

SUMMARY
The goal of this study was to identify individuals at risk of progression and reactivation among household contacts (HHC) of pulmonary TB cases in Vitoria, Brazil. We first evaluated the predictive performance of six published signatures on the transcriptional dataset obtained from peripheral blood mononuclear cell samples from HHC that either progressed to TB disease or not (non-progressors) during a five-year follow-up. The area under the curve (AUC) values for the six signatures ranged from AUC values of 0.670 to 0.461, and the PPVs did not reach the WHO published target product profiles (TPPs). We therefore used as training cohort the earliest time-point samples from the African cohort of adolescents (GSE79362) and applied an ensemble feature selection pipeline to derive a novel 29-gene signature (PREDICT29). PREDICT29 was tested on 16 progressors and 21 non-progressors. PREDICT29 performed better in segregating progressors from non-progressors in the Brazil cohort with the area under the curve (AUC) value of 0.911 and PPV of 20%. This proof of concept study demonstrates that PREDICT29 can predict risk of progression/reactivation to clinical TB disease in recently exposed individuals at least 5 years prior to disease development. Upon validation in larger and geographically diverse cohorts, PREDICT29 can be used to risk-stratify recently infected for targeted therapy.
Keywords: Tuberculosis, Biomarkers, transcriptional signatures, TB risk signature
INTRODUCTION
With one-third of the world’s population estimated to be latently infected with Mycobacterium tuberculosis (Mtb), the World Health Organization’s guidelines on management of latent tuberculosis infection (LTBI) call for better strategies for testing and treating LTBI, particularly pointing out the need for methods to determine risk of LTBI progression to active tuberculosis (TB) disease (1). Administration of preventive treatment to persons with LTBI can reduce the subsequent number of TB disease diagnoses (2, 3). However, in TB endemic regions, mass treatment of all latently infected individuals is neither practical nor cost-effective. Therefore, the identification of Mtb-infected subjects most likely to progress to disease or reactivate several years later, could enable the targeting of anti-TB preventive therapy to those most likely to benefit.
Several blood signatures have been reported to discriminate active TB from LTBI (4–11), but studies centered on predicting the outcome of Mtb infection are limited (12). In a retrospective case-control study conducted in Amsterdam, Netherlands, PBMC samples from HIV-infected drug users with and without TB were analyzed for the expression of 141 genes. The study found that 8 months prior to onset of disease the expression of IL-13 and AIRE could identify individuals that progressed to TB (13). Another large prospective biomarker study in a South African adolescent cohort (GSE79362) identified a 16-gene whole blood signature (henceforth denoted ACS-COR) capable of predicting progression to disease with 53.7% sensitivity and 82.8% specificity, when disease diagnosis was established within 12 months of sample collection (14). Whereas the latter study demonstrated the utility of blood transcriptomics as a biomarker for progression risk, sensitivity was a key limitation to its conclusions (15). Importantly, ASC-COR’s predictive performance, most notably its sensitivity, decreased with increasing time to disease onset –suggesting ASC-COR was likely detecting sub-clinical TB disease that was already present at the time of blood collection. For example, the sensitivity of their approach decreased to 39.3% when disease diagnosis occurred between 361 and 720 days of sample collection (14). Another study consisting of a number of cohorts from Africa (GC6; GSE94438) identified a 4-gene signature (RISK4), but predicted progression only up to two years before onset of disease (16).
Proteomic and metabolic biomarkers with diagnostic potential for distinguishing active from latent TB have also been reported (4–7). Serum-based biomarkers of risk of progression to disease are also being developed. A recent study performed an in-depth proteomic analysis of serum samples from the South African adolescent and GC-6 cohorts on the Somascan platform and discovered a 5-protein signature, TB Risk Model 5 (TRM5) and a 3-protein signature, 3-protein pair-ratio (3PR). Both signatures predicted progression up to a year prior to disease diagnosis. However, the biomarker performed with greatest significance proximal to disease diagnosis. Also, neither signature met the WHO Target Product profile for a progression test (8). Metabolic profiling of serum and plasma samples from the GC6 cohort was successful in generating a TB-specific prognostic metabolic signature that performed well in predicting subclinical TB and progression to active TB as early as 12 months prior to TB diagnosis (9). Circulating miRNAs have also been found to perform well as a short-term predicters of risk (10).
The predictive performance of none of the published biomarker signatures has been evaluated in a Brazilian population. Therefore, the goal of this study was to test the predictive performance of the published transcriptomic signatures in identifying individuals at risk of progression and reactivation among household contacts (HHC) of pulmonary TB cases in Vitoria, Brazil. We found that the published signatures did not perform well in predicting progression to disease in the Brazilian cohort. Using relevant data from published study, we then developed a novel 29-gene biomarker signature that performed significantly better than the published signatures in predicting TB disease risk.
RESULTS
HHC follow up for progression to TB disease
From March 2008 to May 2015, 1203 HHCs (derived from 410 smear positive culture-proven TB index cases) were enrolled in a prospective observational cohort study in Vitória, Brazil. At the time of enrollment, HHCs were screened for tuberculin skin test (TST) reactivity; 573 HHCs had a positive TST (≥10mm), indicating infection with Mtb. Of these TST-positive contacts, 6 (1%) were clinically diagnosed with TB disease at the time of enrollment and excluded from further follow-up for incident TB disease. During post-enrollment follow-up, 27 TST-positive HHC and 1 TST-negative HHC were later diagnosed with TB disease (progressors). In this nested case-control study, baseline (time of enrollment) peripheral blood mononuclear cells (PBMC) samples from only 16 progressors was available for gene expression profiling. These individuals developed TB disease between 11 and 1795 days after enrollment (median 255 days) and baseline blood collection. Five of the 16 progressors were diagnosed within the first three months of sample collection and were defined as co-prevalent cases. Three of the 5 co-prevalent were culture-proven TB. Of the remaining 11 progressors, 5 were diagnosed within 2 years (early progressors) and 6 were diagnosed after 2 years (late progressors) of sample collection. Nine of the 11 progressors were culture-proven TB (Tables S1–S2). All HHCs with suspected TB were evaluated by experienced clinicians from the TB clinics of Núcleo de Doenças Infecciosas (NDI) Vitoria, Brazil. Based on Brazilian TB program guidelines, patients with abnormal CXR suggestive of TB (apical infiltrates, cavities, miliary pattern), systemic symptoms (cough, fever, weight loss) and clinical/radiographic improvement with empiric TB therapy are considered TB by clinical diagnosis.
TST-positive contacts that were not diagnosed with TB disease during long-term follow-up (≥5 years) were considered non-progressors. We selected 21 age- and sex-matched non-progressors as controls for this study. In addition, PBMC from 14 randomly selected TB index patients from the cohort were also studied. PBMC obtained at baseline from the 16 progressors, 21 non-progressors, and 14 TB patients were used for RNA sequencing (RNA-seq) analysis (Tables S1–S2,).
Predicting progressors from non-progressors in the Brazil cohort using existing gene signatures
We first tested the performance of ACS-COR and RISK4 signatures in predicting risk of progression in the Brazil cohort using four methods- ridge logistic regression (glmnet package), SVM, random forest (ranger package), gradient boosting (xgbost package). Co-prevalent cases were not included for prediction analysis. Averaging the ACS-COR signature’s performance across the four models (Table 1), yielded a mean AUC of 0.670 (0.640, 0.700), sensitivity of 0.515 (0.460, 0.571) and a specificity of 0.774 (0.728, 0.820). Similarly, averaging the RISK4 signature’s performance across the four models (Table 1), yielded a mean AUC of 0.461 (0.434, 0.488) with a sensitivity of 0.413 (0.335, 0.491) and a specificity of 0.665 (0.588, 0.743). Evaluation of the predictive performance of four existing TB disease signatures Kaforou27 (6), Sambarey10 (7), Jacobsen3 (5) and Sweeney3 (9) revealed that none of the signatures performed well in classifying progressors from non-progressors with all average AUC values <0.65 (Table 1).
