

Abstract
Background
Sarcopenia and inflammation have been associated with poor survival in patients with cancer. We explored the combined effects of these variables on survival in patients with cancer treated with immunotherapy.
Methods
We performed a retrospective review of 90 patients enrolled on immunotherapy‐based phase I clinical trials at Emory University from 2009 to 2017. Baseline neutrophil‐to‐lymphocyte ratio, monocyte‐to‐lymphocyte ratio, and platelet‐to‐lymphocyte ratio (PLR) were used as surrogates of inflammation. The skeletal muscle index (SMI) was derived from the skeletal muscle density calculated from baseline abdominal computed tomography images. Optimal cutoffs for continuous inflammation biomarkers and SMI were determined by bias‐adjusted log‐rank test. A four‐level risk stratification was used to create low‐risk (PLR <242 and nonsarcopenic), intermediate‐risk (PLR <242 and sarcopenic), high‐risk (PLR ≥242 and nonsarcopenic), and very‐high‐risk (PLR ≥242 and sarcopenic) groups with subsequent association with survival.
Results
Most patients (59%) were male, and the most common cancers were melanoma (33%) and gastrointestinal (22%). Very high‐risk, high‐risk, and intermediate‐risk patients had significantly shorter overall survival (hazard ratio [HR], 8.46; 95% confidence interval [CI], 2.65–27.01; p < .001; HR, 5.32; CI, 1.96–14.43; p = .001; and HR, 4.01; CI, 1.66–9.68; p = .002, respectively) and progression‐free survival (HR, 12.29; CI, 5.15–29.32; p < .001; HR, 3.51; CI, 1.37–9.02; p = .009; and HR, 2.14; CI, 1.12–4.10; p = .022, respectively) compared with low‐risk patients.
Conclusion
Baseline sarcopenia and elevated inflammatory biomarkers may have a combined effect on decreasing survival in immunotherapy‐treated patients in phase I trials. These data may be immediately applicable for medical oncologists for the risk stratification of patients beginning immunotherapeutic agents.
Implications for Practice
Sarcopenia and inflammation have been associated with poor survival in patients with cancer, but it is unclear how to apply this information to patient care. The authors created a risk‐stratification system that combined sarcopenia and platelet‐to‐lymphocyte ratio as a marker of systemic inflammation. The presence of sarcopenia and systemic inflammation decreased progression‐free survival and overall survival in our cohort of 90 patients who received immunotherapy in phase I clinical trials. The data presented in this study may be immediately applicable for medical oncologists as a way to risk‐stratify patients who are beginning treatment with immunotherapy.
Keywords: Sarcopenia, Inflammation, Immunotherapy, Biomarkers, Risk stratification
Short abstract
The interaction between chronic inflammation and body composition is particularly important in the era of immunotherapy, considering that immune checkpoint inhibitors rely on the host immune system for their efficacy. This article reports on the combined effects of inflammation and sarcopenia on clinical outcomes in patients with solid tumors treated with immunotherapy‐based regimens.
Introduction
The effects of body composition on prognosis in patients with cancer is gaining increasing interest as a topic of research. Cachexia is a multifactorial syndrome characterized by weight loss in patients with underlying pathologic states such as cancer 1. This syndrome has been associated with poor responses to cancer treatment, decreased survival, and poor quality of life 2, 3, 4. Cachexia has been linked to systemic inflammation, which is consistent with inflammation being recognized as a hallmark of cancer 5, 6. Although patients with cancer presenting with weight loss and clinical signs of wasting have a poor prognosis, recent statistics from the Centers for Disease Control and Prevention estimate that 40% of cancer diagnoses in the U.S. (55% in women and 24% in men) can be linked to obesity 7. Thus, weight may not be the most effective measure of body composition in predicting outcomes in patients with cancer.
Sarcopenia, decreased levels of skeletal muscle mass and function, has gained interest as a measure of body composition as it relates to human pathology and is defined as a skeletal mass index (SMI) more than two SDs below the mean for a healthy adult (male patients, <55 cm2/m2; female patients, <39 cm2/m2) 1, 8. It can be observed in patients with chronic inflammatory conditions such as autoimmune diseases, and patients with cancer and sarcopenia have been shown to have higher levels of C‐reactive protein (CRP) 9, 10, 11, 12. Chronic inflammation contributes to muscle catabolism via cytokines such as interleukin (IL)‐6, tumor necrosis factor alpha (TNF‐α), and transforming growth factor beta (TGF‐β) 13, 14. Furthermore, sarcopenia at cancer diagnosis is associated with shorter overall survival (OS), cancer‐specific survival, and disease‐free survival 15. Hence, there is an interplay between inflammation and sarcopenia that likely affects clinical outcomes in patients with cancer, although causality is not yet determined.
Sarcopenia may be an effective prognostic body composition indicator, particularly in overweight or obese patients with cancer, given that they are unlikely to be presenting with wasting. The interaction between chronic inflammation and body composition is particularly important in the era of immunotherapy, given that immune checkpoint inhibitors (ICIs) rely on the host immune system for their efficacy 16. We investigated the combined effect of inflammation and sarcopenia on clinical outcomes in patients with solid tumors treated with immunotherapy‐based treatment regimens on phase I clinical trials.
