

Abstract
Aromatic and heterocyclic functionality are ubiquitous in pharmaceuticals. Herein, we disclose a new Mn(PDP)catalyst system using chloroacetic acid additive capable of chemoselectively oxidizing remote tertiary C(sp3)—H bonds in the presence of a broad range of aromatic and heterocyclic moieties. Although catalyst loadings can be lowered to 0.1 mol% under a Mn(PDP)/acetic acid system for aromatic and non-basic nitrogen heterocycle substrates, the Mn(PDP)/chloroacetic acid system generally affords 10–15% higher isolated yields on these substrates and is uniquely effective for remote C(sp3)—H hydroxylations in substrates housing basic nitrogen heterocycles. The demonstrated ability to perform Mn(PDP)/chloroacetic acid C(sp3)—H oxidations in pharmaceutically relevant complex molecules on multi-gram scales will facilitate drug discovery processes via late-stage functionalization.
Keywords: C—H hydroxylation, oxidation, chemoselective, site-selective, late stage functionalization
Graphical Abstract
Introduction
The direct, atomistic change from C(sp3)—H to C(sp3)—O can have a profound impact on the physical and biological properties of pharmaceuticals and complex bioactive molecules. With medicinal chemists moving towards increasing the fraction sp3 (Fsp3) in small molecule therapeutics,[1] methods that enable late stage C(sp3)—H hydroxylation are highly desirable to avoid lengthy de novo syntheses when accessing novel chemical space for molecule derivatization and metabolite synthesis. The discovery of Fe(PDP) catalyst 1 in 2007 demonstrated for the first time that aliphatic C—H bonds of the same bond type (3° or 2°) can be preparatively and predictably distinguished based on small differences in their steric, electronic, and stereoelectronic properties.[2,3] Ligand modifications leading to Fe(CF3-PDP) 2 demonstrated that the site of oxidation can be altered by modifying the catalyst.[4] Using these catalysts and the principles that emerged, there has been an explosion of research in the area of late stage oxidation for diversification of drugs and natural products[3,4,5] and streamlining synthesis[6]. Despite these advances, the majority of methods are limited by chemoselectivity issues when medicinally relevant aromatic and heteroaromatic functionalities are present. The development of methods that overcome this challenge stands to further increase the impact of late stage C—H functionalization in drug discovery (Figure 1.A).
Figure 1.
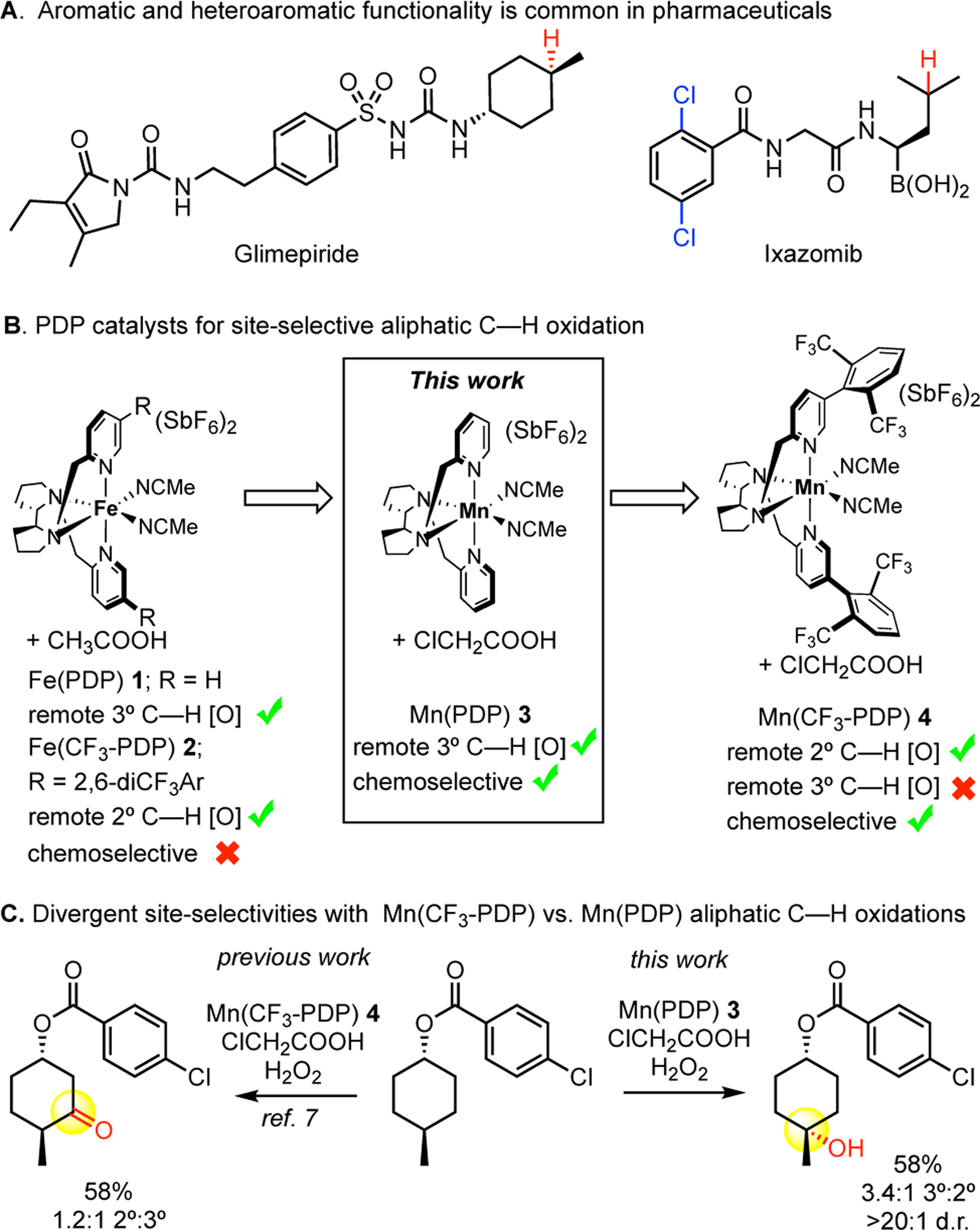
PDP catalysts for site-selective aliphatic C—H oxidation.