Table 1:
Predictive performance of the listed signatures in progressors (co-prevalent cases removed) versus non-progressors in the Brazil Cohort. Receiver operating characteristic (ROC) area-under-curve (AUC), sensitivity, and specificity reported as mean (95% confidence interval) for 50 iterations.
| Signature | AUC | Sensitivity | Specificity |
|---|---|---|---|
| ACS-COR | 0.670 (0.640, 0.700) | 0.515 (0.460, 0.571) | 0.774 (0.728, 0.820) |
| RISK4 | 0.461 (0.434, 0.488) | 0.413 (0.335, 0.491) | 0.665 (0.588, 0.743) |
| Sweeney3 | 0.590 (0.560, 0.620) | 0.427 (0.353, 0.501) | 0.746 (0.676, 0.816) |
| Jacobsen3 | 0.575 (0.546, 0.605) | 0.553 (0.489, 0.616) | 0.633 (0.570, 0.695) |
| Sambarey10 | 0.623 (0.595, 0.651) | 0.632 (0.571, 0.693) | 0.586 (0.525, 0.647) |
| Kaforou27 | 0.629 (0.602, 0.656) | 0.664 (0.603, 0.725) | 0.567 (0.513, 0.620) |
In the Brazil cohort, 573 HHCs were TST-positive, of these 28 were progressors, 6 were diagnosed with TB at the same time as the TB index case, and 539 were non-progressors. In the Brazilian cohort, we assume a rate of TB progression of 4.8%. Based on the sensitivity and specificity of ACS-COR, RISK4, Kaforou27, Sambarey10, Jacobsen3, Sweeney3, in the Brazil cohort PPVs were calculated for these signatures. Importantly the PPVs for none of the 6 signatures reached the WHO published target product profiles (TPPs) (Table 2).
Table 2.
Hypothetical performance of different gene signatures to predict progression to TB disease in the Brazil dataset
| Gene signature | PPV (95% CI) | NPV (95% CI) |
|---|---|---|
| ACS-COR | 0.106 (0.057–0.165) | 0.968 (0.946–0.982) |
| RISK4 | 0.061 (0.035–0.114) | 0.956 (0.933–0.976) |
| Sweeney | 0.080 (0.042,0.136) | 0.961 (0.939,0.976) |
| Jacobsen | 0.072 (0.399,0.113) | 0.964 (0.938,0.980) |
| Sambarey | 0.073 (0.045,0.116) | 0.968 (0.944,0.985) |
| Kaforou | 0.073 (0.046,0.115) | 0.970 (0.942,0.985) |
Thus, these results suggest that neither the existing progression risk signatures nor TB disease signatures offer discriminatory ability between progressors and non-progressors in the Brazil cohort.
Derivation of a 29-gene signature for distinguishing progressors from non-progressors
The ACS-COR progression risk signature was derived using the samples in the GSE79362 cohort most proximal to their TB diagnosis in the initial down-selection of genes, and then the final selection used all available samples, including those at baseline (up to 18 months before progression (14, 17). The signature shows strong predictive performance closer to time of the diagnosis of TB and also distinguishes TB patients from LTBI individuals with high accuracy (14, 18). This suggests that the ACS-COR signature is a biomarker of subclinical TB rather than of long-term risk progression. We, therefore, hypothesized that if we selectively used ‘baseline’ progressor samples from the GSE79362 dataset at the time point furthest from their eventual TB diagnosis, we could derive a risk signature of progression that defines the early host response to infection. This signature would be independent of expression of inflammatory responses that may occur proximal to the clinical expression of TB disease in response to replicating Mtb. We used the existing RNA-seq dataset from the South African adolescent GSE79362 cohort to train a baseline biomarker, using an ensemble feature selection pipeline, and then validated this biomarker on the RNA-seq data from the Brazil cohort. The training RNA-seq dataset contained 46 progressors and 107 matched controls; the samples were collected every six months, ranging from baseline to up to two years per subject (14). For this analysis, available sequencing data for 39 progressor samples from time-points furthest from their TB diagnosis dates as well as 103 non-progressors were used for predictive model training based on gene signatures (Figure 1). Then the Brazilian dataset, which was smaller in size, provided a new independent validation set of progressors and non-progressors for assessing predictive performance of gene signatures. The GC6 cohort (GSE94438) provided a second validation set. This approach of independent training and validation in an ethnically distinct cohort should yield highly robust biomarkers of disease and has not been previously applied to genomic biomarkers of progression.
Figure 1: Analysis strategy used to identify a new gene signature and train predictive models using Africa dataset and quantitatively test predictive performance in Brazil datasets.
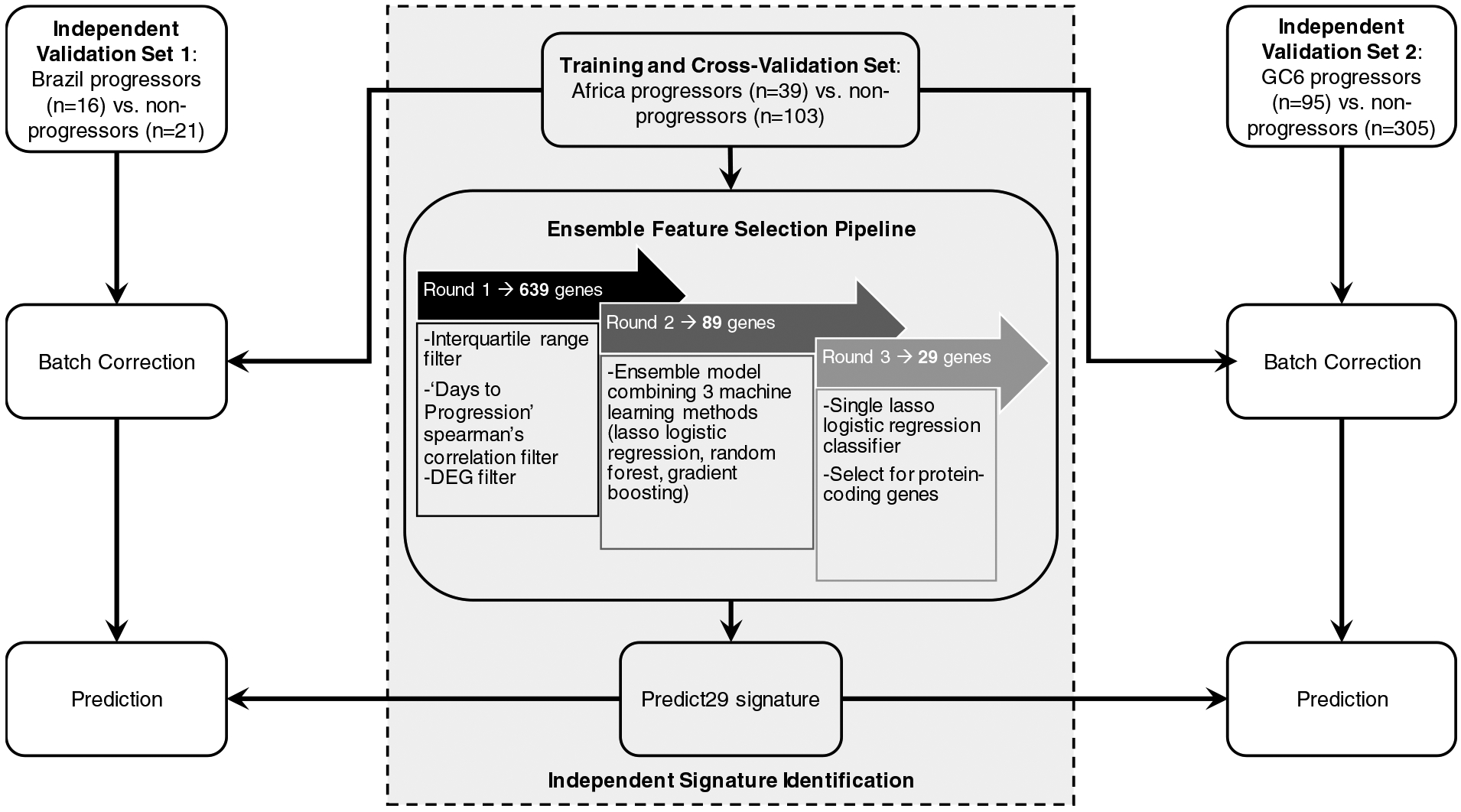
The Africa dataset (ACS-COR; GSE79362) derived from whole blood samples was used to identify a novel 29-gene signature via an ensemble feature selection pipeline: Round 1 led to identification of 639 genes of interest based on expression trends that correlated with progression. Round 2 led to selection of 89 genes based on evaluation using an ensemble model to determine which genes performed most robustly across different models. Round 3 led to final selection of 29-protein coding genes after removing redundant features. Predictive model training was performed using batch-corrected ACS-COR (GSE79362) Africa dataset (Training and Cross-Validation Set), and predictive testing was performed using batch-corrected Brazil progressors vs. non-progressor dataset derived from PBMC samples (Validation Set 1) and GC6-GSE94438 derived from whole blood samples (Validation Set 2).
