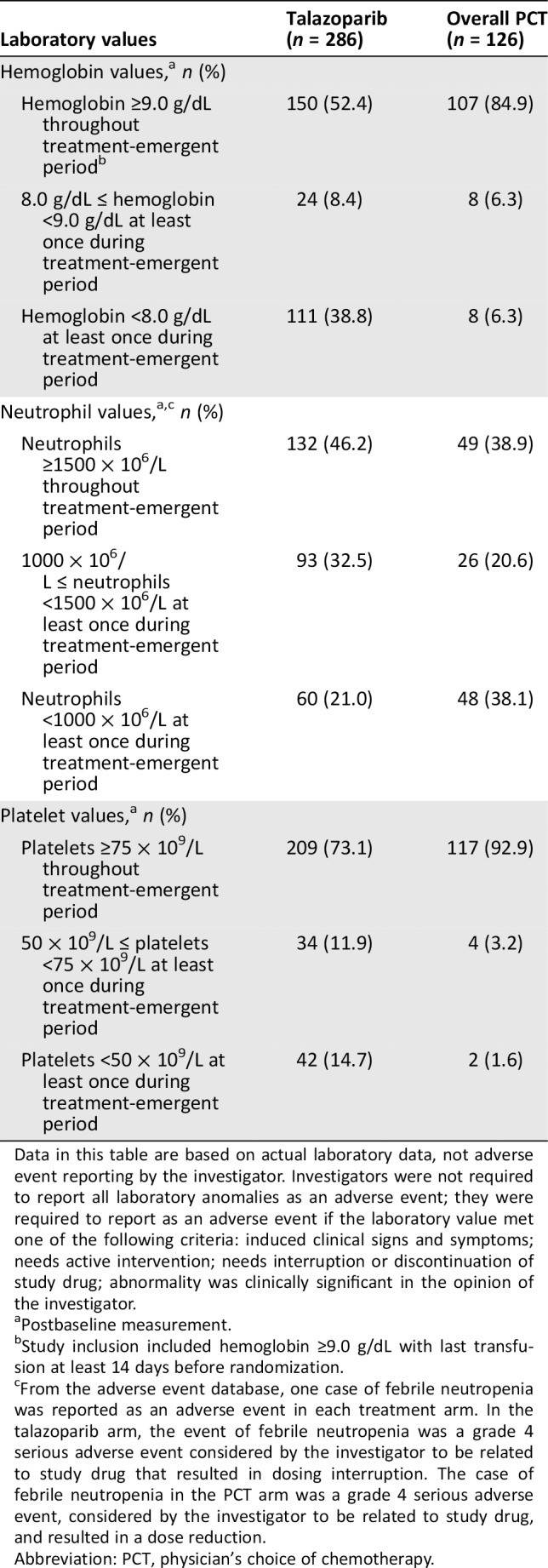
Data in this table are based on actual laboratory data, not adverse event reporting by the investigator. Investigators were not required to report all laboratory anomalies as an adverse event; they were required to report as an adverse event if the laboratory value met one of the following criteria: induced clinical signs and symptoms; needs active intervention; needs interruption or discontinuation of study drug; abnormality was clinically significant in the opinion of the investigator.
Postbaseline measurement.
Study inclusion included hemoglobin ≥9.0 g/dL with last transfusion at least 14 days before randomization.
From the adverse event database, one case of febrile neutropenia was reported as an adverse event in each treatment arm. In the talazoparib arm, the event of febrile neutropenia was a grade 4 serious adverse event considered by the investigator to be related to study drug that resulted in dosing interruption. The case of febrile neutropenia in the PCT arm was a grade 4 serious adverse event, considered by the investigator to be related to study drug, and resulted in a dose reduction.
Abbreviation: PCT, physician's choice of chemotherapy.
Table 2.
TEAEs associated with dose modification in ≥5% of patients in either treatment arm by decreasing frequency in the talazoparib arm (safety population)
| Preferred term | Talazoparib (n = 286), n (%) | Overall PCT (n = 126), n (%) |
|---|---|---|
| Patients with ≥1 TEAE associated with dose modification | 190 (66.4) | 75 (59.5) |
| ANEMIAa | 111 (38.8) | 3 (2.4) |
| NEUTROPENIAa | 65 (22.7) | 29 (23.0) |
| THROMBOCYTOPENIAa | 49 (17.1) | 1 (0.8) |
| LEUKOPENIAa | 22 (7.7) | 6 (4.8) |
| Nausea | 6 (2.1) | 12 (9.5) |
| Diarrhea | 2 (0.7) | 10 (7.9) |
| Palmar‐plantar erythrodysesthesia syndrome | 0 (0.0) | 16 (12.7) |
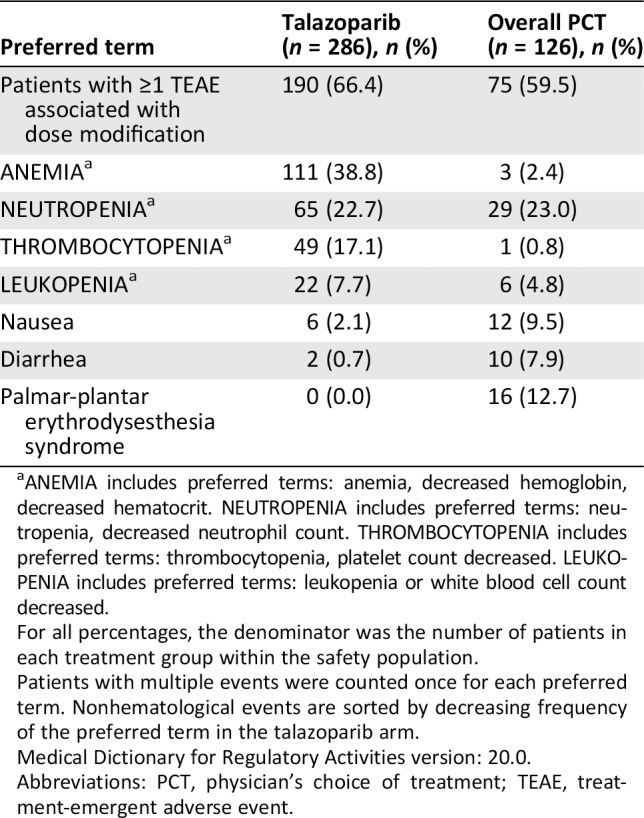
ANEMIA includes preferred terms: anemia, decreased hemoglobin, decreased hematocrit. NEUTROPENIA includes preferred terms: neutropenia, decreased neutrophil count. THROMBOCYTOPENIA includes preferred terms: thrombocytopenia, platelet count decreased. LEUKOPENIA includes preferred terms: leukopenia or white blood cell count decreased.