We recently reported a small molecule Mn(CF3-PDP) catalyst 4 using H2O2 oxidant and chloroacetic acid additive to site-selectively oxidize strong methylene bonds in the presence of more oxidatively labile halogenated aromatic and heteroaromatic functionality.[7] Non-haem Fe(PDP) 1 and Fe(CF3-PDP) 2 catalysts were previously demonstrated to site-selectively hydroxylate strong aliphatic C—H bonds,[2,3,4] but showed no chemoselectivity for π-functionality unless deactivated with strongly electron withdrawing groups (i.e. nitro, trifluoromethyl, triflate) (Figure 1.B). Alternatively, manganese has a lower redox potential[8] which disfavours undesired aromatic oxidation, while the increased basicity of the manganese oxo[9] may promote C—H abstraction. By combining manganese with the sterically bulky CF3-PDP ligand design, which additionally may disfavor the sterically demanding π-system oxidation,[10] Mn(CF3-PDP) 4 showed high chemoselectivity for the oxidation of strong methylene C(sp3)—H bonds. However, the later ligand modification also limits the ability of Mn(CF3-PDP) 4 to oxidize more sterically demanding 3° C—H bonds. Indeed, within substrates containing both 3° and 2° C—H sites available for oxidation, Mn(CF3-PDP) 4 favors oxidation at the more accessible but less electron rich 2° C—H bonds (Figure 1.C).[7] We hypothesized investigating manganese catalyst designs with less sterically demanding ligand frameworks may allow us to access preparative oxidation of 3° C—H bonds in the presence of aromatic and heteroaromatic functionality.
Herein we disclose a chemoselective 3° C—H hydroxylation with a Mn(PDP) 3/chloroacetic acid catalytic system for late-stage functionalization that is tolerant of a wide variety of aromatic and heterocyclic functionality (Figure 1.B).
Results and Discussion
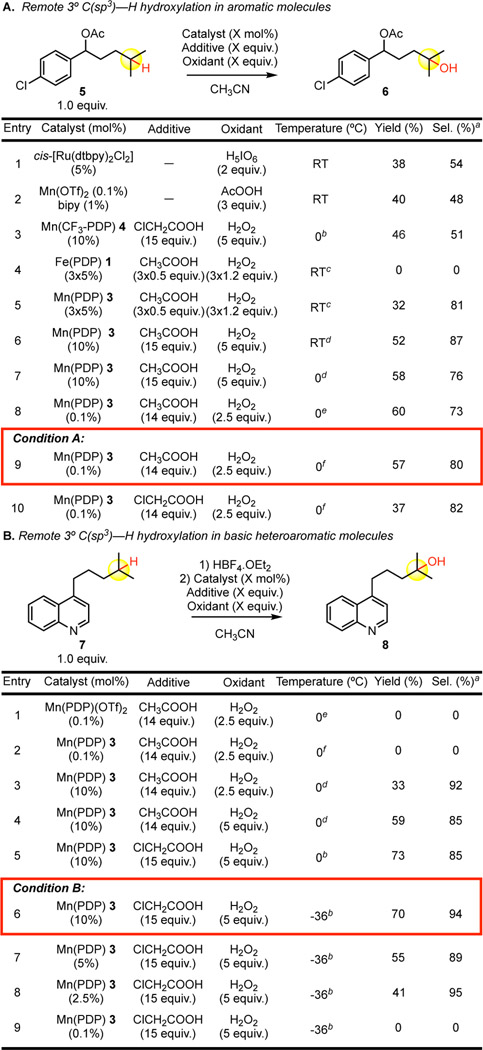
We began by investigating the hydroxylation of tertiary substrate 5 containing a mildly electron-deactivating para-chloro-substituted aromatic ring with catalysts reported to hydroxylate 3° C—H bonds in the presence of benzoate groups. Both cis-[Ru(dtbpy)2Cl2] [11] and Mn(OTf)2/bipy[12] catalysts afforded moderate yields and chemoselectivities under their reported conditions (Table 1.A, entry 1 – 2). Whereas Mn(CF3-PDP) 4 gave a comparable yield and selectivity to these catalyst systems (entry 3), expectedly Fe(PDP) 1 gave no desired 3° C—H hydroxylation product 6 due to competitive aromatic oxidation (entry 4).
Table 1.
Reaction Development.
|
(R,R)-Mn(PDP) and (S,S)-Mn(PDP) used interchangeably. Isolated yields based on the average of 2–3 reactions.
Chemoselectivity (Sel.) = 3° C—H oxidation/total conversion.
Substrate, catalyst and additive with slow addition of H2O2 (in MeCN, 0.4 M) over 3 h.
Iterative addition of 5 mol% catalyst, 0.5 equiv. CH3COOH, 1.2 equiv. H2O2 3 times every 10–15 min.
Substrate, catalyst and additive with slow addition of H2O2 (in MeCN) over 1 h.
Substrate, catalyst, additive with slow addition of H2O2 (in MeCN, 2.5 M) over 1 h and reaction mixture stirred for an additional 1 h.
Substrate, catalyst, additive with slow addition of H2O2 (in MeCN, 0.2 M) over 1 h.
Similar to our previous report, switching from iron to manganese significantly improved chemoselectivity. Mn(PDP) 3 under identical iterative addition conditions gave significantly higher yield (32%) of the desired 3° C—H oxidation product 6 with 81% chemoselectivity (entry 5). Switching from an iterative addition protocol to a single addition of Mn(PDP) 3 catalyst with slow addition of H2O2 oxidant at room temperature further increased the reactivity to a synthetically useful 52% yield of 3° alcohol 6 (entry 6). Reducing the reaction temperature from room temperature to 0 °C further increased the yield for desired 3° oxidation, presumably by avoiding catalase-like decomposition of H2O2 by the manganese catalyst (entry 7).[13] We questioned whether reducing the catalyst loading would be feasible for the oxidation of weaker 3° C—H bonds. Previously reported Mn(PDP)(OTf)2 catalysis for tertiary and secondary C—H oxidations at very low catalyst loadings (0.1 mol%) were known for simple aliphatic substrates, including substrates containing benzoate moieties.[12,14] Using the reported conditions with our Mn(PDP)(SbF6)2 3 catalyst, we found that the desired product 6 was formed in comparable yield and chemoselectivity to the 10 mol% catalyst loading conditions (entry 7 versus 8). By modifying the reaction concentration, we found the chemoselectivity could be further improved to 80% (entry 9). Chloroacetic acid, a key to the high yielding conditions developed below (see Table 1.B), under these low catalyst loading conditions (0.1 mol%), afforded a substantial decrease in yield (entry 10).
