

Abstract
Background:
Acute alcohol produces effects on cerebral metabolism and blood flow (CBF). Alcohol is converted to acetate, which serves as a source of energy for the brain and is an agonist at G protein-coupled receptors distributed in different cell types in the body including neurons. Acetate has been hypothesized to play a role in the CBF response after alcohol ingestion. We tested whether administration of acetate would alter CBF in a pattern similar to or different from that of alcohol ingestion in healthy individuals.
Methods:
Twenty-four healthy participants were assigned by convenience to receive either 0.6 g/kg alcohol orally (n=12) or acetate intravenously (n=12). For each participant, CBF maps were acquired using an arterial spin labeling sequence on a 3T MR scanner after placebo and after drug administration. Whole-brain CBF maps were compared between placebo and drug using a paired t-test, set at a threshold of p<0.05 corrected for multiple comparisons (k ≥142 voxels, ≥3.78 cm3), voxel-level p<0.005. Intoxication was measured after placebo and drug administration with a subjective high assessment survey (SHAS-7).
Results:
Compared to placebo, alcohol and acetate were associated with increased CBF in the medial thalamus. Alcohol, but not acetate, was associated with increased CBF in the right orbitofrontal, medial prefrontal and cingulate cortex, and hippocampus. Plasma acetate levels increased following administration of alcohol and acetate and did not differ between the two arms. Alcohol, but not acetate, was associated with an increase in SHAS-7 scores (p<0.001).
Conclusions:
Increased thalamic CBF associated with either alcohol or acetate administration suggests that the thalamic CBF response after alcohol could be mediated by acetate. Compared to other brain regions, thalamus may differ in its ability to metabolize acetate or expression of receptors responsive to acetate. Increased prefrontal and limbic CBF associated with alcohol may be linked to alcohol’s behavioral effects.
Introduction
Alcohol (ethanol) is the most widely abused drug worldwide and the third leading cause of preventable death in the United States (Mokdad, Marks, Stroup, & Gerberding, 2004). Alcohol affects brain function by modulating the effects of gamma-amino butyric acid (GABA), dopamine, and other neurotransmitters, and it alters brain metabolism and blood flow via direct effects, or through its metabolites (Gundersen et al., 2013, Marxen et al., 2014, Mathew and Wilson, 1986, Schwartz et al., 1993, Tolentino et al., 2011) (for review see (Bjork and Gilman, 2014)). The end product of anaerobic metabolism of ethanol is acetate, a caloric substance that is metabolized through the tricarboxylic acid cycle producing carbon dioxide, water, and reducing equivalents, NADH and FADH. NADH and FADH are, in turn, oxidized to produce ATP. Ethanol is metabolized to acetate in the liver and, for decades, acetate was regarded as simply a source of energy and two carbon units for the synthesis of fats in this organ.
A portion of the acetate produced in the liver enters the circulation and can be used as energy in the brain. Whereas the brain normally relies on glucose for energy, acute alcohol causes a reduction in brain glucose metabolism and an increase in brain acetate metabolism (Pawlosky et al., 2010, Volkow et al., 2013). The significance of this switch in energy substrate is not entirely understood. Acetate utilization by the brain may explain why cognitive function is relatively preserved despite reductions in glucose metabolism in the setting of acute alcohol consumption (Volkow et al., 2013, Volkow et al., 2015, Volkow et al., 2006). In alcohol use disorder (AUD), elevated levels of acetate may contribute to compensatory changes in brain metabolism (Enculescu et al., 2019). Indeed, heavy drinkers tended to have more brain uptake of 11C-acetate compared to occasional social drinkers after acute alcohol (Volkow et al., 2013). An MR spectroscopy study using the isotope [2-13C]-acetate demonstrated greater conversion of brain acetate to glutamate and glutamine in heavy compared to light drinkers (Jiang et al., 2013). In animal investigations, rats chronically fed a diet containing liquid alcohol had higher brain acetate uptake compared to control rats (Hsieh et al., 2018). Together, these studies suggest that chronic alcohol use is associated with an enhanced ability of the brain to utilize acetate.
Among those with AUD who have chronically elevated acetate, cessation of drinking alcohol would cause brain acetate levels to fall. It has been hypothesized that acetate is a form of caloric “reward” and that falling acetate levels may contribute to symptoms of alcohol withdrawal (Jiang et al., 2013, Volkow et al., 2013). In animals, acetate supplementation was shown to decrease alcohol withdrawal tremors (Derr et al., 1981). Notably, acetate is utilized preferentially by astrocytes where it participates in glutamate and GABA cycling (Waniewski and Martin, 1998). Acetate and other short chain fatty acids also activate cell surface G protein-coupled-receptors (GPCRs) through which they may regulate cell function (Milligan et al., 2017). While acetate is important in brain metabolism, neurotransmitter cycling, cell signaling, and possibly alcohol withdrawal, few neuroimaging studies of acetate have been conducted in humans (Jiang et al., 2013, Volkow et al., 2013).
Acute alcohol administration has been shown to increase cerebral blood flow (CBF) (Gundersen, van Wageningen, & Grüner, 2013; Marxen et al., 2014; Mathew & Wilson, 1986; Tolentino et al., 2011). Schwartz et al. studied the relationship between CBF, alcohol, and acetate levels in men and found that blood acetate levels correlated with CBF in a subset of younger participants, suggesting that acetate is involved in the cerebrovascular effects of acute alcohol and is modulated by age (Schwartz et al., 1993). However there have been no studies on the effect of acetate per se on CBF.
Our goal was to investigate the effect of a single, acute administration of either alcohol or acetate on CBF in healthy individuals using MR arterial spin labeling (ASL) methods. We hypothesized that both alcohol and acetate administration would increase CBF in cortical and subcortical regions compared to placebo. We also investigated the subjective effects of alcohol and acetate.
Materials and Methods
Participant recruitment and screening
Potential participants from the Denver, Colorado metropolitan area were recruited through advertisements, community fliers, and clinicaltrials.gov between January 2016 and March 2017. Initial telephone screening was conducted using the following criteria: participants were included who were ages 21–40, English-speaking, and reported prior consumption of at least two standard drinks of alcohol within one hour at some point in their lives. Exclusion criteria included reported alcohol consumption exceeding the level of moderate as defined by the American Dietary Guidelines 2015–2020 (women 1–7 drinks/week; men 1–14 drinks/week), history of psychiatric, neurologic, liver, or kidney problems; reported body mass index (BMI) ≥ 30; self-reported history of head injury resulting in unconsciousness > 15 minutes; self-reported alcohol flushing response. Participants who met inclusion criteria and did not fail exclusion criteria were invited for an initial visit.