For all percentages, the denominator was the number of patients in each treatment group within the safety population.
Patients with multiple events were counted once for each preferred term. Nonhematological events are sorted by decreasing frequency of the preferred term in the talazoparib arm.
Medical Dictionary for Regulatory Activities version: 20.0.
Abbreviations: PCT, physician's choice of treatment; TEAE, treatment‐emergent adverse event.
Endpoints
Endpoints and assessments have been described 8, 9. The primary endpoint was radiographic PFS (blinded independent central review), and secondary and exploratory endpoints included safety, overall survival, objective response rate, duration of response, clinical benefit rate, PROs, and pharmacokinetics.
Safety Assessments
Safety included AEs, concomitant medications, and laboratory values. AEs were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. Treatment‐emergent AEs (TEAEs) were defined as any new AEs that appeared or worsened in severity following study drug start. Not all laboratory anomalies were required to be reported as an AE (Table 1). The treatment‐emergent (TE) period was defined as the time from the first study drug dose through 30 days after the last study drug dose (permanent discontinuation of study drug) or the day before initiation of a new antineoplastic therapy, whichever occurred first. AE data were already reported 8, whereas these analyses evaluated additional aspects of safety for the most common AEs. For analysis of anemia followed by fatigue, thrombocytopenia followed by bleeding event, and neutropenia followed by infections, the second AE (fatigue, bleeding, infection) had to start the same day or later after the first AE (anemia, thrombocytopenia, neutropenia), but the start date of the second AE was ≤ the end date of the first AE.
Relationship Between Talazoparib Exposure and Grade ≥3 Anemia
To examine whether talazoparib exposure in patients with grade ≥3 anemia is higher relative to those without anemia events, a plot was created by comparing time‐varying talazoparib exposure (as assessed by time‐varying average talazoparib concentrations Cavg,t) in patients with versus without anemia events at each time point an event occurred 14.
Patient‐Reported Outcomes
PRO analyses are described in supplemental online data section 2.0 9. The post hoc, exploratory PRO analyses focused on patients with commonly reported AEs: (a) anemia and (b) nausea and/or vomiting while accounting for potential supportive care medication (SCM) as confounders (supplemental online data section 2.0).
Health Resource Utilization
Post hoc, exploratory health resource utilization (HRU) was assessed (serious AE‐associated hospitalization and SCM). While accounting for talazoparib/PCT‐TE period, we analyzed serious adverse event (SAE)–associated hospitalization rates (per 100 patient‐years) and SCM mean utilization ratios (details in supplemental online data section 3.0).
Statistical Analyses
The statistical methodology has been described 8, 9, and most statistical analyses on safety herein were descriptive. The safety population (patients who received any study drug) was used for all safety analyses. PRO and HRU statistics are in supplemental online data sections 2.0 and 3.0, respectively. Statistical methods for the landmark analysis are in supplemental online data section 4.0.
Time‐varying proportional hazards Cox regression models were used to assess the impact of talazoparib dose reduction on PFS by itself as well as in a model adjusting for the effect of other independent variables, with dose reduction as the time‐dependent covariate. All patients and all data from randomization in the talazoparib arm were included. Patients were assigned to the no dose reduction group up to the time of dose reduction and then they were attributed to the dose reduction group. Patients with a dose reduction after disease progression were considered to be in the no dose reduction category. The assumption of proportional hazards was evaluated, and the results supported no departure from that assumption. For the analysis adjusting for multiple variables, additional covariates analyzed within the model were baseline hemoglobin, baseline lactate dehydrogenase, and time from diagnosis (disease‐free interval <12 months, ≥12 months).
Results
Patients
Overall, 431 patients (talazoparib, n = 287; PCT, n = 144) were randomized between October 2013 and April 2017 (intent‐to‐treat: all patients randomized; data cutoff, September 15, 2017). The safety population included patients receiving talazoparib (n = 286) or PCT (n = 126; capecitabine, 44%; eribulin, 40%; gemcitabine, 10%; vinorelbine, 7%). The median (range) duration of the TE period was 7.0 (0.8–36.9) months for talazoparib and 4.5 (0.5–18.3) months for PCT.
Occurrence and Severity of Hematologic Toxicity
Laboratory Data
Percentage of patients with hematologic toxicity based on laboratory data is shown based on three cutoff values per hematologic parameter (Table 1). Based on laboratory data, patients receiving PCT were more likely to have lower neutrophil counts, whereas lower hemoglobin and platelets were more common with talazoparib. The median (range) time to recovery for hemoglobin from the first postbaseline hemoglobin <8.0 g/dL to hemoglobin ≥9.0 g/dL was similar between treatment arms: 8 days (2–47) for talazoparib and 8 days (7–115) for PCT. The median (range) time to recovery of neutrophils from the first postbaseline neutrophil count <1000 × 106/L to ≥1500 × 106/L was 9 (1–32) days for talazoparib and 9 (2–57) days for PCT. The median (range) time to recovery of platelets from the first postbaseline platelets <50 × 109/L to platelet count ≥75 × 109/L was 9 (5–21) days for talazoparib; no median time to recovery was available for PCT.
Hematologic Adverse Event Incidence by Grade
With talazoparib, the most common hematologic AEs were anemia (any grade 52.8%), neutropenia (any grade 34.6%), and thrombocytopenia (any grade 26.9%). PCT had higher rates of neutropenia (any grade 42.9%) and lower rates of anemia (any grade 18.3%) and thrombocytopenia (any grade 7.1%). Hematologic AE incidence by grade is shown in Figure 2A.
Figure 2.