Materials and Methods
Patients and Data
All patients (n = 90) treated on immunotherapy‐based phase I clinical trials at Winship Cancer Institute from 2009 to 2017 with available baseline computed tomography (CT) images were included. CT scans were deemed acceptable if they were performed within 2 months of starting immunotherapy if patients received no other systemic treatment since the scans. Axial images from the middle of the third lumbar vertebrae (mid‐L3) were retrieved from the electronic medical record, a validated muscular measurement source. Two authors (D.J.M., J.M.S.) were trained to correctly identify mid‐L3 on CT and quantify skeletal muscle quantity and density by using the Hounsfield unit threshold (−29 to +150) using SliceOmatic (version 5.0; TomoVision, Magog, Canada) 17. Low intraobserver variation of 1.3% was required to confirm adequate training. Skeletal muscle density was converted to SMI by dividing by height (m) squared 18. Baseline platelet, absolute neutrophil, monocyte, and lymphocyte counts were obtained from the complete blood count within 2 weeks before immunotherapy initiation. Neutrophil‐to‐lymphocyte ratio (NLR), monocyte‐to‐lymphocyte ratio (MLR), and platelet‐to‐lymphocyte ratio (PLR) were then calculated. Other data collected included gender, race, medication allergies, histology, prior lines of systemic therapy, Eastern Cooperative Oncology Group (ECOG) performance status (PS), and number and sites of metastatic disease. Royal Marsden Hospital (RMH) risk groups (albumin <3.5 g/dL, lactate dehydrogenase above the upper limit of normal, more than two metastatic sites) were used to risk‐stratify patients (0–1 risk factors, good risk; 2+ risk factors, poor risk) 19.
The study was approved by the Emory University Institutional Review Board and was conducted in accordance with Good Clinical Practice Guidelines and the Declaration of Helsinki. Informed consent for publication has been obtained and the consent forms are held by the authors. All data generated or analyzed during this study are included in this published article.
Statistical Analysis
OS was calculated from first dose of immunotherapy to date of death or hospice referral. Progression‐free survival (PFS) was measured from first dose to date of clinical or radiographic progression or death. PFS was set as the primary outcome because of the higher number of events at the time of analysis. The nonlinear relationship between each biomarker (NLR, MLR, and PLR) and PFS was examined by the martingale residual plot, and an optimal cutoff value of each biomarker was determined by a bias‐adjusted log‐rank test after searching all possible cuts in terms of PFS 20. An optimal cutoff value for SMI was determined for each gender by the same method described above. A four‐level risk group system was then defined by combining sarcopenia (sarcopenic vs. nonsarcopenic) and inflammation (high vs. low biomarker based on optimal cutoff), as has been done for localized cancer 21. A Cox regression model was carried out to associate the risk group with OS or PFS separately, and the multivariable model controlled for gender, checkpoint indication, number of previous treatments, RMH risk group, age, ECOG PS, race, number of metastatic sites, and histology. All analyses were done in SAS 9.4 and SAS macros developed by Winship Biostatistics and Bioinformatics Shared Resource 22 with significance level set at .05.
Results