Initial visit
During the initial visit, participants provided written informed consent to participate in a protocol approved by the Colorado Multiple Institutional Review Board. They received the computerized Composite International Diagnostic Interview-Substance Abuse Module (CIDI-SAM) (Cottler et al., 1995). Participants who met the DSM-IV criteria for stimulant, opioid, club drug, sedative, hallucinogen or alcohol dependence were excluded from further participation. Alcohol use was assessed with the Alcohol Use Disorders Identification Test (AUDIT), a 10-item screening tool that measures alcohol consumption, drinking behaviors, and alcohol-related problems (Saunders et al., 1993). The highest education level in years completed by the participants was also recorded.
Alcohol or acetate administration and MR scans
After the initial visit, participants returned for a second visit to complete the protocol. They were asked not to drink alcohol for 48 hours preceding the visit, and to eat a low-fat breakfast the morning of the scans. All participants provided urine samples to screen for drug use and women were screened for pregnancy. Participants who tested positive for illicit drugs, alcohol, or pregnancy on the day of the MR scan were excluded. An alcohol level of zero was verified by a BACTrack s80 Select breathalyzer (BACTrac®). All participants received first placebo and then either alcohol or acetate. To eliminate carry-over effects, placebo was always followed by administration of the drug.
Participants in the alcohol arm had one intravenous (IV) catheter placed for phlebotomy. Participants were told they would receive both placebo and alcohol, but were blinded to the content of the study drug at the time of its administration. For placebo, a thin film of alcohol was placed on top of lime-flavored gelatin to provide the smell and flavor of alcohol. For alcohol, participants consumed an estimated dose of 0.6 g/kg of 100 proof vodka in lime-flavored gelatin, predicted to result in a 0.06% blood alcohol concentration (BAC) per Widmark’s equation. Participants were asked to consume the gelatin in 10 minutes. Breath alcohol (BrAC) was measured immediately before and after each of the MRI scans; participants were discharged when BrAC was ≤0.02 g/dl.
Participants in the acetate arm had two IV catheters placed, one for infusion and the other for phlebotomy. Infusates were prepared by the University of Colorado Hospital Research Pharmacy. Participants were told they would receive both placebo and acetate but were blinded to the content of the infusate at the time of its administration. The acetate protocol consisted of 290 mM acetate (Hospira, Inc., Lake Forest, IL) infused at a rate of 6 mg/kg/min for the first 10 minutes, followed by a continuous infusion rate of 3 mg/kg/min for approximately 60 minutes (Jiang et al., 2013). Placebo consisted of saline infused at the same rates as acetate.
Figure 1 shows the order of procedures. In all participants, three blood samples were obtained: i) at baseline, ii) after placebo administration which occurred 10 minutes before the first scan, and iii) after acetate or alcohol administration, both of which occurred 10 minutes before the second scan. Blood samples were drawn into EDTA vacutainer tubes and placed on ice. Samples were centrifuged at 1300g for 15 min at room temperature and the plasma transferred to 1.5 ml tubes and stored at −80°C until analysis. Plasma acetate and alcohol concentrations were measured using commercially available assays (Sigma-Aldrich, St. Louis, MO). Plates were read using Synergy™ HT Multi-Mode Microplate Reader (BioTek® Instruments, Inc., Highland Park, VT). Results were calculated from regression analysis of standard curves.
Figure 1.
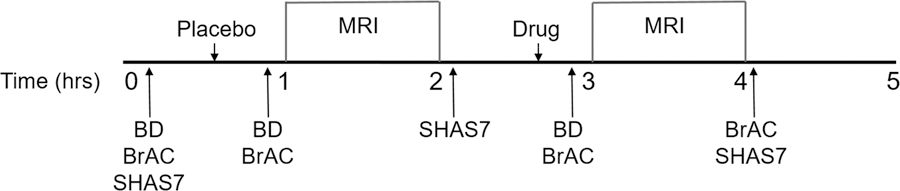
Study timeline. BD=blood draw, BrAC=Breath alcohol concentration measured with breathalyzer, SHAS7=Subjective High Assessment Scale 7.
Subjective High Assessment
The Subjective High Assessment Scale-7 Item (SHAS7) (Schuckit et al., 2000), a visual analog scale assesses subjective responses to seven measures of alcohol intoxication: high, clumsy, confused, dizzy, drunk, general alcohol effects, and difficulty concentrating. SHAS-7 was administered three times: i) at baseline upon arrival to the MR scanner, ii) immediately after the first MR scan, and iii) immediately after the second MR scan (Figure 1). Responses were rated on a scale from 0 (no effect) to 36 (extreme effect).
MR protocol
MR images were acquired on a 3T Siemens Skyra using a 24-channel neurovascular coil. Images acquisition began 10 minutes after placebo administration, acetate infusion, or alcohol administration: i) Structural 3D T1-weighted anatomical images were acquired with the following parameters: TR = 2200 ms; TE = 2.5 ms; flip angle = 8°; FOV = 220 mm2; 176 slices; 0.9 mm thickness; in-plane resolution = 0.9 mm2, ii) Quantitative perfusion images were acquired using a 3D-pulsed arterial spin labelling (pASL) sequence (Siemens WIP 818g) and the following parameters: ascending, interleaved, TR = 4600 ms; TE = 22 ms; flip angle = 28°; FOV = 220 mm2; 4 mm slice thickness with 0 mm gap; in-plane resolution = 1.7 x 1.7 mm2; bolus duration = 700 ms; inversion time = 1990 ms; 16 dynamic contrast measurements; GRAPPA acceleration factor = 2; axial plane angled through the AC-PC line.
ASL perfusion maps were automatically calculated by the vendor and linearly co-registered to each participants’ T1-weighted anatomical images in SPM8. The co-registered ASL maps were masked by the segmented gray matter volume from each participant’s T1-weighted anatomic image and spatially normalized to MNI template space. Normalization was completed following a Unified Segmentation protocol, applying individual participants’ forward deformations from the T1 anatomical normalization to the co-registered ASL volume (Ashburner & Friston, 2005). The resulting perfusion maps were spatially smoothed with a 6 mm FWHM kernel and brought to the second level for statistical analysis.
Statistical analyses
Demographic and behavioral data:
Continuous variables were tested for normality and analyzed using 2-tailed t-tests or analysis of variance (ANOVA) with group (drug arm) as between-participants factor. Categorical variables were analyzed with chi-squared tests. Significance was set at p<0.05. Statistical analyses were carried out using SPSS (IBM SPSS Statistics 25).