Treatment‐emergent adverse events and maximum severity for selected adverse drug reactions. (A): Hematologic. (B): Nonhematologic (safety population). ANEMIA includes preferred terms: anemia, decreased hemoglobin, decreased hematocrit. NEUTROPENIA includes preferred terms: neutropenia, decreased neutrophil count. THROMBOCYTOPENIA includes preferred terms: thrombocytopenia, platelet count decreased. Nonhematologic adverse events (nausea, alopecia, vomiting) are based on a single preferred term, whereas fatigue was inclusive of fatigue and asthenia. There were no occurrences of grade 4 fatigue and asthenia, nausea, and vomiting; alopecia is only graded as grade 1 or 2. The analysis data cutoff date is 15SEP2017. MedDRA Version: 20.0. Adverse event grades are evaluated based on National Cancer Institute‐Common Terminology Criteria (version 4.03). Patients with multiple events for a given preferred term or included in a preferred term are counted once only at the worst severity for the preferred term, and overall, respectively. Patients summarized under missing severity are patients whose severity is all missing for the preferred term.Abbreviation: PCT, physician's choice of chemotherapy.
Onset and Duration of Hematologic AEs Associated with Talazoparib
Cumulative risk of any grade anemia increased more steeply than that of neutropenia and thrombocytopenia with talazoparib treatment; all three generally plateaued by week 25 (supplemental online Fig. 1A). The onset of hematologic toxicities (any grade and grade 3‐4) mostly occurred within the first 16 weeks of talazoparib (Fig. 3A–C). Median (range) time from first talazoparib dose to onset of first grade ≥3 episode of anemia, neutropenia, and thrombocytopenia was 83 (13–961), 50 (1–947), and 36 (11–370) days, respectively (supplemental online Fig. 2A). All hematologic AEs declined over time (Fig. 3A–C). Hematologic AE data by 6‐month intervals further highlight different patterns of occurrence (supplemental online Table 3).
Figure 3.
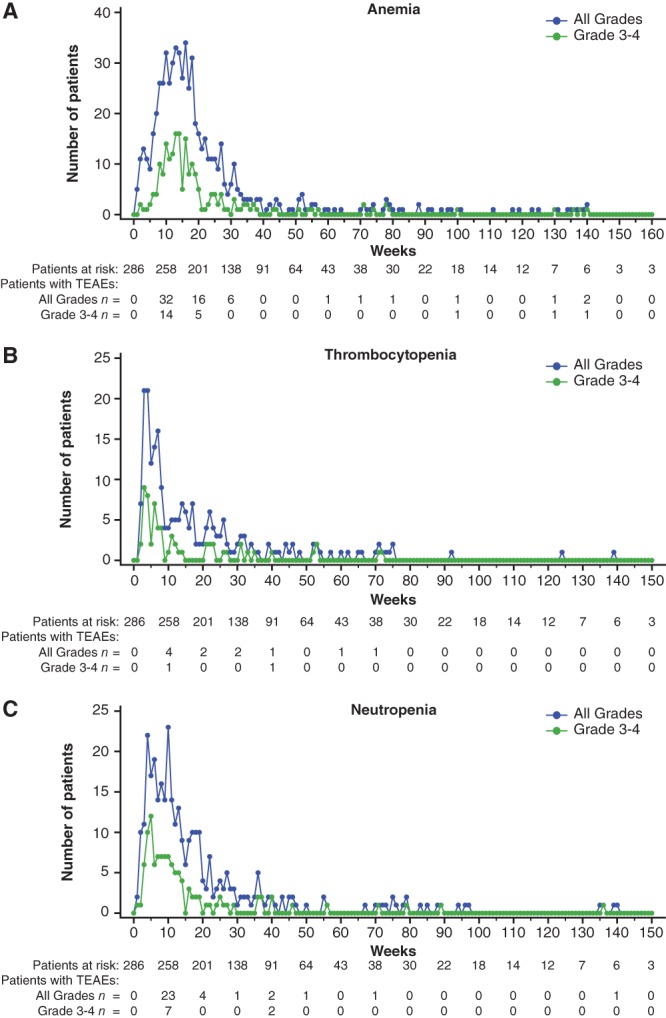
Hematologic treatment‐emergent adverse events (all grades) by week, preferred term (safety population, talazoparib arm).
Abbreviation: TEAE, treatment‐emergent adverse event.
All grade anemia and thrombocytopenia had a slightly shorter median duration with talazoparib (anemia, 21 days; thrombocytopenia, 15 days) than with PCT (anemia, 29 days; thrombocytopenia, 24 days; Fig. 4A–B). The median duration of neutropenia was slightly longer for talazoparib (12 days) versus PCT (8 days; Fig. 4C). Grade 3‐4 hematologic events for anemia and neutropenia were comparable in median duration between the 2 treatment arms (∼7 days), but grade 3‐4 thrombocytopenia was 8 days for talazoparib versus 18 days for PCT.
Figure 4.
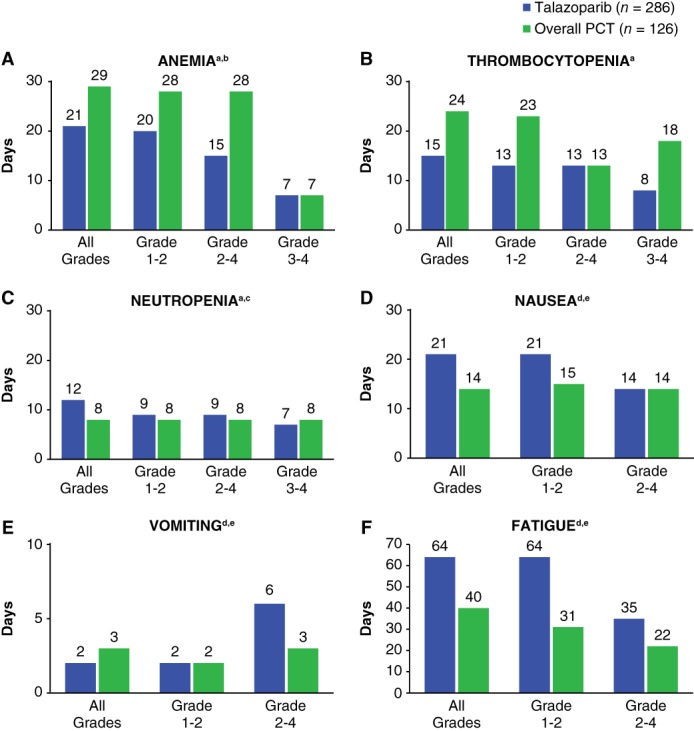
Median duration of selected treatment‐emergent adverse events by episode and grade (safety population).