Baseline demographic and disease characteristics are presented in Table 1. More than one‐half (n = 53, 59%) of patients were male, and 20 patients (22%) were nonwhite. The most common histologies were melanoma (33%), gastrointestinal (22%), lung and head and neck (20%), and breast (12%). Most patients (n = 63, 70%) were ICI‐naïve. Only 31% of patients had received fewer than two lines of prior systemic therapy. Most patients (81%) were RMH good risk. The immunotherapy treatment regimens are shown in Table 1. The median baseline SMI was 47.42, and the median NLR, MLR, and PLR were 3.63, 0.49, and 182.65, respectively. The optimal cutoffs for SMI that defined sarcopenia in our model were 55.97 for male patients and 37.39 for female patients, which closely correspond to previously reported cutoffs 1, 21. NLR, MLR, and PLR were highly correlated (Pearson correlation coefficients ≥0.67, all p < .0001, not shown).
Table 1.
Demographic information and disease characteristics
| Variable | n (%) |
|---|---|
| Gender | |
| Male | 53 (59) |
| Female | 37 (41) |
| Race | |
| White | 70 (78) |
| Black | 16 (18) |
| Asian or unknown | 4 (4) |
| Histology | |
| Melanoma | 30 (33) |
| Gastrointestinal | 20 (22) |
| Lung or head and neck | 18 (20) |
| Breast | 11 (12) |
| Other | 11 (12) |
| Checkpoint Indication | |
| Yes | 49 (54) |
| No | 41 (46) |
| ECOG PS | |
| 0 | 34 (38) |
| 1 | 55 (62) |
| Unknown | 1 (1) |
| Number of prior systemic therapies in the metastatic setting | |
| 0–1 | 28 (31) |
| 2+ | 62 (69) |
| Prior treatment with ICI | |
| Yes | 27 (30) |
| No | 63 (70) |
| RMH risk group | |
| Good | 71 (81) |
| Poor | 17 (19) |
| Unknown | 2 (2) |
| Treatment regimen | |
| FDA‐approved ICI + experimental combination | 46 (51) |
| Anti–PD‐L1 monotherapy | 25 (28) |
| Experimental IO monotherapy | 19 (21) |
| SMI, median (range), cm2/m2 | 47.42 (27.64–71.74) |
| Sarcopenia cutoff, cm2/m2 | |
| For male patients | 55.97 |
| For female patients | 37.39 |
| PLR, median (range) | 182.65 (52.94–1,373.08) |
| Inflammation cutoff | 242 |
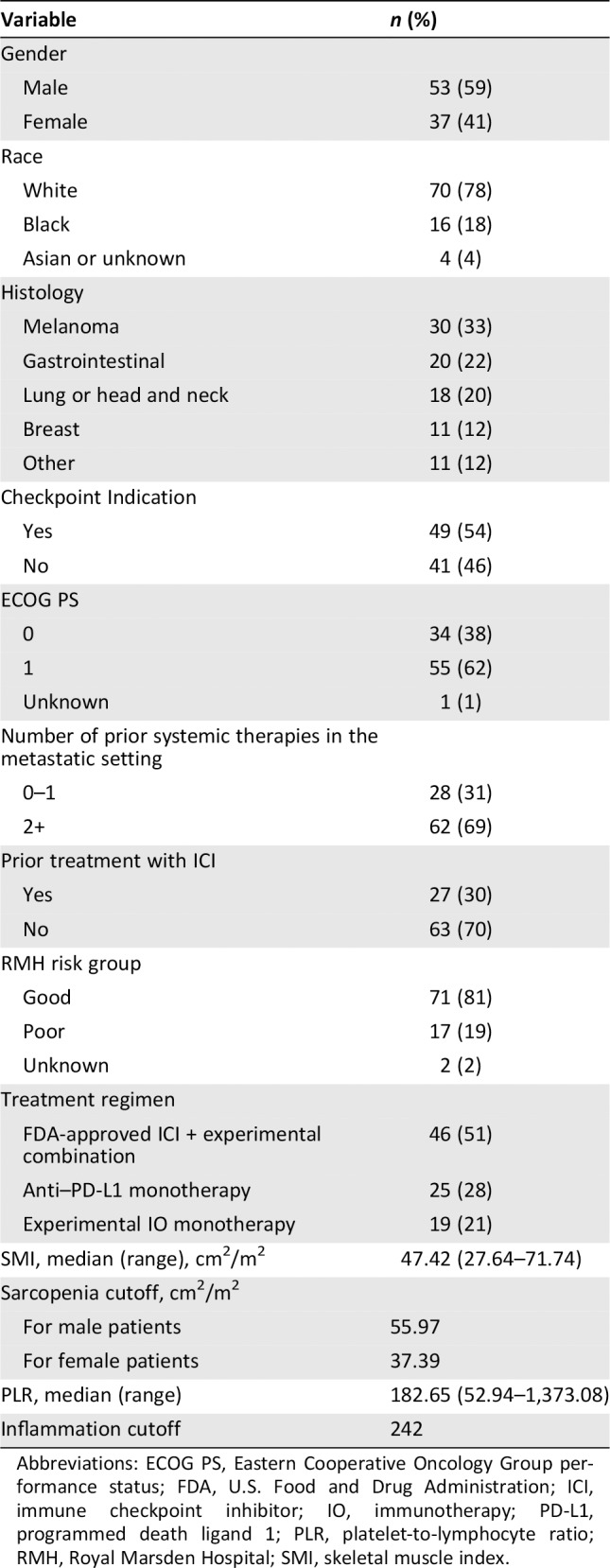
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; FDA, U.S. Food and Drug Administration; ICI, immune checkpoint inhibitor; IO, immunotherapy; PD‐L1, programmed death ligand 1; PLR, platelet‐to‐lymphocyte ratio; RMH, Royal Marsden Hospital; SMI, skeletal muscle index.
The univariable analysis and multivariable analysis (MVA) of the PLR‐based risks groups and survival are shown in Table 2. NLR‐ and MLR‐based risk group analyses are provided in supplemental online Tables 1 and 2. In PLR‐based MVA, very high‐risk patients (PLR ≥242 and sarcopenic) had significantly shorter OS (hazard ratio [HR], 8.46; 95% confidence interval [CI], 2.65–27.01; p < .001) and PFS (HR, 12.29; CI, 5.15–29.32; p < .001) compared with low‐risk patients (PLR <242 and nonsarcopenic). High‐risk (PLR ≥242 and nonsarcopenic) and intermediate‐risk (PLR <242 and sarcopenic) patients also had shorter OS (high‐risk HR, 5.32; CI, 1.96–14.43; p = .001; intermediate‐risk HR, 4.01; CI, 1.66–9.68; p = .002) and shorter PFS (high‐risk HR, 3.51; CI, 1.37–9.02; p = .009; intermediate‐risk HR, 2.14; CI, 1.12–4.10; p = .022) compared with low‐risk patients. The median OS and PFS were longer for low‐risk patients (24.3 months and 4.8 months) than intermediate‐risk (9.4 months and 2.8 months), high‐risk (7.6 months and 1.7 months), and very‐high‐risk patients (4.6 months and 1.6 months) per Kaplan‐Meier estimation (Figs. 1, 2, both p < .005). SMI was negatively correlated with PLR (p = .0387, not shown).
Table 2.