Alcohol and acetate assays and SHAS-7 scores:
Alcohol and acetate levels and SHAS-7 scores were analyzed using repeated measures ANOVA with drug condition (placebo vs drug) as within-participants factor and group (alcohol vs. acetate) as between-participants factor.
Whole brain cerebral blood flow map:
We compared placebo to alcohol and placebo to acetate CBF at the whole brain level using a paired t-test carried out in SPM8. Statistical thresholds were set at p<0.05 cluster-corrected for multiple comparisons, voxel level p<0.005, using 3dClust in AFNI (http://afni.nimh.nih.gov/afni/) (≥ 142 voxels or ≥ 3.78 cm3), consistent with the literature regarding cluster correction (Cox et al., 2017, Eklund et al., 2016).
Post-hoc thalamic CBF and correlation with acetate levels:
Significant clusters from the whole brain analysis were used to define regions-of-interest (ROI). As ROIs were post-hoc, results were used for exploration and not inference. Right and left thalamic CBF was extracted from the ROIs using the MarsBar tool (Brett M., et al. Region of interest analysis using an SPM toolbox [abstract] Presented at the 8th International Conference on Functional Mapping of the Human Brain, June 2–6, 2002, Sendai, Japan). Right and left thalamic CBF were correlated with acetate levels, controlling for the effects of baseline acetate level and age, using Pearson correlation coefficient.
Results
Twenty-eight healthy participants were consented. Four were excluded due to a need for anxiolytics (1), job change (1), elevated body mass index (1), or difficult catheter insertion in different study (1), leaving 24 who completed the study. Alcohol and acetate groups did not differ significantly in sex distribution, education, or AUDIT scores (Table 1).
Table 1.
Demographics and Alcohol use disorder test (AUDIT) scores.
| Acetate N=12 |
Alcohol N=12 |
p-value | |
|---|---|---|---|
| Age (years) | 27.8 ± 4.2 | 31.1 ± 3.9 | 0.06 |
| Education (years) | 17.0 ± 1.7 | 17.5 ±1.9 | 0.51 |
| Gender (%women) (F/M) | 83% (10/2) | 67% (8/4) | 0.35 |
| AUDIT score | 3.9 ± 1.1 | 3.7 ± 1.6 | 0.66 |
Values are mean ± SD
Blood and breath alcohol levels:
Blood (BAC) and breath (BrAc) alcohol levels at baseline and after placebo were zero for all participants. After alcohol administration, there was a significant increase in BAC (0.064 ± 0.036, p<0.001) and BrAc (0.054 ± 0.011, p<0.001) (mean ± SD g/dL).
Plasma acetate levels:
Plasma acetate levels significantly increased compared to placebo, both after alcohol ingestion (0.96 ± 0.20 vs. 1.73 ± 0.50, p<0.001) and acetate administration (0.88 ± 0.11 vs. 1.52 ± 0.47, p<0.001) (placebo vs. drug (mM), mean ± SD) and did not differ from each other (Figure 2).
Figure 2.
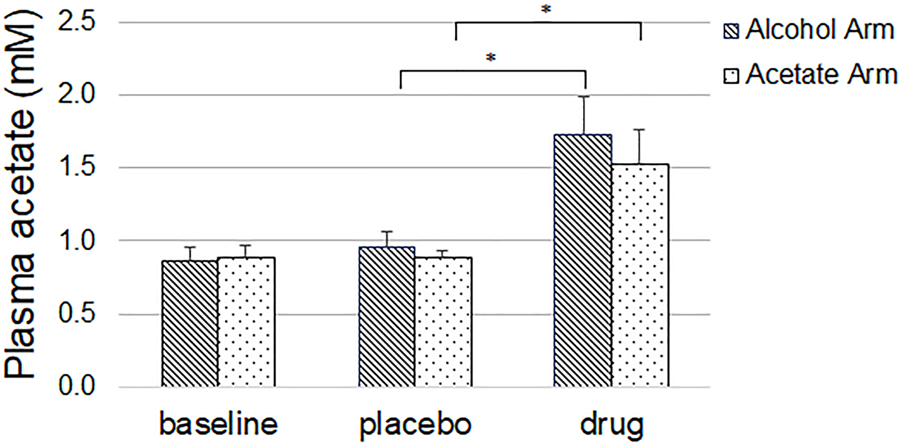
Compared to placebo, alcohol and acetate were associated with a significant increase in acetate levels. Values are mean, error bars are standard deviation
Whole brain cerebral blood flow map:
Figure 3 (upper two rows) shows that compared to placebo, acetate administration was associated with significantly increased CBF in bilateral medial thalamus and left parietal cortex. Figure 3 (lower two rows) shows that compared to placebo, alcohol consumption was associated with significantly increased CBF in bilateral medial thalamus, right anterior and posterior cingulate cortex, right orbitofrontal, ventral medial prefrontal, superior prefrontal cortex, and right hippocampus and putamen. There were no brain regions associated with lower CBF for alcohol or acetate compared to placebo.
Figure 3.
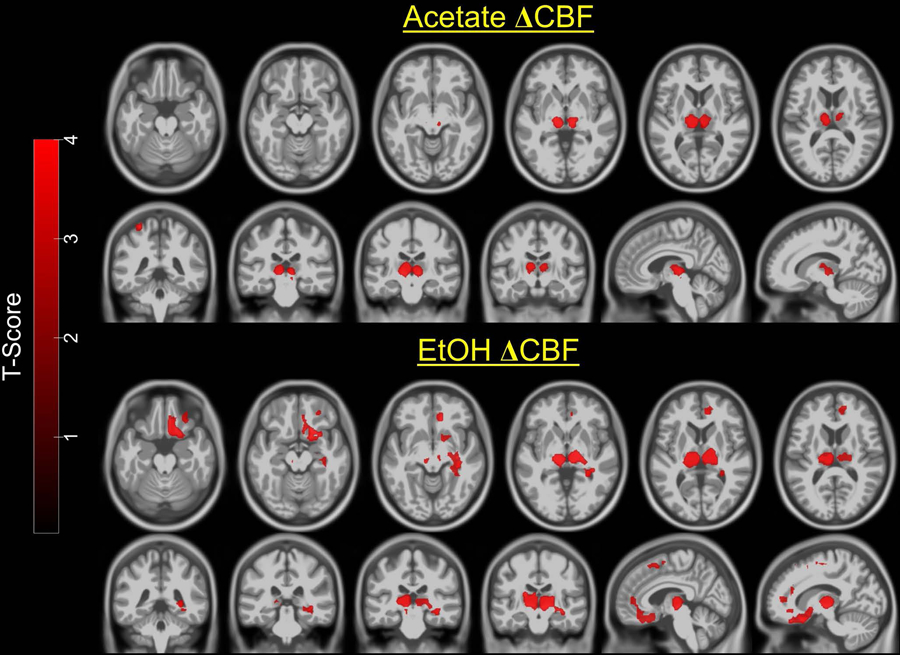
Statistical parametric maps of the paired t-tests overlaid on a standard template. <i>Upper two rows</i>: compared to placebo, acetate was associated with increased CBF in medial thalamus and left parietal cortex. <i>Lower two rows</i>: compared to placebo, alcohol was associated with increased CBF in medial thalamus, right orbitofrontal cortex, medial prefrontal cortex, cingulate cortex, and hippocampus; Images are in neurological convention. Color bar=t-value. Maps were thresholded at p<0.05 cluster-corrected for multiple comparisons, voxel level p<0.005
Post-hoc thalamic cerebral blood flow:
CBF (ml/100g/min) extracted from cluster-defined ROIs are listed in Table 2. Left and right thalamic CBF values did not differ so they were combined into bilateral thalamic CBF. Figure 4 shows the change in bilateral thalamic CBF for each participant in the acetate arm and alcohol arm.