aDuring the study, talazoparib dosing guidance for hematologic and nonhematologic adverse events (AEs) was amended (supplemental online Table 1), which primarily affected management of grade 3 or higher hematologic toxicities. Dose modifications for physician's choice of chemotherapy (PCT) were following the country prescribing information and institutional practice.
bInitial EMBRACA protocol requirements after a grade ≥3 anemia AE (hemoglobin <8 g/dL) required that the talazoparib dose be interrupted until hemoglobin levels recovered to grade 1 (≥10 g/dL) or baseline before resuming talazoparib at the next lower dose level, whereas EMBRACA eligibility criteria were permitted with a hemoglobin value ≥9 g/dL. EMBRACA protocol was later amended as follows: in the case of a grade ≥3 anemia (<8 g/dL), hemoglobin levels must return to grade 1 or meet study eligibility criteria (≥9 g/dL) before talazoparib treatment could resume at the next lower dose level. This requirement potentially facilitated investigators to provide packed red blood cells transfusions and/or antianemic use at a higher hemoglobin level than recommended by current clinical practice and/or international clinical guidelines 15, 16.
cOnly one case of febrile neutropenia was reported in each treatment arm.
dThere were no nonhematologic grade 4 adverse events; the highest grade was grade 3 and occurred in ≤9 patients receiving talazoparib for fatigue and vomiting and 1 patient for nausea. Grade 2‐4 were combined for consistency with panels for hematologic adverse events but only included grade 2‐3 nonhematologic adverse events.
eNonhematologic adverse events (nausea, vomiting) are based on a single preferred term, whereas fatigue was inclusive of fatigue and asthenia. ANEMIA includes preferred terms: anemia, decreased hemoglobin, decreased hematocrit. NEUTROPENIA includes preferred terms: neutropenia, decreased neutrophil count. THROMBOCYTOPENIA includes preferred terms: thrombocytopenia, platelet count decreased.
Overall Incidence and Severity for Select Nonhematologic AEs
Nonhematologic AE Incidence by Grade and Timing
The majority of nonhematologic toxicities were grade 1 or 2 in patients receiving talazoparib. The incidence of some nonhematologic grade 2 toxicities was lower in patients receiving talazoparib than in patients receiving PCT: nausea (14.3% vs. 18.3%), alopecia (2.4% vs. 7.9%), and vomiting (6.6% vs. 10.3%), respectively (Fig. 2B). However, grade 2 fatigue and asthenia was more common with talazoparib than PCT (24.1% vs. 15.9%). Most fatigue (without asthenia) was grade 1 or 2 (29.4% and 19.2%) with talazoparib and PCT (26.2% and 13.5%; all grade, 50.3% with talazoparib and 42.9% with PCT).
Onset and Duration of Nonhematologic AEs
Cumulative risk analysis showed that the risk of select nonhematologic AEs (nausea, fatigue, vomiting, alopecia) all generally increased within the first 4 weeks of receiving talazoparib and plateaued after week 50 (supplemental online Fig. 1B). Median (range) time from first talazoparib dose to first grade 2 episode onset was 54 (1–540) days, 30 (1–681) days, and 46 (2–308) days for fatigue, nausea, and vomiting, respectively (supplemental online Fig. 2B). Nonhematologic AE data by 6‐month intervals is shown in supplemental online Table 3.
Alopecia tended to occur earlier with PCT (peak percentage at 3 weeks; data on file) than with talazoparib (peak percentage at 5 weeks; primarily grade 1 [supplemental online Fig. 3A]). With talazoparib, fatigue (any grade; supplemental online Fig. 3B) was highest during the early treatment cycles of talazoparib (in approximately 13% of patients) but affected ≤6% of patients from week 5 through 24. A similar trend of early occurrence was seen for vomiting and nausea (supplemental online Fig. 3C, D).
Nonhematologic toxicity median duration for the selected AEs is shown (Fig. 4D–F). Median durations of some nonhematologic AEs were slightly longer for talazoparib versus PCT. Grade 2‐4 vomiting had a median duration of 6 days with talazoparib (n = 26) versus 3 days with PCT (n = 15). Grade 2‐4 fatigue/asthenia had a longer median duration with talazoparib (35 days; n = 78) than PCT (22 days; n = 26).
AEs of special interest are shown in supplemental online data section 5.0, and AEs in elderly patients are described in supplemental online data section 6.0.
Concurrent AEs
Overlapping grade 3‐4 hematologic events were infrequent with talazoparib (anemia + neutropenia [5.9%]; anemia + thrombocytopenia [3.1%]; neutropenia + thrombocytopenia [4.9%]). The presence of concurrent AEs was evaluated for anemia (fatigue), thrombocytopenia (bleeding), and neutropenia (infection) based on potential known associations. More patients receiving talazoparib experienced anemia followed by fatigue (13.6%) than those receiving PCT (4.0%). The majority of patients with thrombocytopenia did not have a subsequent bleeding event (97.9% and 99.2% for talazoparib and PCT, respectively). Fewer patients with neutropenia and receiving talazoparib had subsequent infections (3.8%) than those receiving PCT (9.5%). Febrile neutropenia occurred in one patient (0.3%) receiving talazoparib and one patient (0.8%) receiving PCT.
Incidence of Serious AEs
The incidence of SAEs was similar with both treatments. SAEs were reported in 91 (31.8%) patients receiving talazoparib and 37 (29.4%) patients receiving PCT. Study drug‐related SAEs were reported in 26 (9.1%) patients receiving talazoparib and 11 (8.7%) patients receiving PCT. Regardless of the time point examined, few treatment‐related SAEs occurred (supplemental online data section 7.0). The most frequently reported drug‐related SAE with talazoparib was anemia (n = 15; 5.2%; supplemental online Table 4). The most frequently reported drug‐related SAE with PCT was neutropenia (n = 4; 3.2%).
