

Abstract
Altered cellular metabolism is a hallmark of cancer. Metabolic rewiring in cancer cells occurs due to the activation of oncogenes, inactivation of tumor suppressor genes, and/or other adaptive changes in cell signaling pathways. Furthermore, altered metabolism is also reported in tumor-corrupted stromal cells as a result of their interaction with cancer cells or due to their adaptation in the dynamic tumor microenvironment. Metabolic alterations are associated with dysregulation of metabolic enzymes and tumor-stromal metabolic crosstalk is vital for the progressive malignant journey of the tumor cells. Therefore, several therapies targeting metabolic enzymes have been evaluated and/or are being investigated in preclinical and clinical studies. In this review, we discuss some important metabolic enzymes that are altered in tumor and/or stromal cells, and focus on their role in supporting tumor growth. Moreover, we also discuss studies carried out in various cancers to target these metabolic abnormalities for therapeutic exploitation.
INTRODUCTION
The ability to generate energy and biomass is essential for a cell to sustain its growth and survival. Normal cells maintain an intricate balance of metabolic pathways and respond to internal and external cues as per the body’s overall requirement. However, cancer cells, being highly proliferative and often encountering unusual environmental stresses, exhibit adaptive changes in metabolic pathways associated with dysregulation of metabolic enzymes [1, 2]. Indeed, enhanced uptake of glucose by cancer cells was recognized by Warburg about a century ago, and the basic idea of aerobic glycolysis in cancer, termed “Warburg effect” still holds true and has gained further support [1, 3]. Glycolysis is a physiological response to hypoxia in normal cells, but cancer cells tend to constitutively take up more glucose and produce lactate regardless of oxygen availability. Increased glycolytic flux provides quick energy and supplies glycolytic intermediates to secondary pathways to fulfill the metabolic and biosynthetic demands of proliferating cancer cells. This metabolic rewiring primarily takes place in response to the activation of oncogenes and/or inactivation of tumor suppressor genes that alter the expression of metabolic enzymes via transcriptional and post-transcriptional changes [1]. Further, mutations in genes encoding enzymes of key metabolic pathways are also associated with certain hereditary and sporadic forms of cancers [4, 5].
It is now well established that both tumor and tumor-corrupted normal cells (referred to as ‘stromal’ cells) reside within a malignant tumor [6]. Stromal cells include fibroblasts, endothelial cells, immune cells, nerve cells and in many cases microbial cells as well. Incidentally, metabolic rewiring has been observed in these cells of the tumor microenvironment beside cancer cells [7]. It is believed that metabolic changes in the stromal cells result from their functional interactions with the tumor cells and are possibly exploited by the tumor cells for their growth. Data suggest that tumor-stromal metabolic crosstalk is vital for the progressive growth of tumors. It also helps cancer cells in facing the challenges that they encounter during their malignant journey [7]. In this review, we discuss some of the important metabolic enzymes that are altered in tumor cells and stromal cells, and focus on their roles in supporting tumor growth. Moreover, we also discuss preclinical and clinical studies conducted to evaluate the therapeutic efficacy of targeting certain important metabolic enzymes in cancer.
Dysregulation of metabolic enzymes in cancer cells and functional significance
Metabolic enzymes are important nodes of biological metabolic networks that regulate the flux of metabolites as per the cellular and bodily requirements of sustaining growth and physiological homeostasis. This balance is altered in cancer cells, and accordingly, frequent alterations in their expression are reported (Figure 1). Below we discuss some of the important tumor cell-associated metabolic enzymes and their pathobiological significance.
Figure 1: Dysregulation of glucose metabolizing enzymes in cancer cells.