Table 2.
Cerebral blood flow extracted from thalamic clusters
| Oral placebo | Oral alcohol | IV placebo | IV acetate | |
|---|---|---|---|---|
| Right thalamus | 25.0 ± 7.5 | 30.0 ± 9.7 | 26.9 ± 5.7 | 31.7 ± 5.6 |
| Left thalamus | 24.9 ± 6.7 | 29.9 ± 8.9 | 27.3 ± 5.6 | 32.8 ± 6.8 |
Values are mean ± SD; units are cc/100mg/min. Note that CBF values were extracted from cluster-defined ROIs so further statistical testing was not performed.
Figure 4.
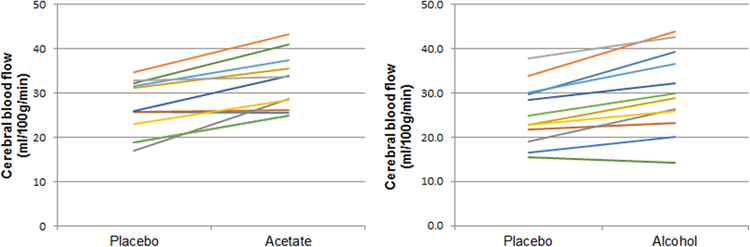
Plots showing bilateral thalamic CBF after placebo and after drug (acetate or alcohol). Each line represents a participant.
Correlations between thalamic CBF and acetate:
There were no significant correlations between change in thalamic CBF and acetate levels.
Subjective High Assessment Scale (SHAS-7):
SHAS-7 total score increased significantly after alcohol compared to placebo (8.4 ± 23.7 vs. 78.8 ± 39.4, p<0.001) but not after acetate compared to placebo (5.4 ± 10.9 vs. 9.7 ± 20.6, ns) (Figure 5)
Figure 5.
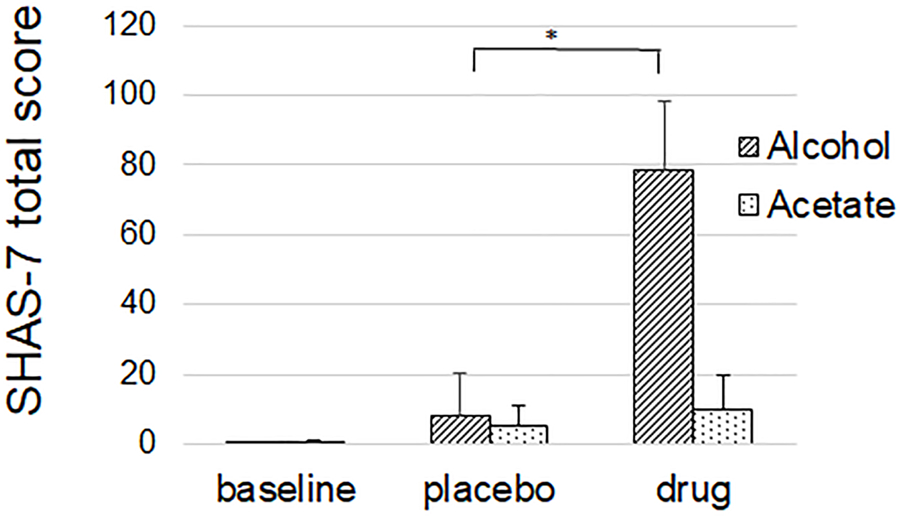
Subjective high assessment score-7 (SHAS-7) was associated with an increase after alcohol, but not after acetate. Values are mean. Error bars are standard deviation.
Discussion
In healthy participants, we observed that consumption of alcohol to attain alcohol levels consistent with intoxication was associated with increased regional cerebral blood flow (CBF) in bilateral thalami, the right hippocampus, and the right prefrontal cortex, compared to placebo. Acetate infusion to achieve a serum acetate level comparable to that observed with alcohol consumption was associated with an increase in CBF in bilateral thalami, but not hippocampi or the prefrontal cortex. Although acetate is metabolized in the brain and may enhance CBF during acute and chronic alcohol consumption, this is the first study to investigate the effects of alcohol and acetate on regional CBF.
Associations of alcohol and CBF
We observed that a single dose of alcohol sufficient to produce mild intoxication was associated with increased CBF to multiple cortical and subcortical brain regions. Increases in cortical CBF were found in right orbitofrontal cortex, cingulate cortex, and ventral medial prefrontal cortex. Early studies on the effect of alcohol on CBF measured with 133Xe-CT (Mathew and Wilson, 1986, Sano et al., 1993) and 15O-water positron emission tomography (PET) (Volkow et al., 1988a) similarly reported increases in CBF primarily in prefrontal and temporal lobe regions. More recent studies using SPECT and MR techniques have reported 4%−20% increases in global and regional CBF (Marxen et al., 2014, Strang et al., 2015, Tolentino et al., 2011, Gundersen et al., 2013) (for review see (Bjork and Gilman, 2014)). Only a few studies observed alcohol-induced decreases in CBF mainly in cerebellum (Volkow et al., 1988b), an area that was not fully imaged in our MR ASL protocol. We observed that CBF increased asymmetrically, favoring the right hemisphere, consistent with other studies, possibly reflecting the time course of blood alcohol levels (BAL) (Newlin et al., 1982, Marxen et al., 2014) or hemispheric functional specialization (Volkow et al., 2008). The magnitude and regional CBF response to alcohol, however, can also vary depending on the dose of alcohol administered, timing of its administration, and individual sensitivity to alcohol (Marxen et al., 2014, Sano et al., 1993, Strang et al., 2015, Volkow et al., 1988a, Tolentino et al., 2011). Although only a few studies have tested for sex effects, differences in cortical CBF response to alcohol have been reported in women compared to men (Rickenbacher et al., 2011).