AEs associated with death were reported in six (2.1%) patients receiving talazoparib and four (3.2%) patients receiving PCT arm (supplemental online data, section 8.0). Of these fatal AE events, two (1 veno‐occlusive liver disease [talazoparib] and 1 sepsis [PCT]) were considered by the investigator to be related to study drug. However, the sponsor considered veno‐occlusive liver disease an unlikely etiology, a consideration supported by two hepatologist consultants to the sponsor who reviewed the case.
Toxicity Management
Discontinuations Caused by AEs
With talazoparib, 17 (5.9%) patients had an all‐causality AE other than disease progression that was associated with permanent discontinuation versus 11 (8.7%) patients receiving PCT. In the talazoparib arm, discontinuation caused by anemia, neutropenia, and thrombocytopenia occurred in 0.7%, 0.3%, and 0.3% of patients, respectively.
Dose Modifications Caused by AEs
AEs associated with dose modifications in ≥5% of patients in either treatment arm are presented by decreasing frequency (Table 2). Sixty‐six percent of patients receiving talazoparib had ≥1 TEAE associated with dose modification.
Detailed information on dosing interruptions (supplemental online data section 9.0, supplemental online Table 5), dose reductions (supplemental online data section 10.0, supplemental online Table 6, supplemental online Fig. 4), and dose modifications (supplemental online data section 11.0 and supplemental online Table 7) is in the supplemental online data.
Examination of the Relationship Between Talazoparib Exposure and Grade ≥3 Anemia
The relationship between talazoparib exposure and grade ≥3 anemia was examined by plotting talazoparib Cavg,t each time an event occurred in patients with events (red dots) versus without events (box and whisker plot; Fig. 5). Talazoparib Cavg,t in patients with grade ≥3 anemia events tended to be higher than the median Cavg,t of patients without events, suggesting that higher talazoparib exposure may be associated with higher risk of anemia (Fig. 5).
Figure 5.

Comparison of talazoparib exposure (Cavg,t) in patients with grade 3 or higher anemia events and patients without events at each event occurrence day.
At each anemia event day, the distribution of Cavg,t for patients without anemia events is represented by the black box plot showing median, 25%/75% quartiles, and whiskers to the last point within 1.5 × interquartile range. Cavg,t for patients with anemia events at each event day is represented by red circles. Black and red dotted lines are the lowess lines for patients without anemia events and with events, respectively. Talazoparib Cavg,t in patients with anemia events tended to be higher than the median Cavg,t of patients without events. Cavg,t was calculated using average daily dose intensity up to each anemia event time divided by the product of talazoparib oral clearance (CL/F) derived from population pharmacokinetic analysis and the dosing interval (i.e., 24 hours).
Impact of Dose Reductions
The impact of dose reductions on PFS was analyzed using Cox regression models with dose reduction as the time‐dependent variable (Table 3). These analyses show that there is a trend toward a slightly less favorable PFS outcome for patients that had a dose reduction versus those that did not. However, the 95% CI crosses between favorable and less favorable outcomes (model with just dose reduction: HR, 1.344; 95% CI, 0.988–1.827; model adjusting for additional independent variables: HR, 1.118; 95% CI, 0.815–1.533). Additional analyses are shown (see supplemental online data section 12.0 and supplemental online Fig. 5). From these analyses it cannot be directly determined whether dose reduction itself may lead to somewhat lower PFS or is just a marker for patients with worse prognosis and hence potentially shorter PFS. The Cox model adjusting for other independent variables showed that baseline lactate dehydrogenase and disease‐free interval seem associated with PFS.
Table 3.
Cox regression models of PFSa including dose reduction as the time‐dependent variable (talazoparib arm only, ITT population)
| Variables | Levels | HR (95% CI) | p value |
|---|---|---|---|
| Model adjusting for dose reduction | |||
| Dose reduction |
1: dose reduction 0: no dose reduction |
1.344b (0.988–1.827) | .0597 |
| Model adjusting for dose reduction and other variables | |||
| Dose reduction |
1: dose reduction 0: no dose reduction |
1.118c (0.815–1.533) | .4905 |
| Baseline hemoglobin, g/L | ─ | 0.990 (0.979–1.002) | .0904 |
| Baseline LDH, μkat/L | ─ | 1.035 (1.021–1.049) | <.0001 |
| Time from diagnosis |
1: <12 mo 2: ≥12 mo |
1.582 (1.175–2.132) | .0025 |
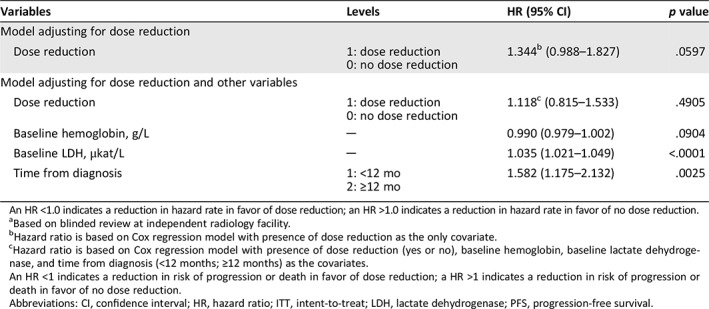
An HR <1.0 indicates a reduction in hazard rate in favor of dose reduction; an HR >1.0 indicates a reduction in hazard rate in favor of no dose reduction.
Based on blinded review at independent radiology facility.
Hazard ratio is based on Cox regression model with presence of dose reduction as the only covariate.
Hazard ratio is based on Cox regression model with presence of dose reduction (yes or no), baseline hemoglobin, baseline lactate dehydrogenase, and time from diagnosis (<12 months; ≥12 months) as the covariates.
An HR <1 indicates a reduction in risk of progression or death in favor of dose reduction; a HR >1 indicates a reduction in risk of progression or death in favor of no dose reduction.
Abbreviations: CI, confidence interval; HR, hazard ratio; ITT, intent‐to‐treat; LDH, lactate dehydrogenase; PFS, progression‐free survival.
Neutropenia and Growth Factor Use
The percentage of patients with ≥1 growth factor administration was 10.1% and 17.5% for talazoparib and PCT, respectively. Grade 3 or 4 myelosuppression‐related AEs reported at a ≥5% lower incidence in the talazoparib arm than the PCT arm were neutropenia (17.8% vs. 24.6%) and decreased neutrophil count (4.2% vs. 10.3%). Growth factors and transfusions were administered as supportive care. Growth factors used were filgrastim, pegfilgrastim, granulocyte colony‐stimulating factor, sargramostim, and lenograstim.