We questioned if these extremely mild oxidation conditions would be effective in more complex molecular settings, particularly substrates containing basic nitrogen functionality. It has been previously demonstrated that such nitrogen functionality requires complexation with a Brønsted acid having a non-coordinating counterion (HBF4) to enable remote aliphatic C—H oxidations with Fe or Mn(PDP) catalysis.[7,15] When a quinoline containing substrate 7 was evaluated with both Mn(PDP)(OTf)2[14] and Mn(PDP)(SbF6)2 3 catalysts, the low 0.1 mol% catalyst loading conditions established in Table 1.A were no longer competent at affording the desired remote 3° C—H oxidation product 8 (Table 1.B, entry 1 – 2). Increasing the catalyst loading from 0.1 mol% to 10 mol% restored reactivity, furnishing tertiary alcohol 8 in an encouraging 33% yield with excellent 92% chemoselectivity (entry 3). Notably, no benzylic oxidation is observed, likely due to the strong inductively withdrawing nature of the protonated quinoline. Increasing the oxidant loading from 2.5 equiv. to 5.0 equiv. provided synthetically useful 59% yield though with slightly diminished chemoselectivity (entry 4). Electron deficient chloroacetic acid additive was demonstrated to be critical for optimal 2° C—H oxidation reactivity with Mn(CF3-PDP) catalyst 4, possibly by increasing the electrophilicity of the postulated manganese(oxo) carboxylate intermediate.[7] Similarly, switching to chloroacetic acid and extending the oxidant addition time further increased the reactivity of Mn(PDP) 3 for 3° C—H oxidation to 73% yield (entry 5). Lowering the temperature from 0 °C to −36 °C gave optimal 94% chemoselectivity while maintaining 70% yield (entry 6). In the majority of cases evaluated, the −36 °C conditions afforded higher yields (vide infra). Under the optimized conditions B, we re-evaluated lowering the catalyst loading. Lowering the catalyst from 10 mol% to 5 mol% maintained the preparative utility of the reaction affording 55% yield of 8 (entry 7). Lowering the catalyst loading further to 2.5 mol% saw a significant diminishment in yield to 41% (entry 8). Re-investigation of 0.1 mol% catalyst 3 loading under optimized conditions B furnished no product (entry 9).
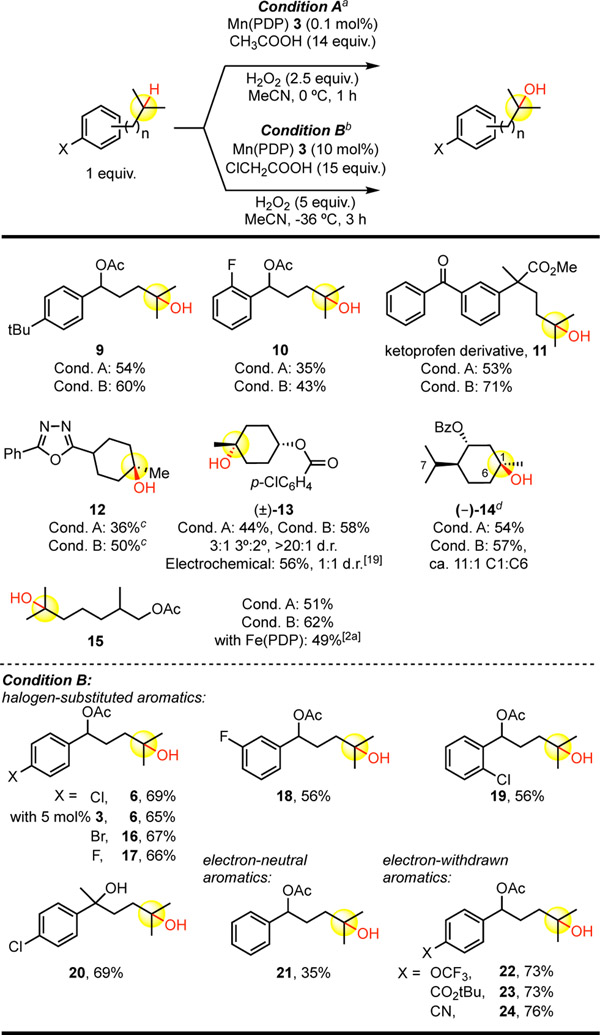
After establishing the optimal conditions for Mn(PDP) 3 catalyzed 3° C—H hydroxylations, we sought to investigate their generality. We initially explored the 0.1 mol% 3 conditions (Table 1.A, entry 9, condition A) versus the 10 mol% 3 conditions (Table 1.B, entry 6, condition B) on aromatic and non-basic heteroaromatic substrates. Although condition A (0.1 mol%) gave useful yields in the majority of cases, a significant improvement was observed with condition B (10 mol%). For example, a derivative of biaryl NSAID ketoprofen afforded ca. 18% higher isolated yield of alcohol 11 under 10 mol% 3 conditions B. Interestingly, a non-basic oxadiazole heterocycle not requiring HBF4 protection furnished alcohol 12 at the low Mn(PDP) 3 loadings of condition A, signifying that the lack of reactivity of these conditions with quinoline substrate 7 may be related to its basicity and/or the Brønsted acid complexation (vide infra). Highlighting the orthogonality of Mn(PDP) 3 catalysis with Mn(CF3-PDP) 4, a trans-cyclohexanol substrate housing competing tertiary and secondary sites preferentially oxidized the tertiary site under both conditions A and B (3°:2° = 3.4:1, 13), whereas Mn(CF3-PDP) 4 favored formation of the methylene ketone (3°:2° = 1:1.2)[7]. Significantly, no erosion in stereochemistry was observed as seen in free radical mediated C—H functionalizations of tertiary sites. Similarly, benzoate-protected menthol furnished preparative yields and excellent 3°:2° selectivity (14), with no observed oxidation at the alternate C7 3° site. Interestingly, dioxirane[16] and oxaziridine[17] oxidants are not reported to give any selectivity on analogous menthol derived substrates whereas radical azidation methods afford C7 products, albeit in poor yields.[18] Archetypical citronellol-derived substrate afforded substantially improved yields of remote tertiary hydroxylated product 15 under Mn(PDP) 3 conditions B relative to its iron counterpart with no observed diminishment in site-selectivity.[2a] We additionally evaluated the more forcing Mn(PDP) 3 oxidation conditions B with a range of mildly electron-withdrawing halogen-substituted aromatic substrates and gratifyingly found uniformly preparative yields for tertiary C—H oxidations (6, 16-24, Table 2). Consistent with previous observations, Table 1.A substrate 5 afford an ca. 10% increase in yield of tertiary hydroxylated product 6 under conditions B (69%) and catalyst 3 could be lowered to 5 mol% with only a small diminishment in yield (65%).