The association between alcohol and increased CBF in bilateral thalamus and subcortical nuclei is consistent with prior reports (Gundersen et al., 2013, Marxen et al., 2014). Using an intravenous alcohol infusion protocol, Marxen modeled the time course of CBF during two phases: the early ramp-up and the later steady-state of 0.6 g/kg breath alcohol level (BrAc). Bilateral medial thalamic CBF increased during this ascending part of the BrAc curve (Marxen et al., 2014). Gunderson reported 20% increases in global CBF with the greatest regional increases in thalamus and frontal lobe that they attributed to vascular anatomy, where large feeding vessels entered the brain (Gundersen et al., 2013).
A major motivation for studying the effects of acute alcohol administration with neuroimaging methods is to advance our understanding of mechanisms by which alcohol induces its behavioral effects. Prior work and reviews on this topic (Bjork and Gilman, 2014, Volkow et al., 2017) suggest that acute alcohol affects brain regions, circuits, and neurotransmitter systems involved in reward, attention and arousal, affect, and cognitive control. Animal studies consistently demonstrate that acute alcohol activates the mesolimbic reward system by increasing dopamine release in the ventral striatum, although human PET dopamine studies have been less consistent (Volkow et al., 2017). With task-based fMRI, alcohol-associated changes in striatal, prefrontal, and limbic regions during reward processing, decision-making, and emotion processing have been widely reported (Claus and Hendershot, 2015, Anderson et al., 2011, Bjork and Gilman, 2014, Gilman et al., 2008). Many studies of alcohol administration and CBF focus on the pharmacokinetics of acute alcohol (Volkow et al., 1988b, Marxen et al., 2014, Tolentino et al., 2011). Surprisingly, few report correlations with behavioral measures (Rickenbacher et al., 2011). One difficulty in interpreting CBF studies is that alcohol itself is vasoactive. Alcohol injected intra-arterially decreases blood flow due to direct vasoconstrictive action on arteries (Fewings et al., 1966). However, alcohol is also a known peripheral vasodilator that decreases vascular resistance in skin, heart, and skeletal muscle and increases capillary flow in these organs. In summary, neuroimaging studies on the effect of alcohol administration have provided evidence of brain-based behavioral changes that are highly complex. To add to the complexity, evidence indicates that the ethanol metabolite acetate may add to the complexity of ethanol’s effect on CBF as discussed below.
Associations of acetate and CBF
Schwartz et al. proposed that acetate plays a role in the cerebrovascular effects of alcohol. Their study of twenty-four healthy men who underwent SPECT brain imaging after consuming 0.6 g/kg alcohol in juice demonstrated a correlation between acetate levels and CBF in a subset of men younger than 37 years. They postulated that acetate increases CBF by increasing adenosine, the metabolite of acetate and known vasodilator. Although alcohol increased average CBF by 4% across participants, alcohol levels correlated negatively with cortical CBF, especially in the older men (mean age 70 years). The authors speculate that paradoxical result involves age-related differences in adenosine sensitivity and vascular compliance may play a role. Accordingly, the vasoconstrictive effects of alcohol are counterbalanced by vasodilatory effects of acetate (via adenosine) and age is hypothesized to tip the balance toward the former (Schwartz et al., 1993). Our study is the first to investigate whether acetate (and its metabolites) has similar effects on CBF as alcohol (and its metabolites). Using the protocol of Jiang et al. (2013), we achieved acetate concentrations equal to that in the alcohol arm (Figure 2). Acetate, but not ethanol, was associated with an increase in CBF in a small region of the parietal cortex. Ethanol, but not acetate, was associated with increased CBF in prefrontal and limbic regions and both ethanol and acetate increased CBF in bilateral medial thalamus. These differences suggest that a majority of the cortical CBF response to alcohol is not mediated by acetate, which is inconsistent with the findings of Schwartz et al. (1993).
Although it is possible that the thalamic CBF response involves acetate, a major question is “Why the thalamus as opposed to other brain regions?” Using 11C-acetate PET, Volkow et al. (2013) found that acute alcohol increased acetate in all brain regions and there was a significant regional effect, with the largest difference between the cerebellum (highest acetate) and thalamus (lowest acetate). The thalamus is well known to be vulnerable to injury in Wernicke’s encephalopathy due to thiamine deficiency, a common comorbidity with AUD. An animal model of thiamine deficiency and alcohol toxicity determined that supplemental acetate reduced microglial activation and neural degeneration in thalamus, suggesting a protective action of acetate in the thalamus (Qin and Crews, 2014). Qin et al. (2014) noted that although acetate serves as an alternative energy source to glucose, acetate transport into thalamus is lower compared to other brain regions. As a result, thalamus may not receive adequate fuel as compared to other regions in the setting of thiamine deficiency and excessive alcohol intake. Thus, an increase in thalamic CBF associated with acetate could be protective. Although at this point speculative, the role of short chain fatty acid receptors, FFA2 and FFA3, for which acetate is an agonist, should also not be ignored as a potential mediator of the action of acetate on thalamic blood flow. FFA2 and FFA3 have been shown to have a role in metabolic, immunologic, and neuronal (sympathetic) function (Milligan et al., 2017). Given that it is metabolized almost exclusively by glia (Waniewski and Martin, 1998), acetate induced glial activation in the thalamus is another possible explanation. Future studies are needed to ascertain the mechanism, but our observation that acetate was associated with an increase in thalamic CBF is consistent with a regional effect.