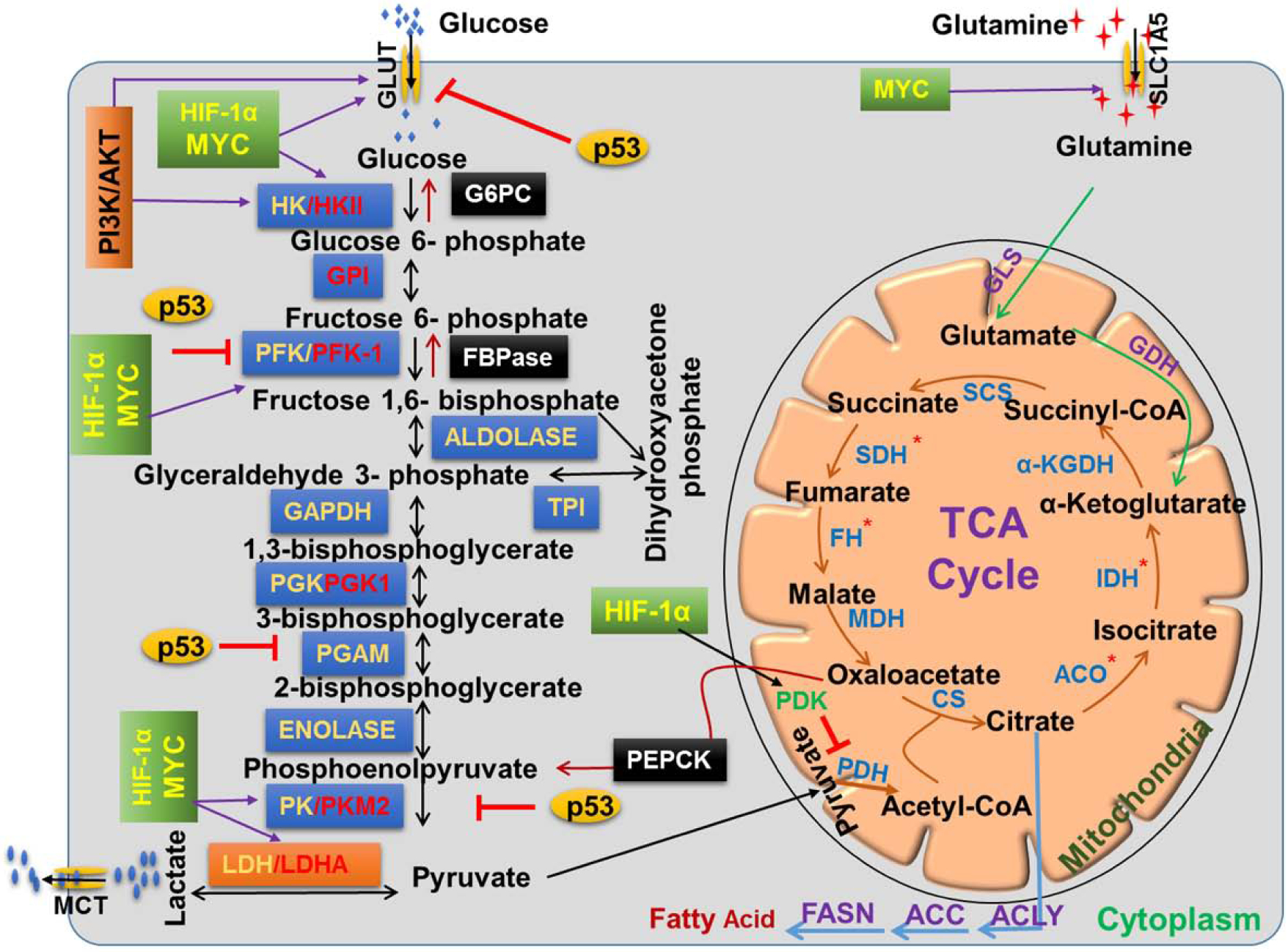
During glycolysis, glucose is converted into pyruvate in a sequential enzymatic reaction. Pyruvate can shuttle to the mitochondria and inter into the TCA cycle for energy generation and/or biomass synthesis or alternatively converted into lactate by LDHA and secreted from cells. Many glycolytic enzymes are highly dysregulated (red in color) in several ways;, p53 and HIF-1α/MYC negatively and positively regulate expression of key glycolytic enzymes in cancer cells. Tumor cells display altered expression in some of the TCA cycle-related enzymes due to genetic mutations/alterations (red asterisk). Gluconeogenesis pathway generates glucose via antagonizing glycolysis. It has enzymatic overlap with glycolysis and utilizes seven out of ten enzymes of glycolysis along with three unique. Glutamine can also generate energy through the TCA cycle after converting into α-Ketoglutarate by enzymes glutaminase (GLS) and glutamate dehydrogenase (GDH). Citrate from the TCA cycle can be secreted out into the cytoplasm and utilized in fatty acid synthesis.