UVA and MVAa of PLR‐based risk groups and survival
| Group | UVA | MVA | ||||||
|---|---|---|---|---|---|---|---|---|
| OS | PFS | OS | PFS | |||||
| HR (CI) | p value | HR (CI) | p value | HR (CI) | p value | HR (CI) | p value | |
|
Group 1: low risk PLR <242 and nonsarcopenic (n = 27) |
— | — | — | — | — | — | — | — |
| Median survival: 24.3 months | Median survival: 4.8 months | |||||||
|
Group 2: intermediate risk PLR <242 and sarcopenic (n = 29) |
1.67 (0.83–3.35) | .148 | 1.40 (0.80–2.44) | .242 | 4.01 (1.66–9.68) | .002b | 2.14 (1.12–4.10) | .022b |
| Median survival: 9.4 months | Median survival: 2.8 months | |||||||
|
Group 3: high risk PLR ≥242 and nonsarcopenic (n = 8) |
2.78 (1.17–6.60) | .021b | 2.72 (1.20–6.15) | .016b | 5.32 (1.96–14.43) | .001b | 3.51 (1.37–9.02) | .009b |
| Median survival: 7.6 months | Median survival: 1.7 months | |||||||
|
Group 4: very high risk PLR ≥242 and sarcopenic (n = 15) |
3.56 (1.65–7.68) | .001b | 5.32 (2.64–10.73) | <.001b | 8.46 (2.65–27.01) | <.001b | 12.29 (5.15–29.32) | <.001b |
| Median survival: 4.6 months | Median survival: 1.6 months | |||||||
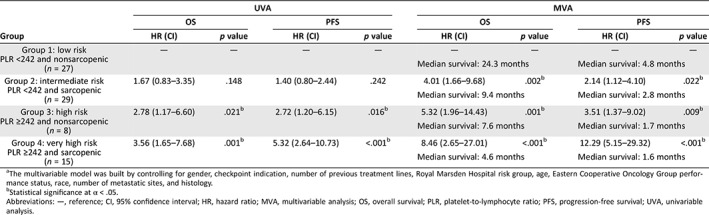
The multivariable model was built by controlling for gender, checkpoint indication, number of previous treatment lines, Royal Marsden Hospital risk group, age, Eastern Cooperative Oncology Group performance status, race, number of metastatic sites, and histology.
Statistical significance at α < .05.
Abbreviations: —, reference; CI, 95% confidence interval; HR, hazard ratio; MVA, multivariable analysis; OS, overall survival; PLR, platelet‐to‐lymphocyte ratio; PFS, progression‐free survival; UVA, univariable analysis.
Figure 1.
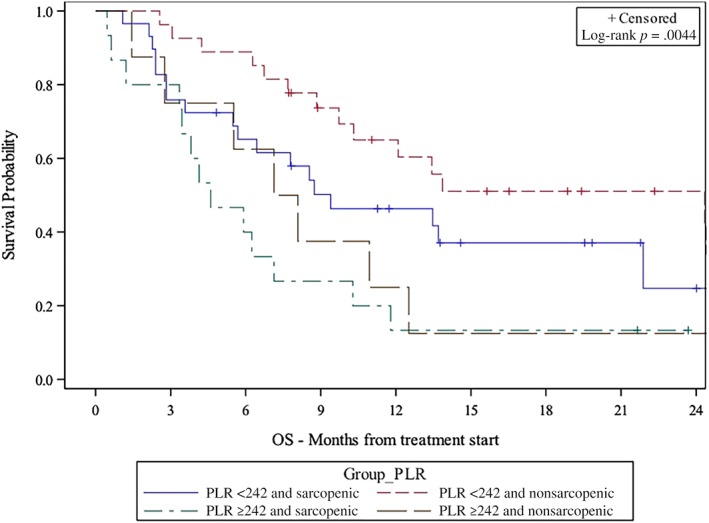
Kaplan‐Meier plot of association between risk group and OS.
Abbreviations: OS, overall survival; PLR, platelet‐to‐lymphocyte ratio.
Figure 2.
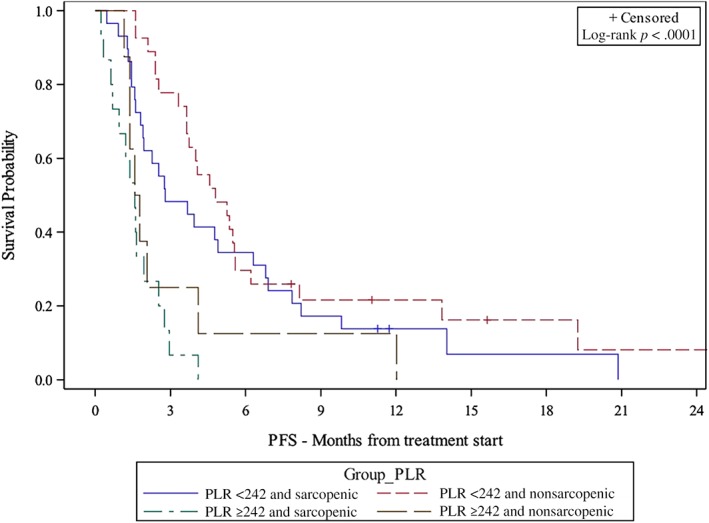
Kaplan‐Meier plot of association between risk group and PFS.
Abbreviations: PFS, progression‐free survival; PLR, platelet‐to‐lymphocyte ratio.
Discussion
We showed that sarcopenia and PLR have a combined effect on decreasing survival in this population of patients treated with immunotherapy on phase I clinical trials. These results build upon previous data showing that sarcopenia and systemic inflammation are independently associated with poor survival in patients with cancer 15, 23. This is the first study, to our knowledge, investigating the combined effect of sarcopenia and inflammation in patients with solid tumors treated with immunotherapy. This study is also novel in that it included heavily pretreated patients enrolled on phase I clinical trials using novel immunotherapeutic agents.