Briefly, initial identification of potential genes (features) of interest involved applying filters for (1) interquartile range, (2) days to progression correlation, and (3) differential gene expression, after which we identified 639 putative biomarker genes. The next step for feature selection used an ensemble model combining random forest, lasso logistic regression, and gradient boosting that resulted in selection of 89 genes. Then, 40 of these 89 genes were selected based on a single lasso logistic regression classifier (Figure 1). Finally, 29 out of these 40 genes were selected based on their mapping to protein-coding genes (Figure 1) and designated as PREDICT29. Predictive performance was also evaluated for models of the existing ACS-COR and PREDICT29 progression signatures in the African training dataset via 10*10 cross validation for unbiased evaluation, and both signatures performed roughly equivalently (Tables S6 and S7).
Several of the genes comprising PREDICT29 signature are associated with innate response. For instance, SH2D1B (SH2 Domain containing 1B) is involved in regulating signal transduction via surface receptors expressed on antigen-presenting cells (APC) (19). CTSA (Cathepsin A) is expressed on human APCs and regulates MHC class II antigen presentation (20). SPSB1 (SplA/Ryanodine Receptor Domain And SOCS Box Containing 1) targets inducible nitric oxide synthase (iNOS) (21, 22). IL31RA (Interleukin 31 Receptor A) is involved in proliferation and function of myeloid cells (23, 24). Lastly, HM13 (Histocompatibility Minor 13) is required to generate lymphocyte HLA-E epitopes from non-classical MHC Class I peptides (25), and HLA-E binding peptides of Mtb induce both cytotoxic and immunoregulatory responses (26, 27). The top four pathways identified from KEGG analysis were Lysosome, Renin-angiotensin system, Glycosphingolipid biosynthesis - globo series, and Vitamin digestion and absorption (Table S8). Ingenuity Pathway analysis predicted Glycolysis 1, Gluconeogenesis 1, Glutathione-mediated detoxification, and tRNA charging as the top pathways (Table S9). None of the top predicted pathways were inflammation related.
Validation of the PREDICT29 signature in predicting progressors from non-progressors in the Brazil and GC6 cohorts
As shown in the heat map (Figure 2A), PREDICT29, discriminated progressors from non-progressors. Consistent with the heat map, the PCA plot also corroborated that progressors and non-progressors segregated into two main clusters (Figure 2B). Genes within PREDICT29 did not overlap with the TB or progression risk signatures tested, except for four genes (SRBD1, WARS, APOL6, and TCN2) that also were part of the signature shown to differentiate TB from LTBI in the original work by Berry and colleagues (4).
Figure 2: Expression of the PREDICT29 signature in Brazilian progressors and non-progressors.
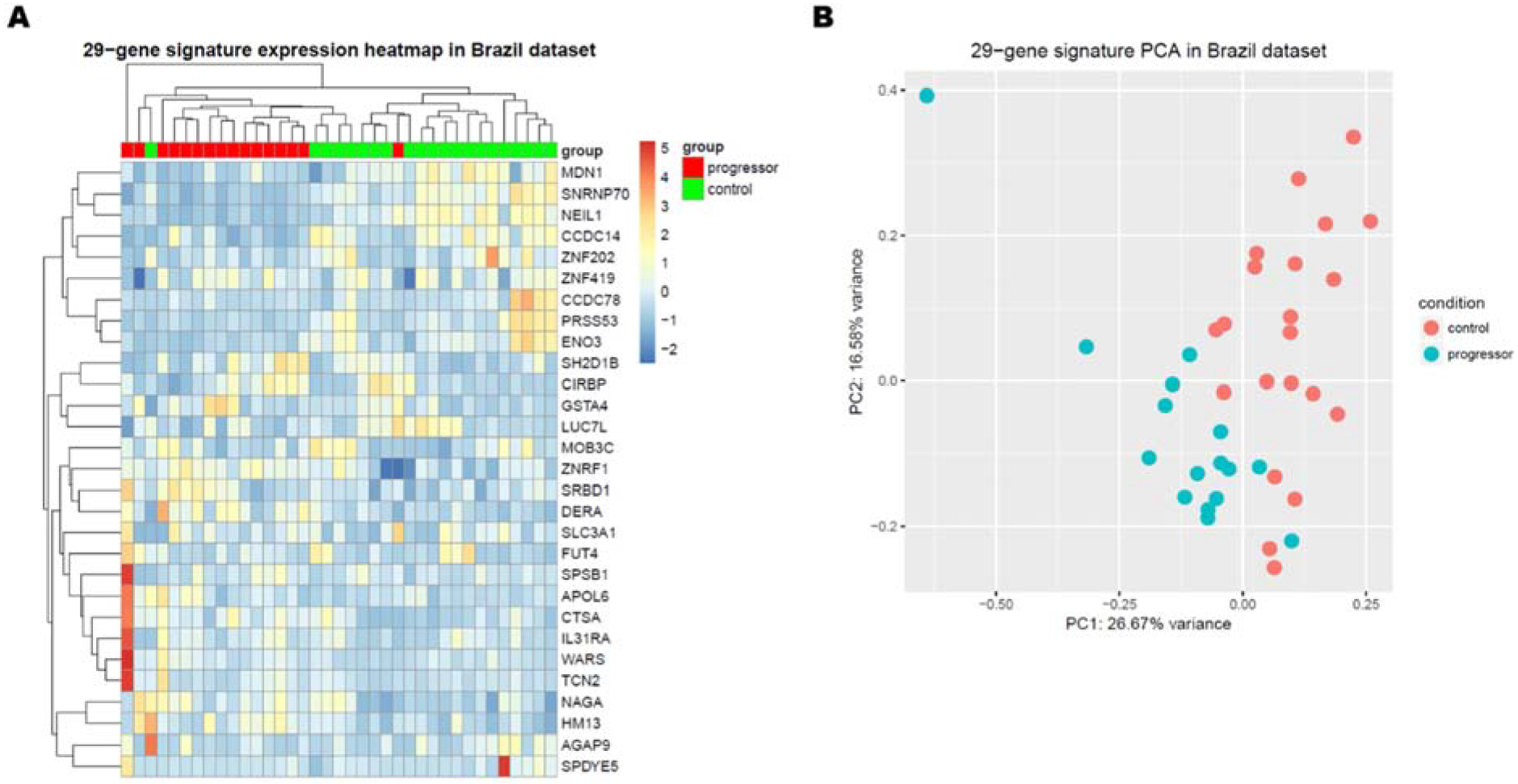
(A) Gene expression heatmap of the PREDICT29 signature. (B) Principal Component Analysis plot of the PREDICT29 signature. Progressors with co-prevalent cases (n=16), non-progressors (n=21).
We next tested the performance of PREDICT29 using various predictive model methods. PREDICT29 performed well across all four models used, suggesting its reproducibility and versatility across modeling methods. When averaging performance across the four models tested, the PREDICT29 signature resulted in a mean AUC 0.911 (0.894, 0.928), sensitivity of 0.742 (0.704, 0.780) and specificity of 0.848 (0.816, 0.880) (Table 3A; Figure 3B). There was no significant change in performance when co-prevalent cases were included in the analysis (Figure 3A). In terms of segregating active TB from LTBI, PREDICT29 performed with a mean AUC of 0.757 (0.732, 0.782), sensitivity of 0.643 (0.597, 0.688) and specificity of 0.773 (0.733, 0.813) (Table S10; Figure 3B), Furthermore, there was no correlation between PREDICT29 and age (Table S11).
Table 3-.
Predictive performance of PREDICT29 in Brazil (A) and GC6 (B) Cohorts.
| (A) Brazil | |||
|---|---|---|---|
| Model used | AUC | Sensitivity | Specificity |
| glmnet | 0.929 (0.914, 0.944) | 0.741 (0.703, 0.779) | 0.867 (0.836, 0.897) |
| SVM | 0.915 (0.900, 0.930) | 0.756 (0.716, 0.796) | 0.836 (0.807, 0.866) |
| ranger | 0.867 (0.843, 0.891) | 0.679 (0.641, 0.717) | 0.830 (0.795, 0.866) |
| XGBoost | 0.932 (0.919, 0.945) | 0.794 (0.757, 0.830) | 0.860 (0.828, 0.893) |
| AVERAGE | 0.911 (0.894, 0.928) | 0.742 (0.704, 0.780) | 0.848 (0.816, 0.880) |
| (B) GC6 | |||
| Model used | AUC | Sensitivity | Specificity |
| glmnet | 0.664 (0.655, 0.674) | 0.497 (0.459, 0.535) | 0.787 (0.750, 0.824) |
| SVM | 0.685 (0.676, 0.695) | 0.535 (0.516, 0.554) | 0.780 (0.766, 0.795) |
| ranger | 0.683 (0.673, 0.693) | 0.649 (0.616, 0.683) | 0.651 (0.623, 0.678) |
| XGBoost | 0.688 (0.677, 0.699) | 0.549 (0.531, 0.567) | 0.803 (0.787, 0.819) |
| AVERAGE | 0.680 (0.670, 0.690) | 0.558 .531, 0.585) | 0.755 (0.732, 0.779) |
Figure 3: Receiver operating characteristic (ROC) curves for predicting clinical classification in Brazilian cohort using PREDICT29.