a. Enzymes of the glycolytic pathway
Glycolysis is a universal metabolic pathway that occurs, albeit with variations, in all organisms, whether aerobic or anaerobic. The process is completed in 10 enzymatic steps, of which 3 are irreversible due to high Gibbs free energy [7]. An overexpression of glycolytic enzymes has been reported in several cancers and associated with multiple oncogenic or tumor suppressor transcription factors such as Myc, STAT3, NFκB, HIF1α, p53, etc. [8]. The activation of these transcription factors occurs either due to direct genetic alterations or because of the changes in their upstream signaling pathways. The major enzymes with altered expression are:
Hexokinase:
Hexokinases (HK) help in the conversion of glucose to glucose-6-phosphate and have four important mammalian isozymes- HK I-IV- varying in their substrate affinity, physiological function, and subcellular localization [9]. All consist of two similar 50kDa halves, but only HK II has active functional sites in both the halves. Moreover, HK II is the most commonly upregulated hexokinase in nearly all cancer types and has normal expression in the muscle and heart [9]. HK II promoter is responsive to glucose, insulin, and glucagon and contains functional response elements in the distal and proximal regions with active binding sites for multiple oncogenic transcription factors, including NFκB, MYC, HIF-1α, STAT3 [10, 11]. While the upregulation of HK II by glucagon seems paradoxical, it benefits the cancer cells by allowing its synthesis regardless of the metabolic state of the individual. In some tumors, HK II promoter hypomethylation is also reported leading to its upregulation [12]. Interestingly, a non-canonical function of HK II in resisting mitochondria-induced apoptosis in cancer cells has also been demonstrated. AKT-mediated phosphorylation of HK II supports its binding to mitochondrial VDAC, which reduces the availability of the latter for interaction with pro-apoptotic proteins, thus disrupting the formation of mitochondrial permeability transition pore complex [13].
Glucose-6-phosphate isomerase:
Glucose-6-phosphate isomerase (GPI) is overexpressed in many cancers and associated with poor prognosis [14]. It is primarily involved in the interconversion of glucose-6-phosphate to fructose-6-phosphate during glycolysis or gluconeogenesis. GPI is also secreted in extracellular spaces and referred to as the autocrine motility factor (AMF) since it promotes the migratory potential of cancer cells [14]. In breast cancer cells, GPI and HER2 interact in a positive feedback loop to promote tumor progression [15]. Additionally, GPI/AMF upregulates NF-κB and downregulates the miR-200 family to support EMT. Notably, the release of GPI is exclusively observed from the cancer cells suggesting therapeutic and biomarker opportunities.
Phosphofructokinase and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase:
Phosphofructokinase-1 (PFK-1) mediates the second committed step in the glycolytic pathway. Its activity is stimulated by low ADP/AMP and fructose 2,6 bisphosphate (F2,6BP), whereas citrate, long-chain fatty acids, lactate, and ATP act as strong inhibitors in a negative feedback loop [4]. PFK-1 is significantly upregulated in cancer and promotes a glycolytic phenotype. F2,6BP is the most important allosteric activator of PFK-1 that promotes glycolytic flux even in the presence of ATP [16]. The synthesis and degradation of F2,6BP are carried out by a single enzyme, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, having both kinase and phosphatase activities. Four mammalian isoforms (PFKFB 1–4) are known, of which PFKFB3 is the most commonly upregulated. Hypoxia upregulates PFKFB3 through direct HIF-1α-mediated transcriptional activation. It also has the highest kinase/phosphatase activity ratio (740/1), leading to elevated F2,6BP levels and sustained high glycolytic rates through FBP1 activation [17].