Table 2.
Mn(PDP)-Catalyzed 3° C(sp3)—H Hydroxylations in Aromatic and non-Basic Heteroaromatic Compounds.
|
(R,R)-MnPDP and (S,S)-Mn(PDP) used interchangeably. Isolated yields based on the average of 2–3 reactions.
Substrate, 0.1 mol% Mn(PDP) 3, 14 equiv. CH3COOH additive with slow addition of 2.5 equiv. H2O2 (in MeCN, 0.2 M) over 1 h at 0 °C.
Substrate, 5–10 mol% Mn(PDP) 3, 15 equiv. ClCH2COOH additive with slow addition of 5.0 equiv. H2O2 (in MeCN, 0.4 M) over 3 h at −36 °C.
Starting material recycled once.
(S,S)-Mn(PDP) required for optimal yield.
Consistent with previous observations, Mn(PDP) 3 C—H hydroxylations of substrates housing basic nitrogen moieties requiring Brønsted acid protection were uniquely effective under conditions B, with no desired product being observed under the low catalyst loading (0.1 mol%) conditions A (Table 3). The temperature influence on yield was substrate dependent: whereas a benzimidazole substrate afforded slightly higher yield of 25 under the 0 °C conditions, the analogous imidazole substrate gave ca. 16% higher yield of 26 at −36 °C. A challenging tetrahydroisoquinoline substrate evaluated under Mn(PDP) 3/chloroacetic acid catalysis at −36 °C afforded a modest 27% yield of the desired remote 3° alcohol 27 with only a slight diminishment in yield at 0 °C. Such substrates containing electron neutral aromatic moieties have not previously been demonstrated in methylene C(sp3)—H oxidations with Mn(CF3-PDP) 4 catalysis due to competing aromatic oxidation. A ketobemidone analogue containing a 4-chloroarylpiperidine pharmacophore afforded remote oxidation product 28 in 68% yield under condition B.
Table 3.
Mn(PDP)-Catalyzed 3° C(sp3) —H Hydroxylations in Basic Heteroaromatic Compounds.
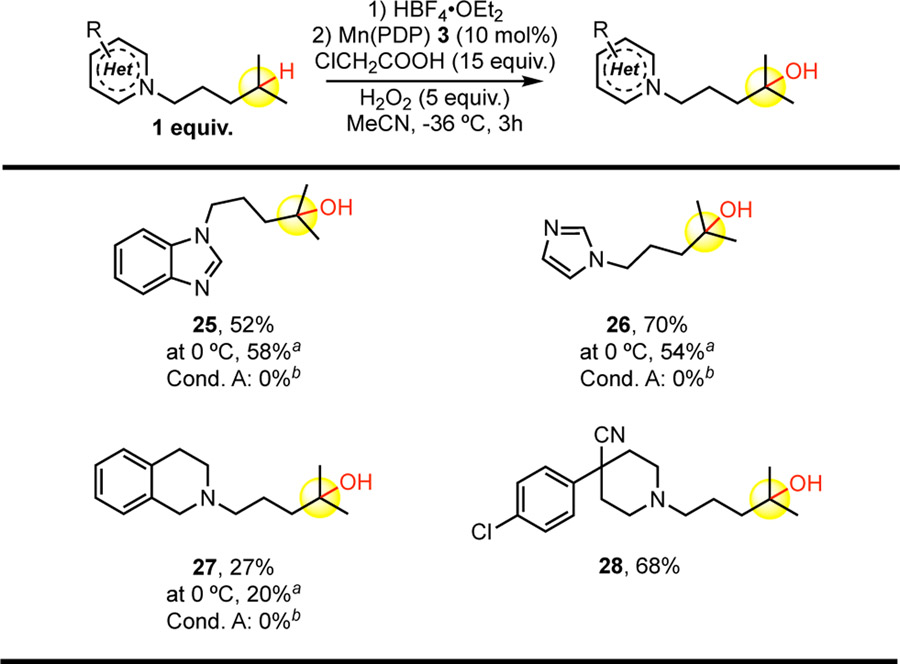
|
(R,R)-Mn(PDP) and (S,S)-Mn(PDP) used interchangeably. Isolated yields based on the average of 2–3 reactions.
Reactions runs at 0 °C instead of −36 °C.
Substrate, 0.1 mol% catalyst, 14 equiv. CH3COOH additive with slow addition of 2.5 equiv. H2O2 (in MeCN, 0.2 M) over 1 h at 0 °C.
We hypothesized that analogous to Fe(PDP) 1 hydroxylations, Mn(PDP) 3 oxidations proceed via a high valent metal oxidant that effects C—H hydroxylations through a mechanism that does not involve the generation of long-lived carbon centered radical species.[3] To probe this, we evaluated the oxidation of enantiomerically enriched substrate 29 under the general Mn(PDP) 3 catalysis condition B. Consistent with oxidation results with other chiral substrates (e.g. 13, 14, 34), Mn(PDP) 3 oxidation of 29 proceeds with stereoretention to afford 3° alcohol 30 (Scheme 1). This result is in contrast to electrochemical[19] and iron and manganese porphyrin catalyzed C—H functionalizations which proceed via long lived radicals[18,20,21] where ablation of stereochemistry at tertiary sites is observed.
Scheme 1.