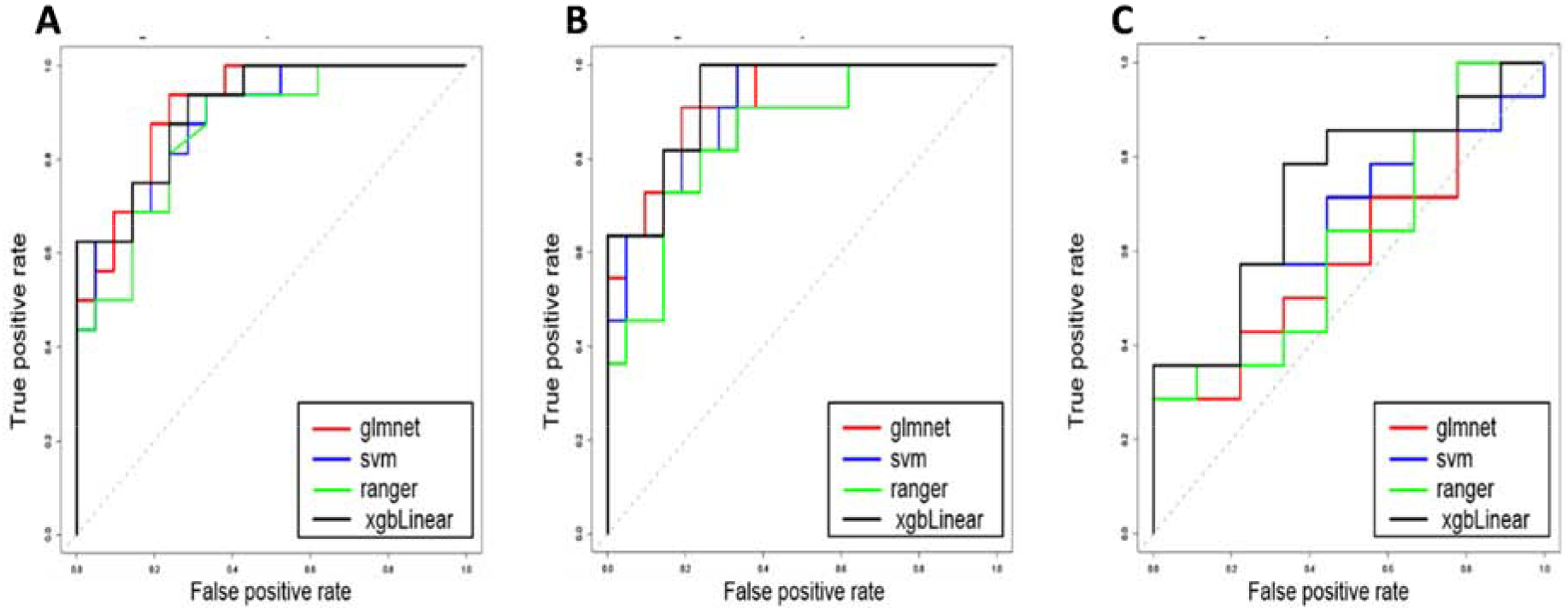
(A) PREDICT29 predictive performance of progressors (n=16) and non-progressors (n=21). (B) PREDICT29 predictive performance of progressors (n=11) and non-progressors (n=21). (C) PREDICT29 predictive performance of active TB patients (n=14) versus non-progressors (n=21). Different colored lines represent the four modeling methods used: glmnet (ridge logistic regression), svm (support vector machine), ranger (random forest), and xgbLinear (gradient boosting).
GC6 is also an HIV-negative African cohort of exposed HHC, but unlike the Brazil cohort, disease progression was followed for only 2 years (Table S12). The performance of PREDICT29 was also validated in this cohort. When averaging performance across the four models tested, the PREDICT29 signature resulted in a mean AUC of 0.680 (0.670, 0.690) with a sensitivity of 0.558 (0.531, 0.585) and specificity of 0.755 (0.732, 0.779) (Table 3B). These data indicate that PREDICT29 performed less better in the GC6 short-term risk cohort compared with the Brazil cohort.
Several of the HHC progressed > 2 years after exposure which might reflect transmission that occurred outside of the household. We, therefore, performed RFLP analysis to determine Mtb strain match between the strain derived from the index case and that obtained from the secondary case (progressors). RFLP data was available on seven of the progressors, and it showed that all of them were infected by the same strain as the index case (Table S2). These data suggest that transmission is occurring in the household and transcriptional changes occurring at the time of Mtb exposure may contribute to the PREDICT29 risk signature.
Next, we determined if PREDICT29, in comparison to the currently available transcriptional signatures, could more reliably predict disease progression. Based on the sensitivity and specificity in the Brazil cohort the calculated PPV for PREDICT29 was 20.2% (95% CI 13.1–29.4%) and NPV was 98.5% (95% CI 96.9–99.3%) (Table 4). However, in the GC6 cohort, the calculated PPV for PREDICT29 was 4.1% (95% CI 2.3–7.4) and NPV was 98.7% (95% CI 97.6–99.4). (Table 4). PPV of PREDICT29 reached the WHO published target product profiles (TPPs).
Table 4-.
Hypothetical performance of PREDICT29 in Brazil and GC6 cohorts
| Cohort | PPV (95% CI) | NPV (95% CI) |
|---|---|---|
| Brazil | 0.202 (0.131–0.294) | 0.984 (0969–0993) |
| GC6 | 0.041 (0.023–0.074) | 0.987 (0.976–0.994) |
DISCUSSION
In this study, we developed a predictive blood-based signature that accurately determined an individual’s risk of progression from Mtb infection to disease. Evaluation of predictive performance of the existing ACS-COR and RISK4 signatures for TB progression risk and four TB signatures demonstrated their moderate ability to distinguish progressors from non-progressors in the Brazil cohort. However, PREDICT29 signature offered superior performance in predicting progressors in the Brazil dataset. Although the same RNA-seq prospective African cohort dataset was used to derive both the ACS-COR signature and the PREDICT29 signature, markedly different signatures resulted, largely attributable to the differing methodologies of signature derivation. ACS-COR signature improved closer to TB diagnosis, during which TB disease-related inflammatory processes are occurring although clinical manifestations of the disease may not yet be present (17, 28, 29). Consistent with this, the ACS-COR signature performed well in distinguishing active TB disease from LTBI in multiple datasets (14, 18). Unlike ACS-COR (17), the PREDICT29 signature’s lack of inflammatory gene or pathway enrichment suggests specific detection of progression risk at early time-points prior to eventual TB diagnosis many years later. PREDICT29 signature appears to capture processes occurring in the early host response to Mtb that could dictate successful long-term pathogen control or permissiveness resulting in progressive disease. The advantage of our analytical approach is that it allowed us to develop PREDICT29 that measures risk of progression to TB disease years ahead of the onset of infectiousness. Thus, ACS-COR and PREDICT29 are likely biomarkers for different phenomena with differences in performance in different cohorts based on time after Mtb exposure and time to disease progression. The limited overlap with ACS-COR and PREDICT29, therefore, is not unexpected.
In a study that directly compared publicly available gene expression datasets (30) found that the previously reported 3-gene signature (31) performed with high accuracy for diagnosis of tuberculosis and in predicting progression of LTBI to TB disease prior to sputum conversion. It is important to note that in this study the LTBI progressed to disease within 6 months of baseline evaluation and thus the 3-gene signature is likely detecting subclinical disease in asymptomatic individuals rather than truly predicting risk of progression in recently infected. Consistent with our assessment that PREDICT29 captures early changes in the host following infection, the 3-gene signature did not perform well in the Brazil dataset in predicting risk of TB progression in the recently exposed HHC.
Two studies have shown that the diagnostic performance of the ACS-11 gene signature (derived from 16 gene ACS-COR) was maintained in cryopreserved PBMC samples (1, 2). This suggests that lack of neutrophils in the PBMC is not affecting the performance of TB signatures. Furthermore, transcriptional module enrichment analysis of the whole blood transcriptional data from progressors and non-progressors in the African cohort of adolescents (GSE79362) implicated monocytes as contributing to the gene signature (3). Supporting a role for monocytes, the authors of this study also found that progressors have increased numbers of monocytes in peripheral blood and the isolated monocytes had significantly elevated expression of the risk signature genes (ACS-COR) (3). These results strongly indicate that the performance of ACS-COR and other signatures on PBMC samples should not be affected by the lack of neutrophils in these samples. Nevertheless, longitudinal studies comparing PBMC and whole blood should be conducted to fully address the performance of available prognostic and diagnostic signatures on different sample types and the contribution of specific cell types to a given signature.