The effect of sarcopenia and inflammation on clinical outcomes has been explored in patients with malignancies. In a previous analysis, we showed that increased NLR, MLR, and PLR were associated with worse clinical outcomes in this group of patients treated with immunotherapy on phase I clinical trials 24. A study of 117 male patients with small cell lung cancer treated with chemotherapy or chemo‐radiotherapy showed that sarcopenia and high NLR were independently associated with shorter OS and PFS 25. Patients with melanoma with sarcopenia were found to be more likely to experience ipilimumab‐related toxicity, whereas a cohort of patients with advanced non‐small cell lung cancer treated with nivolumab or pembrolizumab appeared to have poorer survival outcomes and response rates if they were sarcopenic 26, 27. These clinical findings in immunotherapy‐treated populations in particular may be explained by the interplay between skeletal muscle and the immune system. Chronic inflammation has been posited to play a role in both the development of sarcopenia and resistance to immune checkpoint inhibitors. The same cytokines implicated in sarcopenia such as TGF‐β and IL‐6 have also been cited as mediators of T‐cell exhaustion 28. Increased TGF‐β signaling in urothelial cell tumors of patients unresponsive to the anti–programmed death ligand 1 agent atezolizumab has been correlated with the sequestering of CD8+ T cells in the peritumoral stroma rather than in the tumor itself 29. It is also known that skeletal muscle produces anti‐inflammatory cytokines with increased production during times of exercise. IL‐15 is one such cytokine, also known as myokine, and is known to decrease the activity of proinflammatory TNF‐α during cachexia 30. In addition, skeletal muscle undergoes necrosis in response to injury and releases intracellular contents, including chemotactic factors, which recruit immune cells to mediate regeneration of skeletal muscle 31. Thus, the decreased muscle mass of sarcopenia may have a direct link to immune dysregulation, resistance to ICIs, and poorer outcomes in patients with cancer treated with these agents.
The inclusion of both sarcopenia and inflammatory biomarkers in these risk groups better accounts for the multifactorial contribution of prognostic indicators in oncology patients. Risk groups provide clinicians with a tool to assess the clinical outcomes of individual patients before they begin treatment. The data presented in this study may be immediately applicable for medical oncologists. For example, these results suggest that sarcopenia may be an important consideration in further risk‐stratifying patients without laboratory signs of inflammation such as a low PLR. Furthermore, recent validated work has shown that sarcopenia can be measured in the clinic setting using standard picture archiving and communication system tools without the need for advanced software 21, making sarcopenia an easily determined prognostic marker.
The utility of sarcopenia as a biomarker of response to immunotherapy is particularly appealing in patients who are overweight or obese. Although increased body mass index (BMI) has been shown to be protective for some patients with cancer 32, 33, there is evidence suggesting that obesity contributes to oncologic development and progression via chronic inflammation 34. It suffices to say that BMI is an imperfect biomarker of body composition as it relates to cancer outcomes 35. Recently, Young et al. (2019) examined the role of skeletal muscle mass and adiposity on clinical outcomes in a cohort of patients with melanoma who received anti–programmed cell death protein 1 monotherapy. Interestingly, high skeletal muscle gauge (SMG), a composite of muscle area and density, was the best predictor of clinical outcomes, as the cohort with high SMG and high adiposity achieved superior OS and PFS, whereas patients with low SMG and high adiposity had the worst outcomes 36. Therefore, a simple calculation of BMI that relies heavily on adiposity may understandably fall short as a prognostic marker.
Sarcopenia may be a better predictor of clinical outcomes in obese and overweight patients, which is supported by two patients with lung cancer presented in Figure 3. The first patient had a BMI of 26.5 and an SMI of 59.6 (Fig. 3A). His baseline NLR, MLR, and PLR were 4.26, 0.60, and 174.45, respectively. He maintained a best response of stable disease without progression for 11 months. The second patient (Fig. 3B) had a similar BMI (26.0) and lower baseline inflammatory markers but had a lower SMI (42.8). This patient had similar inflammatory markers as the first patient (NLR, 4.25; MLR, 0.55; and PLR, 142.11). This patient experienced progressive disease (PD) on their first restaging scans after starting immunotherapy. This suggests that sarcopenia may be a more valuable measure of body composition than BMI in this subset of patients. The fact that these patients had comparable inflammatory markers also highlights the importance of using SMI along with an inflammatory marker such as PLR when risk‐stratifying oncology patients treated with immunotherapy.
Figure 3.
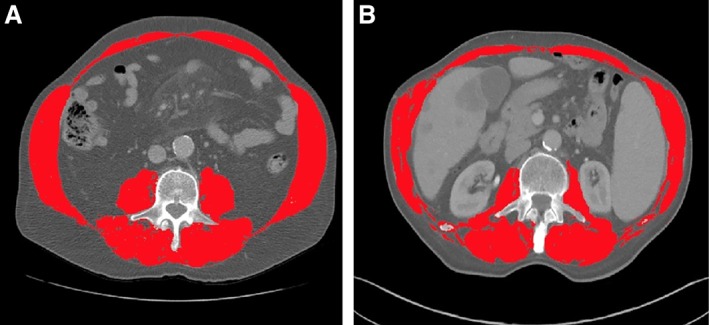
Segmented computed tomography images comparing clinical outcomes of two overweight patients (25 ≤ body mass index [BMI] < 30). (A): Baseline BMI, 26.5. Baseline skeletal muscle index (SMI), 59.6. Best response to immunotherapy: stable disease maintained for 11 months. (B): Baseline BMI, 26.0. Baseline SMI, 42.8. Best response to immunotherapy: progressive disease on first reimaging scan.