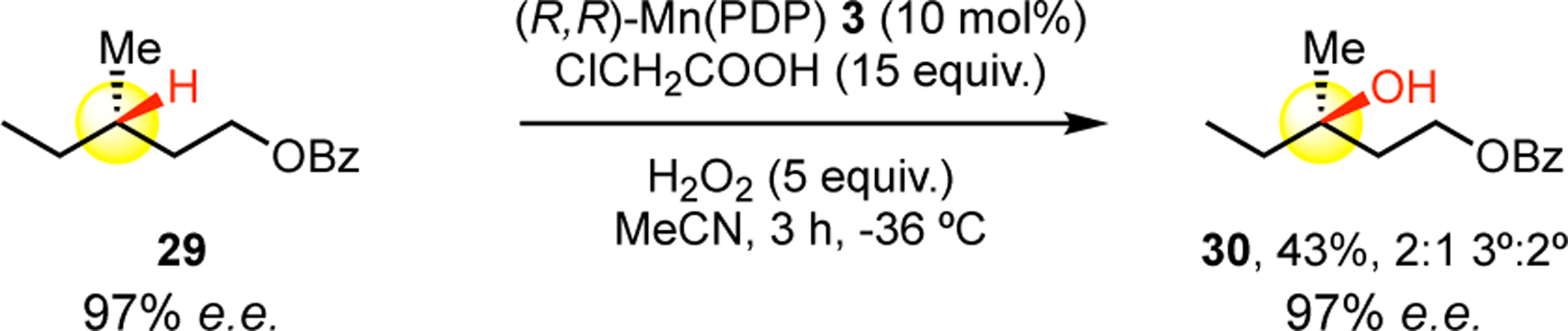
Stereoretention Study.
The chemoselectivity and reactivity observed with Mn(PDP) 3 catalysis in the presence of pharmaceutically relevant aromatic and heteroaromatic moieties provides an opportunity for late-stage diversification of drug scaffolds. Evaluation of an efavirenz derivative 31, the parent compound being a WHO essential medicine for HIV, afforded tertiary hydroxylated product 32 in 53% isolated yield under condition B with no protection of the non-basic carbamate nitrogen (Figure 2.A). In addition to demonstrating high chemoselectivity for the π-systems of an aromatic and alkyne moiety, Mn(PDP) 3 tertiary oxidation also displayed preferential reactivity for tertiary C—H bond oxidation versus oxidation alpha to the heterocyclic nitrogen. Previous Mn(CF3-PDP) 4 methylene oxidation of a hexanoyl efavirenz derivative had also shown π-system tolerance when the alpha-nitrogen site was blocked with a carbonyl moiety (vide infra, Fig. 3.B).[7] Warming the reaction to 0 °C or using the low Mn(PDP) 3 catalyst loading condition A (0.1 mol%) afforded product 32 with comparable diminishments in yield to those noted in Table 2.
Figure 2.
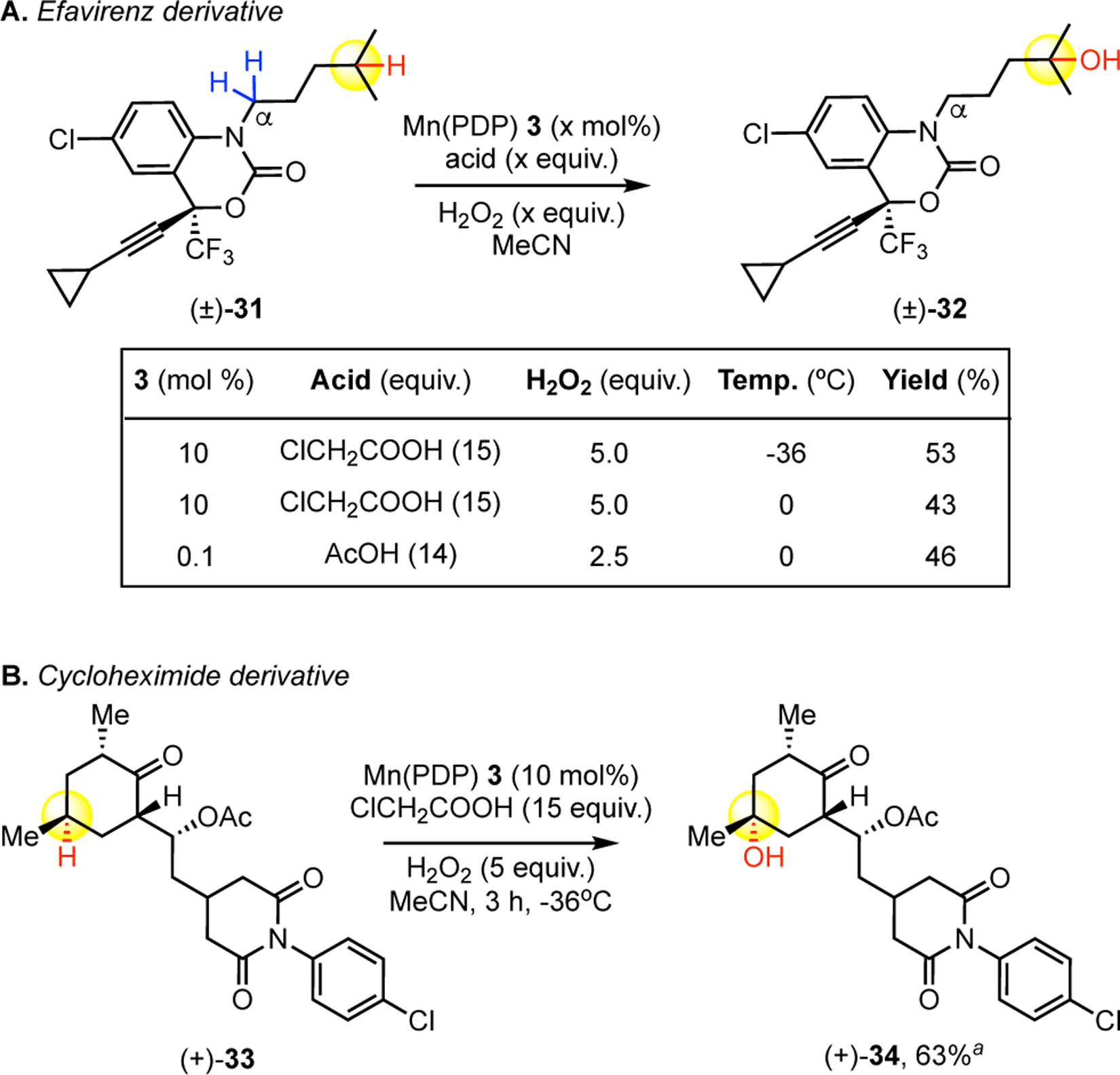
Late Stage C—H Oxidation of Pharmaceutical Derivatives. a)Starting material recycled once.