It was unexpected to find that PREDICT29 did not perform well on the GC6 which consists of South African, Gambian and Ethiopian HHC cohorts who were followed for 2 years for disease progression. Site-specific biomarker of risk of progression derived from the RNA-seq data separately for the South African and Gambian cohorts showed that the Gambia signature did not validate in the South Africa cohort and similarly the South African signature had poor performance when tested on the Gambian cohort. These studies reveal that site-specific differences could have affected PREDICT29’s performance when tested on the GC6 cohort. It is also possible that the extent of infectiousness of the index case and amount of exposure of the HHCs to the index case in the GC6 and Brazil cohorts was different, resulting in GC6 HHCs being at a more advanced stage in the spectrum of LTBI progression from infection to subclinical TB to preclinical and clinical TB than the Brazil Cohort. Thus, differences in gene expression in the baseline samples in the two cohorts may explain the poor performance of PREDICT29 in GC6.
In the Brazil cohort, PREDICT29 showed a PPV of 20% while retaining a very high NPV. A meta-analysis conducted to assess the PPV and NPV of IGRAs and TST for predicting progression to active TB was 2.7% and 1.5%, respectively. In high-risk groups, PPV increased to 6.8% and PPD to 2.4% (32). The performance characteristics of PREDICT29, therefore, exceeds the WHO recommended optimum target product profile that is a PPV of 16% (12). In determining the target PPV and NPV, the WHO expert committee takes into account the clinical and public health benefits (including cost-effectiveness) of introduction of a new diagnostic. Once the target is achieved or exceeded the WHO would consider recommending implementation.
PREDICT29 has the potential to provide a novel long sought clinical screening tool for risk stratification of recently infected individuals. Since subclinical TB can be a significant source of transmission in the community (33, 34), a test that can identify recently infected at risk of progression is significant. Since the sample size of progressors in the Brazil cohort was limited, we argue that this is a proof-of-concept study demonstrating that PREDICT29 outperforms other biomarkers. Clearly further investigation and validation of PREDICT29 is warranted in larger and geographically diverse cohorts and countries with diverse TB incidence to ascertain its prognostic accuracy and generalizability as a biomarker signature of TB disease risk in recently infected individuals. These studies should also include head-to-head comparisons of PREDICT29 with the published signatures to determine if the different signatures predict different stages in the spectrum from infection to clinical disease. Combining existing transcriptomic and metabolomic signatures was reported to significantly enhance prediction of risk of progression TB (11). Future studies should also consider this approach to develop a highly sensitive and globally applicable TB risk biomarker.
METHODS
Household contact study design and subject inclusion criteria
Subject groups:
A household contact (HHC) study of index TB cases and their household contacts was conducted as previously described (35, 36). Briefly, index cases were eligible for enrollment if they were consenting adult (≥ 18 years old) pulmonary TB cases living in a household with 3 or more contacts and had a 2+ or greater sputum acid fast bacilli (AFB) smear, positive culture for Mtb, and a history of cough ≥ 3 months. Enrolled household contacts (HHCs) included consenting individuals of all ages that had close contact with the index case for at least 3 months. Close contact was defined as meeting at least one of the following criteria: sleeping under the same roof ≥ 5 days/week, sharing meals ≥ 5 days/week, watching TV nights or weekends, or other significant contact, such as visiting the household > 18 days/month.
At the time of enrollment, all HHCs underwent tuberculin skin testing and were screened for active TB based on symptoms. HHCs enrolled into the study were HIV-uninfected. Baseline blood samples were collected and peripheral blood mononuclear cells (PBMC) isolated and stored in liquid nitrogen. Additional subject data, including age, gender, socioeconomic information, and health history, as well as household environmental evaluation were also collected Tables S1 and S2. Follow up of the HHCs was by TB program and secondary TB cases was diagnosed through the National registry. However, the NDI program assured capture of all cases that were culture positive. Sputum from TB suspects in program was cultured at the NDI. The screening process included a household visit for symptom screening and TST/IGRA placement. HHCs with symptoms suggestive of TB (cough or systemic symptoms) and these with positive TST/IGRA were referred to the National TB Program for evaluation. According to the Brazilian TB Guidelines, TB suspects undergo 2–3 sputum sampling for AFB smear and culture in solid media (Ogawa-Kudow) along with a chest radiograph. Microbiologically proven TB is defined as cases with suggestive symptoms and positive sputum AFB smear or culture. Clinical TB cases are defined as these with suggestive symptoms (cough for more than 2 weeks with systemic symptoms) and a chest radiograph with either an upper lobe infiltrate, presence of cavities or miliary pattern and response to TB treatment. TB is a reportable disease in Vitoria, all smear-culture positive TB cases are processed in the Núcleo de Doenças Infecciosas (NDI) microbiology laboratory, similarly all clinical TB cases are reported to the National notifiable disease database system.
Individuals that were diagnosed with TB disease during follow-up were classified as progressors. Individuals that did not develop TB during long-term follow-up (≥4 years) were considered non-progressors. Genotypic analysis by IS6110 RFLP was performed according to a standardized method (37) on the index and secondary case (progressors) Mtb isolates.
Sample collection and PBMC preparation
Eight weeks post-enrollment, baseline peripheral blood samples were collected from eligible subjects using BD Vacutainer tubes (BD 367874), and peripheral blood mononuclear cells (PBMC) were isolated by Ficoll gradient separation method using Histopaque-1077 (Sigma-Aldrich 10771). PBMC were cryopreserved using 90% heat-inactivated fetal bovine serum (GIBCO #12657–029, South America origin) and 10% DMSO (Sigma-Aldrich #D2650) for storage in liquid nitrogen and cryoshipment from Brazil to the United States.
Cryopreserved PBMC were flash thawed in a 37°C water bath and added drop-wise to 10 mL of pre-warmed cell culture medium, consisting of RPMI 1640 (Corning #15040CM), 10% defined fetal bovine serum (GE Healthcare Life Sciences #SH30070.03, U.S. Origin), 1% penicillin-streptomycin (Corning #30002CI), 1% L-glutamine (Corning #25005CI), and 1% HEPES buffer (Corning #25–060-CI). Cells were pelleted and rinsed in dPBS (Corning #21–031-CV) prior to immediate re-suspension in 1 mL of Ambion TRIzol Reagent (ThermoFisher Scientific #15596018) for total RNA extraction.
RNA extraction and sequencing
Total RNA was extracted from PBMC using TRIzol Reagent (ThermoFisher Scientific #15596018) as per standard protocols recommended by the manufacturer. Total RNA quality and quantity was assessed using the Agilent 2100 Bioanalyzer (Agilent, CA, USA) prior to downstream processing. Total RNA was enriched for mRNA via poly(A) tail enrichment via a single round of amplification using MessageAmp II aRNA Amplification Kit (ThermoFisher Scientific #AM1751). Amplified mRNA was re-assessed for quality on the Agilent 2100 Bioanalyzer prior to preparation of cDNA libraries. Strand-specific cDNA libraries for sequencing on the Illumina platform were prepared using a modified version of the low-input Illumina TruSeq RNA Sample Preparation protocol (Illumina Inc., CA, USA). Briefly, amplified mRNA was fragmented, purified, and ligated to adaptors at 3’ and 5’ ends prior to reverse transcription and 15 cycles of PCR amplification. Resultant cDNA libraries were purified using AMPure XP beads and subsequently quantified. Sequencing was performed using lllumina HiSeq 2500 at an approximate depth of 40 million 50 basepair (bp) single-end reads per sample.
RNA sequencing data processing and analysis
Quality control and data processing
Raw RNA sequencing data derived from our Brazilian cohort samples (GSE112104) as well as those obtained from the Zak et al. (2016) Africa dataset from GEO (GSE79362) (38), and the GC6 African dataset from GEO (GSE94438) (16) were processed using the same pipeline. Raw sequencing files were assessed for quality control using FastQC (39) and MultiQC (40). On average, the Brazil samples had a mean Phred score of ⩾ 30 for each base pair (bp) position, the GSE79362 samples had a mean Phred score of ⩾ 25 for each bp position, and the GSE94438 samples had a mean Phred score of ⩾ 36.2 for each bp position suggesting high quality data with approximately 99.9% base call accuracy. Resultant short sequence reads were aligned to human genome hg19 using Rsubread (36). The average alignment rate was 71.9% for the Brazil dataset and 88.3% for the GSE79362 Africa dataset, and 64.5% for the GSE94438 Africa dataset. Furthermore, the FASTQC software also estimates the average duplication rates in the RNA-seq data to be 30.1% for the Brazil dataset and 49.5% for the GSE79362 Africa dataset, and 59.1% for the GSE94438 Africa dataset. We note that the average alignment percentages for the GC6 dataset GSE94438 was lower and the duplication percentages were higher than those of the Brazil and GSE79362, datasets. In particular, 394 of the 405 GC6 samples failed the FastQC duplication percentage threshold.