In this study, sarcopenia was useful in distinguishing low‐ and intermediate‐risk patients without laboratory signs of inflammation (PLR <242). This highlights another clinical situation in which sarcopenia may be a useful measure for clinicians. The clinical utility of sarcopenia in this situation is illustrated in Figure 4, which compares the baseline SMI of two patients who had PLR <242. One patient who had a baseline PLR of 160.19 at baseline and an SMI of 66.38 (Fig. 4A). This patient received an immunotherapy‐based experimental treatment regimen on the phase I clinical trial as sixth‐line systemic therapy and has sustained a partial response for 16 months. The second patient had a baseline PLR of 192.95 and an SMI of 31.71 (Fig. 4B). Unfortunately, this patient experienced PD as best response on an immunotherapy‐based experimental treatment regimen and passed away 12 months after the first dose of immunotherapy. These two patients highlight a second clinical situation where sarcopenia can be used as a biomarker of response to immunotherapy.
Figure 4.
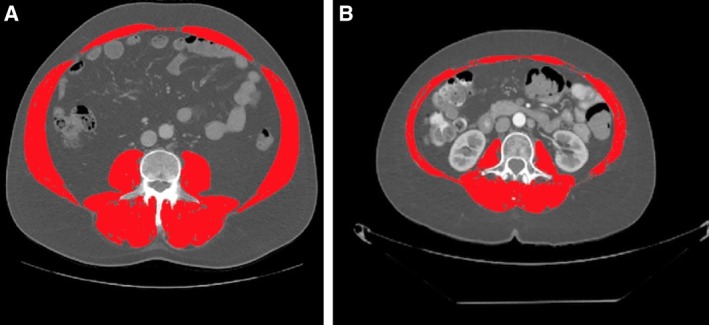
Segmented computed tomography images comparing clinical outcomes of two patients without inflammation (platelet‐to‐lymphocyte ratio [PLR] <242). (A): Baseline PLR, 160.19. Baseline skeletal muscle index (SMI), 66.38. Best response to immunotherapy: partial response. (B): Baseline PLR, 192.95. Baseline SMI, 31.71. Best response to immunotherapy: progressive disease.
Despite the novelty of this study, there are several limitations that should be noted. First, this is a retrospective study and is subject to selection bias. We attempted to mitigate the effect of selection bias by including all patients with available clinical data and baseline CT images who were treated on phase I clinical trials at our center, regardless of their primary malignancy or baseline characteristics. We did not investigate some markers of inflammation, such as CRP, given that it is not routinely collected. Finally, we only included skeletal muscle as a surrogate of body composition and did not include other markers.
Conclusion
In this study of patients with advanced stage cancer treated with immunotherapy‐based treatment regimens on phase I clinical trials, sarcopenia and inflammation had a combined effect on decreasing survival. The inclusion of both sarcopenia and inflammatory biomarkers better accounts for the multifactorial contribution of prognostic indicators in patients with advanced cancer. The data presented in this study may be immediately applicable for medical oncologists, given that these risk groups may be used for risk stratification for patients who are beginning treatment with immunotherapy. Future studies should further elucidate the biological relationship between body composition and inflammation in patients with cancer treated with immunotherapeutic agents.
Author Contributions
Conception/design: Mehmet Asim Bilen, Dylan J. Martini, Yuan Liu
Provision of study material or patients: Mehmet Asim Bilen, Colleen Lewis, Hannah Collins, Bradley C. Carthon, Mehmet Akce, Walid L. Shaib, Olatunji B. Alese, Rathi N. Pillai, Conor E. Steuer, Christina S. Wu, David H. Lawson, Ragini R. Kudchadkar, Bassel F. El‐Rayes, Suresh S. Ramalingam, Taofeek K. Owonikiki, R. Donald Harvey, Viraj A. Master
Collection and/or assembly of data: Dylan J. Martini, Yuan Liu, Julie M. Shabto, Milton Williams, Amir I. Khan
Data analysis and interpretation: Mehmet Asim Bilen, Dylan J. Martini, Yuan Liu, Julie M. Shabto, Jacqueline T. Brown, Milton Williams, Amir I. Khan, Alexandra Speak, Colleen Lewis, Hannah Collins, Haydn T. Kissick, Bradley C. Carthon, Mehmet Akce, Walid L. Shaib, Olatunji B. Alese, Rathi N. Pillai, Conor E. Steuer, Christina S. Wu, David H. Lawson, Ragini R. Kudchadkar, Bassel F. El‐Rayes, Suresh S. Ramalingam, Taofeek K. Owonikoko, R. Donald Harvey, Viraj A. Master
Manuscript writing: Mehmet Asim Bilen, Dylan J. Martini, Yuan Liu, Julie M. Shabto, Jacqueline T. Brown, Milton Williams, Amir I. Khan, Alexandra Speak, Colleen Lewis, Hannah Collins, Haydn T. Kissick, Bradley C. Carthon, Mehmet Akce, Walid L. Shaib, Olatunji B. Alese, Rathi N. Pillai, Conor E. Steuer, Christina S. Wu, David H. Lawson, Ragini R. Kudchadkar, Bassel F. El‐Rayes, Suresh S. Ramalingam, Taofeek K. Owonikoko, R. Donald Harvey, Viraj A. Master
Final approval of manuscript: Mehmet Asim Bilen, Dylan J. Martini, Yuan Liu, Julie M. Shabto, Jacqueline T. Brown, Milton Williams, Amir I. Khan, Alexandra Speak, Colleen Lewis, Hannah Collins, Haydn T. Kissick, Bradley C. Carthon, Mehmet Akce, Walid L. Shaib, Olatunji B. Alese, Rathi N. Pillai, Conor E. Steuer, Christina S. Wu, David H. Lawson, Ragini R. Kudchadkar, Bassel F. El‐Rayes, Suresh S. Ramalingam, Taofeek K. Owonikoko, R. Donald Harvey, Viraj A. Master