Disentangling effects of alcohol and its metabolites
Numerous studies have attempted to separate the behavioral and pharmacological effect of alcohol from its metabolites. Most of the ethanol is metabolized in the liver to acetaldehyde and acetate. While both enter the circulation only acetate freely diffuses into the brain (as does ethanol). However, it has been shown that some ethanol is metabolized in the brain via catalase (Cohen et al., 1980) and cytochrome P-4502E1 (CYP2E1) to produce acetaldehyde. To what extent are brain effects due to alcohol or its biologically active metabolites? On one extreme, the hypothesis that alcohol is merely a “prodrug” for the effects of acetaldehyde has been debated (Quertemont et al., 2005). However, the amount of acetaldehyde produced through the catalase and brain CYP2E1 system is difficult to measure in humans and the behavioral significance remains unknown. Although we cannot exclude contributions from acetaldehyde on the results seen after ethanol, acetaldehyde would not be a candidate for mediating acetate’s actions since acetate is not metabolized to acetaldehyde. In addition, the route of administration differed in the two arms, with oral alcohol stimulating the enteric neural system whereas intravenous acetate would not. Further studies are needed to determine the role of the “gut-brain” axis in the CNS response.
Acetate as possible therapeutic substance in alcohol withdrawal
Emerging evidence suggests that heavy alcohol exposure facilitates the utilization of acetate for metabolic activity in brain and other organs. Compared to social drinkers, heavy drinkers have higher circulating acetate levels (Nuutinen et al., 1985), higher brain uptake of acetate following alcohol consumption (Volkow et al., 2013), and faster turnover of acetate in the brain (Jiang et al., 2013). Proteomics have demonstrated lower abundance of glycolytic enzymes and an increase in reducing equivalents in brains of individuals with AUD, suggesting greater acetate metabolism in AUD (Enculescu et al., 2019). Animals exposed chronically to alcohol demonstrated higher acetate turnover compared to controls (Hsieh et al., 2018). This compensatory increase in acetate metabolism could contribute to alcohol dependence. It has been proposed that high brain acetate levels in heavy drinkers represent a form of caloric “reward” that perpetuates drinking, and that falling acetate levels associated with sobriety may contribute to withdrawal. Indeed, acetate supplementation was shown to decrease alcohol withdrawal tremors in rats (Derr et al., 1981).
Acetate also has anti-inflammatory properties that may be of benefit in alcohol use disorder. Acetate decreased activated microglia and inflammatory markers in models of experimentally induced neuro-inflammation (Reisenauer et al., 2011, Brissette et al., 2012, Soliman et al., 2012). An anti-inflammatory action is thought to be an additional mechanism for the reduced thalamic degeneration in the alcohol and thiamine-deficiency model discussed earlier (Qin and Crews, 2014). Acetate and alcohol also increase the formation of adenosine (Carmichael et al., 1988) and in addition to cerebrovascular effects mentioned above adenosine may play a role in regulating inflammatory processes through A2A receptors on immune cells (Sitkovsky et al., 2004).
Subjective effects of acute alcohol and acetate
In this investigation, alcohol consumption was associated with feelings of mild intoxication, as expected, but acetate was not. No prior study comparing the behavioral sequelae of acetate to alcohol have been conducted in humans. Our results provide evidence that the intoxicating effects of alcohol appear to be associated with cortical function and that acetate is not associated with feelings of intoxication, at least at the levels generated in our protocol in healthy individuals. However, in animals, acetate was associated with decreases in motor activity and coordination (Correa et al., 2003, Israel et al., 1994). Future studies focusing on withdrawal symptoms, motor function, and coordination are needed to fully understand the effect of acetate in humans.
Limitations
Our study is not without limitations. The assignment to alcohol or acetate was done for convenience, rather than by randomization, leaving us only able to assess association, rather than causality. Second, the sample size of this pilot study was modest. Third, to limit costs and eliminate drug carry over effects, we fixed the drug administration order, so that placebo always preceded active drug. Hence, order effects cannot be excluded. We believe order effects are unlikely, given the magnitude of drug-associated changes observed and evidence of good test-retest reliability of the ASL CBF method reported previously (Chen et al., 2011). Fourth, since ethanol and acetate were measured in the early phases of absorption and metabolism, the blood levels may not reflect levels in the brain at the time of the measurement of CBF or subjective intoxication. Fifth, more study participants were women than men, although the numbers were not statistically different. Sex differences in ethanol metabolism, behavioral response, progression to alcohol use disorder have been reported (Erol and Karpyak, 2015) and the phase of menstrual cycle has been hypothesized to play a role in alcohol consumption (Carroll et al., 2015). The preponderance of early literature in male-only participants and one study showing sex differences in CBF response to alcohol underscore the need to consider sex effects (Rickenbacher et al., 2011). Also, we did not control for genetic variations in ethanol metabolism although we excluded participants who reported a flushing response to alcohol (none did). The sample size of this pilot study precludes inferences about sex or genetic effects on the CBF response which will require a larger study. Finally, we are aware that the CBF values we report here are somewhat lower than others in the literature. For this study, we used the reconstruction engine provided with the ASL work-in-progress by Siemens (WIP 818g) that utilizes the calculation of CBF as given in Wang et al. (Wang et al., 2005). It includes an inverse dependence on α, the inversion efficiency, which was not directly measured. However, a 5% error in α would result in a difference in values close to the reported literature values. This amounts to a scaling factor that was uniform across all runs.
Conclusion
In conclusion, acetate does not appear to mediate the increase in cortical CBF seen after acute alcohol administration; however, acetate may well be responsible for changes in thalamic CBF after alcohol ingestion. Compared to other brain regions, the thalamus may differ in its ability to metabolize acetate or its expression of receptors responsive to acetate. As the thalamus is vulnerable in alcohol-related injury, future studies to understand these mechanisms are needed. This is the first study comparing acetate to alcohol on brain function in humans, an important step given acetate’s diverse physiological effects and potential therapeutic applications.
Acknowledgements
The authors would like to acknowledge Siemens for sharing their Investigational Software for ASL, Jenny Yu, MS, for technical help on alcohol and acetate analyses, and Debra Singel, RT, for running the MR scanner.
Role of funding source
Funding for this study was provided by the National Institute of Alcohol Abuse and Alcoholism (NIAAA) T32AA7464 (PH) to DJY; R24AA013162 (BT and PH); R24AA019661 (ELB); Brain & Behavior Research Foundation (DJY); Colorado Clinical and Translational Sciences Institute (CCTSI) UL1TR001082 (JT, ELB) and microgrant (DJY); R01GM121081, R25GM111901, and R25GM11901–04S1 (DHG). None of the funding entities had a role in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Footnotes
Conflict of Interest
Dr. Tabakoff is founder and CEO of Lohocla Research Corporation and Dr. Hoffman is an employee of Lohocla Research Corporation (U44AA024905, UG3DA047680). The reported work is independent of, and in no conflict with work performed for Lohocla. All other authors report no conflict of interest.