Health Resource Utilization
Within the safety population who experienced anemia, 59 out 151 (39.1%) patients in the talazoparib arm and 9 out of 23 (39.1%) patients in the PCT arm received ≥1 concomitant SCM (supplemental online Table 8). In patients receiving talazoparib, 38.1% (109/286 [safety population]) required packaged red blood cells (PRBC) transfusions (median, 2 transfusions per patient in 109 patients), whereas 5.6% (7/126) of the PCT‐treated patients required PRBC transfusions (median, 1.0 transfusion per patient in 7 patients; supplemental online Table 8). Initial EMBRACA protocol requirements after a grade ≥3 anemia AE are described in detail in the supplemental online Table 1 footnote. This requirement potentially facilitated investigators to provide PRBC transfusions and/or antianemic use at a higher hemoglobin level than recommended by current clinical practice and international clinical guidelines 15, 16. The median time between measurement of hemoglobin levels and first transfusion was approximately one week. The hemoglobin levels at the time of each transfusion to resume talazoparib dosing are shown (supplemental online Fig. 6A–C). Overall, 41.3% of patients had hemoglobin levels at approximately 8.5 g/dL before their first transfusion and 17.4% had a hemoglobin level of 9.5 g/dL before their first transfusion (supplemental online Fig. 6A).
After accounting for the TE period among the EMBRACA safety population, across all categories of SCM (except for platelet transfusion), the mean SCM utilization ratios for talazoparib‐treated patients were lower than patients treated with PCT (supplemental online Table 9).
SAE‐Associated Hospitalization
Overall, among the EMBRACA safety population, 29.4% and 26.2% of patients in the talazoparib and PCT arms, respectively, had recorded SAE‐associated hospitalizations during EMBRACA. After accounting for the different treatment exposure periods of the respective treatment arms, patients treated with talazoparib had lower SAE‐associated hospitalization rates versus PCT (46.8 vs. 71.9 hospitalizations per 100 patient‐years, respectively).
Post Hoc, Exploratory Patient‐Reported Outcomes
Overall differences were observed in patient‐reported GHS/QoL (10.0; 95% CI, −0.1 to 20.2) and fatigue symptoms (−11.5; 95% CI, −24.1 to 1.0), both favoring talazoparib, among patients with anemia who did not receive PRBC transfusion and/or antianemic medication at any point during the study. There was also a delay in TTD in GHS/QoL (HR, 0.38; 95% CI, 0.12–1.21) favoring talazoparib. A delay in TTD in fatigue was not observed in the talazoparib arm (HR, 1.54; 95% CI, 0.30–8.04; supplemental online Fig. 7).
Overall differences were observed in patient‐reported GHS/QoL (6.5; 95% CI, 0.1–12.9) and nausea and vomiting symptoms (−3.9; 95% CI, −8.8–1.0), both favoring talazoparib, among patients who reported nausea and vomiting and who did not receive antiemetic and/or antinauseant medication at any point during study. There was also a delay in TTD in GHS/QoL (HR, 0.31; 95% CI, 0.14–0.68) and nausea and vomiting symptoms (HR, 0.44; 95% CI, 0.17–1.11) favoring talazoparib (supplemental online Fig. 7).
Similar favorable PRO results (data not shown) favoring talazoparib were observed among patients with (a) anemia AEs and who received PRBC transfusion(s) and/or antianemic medication(s) at any point during EMBRACA, or (b) nausea and/or vomiting AEs and who received antiemetic and/or antinauseant medication(s) at any point during EMBRACA.
Discussion
The phase III EMBRACA trial demonstrated improved PFS associated with talazoparib compared with PCT for patients with gBRCA‐mutated HER2‐negative, locally advanced/metastatic breast cancer. These results supported the U.S. FDA and European Medicines Agency's approval of talazoparib in this setting 8, 9, 12, 13. The most common (≥20%) AEs of any grade reported for talazoparib included fatigue, anemia, nausea, neutropenia, headache, thrombocytopenia, vomiting, alopecia, diarrhea, and decreased appetite. Herein, we provide a detailed exploration of safety, further PRO analyses in those that experienced common AEs, and HRU outcomes. We aim to provide clinicians with a clearer understanding of the side effect profile, toxicity management, and patient experience of talazoparib.
Talazoparib was generally well tolerated by patients enrolled in EMBRACA. As expected, there were hematologic toxicities and low‐grade nonhematologic toxicities. In the talazoparib arm, 17 (5.9%) patients had an AE other than disease progression that was associated with permanent study drug discontinuation versus 11 (8.7%) patients in the PCT arm. The majority of frequently reported AEs with talazoparib were consistent with what is commonly observed with other PARP inhibitors 17, 18.
Hematologic AEs were rarely grade 4 in severity with talazoparib. Serious hematologic AEs were relatively uncommon (≤5.2%). Former exposure‐safety analyses using pooled data from ABRAZO and EMBRACA demonstrated that higher talazoparib exposure was associated with a higher risk of grade ≥3 anemia or thrombocytopenia; a similar trend was also observed with grade ≥3 neutropenia 14. This is consistent with the analysis of the relationship between talazoparib exposure and grade ≥3 anemia in EMBRACA (Fig. 5). As these 3 safety endpoints correlate with talazoparib exposure levels, lowering the exposure by dosing interruption or dose reduction will lead to a lower occurrence of these events. Titration to a lower dose should occur only if necessitated by AEs.
Exposure‐efficacy analysis from EMBRACA showed that higher exposure was associated with longer PFS, indicating that the maximum tolerated dose (1 mg once daily) will provide the highest tolerable exposure that will lead to the best PFS outcomes 18. The Cox regression models suggest that there could be a potential trend for slightly less favorable PFS in patients who dose reduce, although the 95% CIs included 1.0. In the model adjusting for additional independent variables in addition to dose reduction, a relationship with PFS was observed for lactate dehydrogenase and disease‐free interval. The Cox analyses cannot adjust for the lack of randomization to the dose reduction or no dose reduction groups. Therefore, whether dose reduction itself may lead to lower PFS or is just a marker for patients with worse prognosis and hence potentially shorter PFS is unknown.