Figure 3.
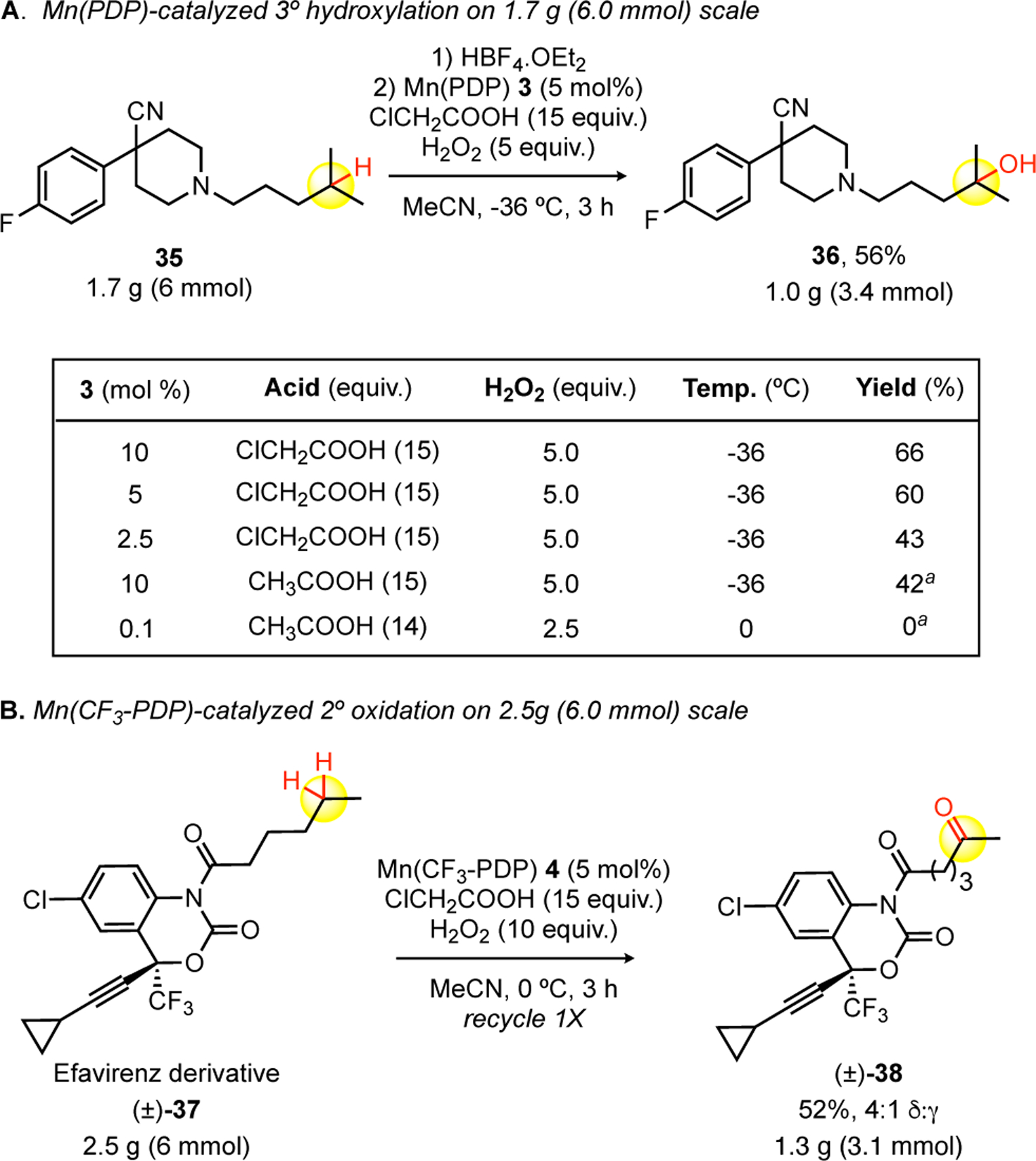
Scale-up C—H Oxidations of Bioactive Molecules. a)1 h addition of H2O2 (in MeCN) instead of 3 h addition.
Additionally, we evaluated an aryl-substituted cycloheximide derivative (+)-33, the parent compound having broad antimicrobial activity, for remote tertiary oxidation on the cyclohexanone core. Consistent with previous observations, the imide functionality was tolerated with no Brønsted acid complexation.[15] Notably, Mn(PDP) 3 catalysis using general condition B was tolerant of the newly introduced aniline moiety, albeit electronically deactivated, furnishing the tertiary hydroxylated product (+)-34 in excellent 63% yield (Figure 2.B).