References:
- ANDERSON BM, STEVENS MC, MEDA SA, JORDAN K, CALHOUN VD & PEARLSON GD 2011. Functional imaging of cognitive control during acute alcohol intoxication. Alcohol Clin Exp Res, 35, 156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BJORK JM & GILMAN JM 2014. The effects of acute alcohol administration on the human brain: insights from neuroimaging. Neuropharmacology, 84, 101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRISSETTE CA, HOUDEK HM, FLODEN AM & ROSENBERGER TA 2012. Acetate supplementation reduces microglia activation and brain interleukin-1beta levels in a rat model of Lyme neuroborreliosis. J Neuroinflammation, 9, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARMICHAEL FJ, SALDIVIA V, VARGHESE GA, ISRAEL Y & ORREGO H 1988. Ethanol-induced increase in portal blood flow: role of acetate and A1- and A2-adenosine receptors. Am J Physiol, 255, G417–23. [DOI] [PubMed] [Google Scholar]
- CARROLL HA, LUSTYK MK & LARIMER ME 2015. The relationship between alcohol consumption and menstrual cycle: a review of the literature. Arch Womens Ment Health, 18, 773–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Y, WANG DJ & DETRE JA 2011. Test-retest reliability of arterial spin labeling with common labeling strategies. J Magn Reson Imaging, 33, 940–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLAUS ED & HENDERSHOT CS 2015. Moderating effect of working memory capacity on acute alcohol effects on BOLD response during inhibition and error monitoring in male heavy drinkers. Psychopharmacology (Berl), 232, 765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN G, SINET PM & HEIKKILA R 1980. Ethanol oxidation by rat brain in vivo. Alcohol Clin Exp Res, 4, 366–70. [DOI] [PubMed] [Google Scholar]
- CORREA M, ARIZZI MN, BETZ A, MINGOTE S & SALAMONE JD 2003. Open field locomotor effects in rats after intraventricular injections of ethanol and the ethanol metabolites acetaldehyde and acetate. Brain Res Bull, 62, 197–202. [DOI] [PubMed] [Google Scholar]
- COTTLER LB, SCHUCKIT MA, HELZER JE, CROWLEY T, WOODY G, NATHAN P & HUGHES J 1995. The DSM-IV field trial for substance use disorders: major results. Drug Alcohol Depend, 38, 59–69; discussion 71–83. [DOI] [PubMed] [Google Scholar]
- COX RW, CHEN G, GLEN DR, REYNOLDS RC & TAYLOR PA 2017. FMRI Clustering in AFNI: False-Positive Rates Redux. Brain Connect, 7, 152–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DERR RF, DRAVES K & DERR M 1981. Abatement by acetate of an ethanol withdrawal syndrome. Life Sci, 29, 1787–90. [DOI] [PubMed] [Google Scholar]
- EKLUND A, NICHOLS TE & KNUTSSON H 2016. Cluster failure: Why fMRI inferences for spatial extent have inflated false-positive rates. Proceedings of the National Academy of Sciences of the United States of America, 113, 7900–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCULESCU C, KERR ED, YEO KYB, SCHENK G, FORTES MRS & SCHULZ BL 2019. Proteomics Reveals Profound Metabolic Changes in the Alcohol Use Disorder Brain. ACS Chem Neurosci. [DOI] [PubMed]
- EROL A & KARPYAK VM 2015. Sex and gender-related differences in alcohol use and its consequences: Contemporary knowledge and future research considerations. Drug Alcohol Depend, 156, 1–13. [DOI] [PubMed] [Google Scholar]
- FEWINGS JD, HANNA MJ, WALSH JA & WHELAN RF 1966. The effects of ethyl alcohol on the blood vessels of the hand and forearm in man. Br J Pharmacol Chemother, 27, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILMAN JM, RAMCHANDANI VA, DAVIS MB, BJORK JM & HOMMER DW 2008. Why we like to drink: a functional magnetic resonance imaging study of the rewarding and anxiolytic effects of alcohol. J Neurosci, 28, 4583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUNDERSEN H, VAN WAGENINGEN H & GRÜNER R 2013. Alcohol-induced changes in cerebral blood flow and cerebral blood volume in social drinkers. Alcohol and Alcoholism (Oxford, Oxfordshire), 48, 160–165. [DOI] [PubMed] [Google Scholar]
- HSIEH YJ, WU LC, KE CC, CHANG CW, KUO JW, HUANG WS, CHEN FD, YANG BH, TAI HT, CHEN SC & LIU RS 2018. Effects of the Acute and Chronic Ethanol Intoxication on Acetate Metabolism and Kinetics in the Rat Brain. Alcohol Clin Exp Res, 42, 329–337. [DOI] [PubMed] [Google Scholar]
- ISRAEL Y, ORREGO H & CARMICHAEL FJ 1994. Acetate-mediated effects of ethanol. Alcohol Clin Exp Res, 18, 144–8. [DOI] [PubMed] [Google Scholar]
- JIANG L, GULANSKI BI, DE FEYTER HM, WEINZIMER SA, PITTMAN B, GUIDONE E, KORETSKI J, HARMAN S, PETRAKIS IL, KRYSTAL JH & MASON GF 2013. Increased brain uptake and oxidation of acetate in heavy drinkers. Journal of Clinical Investigation, 123, 1605–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARXEN M, GAN G, SCHWARZ D, MENNIGEN E, PILHATSCH M, ZIMMERMANN US, GUENTHER M & SMOLKA MN 2014. Acute effects of alcohol on brain perfusion monitored with arterial spin labeling magnetic resonance imaging in young adults. J Cereb Blood Flow Metab, 34, 472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHEW RJ & WILSON WH 1986. Regional cerebral blood flow changes associated with ethanol intoxication. Stroke, 17, 1156–9. [DOI] [PubMed] [Google Scholar]
- MILLIGAN G, SHIMPUKADE B, ULVEN T & HUDSON BD 2017. Complex Pharmacology of Free Fatty Acid Receptors. Chem Rev, 117, 67–110. [DOI] [PubMed] [Google Scholar]
- NEWLIN DB, GOLDEN CJ, QUAIFE M & GRABER B 1982. Effect of alcohol ingestion on regional cerebral blood flow. Int J Neurosci, 17, 145–50. [DOI] [PubMed] [Google Scholar]
- NUUTINEN H, LINDROS K, HEKALI P & SALASPURO M 1985. Elevated blood acetate as indicator of fast ethanol elimination in chronic alcoholics. Alcohol, 2, 623–626. [DOI] [PubMed] [Google Scholar]