Population pharmacokinetic analyses showed that dose adjustment is not necessary based on the patient's age, weight, race, sex, mild renal impairment, mild hepatic impairment, or acid‐reducing agents 19. Patients with moderate renal impairment (30 mL/min ≤ creatinine clearance <60 mL/min) had 37.1% lower talazoparib clearance and a 59% increase in exposure 19. Concomitant administration of strong inhibitors of the efflux transporter P‐glycoprotein (P‐gp) increased talazoparib exposure by 45% 19. In patients with moderate renal impairment or who are using strong P‐gp inhibitors, the talazoparib starting dose is recommended to be 0.75 mg once daily 11.
Among patients with an AE associated with permanent discontinuation of talazoparib, only 1.4% of patients discontinued from the study due to hematologic toxicities; of these, 0.7% was due to anemia. Dose reductions for talazoparib were strictly defined during the study (see Materials and Methods and supplemental online Table 1), whereas PCT followed local prescribing information and institutional practice. Per protocol, blood transfusions were implemented to counteract anemia. In the talazoparib arm, 109 (38.1%) patients ultimately required PRBC transfusions and the median number of PRBC transfusions per patient was 2. In the PCT arm, 7 (5.6%) patients required a median of 1 PRBC transfusion per patient. The higher rate of blood transfusions for the talazoparib arm may be due to the initial study requirements, which mandated hemoglobin values to recover to grade 1 (≥10 g/dL) or baseline before talazoparib could be resumed at a lower dose level after a dosing interruption (supplemental online Tables 1 and 2). The amended talazoparib dosing protocol (supplemental online Table 1) potentially facilitated investigators to provide PRBC transfusions and/or antianemic use at a higher hemoglobin level than recommended by current clinical practice/international clinical guidelines 15, 16. The rate of PRBC transfusions declined by approximately 11% after the amendment (supplemental online Table 1).
Among patients with reported anemia or nausea and vomiting AE, the overall improvement and delay in TTD in patient‐reported GHS/QoL favoring talazoparib indicate that from a patient's perspective, the observed anemia or nausea and vomiting AE rates did not detrimentally impact the QoL in patients treated with talazoparib compared with PCT.
The lower SAE‐associated hospitalization rates and lower mean utilization ratios in a majority of SCM types observed among talazoparib‐treated patients versus PCT further support the favorable patient experience and positive risk‐benefit profile of talazoparib.
Conclusions
Treatment with talazoparib was generally safe and well tolerated. Clinical trial data indicate a consistent and therefore predictable safety profile for talazoparib. Talazoparib‐induced AEs can be readily managed by dose modifications and supportive measures (including PRBC transfusion) with a favorable HRU rate. Positive PROs of talazoparib‐treated patients with reported anemia or nausea and vomiting AE suggests that these AEs are manageable and that patients’ QoL improved versus PCT‐treated patients. Altogether, the findings of this study support the incorporation of talazoparib in clinical practice as a favorable treatment option for patients with locally advanced or metastatic breast cancer with gBRCA mutation.
Data Sharing
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (a) for indications that have been approved in the U.S. and/or E.U. or (b) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Author Contributions
Conception/design: Sara A. Hurvitz, Hope S. Rugo, Joanne L. Blum, Jennifer K. Litton
Provision of study material or patients: Sara A. Hurvitz, Anthony Gonçalves, Hope S. Rugo, Kyung‐Hun Lee, Louis Fehrenbacher, Lida A. Mina, Sami Diab, Joanne L. Blum, Jennifer K. Litton, Johannes Ettl
Collection and/or assembly of data: Mohamed Elmeliegy, Liza DeAnnuntis, Eric Gauthier, Akos Czibere, Iulia Cristina Tudor, Ruben G.W. Quek
Data analysis and interpretation: Sara A. Hurvitz, Anthony Gonçalves, Hope S. Rugo, Kyung‐Hun Lee, Louis Fehrenbacher, Lida A. Mina, Sami Diab, Joanne L. Blum, Jayeta Chakrabarti, Mohamed Elmeliegy, Liza DeAnnuntis, Eric Gauthier, Akos Czibere, Iulia Cristina Tudor, Ruben G.W. Quek, Jennifer K. Litton, Johannes Ettl
Manuscript writing: Sara A. Hurvitz, Anthony Gonçalves, Hope S. Rugo, Kyung‐Hun Lee, Louis Fehrenbacher, Lida A. Mina, Sami Diab, Joanne L. Blum, Jayeta Chakrabarti, Mohamed Elmeliegy, Liza DeAnnuntis, Eric Gauthier, Akos Czibere, Iulia Cristina Tudor, Ruben G.W. Quek, Jennifer K. Litton, Johannes Ettl
Final approval of manuscript: Sara A. Hurvitz, Anthony Gonçalves, Hope S. Rugo, Kyung‐Hun Lee, Louis Fehrenbacher, Lida A. Mina, Sami Diab, Joanne L. Blum, Jayeta Chakrabarti, Mohamed Elmeliegy, Liza DeAnnuntis, Eric Gauthier, Akos Czibere, Iulia Cristina Tudor, Ruben G.W. Quek, Jennifer K. Litton, Johannes Ettl
Disclosures
Sara Hurvitz: Ambrx, Amgen, Bayer, Daiichi‐Sankyo, Genentech/Roche, GSK, Immunomedics, Lilly, Macrogenics, Novartis, Pfizer, OBI Pharma, Pieris, PUMA, Radius, Sanofi, Seattle Genetics, Dignitana (RF), Pfizer, Roche (other: medical writing); Anthony Gonçalves: Pfizer, AstraZeneca, Novartis, Roche, Celgene, Amgen, Boehringer, MSD (other: travel, accommodations, and meeting registration support); Hope S. Rugo: University of California San Francisco from Eisai, Roche/Genentech, Eli Lilly & Co, Macrogenics, Merck, Novartis, OBI Pharma, Odonate, Immunomedics, Daichi, Pfizer (RF), Mylan, Pfizer, Novartis (other: travel); Joanne L. Blum: Biomarin, Medivation, Pfizer, Novartis (C/A), Dava Oncology, Genomic Health, Research to Practice (H); Jayeta Chakrabarti: Pfizer Ltd. (E, OI); Mohamed Elmeliegy: Pfizer (E, OI); Liza DeAnnuntis: Pfizer (E, OI); Eric Gauthier: Pfizer Inc. (E, OI); Akos Czibere: Pfizer (E, OI); Iulia Cristina Tudor: Pfizer Inc. (E, OI); Ruben G.W. Quek: Pfizer Inc. (E, OI), Amgen (OI); Jennifer K. Litton: Pfizer, Novartis, EMD Serono, AstraZeneca, GlaxoSmithKline, Genentech (RF), AstraZeneca, Pfizer (both uncompensated, SAB), UptoDate, Med Learning Group, Physician's Education Resource (other: royalties, CME Speaker); Johannes Ettl: Eli Lilly & Co, Novartis, Pfizer, Roche, Eisai (C/A), Celgene (RF), AstraZeneca, Pfizer, Roche, Tesaro, Teva (H), Celgene, Novartis, Pfizer (other: travel).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figures
Supplemental Tables
Acknowledgments
The authors would like to thank the patients and their families/caregivers for their participation, as well as the study centers and patient advocacy groups who supported this study. Editorial and medical writing support was provided to all authors by Edwin Thrower, Ph.D., Gautam Bijur, Ph.D., Paula Stuckart, and Dena McWain of Ashfield Healthcare Communications and was funded by Pfizer.