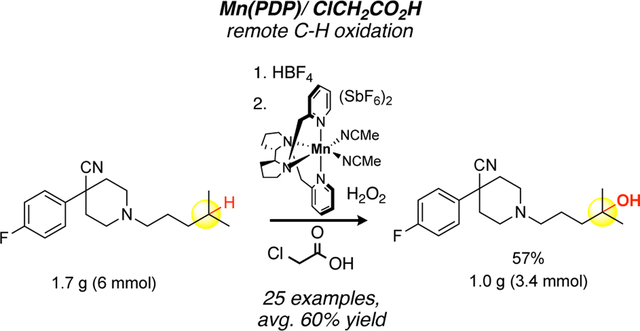
Given the ability of Mn(PDP) 3 and Mn(CF3-PDP) 4 to catalyze chemoselective tertiary and methylene C—H oxidations to access metabolites and perform late-stage functionalization on medicinally relevant candidates, we wanted to evaluate the ability to perform these oxidation reactions on scales that facilitate drug discovery processes. We examined both Mn(PDP) 3 and Mn(CF3-PDP) 4 catalysts for 6.0 mmol scale tertiary and methylene oxidations of pharmaceutically relevant substrates. Compound 35, with a 4-fluoroarylpiperidine, a pharmacophore found in opioids, such as ketobemidone and haloperidol, was initially evaluated on a 0.3 mmol scale to explore lower Mn(PDP) 3 catalyst loadings under condition B that would be particularly relevant for large scale oxidations (Figure 3.A). Using Mn(PDP) 3 condition B (10 mol%), remote oxidation product 36 was isolated in an optimal 66% yield. The catalyst loading of Mn(PDP) 3 can be reduced to 5 mol% while maintaining a preparatively useful 60% yield. Underscoring the significance of the chloroacetic acid additive, a switch to acetic acid furnished a comparable diminishment in yield to decreasing the catalyst loading to 2.5 mol% (42% vs 43%). Expectedly, the 0.1 mol% catalyst loading condition A afforded no product with this basic piperidine substrate. From these studies, we concluded the optimal conditions for reaction scale-up would use 5 mol% catalyst loading with chloroacetic acid additive. Following piperidine protection using the HBF protection strategy,[15] a 1.7 g (6.0 mmol) C—H hydroxylation of 35 using 5 mol% Mn(PDP) 3 afforded 1.0 g (3.4 mmol) of tertiary hydroxyl product 36 in 56% yield. We additionally examined the previously reported Mn(CF3-PDP) 4 methylene oxidation of HIV-1 drug efavirenz derivative 37 (Figure 3.B). A similar reduction in Mn(CF3-PDP) 4 loading from 10 mol% to 5 mol% on a small scale (0.2 mmol) oxidation resulted in only minor reduction in yield (58% → 50%, see Supporting Information). On a 2.5-gram scale (6 mmol), Mn(CF3-PDP) 4 at 5 mol% afforded ca. 41% yield of 38 with 27% recovered starting material 37 (see Supporting Information). A simple recycle of the recovered starting material 37 afforded 1.3 grams (3.1 mmol) of remote oxidation product 38 in 52% overall yield.
Conclusion
We describe the development of the first general conditions for chemoselective Mn(PDP) 3 catalyzed tertiary C(sp3)—H hydroxylations in substrates containing a broad range of aromatic and heteroaromatic functionality. Systematic evaluation of the low loading conditions[14] for Mn(PDP) 3 (0.1 mol%) using acetic acid additive illuminated that these conditions afford moderate to good hydroxylation yields for simple halogenated aromatics and non-basic heterocyclic substrates but prove ineffective for molecules containing basic-nitrogen heterocycles. The general oxidation conditions reported in this work using Mn(PDP) 3 catalysis at higher catalyst loadings (5–10 mol%) in combination with chloroacetic acid additive are uniquely effective in the remote C—H hydroxylation of medicinally important substrates housing basic nitrogen functionality that must be masked with Brønsted acid complexation prior to C—H oxidation. Moreover, these conditions afford 10–15% higher yields in halogenated aromatic and non-basic heteroaromatic substrates. Given the relative abundance of manganese and the ease of preparation of Mn(PDP) 3 catalyst, we believe these general conditions will find widespread use in late stage diversification of pharmaceutically relevant molecules and the rapid identification of metabolites.
Experimental Section
General Procedure for C—H Oxidation Condition A Using 0.1 mol% Mn(PDP) Catalyst/AcOH
A 40 mL vial was charged with substrate (0.3 mmol, 1.0 equiv), Mn(PDP) 3 (5 mM stock solution in MeCN, 60 μL, 0.3 μmol, 0.001 equiv.), CH3COOH (0.24 mL, 4.2 mmol, 14.0 equiv.) and a stir bar. Acetonitrile (MeCN, 0.6 mL, 0.5 M) was added and the vial was sealed with a screw cap fitted with a PTFE/silicone septum. The vial was cooled to 0 °C with an ice/water bath. A separate solution of H2O2 (51.0 mg, 0.75 mmol, 2.5 equiv., 50% wt. in H2O, purchased from Sigma Aldrich) in MeCN (3.75 mL, 0.2 M) was loaded into a 10 mL syringe fitted with a 25G needle and added dropwise to the stirring reaction via syringe pump over 1 h (3.75 mL h−1 addition rate) while maintaining the reaction vial at 0 °C. Upon completion of addition, the reaction was concentrated in vacuo to a minimum amount of solvent. The residue was dissolved in DCM and washed with sat. NaHCO3 solution. The aqueous layer was extracted with DCM twice. The combined organic layer was dried with Na2SO4, filtered and concentrated. The crude mixture was purified by flash column chromatography to afford the desired oxidation product.
General Procedure for C—H Oxidation Condition B Using 10 mol% Mn(PDP) Catalyst/ClCH2COOH
A 40 mL vial was charged with substrate (0.3 mmol, 1.0 equiv.), Mn(PDP) 3 (27.9 mg, 0.03 mmol, 10 mol%), ClCH2CO2H (425 mg, 4.5 mmol, 15.0 equiv.) and a stir bar. Acetonitrile (MeCN, 0.6 mL, 0.50 M) was added along the wall to ensure all compounds were washed beneath the solvent level and the vial was sealed with a screw cap fitted with a PTFE/silicone septum. The vial was cooled to −36 °C with a 1,2-dichloroethane/dry ice bath. A separate solution of H2O2 (102 mg, 1.5 mmol, 5.0 equiv., 50% wt. in H2O, purchased from Sigma-Aldrich) in MeCN (3.75 mL, 0.4 M) was loaded into a 10 mL syringe fitted with a 25 G needle and added dropwise to the stirring reaction via a syringe pump over 3 h (1.25 mL h−1 addition rate) while maintaining the reaction vial at −36 °C. Upon completion, the reaction was concentrated in vacuo to a minimum amount of solvent. The residue was dissolved in DCM and washed with sat. NaHCO3 solution (caution: CO2 released) to remove ClCH2CO2H. The aqueous layer was extracted with DCM twice. The combined organic layer was dried with Na2SO4, filtered and concentrated. The crude mixture was purified by flash column chromatography to afford the desired oxidation product. If this method gave low conversion, an alternative addition protocol was used. See Supporting Information Methods C and D.
Detailed experimental procedures and characterization data for all new compounds are described in the supporting information.