- PAWLOSKY RJ, KASHIWAYA Y, SRIVASTAVA S, KING MT, CRUTCHFIELD C, VOLKOW N, KUNOS G, LI TK & VEECH RL 2010. Alterations in brain glucose utilization accompanying elevations in blood ethanol and acetate concentrations in the rat. Alcohol Clin Exp Res, 34, 375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QIN L & CREWS FT 2014. Focal thalamic degeneration from ethanol and thiamine deficiency is associated with neuroimmune gene induction, microglial activation, and lack of monocarboxylic acid transporters. Alcohol Clin Exp Res, 38, 657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUERTEMONT E, ERIKSSON CJ, ZIMATKIN SM, PRONKO PS, DIANA M, PISANO M, RODD ZA, BELL RR & WARD RJ 2005. Is ethanol a pro-drug? Acetaldehyde contribution to brain ethanol effects. Alcohol Clin Exp Res, 29, 1514–21. [DOI] [PubMed] [Google Scholar]
- REISENAUER CJ, BHATT DP, MITTENESS DJ, SLANCZKA ER, GIENGER HM, WATT JA & ROSENBERGER TA 2011. Acetate supplementation attenuates lipopolysaccharide-induced neuroinflammation. J Neurochem, 117, 264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RICKENBACHER E, GREVE DN, AZMA S, PFEUFFER J & MARINKOVIC K 2011. Effects of alcohol intoxication and gender on cerebral perfusion: an arterial spin labeling study. Alcohol, 45, 725–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANO M, WENDT PE, WIRSEN A, STENBERG G, RISBERG J & INGVAR DH 1993. Acute effects of alcohol on regional cerebral blood flow in man. J Stud Alcohol, 54, 369–76. [DOI] [PubMed] [Google Scholar]
- SAUNDERS JB, AASLAND OG, BABOR TF, DE LA FUENTE JR & GRANT M 1993. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO Collaborative Project on Early Detection of Persons with Harmful Alcohol Consumption--II. Addiction, 88, 791–804. [DOI] [PubMed] [Google Scholar]
- SCHUCKIT MA, SMITH TL, KALMIJN J, TSUANG J, HESSELBROCK V & BUCHOLZ K 2000. Response to alcohol in daughters of alcoholics: a pilot study and a comparison with sons of alcoholics. Alcohol Alcohol, 35, 242–8. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ JA, SPEED NM, GROSS MD, LUCEY MR, BAZAKIS AM, HARIHARAN M & BERESFORD TP 1993. Acute effects of alcohol administration on regional cerebral blood flow: the role of acetate. Alcoholism, Clinical and Experimental Research, 17, 1119–1123. [DOI] [PubMed] [Google Scholar]
- SITKOVSKY MV, LUKASHEV D, APASOV S, KOJIMA H, KOSHIBA M, CALDWELL C, OHTA A & THIEL M 2004. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol, 22, 657–82. [DOI] [PubMed] [Google Scholar]
- SOLIMAN ML, SMITH MD, HOUDEK HM & ROSENBERGER TA 2012. Acetate supplementation modulates brain histone acetylation and decreases interleukin-1beta expression in a rat model of neuroinflammation. J Neuroinflammation, 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRANG NM, CLAUS ED, RAMCHANDANI VA, GRAFF-GUERRERO A, BOILEAU I & HENDERSHOT CS 2015. Dose-dependent effects of intravenous alcohol administration on cerebral blood flow in young adults. Psychopharmacology (Berl), 232, 733–44. [DOI] [PubMed] [Google Scholar]
- TOLENTINO NJ, WIERENGA CE, HALL S, TAPERT SF, PAULUS MP, LIU TT, SMITH TL & SCHUCKIT MA 2011. Alcohol Effects on Cerebral Blood Flow in Subjects With Low and High Responses to Alcohol. Alcoholism: Clinical and Experimental Research, 35, 1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOLKOW ND, KIM SW, WANG GJ, ALEXOFF D, LOGAN J, MUENCH L, SHEA C, TELANG F, FOWLER JS, WONG C, BENVENISTE H & TOMASI D 2013. Acute alcohol intoxication decreases glucose metabolism but increases acetate uptake in the human brain. NeuroImage, 64, 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOLKOW ND, MA Y, ZHU W, FOWLER JS, LI J, RAO M, MUELLER K, PRADHAN K, WONG C & WANG GJ 2008. Moderate doses of alcohol disrupt the functional organization of the human brain. Psychiatry Res, 162, 205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOLKOW ND, MULLANI N, GOULD L, ADLER SS, GUYNN RW, OVERALL JE & DEWEY S 1988a. Effects of acute alcohol intoxication on cerebral blood flow measured with PET. Psychiatry Research, 24, 201–209. [DOI] [PubMed] [Google Scholar]
- VOLKOW ND, MULLANI N, GOULD L, ADLER SS, GUYNN RW, OVERALL JE & DEWEY S 1988b. Effects of acute alcohol intoxication on cerebral blood flow measured with PET. Psychiatry Res, 24, 201–9. [DOI] [PubMed] [Google Scholar]
- VOLKOW ND, WANG GJ, FRANCESCHI D, FOWLER JS, THANOS P, MAYNARD L, GATLEY SJ, WONG C, VEECH RL, KUNOS G & KAI LI T 2006. Low doses of alcohol substantially decrease glucose metabolism in the human brain. NeuroImage, 29, 295–301. [DOI] [PubMed] [Google Scholar]
- VOLKOW ND, WANG GJ, SHOKRI KOJORI E, FOWLER JS, BENVENISTE H & TOMASI D 2015. Alcohol decreases baseline brain glucose metabolism more in heavy drinkers than controls but has no effect on stimulation-induced metabolic increases. J Neurosci, 35, 3248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOLKOW ND, WIERS CE, SHOKRI-KOJORI E, TOMASI D, WANG GJ & BALER R 2017. Neurochemical and metabolic effects of acute and chronic alcohol in the human brain: Studies with positron emission tomography. Neuropharmacology, 122, 175–188. [DOI] [PubMed] [Google Scholar]
- WANG J, ZHANG Y, WOLF RL, ROC AC, ALSOP DC & DETRE JA 2005. Amplitude-modulated continuous arterial spin-labeling 3.0-T perfusion MR imaging with a single coil: feasibility study. Radiology, 235, 218–28. [DOI] [PubMed] [Google Scholar]
- WANIEWSKI RA & MARTIN DL 1998. Preferential Utilization of Acetate by Astrocytes Is Attributable to Transport. The Journal of Neuroscience, 18, 5225–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]