Disclosures
Mehmet Asim Bilen: Exelixis, Nektar, Sanofi (C/A), Bayer, Bristol‐Myers Squibb, Genentech/Roche, Incyte, Nektar, AstraZenecca, Tricon Pharmaceuticals, Peleton, Pfizer (RF); Bradley C. Carthon: Astellas Medivation, Pfizer, Blue Earth Diagnostics (C/A), Bristol‐Myers Squibb (other—travel); Walid L. Shaib: ArQule, Eli Lilly & Co. (RF); Rathi N. Pillai: Natera, AstraZeneca (C/A), Bristol‐Myers Squibb (RF), Genentech/Roche, Takeda, Novartis, Clovis Oncology (other—travel); Conor E. Steuer: Abbvie, Merck, Bergen Bio, Armo, Eli Lilly & Co. (C/A); Christina S. Wu: BioTheranostics (H), Amgen, Bristol‐Myers Squibb, Vaccinex, Boston Biomedical (RF); Ragini R. Kudchadkar: Bristol‐Myers Squibb, Novartis, Array BioPharma (C/A), Bristol‐Myers Squibb (H), Merck (RF); Bassel F. El‐Rayes: Merrimack, BTG, Bayer, Loxo, RTI Health Solutions (C/A), Lexicon, RTI Health Solutions, Bayer (H), Taiho Pharmaceutical, Bristol‐Myers Squibb, Boston Biomedical, Cleave Biosciences, Genentech, AVEO, Pfizer, Novartis, Hoosier Cancer Research Network, Five Prime Therapeutics, PPD Inc., Merck, ICON Clinical Research (RF), Lexion, Bristol‐Myers Squibb (other—speakers’ bureau); Suresh S. Ramalingam: Amgen, AstraZeneca, Bristol‐Myers Squibb, Merck, Genentech/Roche, Tesaro, Eli Lilly & Co. (C/A), Amgen, Advaxis, AstraZeneca, Bristol‐Myers Squibb, Takeda, Genmab, Tesaro (RF—institution); Taofeek K. Owonikoko: Novartis, Bristol‐Myers Squibb, MedImmune (C/A); R. Donald Harvey: Bristol‐Myers Squibb, Genentech, Takeda (C/A), Abbvie, Amgen, Arqule, AstraZeneca, Bristol‐Myers Squibb, Boston Biomedical, Calithera, Celgene, Cleave, Corvus, Eli Lilly & Co., Five Prime Therapeutics, Genmab, Halozyme, Ignyta, Incyte, Merck, Nektar, Pfizer, Regeneron, Rgenix, Sanofi, Syndax, Takeda, Tesaro, Vertex, Xencor (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Table S1: UVA and MVAa of NLR‐based risk groups and survival
Supplemental Table S2: UVA and MVAa of MLR‐based risk groups and survival
Acknowledgments
Preliminary data on the combined effect of sarcopenia and inflammation were presented at the European Society for Medical Oncology 2018 Congress in Munich, Germany. Data included in this analysis on the prognostic and predictive impact of neutrophil‐to‐lymphocyte ratio, monocyte‐to‐lymphocyte ratio, and platelet‐to‐lymphocyte ratio have been published in Cancer (PMID: 30329148) 24. This work was supported by the National Institutes of Health/National Cancer Institute and the Biostatistics and Bioinformatics Shared Resource of the Winship Cancer Institute of Emory University under award number P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Fearon K, Strasser F, Anker SD et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 2. Aoyagi T, Terracina KP, Raza A et al. Cancer cachexia, mechanism and treatment. World J Gastrointest Oncol 2015;7:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dewys WD, Begg C, Lavin PT et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med 1980;69:491–497. [DOI] [PubMed] [Google Scholar]
- 4. Couch M, Lai V, Cannon T et al. Cancer cachexia syndrome in head and neck cancer patients: Part I. Diagnosis, impact on quality of life and survival, and treatment. Head Neck 2007;29:401–411. [DOI] [PubMed] [Google Scholar]
- 5. Colotta F, Allavena P, Sica A et al. Cancer‐related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009;30:1073–1081. [DOI] [PubMed] [Google Scholar]
- 6. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70. [DOI] [PubMed] [Google Scholar]
- 7. Byers T, Mouchawar J, Marks J et al. The American Cancer Society challenge goals. How far can cancer rates decline in the U.S. by the year 2015? Cancer 1999;86:715–727. [PubMed] [Google Scholar]
- 8. Santilli V, Bernetti A, Mangone M et al. Clinical definition of sarcopenia. Clin Cases Miner Bone Metab 2014;11:177–180. [PMC free article] [PubMed] [Google Scholar]
- 9. Dogan SC, Hizmetli S, Hayta E et al. Sarcopenia in women with rheumatoid arthritis. Eur J Rheumatol 2015;2:57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schneider SM, Al‐Jaouni R, Filippi J et al. Sarcopenia is prevalent in patients with Crohn's disease in clinical remission. Inflamm Bowel Dis 2008;14:1562–1568. [DOI] [PubMed] [Google Scholar]
- 11. Zhang G, Li X, Sui C et al. Incidence and risk factor analysis for sarcopenia in patients with cancer. Oncol Lett 2016;11:1230–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bano G, Trevisan C, Carraro S et al. Inflammation and sarcopenia: A systematic review and meta‐analysis. Maturitas 2017;96:10–15. [DOI] [PubMed] [Google Scholar]