Before initiation of the study, the trial protocol was approved by an independent ethics committee at each site, and all enrolled patients provided written informed consent. This study was conducted in conformance with the principles of the Declaration of Helsinki and conducted using Good Clinical Practice according to International Council for Harmonisation Tripartite Guidelines.
This study was sponsored by Medivation, which was acquired by Pfizer in September 2016.
Pfizer, Inc. was involved in the study design, data collection, analysis, interpretation, and funding for editorial and medical writing support. All authors had full access to all data in the study and final responsibility for the decision to submit for publication.
Iulia Cristina Tudor is currently affiliated with Corcept Therapeutics Inc, Menlo Park, California, USA.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017;355:1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bryant HE, Schultz N, Thomas HD et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 2005;434:913–917. [DOI] [PubMed] [Google Scholar]
- 3. Farmer H, McCabe N, Lord CJ et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–921. [DOI] [PubMed] [Google Scholar]
- 4. Hay T, Clarke AR. DNA damage hypersensitivity in cells lacking BRCA2: A review of in vitro and in vivo data. Biochem Soc Trans 2005;33:715–717. [DOI] [PubMed] [Google Scholar]
- 5. Farres J, Martín‐Caballero J, Martínez C et al. PARP‐2 is required to maintain hematopoiesis following sublethal gamma‐irradiation in mice. Blood 2013;122:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Bono J, Ramanathan RK, Mina L et al. Phase I, dose‐escalation, two‐part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov 2017;7:620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turner NC, Telli ML, Rugo HS et al. A phase II study of talazoparib after platinum or cytotoxic nonplatinum regimens in patients with advanced breast cancer and germline BRCA1/2 mutations (ABRAZO). Clin Cancer Res 2019;25:2717–2724. [DOI] [PubMed] [Google Scholar]
- 8. Litton JK, Rugo HS, Ettl J et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med 2018;379:753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ettl J, Quek RGW, Lee KH et al. Quality of life with talazoparib versus physician's choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: Patient‐reported outcomes from the EMBRACA phase III trial. Ann Oncol 2018;29:1939–1947. [DOI] [PubMed] [Google Scholar]
- 10. Gera R, Mokbel K, El Hage Chehade H et al. Locoregional therapy targeted at the primary tumour improves overall survival in patients with stage IV metastatic breast cancer: A systematic review and meta‐analysis with 185942 patients. Ann Oncol 2019;30(suppl 3):164 P. [Google Scholar]
- 11. Talzenna (talazoparib) [package insert]. New York, NY: Pfizer Inc.; 2018.
- 12. Food U.S. & Drug Administration. FDA approves talazoparib for gBRCAm HER2‐negative locally advanced or metastatic breast cancer 2018. Available at https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm623540.htm. Accessed June 11, 2019. [Google Scholar]
- 13. Pfizer, Inc . European Commission approves TALZENNA® (talazoparib) for patients with inherited (germline) BRCA‐mutated locally advanced or metastatic breast cancer. Available at https://www.pfizer.com/news/press-release/press-release-detail/european_commission_approves_talzenna_talazoparib_for_patients_with_inherited_germline_brca_mutated_locally_advanced_or_metastatic_breast_cancer. Accessed June 21, 2019.
- 14. Elmeliegy M, Yu Y, Litton JK et al. Exposure‐safety analyses in breast cancer patients with germline BRCA1/2 mutations receiving talazoparib (TALA) in EMBRACA and ABRAZO trials. Ann Oncol 2018;29(suppl 8):viii90–viii121. [Google Scholar]
- 15. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Management of cancer‐ and chemotherapy‐induced anemia 2019. Available at https://www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/growthfactors.pdf. Accessed June 11, 2019. [Google Scholar]
- 16. Aapro M, Beguin Y, Bokemeyer C et al. Management of anaemia and iron deficiency in patients with cancer: ESMO Clinical Practice Guidelines. Ann Oncol 2018;29(suppl 4):iv96–iv110. [DOI] [PubMed] [Google Scholar]
- 17. Tutt A, Robson M, Garber JE et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof‐of‐concept trial. Lancet 2010;376:235–244. [DOI] [PubMed] [Google Scholar]
- 18. Yu Y, Elmeliegy M, Litton JK et al. Exposure‐efficacy progression‐free survival (PFS) analyses of breast cancer patients with germline BRCA1/2 mutations receiving talazoparib in the phase 3 EMBRACA trial. Ann Oncol 2018;29(suppl 8):viii90–viii121. [Google Scholar]
- 19. Yu Y, Durairaj C, Shi H et al. Population pharmacokinetic analyses for talazoparib (TALA) in cancer patients. Ann Oncol 2018;29(suppl 8):viii133–viii48. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figures
Supplemental Tables