In addition, we note that the Brazil samples consisted of two batches of RNA-seq samples. The first batch (reported above) consisted of 16 progressors and 21 non-progressors produced high-quality RNA-sequencing data with a clear distinction between the progressors and non-progressors. The second batch consisted of 10 progressors and 28 non-progressors. However, these samples were excluded from this analysis because their data quality were low. For example, the average alignment percentage was merely 49.2% and the duplication rate was 55.9%, across the entire batch, suggesting that these samples in this batch were low-quality and should be removed from the analysis.
Differential expression analysis
All of our analysis code is available at the following GitHub repository (https://github.com/compbiomed/TBP). DESeq2 (41) was used for differential expression analysis between progressors and non-progressors using raw read counts outputted by Rsubread. All default parameters were used with the model design incorporating both subjects’ gender and TB condition as variables as follows: “design = ~ condition + gender”. Differential expression was contrasted based on TB condition as progressors over non-progressors as follows: “contrast = c(“condition”, “progressors”, “non-progressors”)”.
Signature identification by ensemble feature selection
All of our biomarker development code is available through the GitHub repository named above. Raw read count data for the African dataset were pre-processed by filtering out genes with low read counts (maximum read count < 5) and log2 normalization using the ‘rlog’ function in DESeq2 (41). In order to remove additional unwanted noise from the dataset, combat (42) was used. To initially identify potential genes (features) of interest (Round 1): genes were ranked by interquartile range, and the top 80% of genes were selected. From this list, genes having ≥0.1 Spearman’s correlation between expression level and ‘days to progression’ in the African dataset were selected. Finally, genes with an adjusted p-value <0.1 in differential expression analysis between progressors and non-progressors were selected. Round 1 led to identification of 639 initial genes.
For feature selection (Round 2): using an ensemble feature selection procedure, feature weights of the previously selected genes for three machine learning modeling methods (lasso logistic regression (glmnet-lasso (43)), gradient boosting (XGBoost (44)), and random forest (ranger (45)) were determined by applying a 5-fold cross-validation training process and normalizing weights between 0 and 1, and averaging the weights obtained from the three models. This procedure was repeated 100 times, and for each time after obtaining the gene weights, one run of ridge logistic regression was performed using leave-one-out cross-validation in order to obtain a best-normalized feature weights cut-off, and then all genes with weights higher than the cut-off were selected. This process led to identification of 100 gene signatures. Genes were ranked by appearing times in the 100 signatures, and the top 89 genes (average signature length) were selected based on the number of times they appeared across these 100 signatures. Thus, Round 2 led to selection of 89 genes.
For feature dimension reduction (Round 3): to further reduce the number of genes comprising a signature, glmnet-lasso was used to perform 100 iterations of 5-fold cross-validation lasso logistic regression in order to obtain 100 gene signatures (on average, 40 genes). Based on the number of times each gene appeared in the 100 signatures, genes were ranked, and the top 40 genes were selected. Round 3 led to selection of 40 genes. Final filtering was performed by selecting only protein-coding genes based on protein coding information available in BioMart (46). This final filtering step led to a final signature of 29 protein-coding genes (Tables S3–S5 and Figure 1).
Signature-based modeling and evaluation of predictive performance
Raw read counts from Rsubread for the both the Africa dataset and Brazil dataset were pre-processed by filtering out genes with low read counts (maximum read count < 5) and log2 normalization using the ‘rlog’ function in DESeq2 on the combined dataset containing both Africa and Brazil data. ComBat and BatchQC (47, 48) were used to correct for batch differences between the Africa and Brazil datasets. This batch correction procedure was performed separately for the Africa + Brazil progressors vs. non-progressors data as well as the Africa + Brazil TB vs. non-progressors data. Batch-corrected data was used for quantitative evaluation of predictive performance for the gene signatures.
Using the caret package (49), classification models were derived using four different widely used modeling methods: “glmnet” (ridge logistic regression), “svmLinear” (support vector machine with linear kernel), “ranger” (random forest), and “xgblinear” (gradient boosting). Caret was used to train four classification models based on the batch-corrected Africa dataset gene expression of either the PREDICT29 or ACS-COR. During this training process, parameters were tuned by a 10-repeats 10-fold cross-validation. The final classification models were used to predict in the batch-corrected Brazil and GC6 dataset.
For a single iteration of predictive evaluation (done separately for progressors vs. non-progressors in the Brazil dataset and GC6 datasets: bootstrapping was performed on the Brazil and GC6 dataset to obtain a new dataset containing the same total number of samples as the starting Brazil and GC6 datasets. The classification models trained on the Africa dataset were used to predict progressors and non-progressors in the bootstrapped Brazil and GC6 datasets. Performance was evaluated by generating receiver operating characteristic (ROC) curves and computing area-under-curve (AUC) values using the ROCR package in R (50). Additionally, optimal sensitivity and specificity values were obtained by using the probability threshold that yielded a maximized sum of sensitivity and specificity. This bootstrapping and evaluation process was repeated for 50 iterations for each Brazil and GC6 datasets. Thus, mean AUC, sensitivity, and specificity values with corresponding 95% confidence intervals were calculated based on the 50 iterations of results. As mentioned above, all our code is available at (https://github.com/compbiomed/TBP).
A distinctive feature of our methodology was that we adopted a classic approach for ensemble feature selection that has been used successfully in cancer biomarker studies (51, 52). We also used multiple biomarker/machine learning approaches in order to demonstrate the robustness of our signature gene set, regardless of the methods used to train the classification model. On the other hand, Zak et al. (38) employed a single, paired-SVM method that involved measuring gene expression abundance at the level of splice junction counts by quantifying frequency of mRNA splicing events (14), which only uses 16.9% of the available RNA-seq data. Thus, different methodology for RNA-Seq data analysis enabled the derivation of PREDICT29 that had significantly improved performance over ACS-COR in predicting risk of progression/reactivation.
Data visualization
The heatmap was generated using the pheatmap package in R (53). ROC curves were plotted using the plotting functions in R.
Data and Materials Availability
We have created a secure token to allow review of record of the RNA-seq data - GSE112104 if required by the reviewers. Below is the link.
To review GEO accession GSE112104:
Enter token uxstcqkcvhkpnwh into the box.
Study Approval
The study was approved by the Comite de Ética em Pesquisa do Hospital Universitário Cassiano Antonio de Morais, and the Institutional Review Boards of Rutgers Biomedical Health Sciences (formerly UMDNJ) and Boston University School of Medicine. Written informed consent and assent in Portuguese were obtained from all study participants as per the consent procedure approved by IRBs from all participating institutions.