- 13. Londhe P, Guttridge DC. Inflammation induced loss of skeletal muscle. Bone 2015;80:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schaap LA, Pluijm SM, Deeg DJ et al. Inflammatory markers and loss of muscle mass (sarcopenia) and strength. Am J Med 2006;119:526.e9–e17. [DOI] [PubMed] [Google Scholar]
- 15. Shachar SS, Williams GR, Muss HB et al. Prognostic value of sarcopenia in adults with solid tumours: A meta‐analysis and systematic review. Eur J Cancer 2016;57:58–67. [DOI] [PubMed] [Google Scholar]
- 16. Seidel JA, Otsuka A, Kabashima K. Anti‐PD‐1 and anti‐CTLA‐4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front Oncol 2018;8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mitsiopoulos N, Baumgartner RN, Heymsfield SB et al. Cadaver validation of skeletal muscle measurement by magnetic resonance imaging and computerized tomography. J Appl Physiol 1998;85:115–122. [DOI] [PubMed] [Google Scholar]
- 18. Baumgartner RN, Koehler KM, Gallagher D et al. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 1998;147:755–763. [DOI] [PubMed] [Google Scholar]
- 19. Arkenau HT, Olmos D, Ang JE et al. Clinical outcome and prognostic factors for patients treated within the context of a phase I study: The Royal Marsden Hospital experience. Br J Cancer 2008;98:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mandrekar JN, Mandrekar SJ, Cha SS. Cutpoint determination methods in survival analysis using SAS. Paper 261‐28 presented at: 28th SAS Users Group International Conference (SUGI); Seattle, WA; March 30 to April 2, 2003. [Google Scholar]
- 21. Feliciano EMC, Kroenke CH, Meyerhardt JA et al. Association of systemic inflammation and sarcopenia with survival in nonmetastatic colorectal cancer: Results from the C SCANS study. JAMA Oncol 2017;3:e172319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y, Nickleach DC, Zhang C et al. Carrying out streamlined routine data analyses with reports for observational studies: Introduction to a series of generic SAS macros. F1000Res 2018;7:1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Templeton AJ, McNamara MG, Seruga B et al. Prognostic role of neutrophil‐to‐lymphocyte ratio in solid tumors: A systematic review and meta‐analysis. J Natl Cancer Inst 2014;106:dju124. [DOI] [PubMed] [Google Scholar]
- 24. Bilen MA, Martini DJ, Liu Y et al. The prognostic and predictive impact of inflammatory biomarkers in patients who have advanced‐stage cancer treated with immunotherapy. Cancer 2019;125:127–134. [DOI] [PubMed] [Google Scholar]
- 25. Go SI, Park MJ, Song HN et al. Sarcopenia and inflammation are independent predictors of survival in male patients newly diagnosed with small cell lung cancer. Support Care Cancer 2016;24:2075–2084. [DOI] [PubMed] [Google Scholar]
- 26. Daly LE, Power DG, O'Reilly A et al. The impact of body composition parameters on ipilimumab toxicity and survival in patients with metastatic melanoma. Br J Cancer 2017;116:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shiroyama T, Nagatomo I, Koyama S et al. Impact of sarcopenia in patients with advanced non‐small cell lung cancer treated with PD‐1 inhibitors: A preliminary retrospective study. Sci Rep 2019;9:2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mariathasan S, Turley SJ, Nickles D et al. TGFbeta attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pajak B, Orzechowska S, Pijet B et al. Crossroads of cytokine signaling‐‐the chase to stop muscle cachexia. J Physiol Pharmacol 2008;59(suppl 9):251–264. [PubMed] [Google Scholar]
- 31. Yang W, Hu P. Skeletal muscle regeneration is modulated by inflammation. J Orthop Translat 2018;13:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsang NM, Pai PC, Chuang CC et al. Overweight and obesity predict better overall survival rates in cancer patients with distant metastases. Cancer Med 2016;5:665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schlesinger S, Siegert S, Koch M et al. Postdiagnosis body mass index and risk of mortality in colorectal cancer survivors: A prospective study and meta‐analysis. Cancer Causes Control 2014;25:1407–1418. [DOI] [PubMed] [Google Scholar]
- 34. Berger NA. Obesity and cancer pathogenesis. Ann NY Acad Sci 2014;1311:57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Strulov Shachar S, Williams GR. The obesity paradox in cancer ‐ moving beyond BMI. Cancer Epidemiol Biomarkers Prev 2017;26:13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Young A, Quach HT, Davis EJ et al. Impact of body composition on outcomes from anti‐programmed death‐1 (PD‐1) treatment. J Clin Oncol 2019;37(suppl 15):9516A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Table S1: UVA and MVAa of NLR‐based risk groups and survival
Supplemental Table S2: UVA and MVAa of MLR‐based risk groups and survival