Crystallographic data for (S,S)-Mn(PDP) 3 can be obtained free of charge from www.ccdc.cam.ac.uk/structures/ with deposit number CCDC 1869257.
Crystallographic data for (S,S)-Mn(CF3-PDP) 4 can be obtained free of charge from www.ccdc.cam.ac.uk/structures/ with deposit number CCDC 1964541.
Supplementary Material
Acknowledgements
The authors thank L. Zhu and D. Olsen for assistance with NMR spectroscopy, D. Gray and T. Woods for X-Ray crystallographic studies.
Funding Sources
Financial Support for this work was provided by the NIH NIGMS Maximizing Investigator’s Research Award MIRA (R35 GM122525). RKC was partially funded by a Sir Keith Murdoch Scholarship from the American Australian Association.
Footnotes
This paper is dedicated to Eric N. Jacobsen on the occasion of his 60th birthday for his inspirational work on the discovery and study of practical reactions that illuminate new selectivity principles in catalysis.
Competing Interest
The University of Illinois has filed a patent application on the Mn(CF3-PDP) catalyst for C—H oxidation in aromatic molecules.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- [1].Lovering F, Bikker J, Humblet C, J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- [2].a) Chen MS, White MC, Science 2007, 318, 783–787; [DOI] [PubMed] [Google Scholar]; b) Chen MS, White MC, Science 2010, 327, 566–571. [DOI] [PubMed] [Google Scholar]
- [3].White MC, Zhao J, J. Am. Chem. Soc 2018, 140, 13988–14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gormisky PE, White MC, J. Am. Chem. Soc 2013, 135, 14052–14055. [DOI] [PubMed] [Google Scholar]
- [5].a) White MC, Science 2012, 335, 807–809; [DOI] [PubMed] [Google Scholar]; b) Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW, Chem. Soc. Rev 2016, 45, 546–576; [DOI] [PubMed] [Google Scholar]; c) Yamaguchi J, Yamaguchi AD, Itami K, Angew. Chem. Int. Ed 2012, 51, 8960–9009; [DOI] [PubMed] [Google Scholar]; d) McMurray L, O’Hara F, Gaunt MJ, Chem. Soc. Rev 2011, 40, 1885–1898; [DOI] [PubMed] [Google Scholar]; e) Blakemore DC, Castro L, Churcher I, Rees DC, Thomas AW, Wilson DM, Wood A, Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- [6].a) Fraunhoffer KJ, Bachovchin DA, White MC, Org. Lett 2005, 7, 223–226. [DOI] [PubMed] [Google Scholar]; b) Stang EM, White MC, Nature Chem 2009, 1, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rasik CM, Brown MK, Angew. Chem. Int. Ed 2014, 53, 14522–14526; [DOI] [PubMed] [Google Scholar]; d) Hung K, Condakes ML, Morikawa T, Maimone TJ, J. Am. Chem. Soc 2016, 138, 16616–16619; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ye Q, Qu P, Snyder SA, J. Am. Chem. Soc 2017, 139, 18428–18431; [DOI] [PubMed] [Google Scholar]; f) Burns AS, Rychnovsky SD, Am. Chem. Soc 2019, 141, 13295–13300; [DOI] [PubMed] [Google Scholar]; g) Wein LA, Wurst K, Angyal P, Weisheit L, Magauer T, J. Am. Chem. Soc 2019, DOI: 10.1021/jacs.9b11646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhao J, Nanjo T, de Lucca EC Jr., White MC, Chem. 2019, 11, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jeon S, Bruice TC, Inorg. Chem 1992, 31, 4843–4848. [Google Scholar]
- [9].a) Green MT, Dawson JH, Gray HB, Science 2004, 304, 1653–1656; [DOI] [PubMed] [Google Scholar]; b) Yosca TH, Rittle J, Krest CM, Onderko EL, Silakov A, Calixto JC, Behan RK, Green MT, Science 2013, 342, 825–829; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mayer JM, Acc. Chem. Res 1998, 31, 441–450; [Google Scholar]; d) Borovik AS, Chem. Soc. Rev 2011, 40, 1870–1874; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chen J, Cho KB, Lee YM, Kwon YH, Nam W, Chem. Commun 2015, 51, 13094–13097; [DOI] [PubMed] [Google Scholar]; f) Lansky DE, Goldberg DP, Inorg. Chem 2006, 45, 5119–5125. [DOI] [PubMed] [Google Scholar]
- [10].Guroff G, Renson J, Udenfriend S, Daly JW, Jerina DM, Witkop B, Science 1967, 157, 1524–1530. [DOI] [PubMed] [Google Scholar]
- [11].Mack JBC, Gipson JD, Du Bois J, Sigman MS, J. Am. Chem. Soc 2017, 139, 9503–9506. [DOI] [PubMed] [Google Scholar]
- [12].Adams AM, Du Bois J, Malik HA, Org. Lett 2015, 17, 6066–6069. [DOI] [PubMed] [Google Scholar]
- [13].Palucki M, Hanson P, Jacobsen EN, Tetrahedron Lett. 1992, 33, 7111–7114. [Google Scholar]
- [14].Ottenbacher RV, Samsonenko DG, Talsi EP, Bryliakov KP, Org. Lett 2012, 14, 4310–4313. [DOI] [PubMed] [Google Scholar]
- [15].Howell JM, Feng K, Clark JR, Trzepkowski LJ, White MC, J. Am. Chem. Soc 2015, 137, 14590–14593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen K, Richter JM, Baran PS, J. Am. Chem. Soc 2008, 130, 7247–7249. [DOI] [PubMed] [Google Scholar]
- [17].Litvinas ND, Brodsky BH, Du Bois J, Angew. Chem. Int. Ed 2009, 48, 4513–4516. [DOI] [PubMed] [Google Scholar]
- [18].Sharma A, Hartwig JF, Nature 2015, 517, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kawamata Y, Yan M, Liu Z, Bao DH, Chen J, Starr JT, Baran PS, J. Am. Chem. Soc 2017, 139, 7448–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang X, Bergsten TM, Groves JT, J. Am. Chem. Soc 2015, 137, 5300–5303. [DOI] [PubMed] [Google Scholar]
- [21].Groves JT, Nemo TE, J. Am. Chem. Soc 1983, 105, 6243–6248. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.