Supplementary Material
ACKNOWLEDGEMENTS
The work was funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health grants U19AI111276 and U01AI065663, and NIAID training grant T32AI125185 to SL. The study sponsors had no involvement in the study design, in the collection, analysis and interpretation of data; in the writing of the manuscript; or in the decision to submit the manuscript for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
References
- 1.World Health Organization. Guidelines on the management of latent tuberculosis infection.2015. [PubMed]
- 2.Lobue P, Menzies D. Treatment of latent tuberculosis infection: An update. Respirology. 2010;15(4):603–22. doi: 10.1111/j.1440-1843.2010.01751.x. [DOI] [PubMed] [Google Scholar]
- 3.Denholm JT, McBryde ES. The use of anti-tuberculosis therapy for latent TB infection. Infection and Drug Resistance. 2010;3:63–72. Epub 2010 Jul 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466(7309):973–7. Epub 2010/08/21. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobsen M, Repsilber D, Gutschmidt A, Neher A, Feldmann K, Mollenkopf HJ, Ziegler A, Kaufmann SH. Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. J Mol Med. 2007;85(6):613–21. Epub 2007/02/24. doi: 10.1007/s00109-007-0157-6. [DOI] [PubMed] [Google Scholar]
- 6.Kaforou M, Wright VJ, Oni T, French N, Anderson ST, Bangani N, Banwell CM, Brent AJ, Crampin AC, Dockrell HM, Eley B, Heyderman RS, Hibberd ML, Kern F, Langford PR, Ling L, Mendelson M, Ottenhoff TH, Zgambo F, Wilkinson RJ, Coin LJ, Levin M. Detection of tuberculosis in HIV-infected and -uninfected African adults using whole blood RNA expression signatures: a case-control study. PLoS Med. 2013;10(10):e1001538. doi: 10.1371/journal.pmed.1001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sambarey A, Devaprasad A, Mohan A, Ahmed A, Nayak S, Swaminathan S, D’Souza G, Jesuraj A, Dhar C, Babu S, Vyakarnam A, Chandra N. Unbiased Identification of Blood-based Biomarkers for Pulmonary Tuberculosis by Modeling and Mining Molecular Interaction Networks. EBioMedicine. 2017;15:112–26. doi: 10.1016/j.ebiom.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maertzdorf J, McEwen G, Weiner J 3rd, Tian S, Lader E, Schriek U, Mayanja-Kizza H, Ota M, Kenneth J, Kaufmann SH. Concise gene signature for point-of-care classification of tuberculosis. EMBO Mol Med. 2016;8(2):86–95. doi: 10.15252/emmm.201505790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sweeney TE, Braviak L, Tato CM, Khatri P. Genome-wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis. Lancet Respir Med. 2016;4(3):213–24. doi: 10.1016/S2213-2600(16)00048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutherland JS, Loxton AG, Haks MC, Kassa D, Ambrose L, Lee JS, Ran L, van Baarle D, Maertzdorf J, Howe R, Mayanja-Kizza H, Boom WH, Thiel BA, Crampin AC, Hanekom W, Ota MO, Dockrell H, Walzl G, Kaufmann SH, Ottenhoff TH, consortium GBfT. Differential gene expression of activating Fcgamma receptor classifies active tuberculosis regardless of human immunodeficiency virus status or ethnicity. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2014;20(4):O230–8. doi: 10.1111/1469-0691.12383. [DOI] [PubMed] [Google Scholar]
- 11.Roe JK, Thomas N, Gil E, Best K, Tsaliki E, Morris-Jones S, Stafford S, Simpson N, Witt KD, Chain B, Miller RF, Martineau A, Noursadeghi M. Blood transcriptomic diagnosis of pulmonary and extrapulmonary tuberculosis. JCI Insight. 2016;1(16):e87238. doi: 10.1172/jci.insight.87238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petruccioli E, Scriba TJ, Petrone L, Hatherill M, Cirillo DM, Joosten SA, Ottenhoff TH, Denkinger CM, Goletti D. Correlates of tuberculosis risk: predictive biomarkers for progression to active tuberculosis. Eur Respir J. 2016;48(6):1751–63. doi: 10.1183/13993003.01012-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sloot R, Schim van der Loeff MF, van Zwet EW, Haks MC, Keizer ST, Scholing M, Ottenhoff TH, Borgdorff MW, Joosten SA. Biomarkers Can Identify Pulmonary Tuberculosis in HIV-infected Drug Users Months Prior to Clinical Diagnosis. EBioMedicine. 2015;2(2):172–9. doi: 10.1016/j.ebiom.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Hussey GD, Abrahams D, Kafaar F, Hawkridge T, Verver S, Hughes EJ, Ota M, Sutherland J, Howe R, Dockrell HM, Boom WH, Thiel B, Ottenhoff THM, Mayanja-Kizza H, Crampin AC, Downing K, Hatherill M, Valvo J, Shankar S, Parida SK, Kaufmann SHE, Walzl G, Aderem A, Hanekom WA. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. The Lancet. 2016. doi: 10.1016/s0140-6736(15)01316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levin M, Kaforou M. Predicting active tuberculosis progression by RNA analysis. The Lancet. 2016. doi: 10.1016/s0140-6736(16)00165-3. [DOI] [PubMed] [Google Scholar]
- 16.Suliman S, Thompson E, Sutherland J, Weiner Rd J, Ota MOC, Shankar S, Penn-Nicholson A, Thiel B, Erasmus M, Maertzdorf J, Duffy FJ, Hill PC, Hughes EJ, Stanley K, Downing K, Fisher ML, Valvo J, Parida SK, van der Spuy G, Tromp G, Adetifa IMO, Donkor S, Howe R, Mayanja-Kizza H, Boom WH, Dockrell H, Ottenhoff THM, Hatherill M, Aderem A, Hanekom WA, Scriba TJ, Kaufmann SH, Zak DE, Walzl G, and the GC, groups ACScs. Four-gene Pan-African Blood Signature Predicts Progression to Tuberculosis. Am J Respir Crit Care Med. 2018. doi: 10.1164/rccm.201711-2340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scriba TJ, Penn-Nicholson A, Shankar S, Hraha T, Thompson EG, Sterling D, Nemes E, Darboe F, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Johnson JL, Boom WH, Hatherill M, Valvo J, De Groote MA, Ochsner UA, Aderem A, Hanekom WA, Zak DE, other members of the ACScst. Sequential inflammatory processes define human progression from M. tuberculosis infection to tuberculosis disease. PLoS Pathog. 2017;13(11):e1006687. doi: 10.1371/journal.ppat.1006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leong S, Zhao Y, Joseph NM, Hochberg NS, Sarkar S, Pleskunas J, Hom D, Lakshminarayanan S, Horsburgh CR, Jr., Roy G, Ellner JJ, Johnson WE, Salgame P. Existing blood transcriptional classifiers accurately discriminate active tuberculosis from latent infection in individuals from south India. Tuberculosis (Edinb). 2018;109:41–51. doi: 10.1016/j.tube.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Morra M, Lu J, Poy F, Martin M, Sayos J, Calpe S, Gullo C, Howie D, Rietdijk S, Thompson A, Coyle AJ, Denny C, Yaffe MB, Engel P, Eck MJ, Terhorst C. Structural basis for the interaction of the free SH2 domain EAT-2 with SLAM receptors in hematopoietic cells. Embo j. 2001;20(21):5840–52. Epub 2001/11/02. doi: 10.1093/emboj/20.21.5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reich M, Spindler KD, Burret M, Kalbacher H, Boehm BO, Burster T. Cathepsin A is expressed in primary human antigen-presenting cells. Immunology letters. 2010;128(2):143–7. Epub 2009/12/04. doi: 10.1016/j.imlet.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 21.Kuang Z, Lewis RS, Curtis JM, Zhan Y, Saunders BM, Babon JJ, Kolesnik TB, Low A, Masters SL, Willson TA, Kedzierski L, Yao S, Handman E, Norton RS, Nicholson SE. The SPRY domain-containing SOCS box protein SPSB2 targets iNOS for proteasomal degradation. J Cell Biol. 2010;190(1):129–41. Epub 2010/07/07. doi: 10.1083/jcb.200912087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishiya T, Matsumoto K, Maekawa S, Kajita E, Horinouchi T, Fujimuro M, Ogasawara K, Uehara T, Miwa S. Regulation of inducible nitric-oxide synthase by the SPRY domain- and SOCS box-containing proteins. J Biol Chem. 2011;286(11):9009–19. Epub 2011/01/05. doi: 10.1074/jbc.M110.190678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghilardi N, Li J, Hongo JA, Yi S, Gurney A, de Sauvage FJ. A novel type I cytokine receptor is expressed on monocytes, signals proliferation, and activates STAT-3 and STAT-5. J Biol Chem. 2002;277(19):16831–6. Epub 2002/03/06. doi: 10.1074/jbc.M201140200. [DOI] [PubMed] [Google Scholar]
- 24.Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, Haugen HS, Maurer M, Harder B, Johnston J, Bort S, Mudri S, Kuijper JL, Bukowski T, Shea P, Dong DL, Dasovich M, Grant FJ, Lockwood L, Levin SD, LeCiel C, Waggie K, Day H, Topouzis S, Kramer J, Kuestner R, Chen Z, Foster D, Parrish-Novak J, Gross JA. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5(7):752–60. Epub 2004/06/09. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- 25.Lemberg MK, Bland FA, Weihofen A, Braud VM, Martoglio B. Intramembrane proteolysis of signal peptides: an essential step in the generation of HLA-E epitopes. J Immunol. 2001;167(11):6441–6. Epub 2001/11/21. [DOI] [PubMed] [Google Scholar]
- 26.Joosten SA, van Meijgaarden KE, van Weeren PC, Kazi F, Geluk A, Savage ND, Drijfhout JW, Flower DR, Hanekom WA, Klein MR, Ottenhoff TH. Mycobacterium tuberculosis peptides presented by HLA-E molecules are targets for human CD8 T-cells with cytotoxic as well as regulatory activity. PLoS Pathog. 2010;6(2):e1000782. doi: 10.1371/journal.ppat.1000782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harriff MJ, Wolfe LM, Swarbrick G, Null M, Cansler ME, Canfield ET, Vogt T, Toren KG, Li W, Jackson M, Lewinsohn DA, Dobos KM, Lewinsohn DM. HLA-E Presents Glycopeptides from the Mycobacterium tuberculosis Protein MPT32 to Human CD8(+) T cells. Sci Rep. 2017;7(1):4622 Epub 2017/07/06. doi: 10.1038/s41598-017-04894-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robertson BD, Altmann D, Barry C, Bishai B, Cole S, Thomas D, Duncan K, Dye C, Ehrt S, Esmail H, Flynn J, Hafner R, Handley G, Hanekom W, van Helden P, Kaplan G, Kaufmann SHE, Kim P, Lienhardt C, Mizrahi V, Rubin E, Schnappinger D, Sherman D, Thole J, Vandal O, Walzl G, Warner D, Wilkinson R, Young D. Detection and treatment of subclinical tuberculosis. Tuberculosis. 2012;92(6):447–52. doi: 10.1016/j.tube.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, Ginsberg A, Swaminathan S, Spigelman M, Getahun H, Menzies D, Raviglione M. Tuberculosis. Nat Rev Dis Primers. 2016;2:16076. doi: 10.1038/nrdp.2016.76. [DOI] [PubMed] [Google Scholar]
- 30.Warsinske H, Vashisht R, Khatri P. Host-response-based gene signatures for tuberculosis diagnosis: A systematic comparison of 16 signatures. PLoS Med. 2019;16(4):e1002786. doi: 10.1371/journal.pmed.1002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warsinske HC, Rao AM, Moreira FMF, Santos PCP, Liu AB, Scott M, Malherbe ST, Ronacher K, Walzl G, Winter J, Sweeney TE, Croda J, Andrews JR, Khatri P. Assessment of Validity of a Blood-Based 3-Gene Signature Score for Progression and Diagnosis of Tuberculosis, Disease Severity, and Treatment Response. JAMA Netw Open. 2018;1(6):e183779. doi: 10.1001/jamanetworkopen.2018.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diel R, Loddenkemper R, Nienhaus A. Predictive value of interferon-gamma release assays and tuberculin skin testing for progression from latent TB infection to disease state: a meta-analysis. Chest. 2012;142(1):63–75. doi: 10.1378/chest.11-3157. [DOI] [PubMed] [Google Scholar]
- 33.Drain PK, Bajema KL, Dowdy D, Dheda K, Naidoo K, Schumacher SG, Ma S, Meermeier E, Lewinsohn DM, Sherman DR. Incipient and Subclinical Tuberculosis: a Clinical Review of Early Stages and Progression of Infection. Clin Microbiol Rev. 2018;31(4). doi: 10.1128/CMR.00021-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dowdy DW, Basu S, Andrews JR. Is passive diagnosis enough? The impact of subclinical disease on diagnostic strategies for tuberculosis. Am J Respir Crit Care Med. 2013;187(5):543–51. doi: 10.1164/rccm.201207-1217OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ribeiro-Rodrigues R, Kim S, Coelho da Silva FD, Uzelac A, Collins L, Palaci M, Alland D, Dietze R, Ellner JJ, Jones-Lopez E, Salgame P. Discordance of tuberculin skin test and interferon gamma release assay in recently exposed household contacts of pulmonary TB cases in Brazil. PLoS One. 2014;9(5):e96564 Epub 2014/05/14. doi: 10.1371/journal.pone.0096564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones-Lopez EC, Kim S, Fregona G, Marques-Rodrigues P, Hadad DJ, Molina LP, Vinhas S, Reilly N, Moine S, Chakravorty S, Gaeddert M, Ribeiro-Rodrigues R, Salgame P, Palaci M, Alland D, Ellner JJ, Dietze R. Importance of cough and M. tuberculosis strain type as risks for increased transmission within households. PLoS One. 2014;9(7):e100984. doi: 10.1371/journal.pone.0100984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinhas SA, Jones-Lopez EC, Ribeiro Rodrigues R, Gaeddert M, Peres RL, Marques-Rodrigues P, de Aguiar PPL, White LF, Alland D, Salgame P, Hom D, Ellner JJ, Dietze R, Collins LF, Shashkina E, Kreiswirth B, Palaci M. Strains of Mycobacterium tuberculosis transmitting infection in Brazilian households and those associated with community transmission of tuberculosis. Tuberculosis (Edinb). 2017;104:79–86. doi: 10.1016/j.tube.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Hussey GD, Abrahams D, Kafaar F, Hawkridge T, Verver S, Hughes EJ, Ota M, Sutherland J, Howe R, Dockrell HM, Boom WH, Thiel B, Ottenhoff TH, Mayanja-Kizza H, Crampin AC, Downing K, Hatherill M, Valvo J, Shankar S, Parida SK, Kaufmann SH, Walzl G, Aderem A, Hanekom WA, Acs, groups GCcs. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet. 2016. doi: 10.1016/S0140-6736(15)01316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wingett SW, Andrews S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res. 2018;7:1338. doi: 10.12688/f1000research.15931.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32(19):3047–8. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leek JT. svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014;42(21). doi: 10.1093/nar/gku864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33(1):1–22. Epub 2010/09/03. [PMC free article] [PubMed] [Google Scholar]
- 44.Chen T, Guestrin C. XGBoost: A Scalable Tree Boosting System Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; San Francisco, California, USA: 2939785: ACM; 2016. p. 785–94. [Google Scholar]
- 45.Wright MN, Ziegler A. ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. Journal of Statistical Software. 2017;77(1). doi: 10.18637/jss.v077.i01. [DOI] [Google Scholar]
- 46.Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G, Bardou P, Beck T, Blake A, Bonierbale M, Brookes AJ, Bucci G, Buetti I, Burge S, Cabau C, Carlson JW, Chelala C, Chrysostomou C, Cittaro D, Collin O, Cordova R, Cutts RJ, Dassi E, Di Genova A, Djari A, Esposito A, Estrella H, Eyras E, Fernandez-Banet J, Forbes S, Free RC, Fujisawa T, Gadaleta E, Garcia-Manteiga JM, Goodstein D, Gray K, Guerra-Assuncao JA, Haggarty B, Han DJ, Han BW, Harris T, Harshbarger J, Hastings RK, Hayes RD, Hoede C, Hu S, Hu ZL, Hutchins L, Kan Z, Kawaji H, Keliet A, Kerhornou A, Kim S, Kinsella R, Klopp C, Kong L, Lawson D, Lazarevic D, Lee JH, Letellier T, Li CY, Lio P, Liu CJ, Luo J, Maass A, Mariette J, Maurel T, Merella S, Mohamed AM, Moreews F, Nabihoudine I, Ndegwa N, Noirot C, Perez-Llamas C, Primig M, Quattrone A, Quesneville H, Rambaldi D, Reecy J, Riba M, Rosanoff S, Saddiq AA, Salas E, Sallou O, Shepherd R, Simon R, Sperling L, Spooner W, Staines DM, Steinbach D, Stone K, Stupka E, Teague JW, Dayem Ullah AZ, Wang J, Ware D, Wong-Erasmus M, Youens-Clark K, Zadissa A, Zhang SJ, Kasprzyk A. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015;43(W1):W589–98. doi: 10.1093/nar/gkv350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 48.Manimaran S, Selby HM, Okrah K, Ruberman C, Leek JT, Quackenbush J, Haibe-Kains B, Bravo HC, Johnson WE. BatchQC: interactive software for evaluating sample and batch effects in genomic data. Bioinformatics. 2016;32(24):3836–8. doi: 10.1093/bioinformatics/btw538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuhn M Building Predictive Models in R Using the caret Package. J Stat Softw. 2008;28(5):1–26. doi: 10.18637/jss.v028.i05.27774042 [DOI] [Google Scholar]
- 50.Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21(20):3940–1. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 51.Abeel T, Helleputte T, Van de Peer Y, Dupont P, Saeys Y. Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics. 2010;26(3):392–8. Epub 2009/11/28. doi: 10.1093/bioinformatics/btp630. [DOI] [PubMed] [Google Scholar]
- 52.Moon H, Ahn H, Kodell RL, Baek S, Lin CJ, Chen JJ. Ensemble methods for classification of patients for personalized medicine with high-dimensional data. Artificial intelligence in medicine. 2007;41(3):197–207. Epub 2007/08/28. doi: 10.1016/j.artmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Kolde R pheatmap: Pretty Heatmaps. R package version 1.0.8. http://CRANR-projectorg/package=pheatmap. 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.