Phosphoglycerate kinase:
Phosphoglycerate kinase (PGK1) mediates one of the only two ATP generating steps in the glycolytic pathway. It is highly expressed in cancers and considered a major positive regulator of the Warburg effect [8]. EGFR activation, mutant KRAS/B-Raf, hypoxic stress, etc., induce phosphorylation of PGK1 via activated ERK1/2. This leads to its mitochondrial translocation, where it phosphorylates pyruvate dehydrogenase kinase (PDK1). Activation of PDK1 suppresses the activity of pyruvate dehydrogenase complex (PDC). As a result, the conversion of pyruvate to Acetyl-CoA is inhibited suppressing the TCA cycle [18]. Additionally, PGK1 also regulates autophagy in cancer cells under stress conditions such as glutamine-deprivation and hypoxia. ARD1 acetylates PGK1 at Lys388 under stress conditions, which then phosphorylates Beclin at Ser30, which is an important initial step for the formation of autophagosomes [19]. We have also recently observed that IKK-epsilon, a member of IKK family of kinases, promotes pancreatic tumor growth by reprogramming glucose metabolism. Metabolic reprogramming is mediated through the altered expression of several metabolic enzymes, including PGK1, HKII, etc., via IKK-epsilon-induced stabilization of c-MYC [20].
Pyruvate kinase:
Pyruvate kinase (PK) is the second ATP generating enzyme in glycolysis and has four major mammalian isoforms (PKM1, PKM2, PKL, and PKR). PKM2 is the embryonic isoform of PK that is widely expressed in cancer [21]. PKM2 expression cannot be attributed to fast-dividing cells since quiescent T cells and white adipose tissue also express PKM2. Despite reported overexpression of PKM2 in several cancers, its reduced expression and/or activity is shown to support cancer cell proliferation [22]. PKM2 activity is regulated by allosteric effectors (succinyl-5-aminoimidazole-4-carboxamide-1-ribose 5′-phosphate, fructose-1,6-bisphosphate, phenylalanine, cysteine, serine) and posttranslational modifications. In addition, its activity is affected by its oligomeric state, dimer or tetramer. The tetrameric form is active whereas dimeric form is inactive that diverts the glycolytic flux towards increased biomass production by blocking phosphoenolpyruvate utilization. It is also reported that the inactive dimeric PKM2 translocates into the nucleus and supports tumor cell proliferation by transcriptionally activating the expression of oncogenes [23]. Cancer cells withstand oxidative stress by promoting ROS-mediated inhibition of the tetrameric form of PKM2 that activates the pentose phosphate pathway and generates reducing cellular environment [24]. Substrate (phosphoenolpyruvate) binding affinity of PKM2 is reduced upon phosphorylation by PP60v-src-tyrosine kinase, which leads to the accumulation of different glycolytic intermediates/phosphometabolites [25]. PKM2 transfers phosphate groups directly from phosphoenolpyruvate to serine, threonine, or tyrosine residues of recipient proteins, and its important substrates include H3 histone and STAT3 among others [26].
Lactate dehydrogenase:
Lactate dehydrogenase (LDHA) is an important determinant of glycolytic ability and multiple mechanisms have been attributed to its upregulated expression in cancer cells. For example, the LDHA promoter contains DNA-binding elements for different oncogenic transcription factors, including c-MYC, HIF-1α, CREB, AP-1, STAT3, which mediate its transcriptional upregulation [27]. In our recent studies, we observed that MYB, an oncogenic transcription factor, also positively regulated the expression of LDHA along with GLUT1 in pancreatic cancer cells [28]. This is signficant considering our earlier findings on MYB that demonstrated its role in pancreatic tumor growth, metastasis, and desmoplasia [29, 30]. In addition, LDHA is also a target of several tumor suppressor miRNAs [31–33]. Direct repression of LDHA by miR-34a enhances the sensitivity of advanced colon cancer cells to 5-fluorouracil [31]. LDHA silencing also reduces the survival of cancer cells under both normoxia and hypoxia due to a decrease in ATP levels. The release of lactate from the cancer cells benefits the cancer cells by inducing an immunosuppressive microenvironment [34]. Acidic environment caused by released lactate downregulates nuclear factor of activated T cells (NFAT), which then causes an upregulation of IFN-γ transcripts in competent immune cells (CD8+T and NK cell) [34]. Lactic acid produced by tumor cells also activates IL-23 expression both at transcriptional and protein levels in infiltrated immune cells. This, in turn, promotes tumor growth, metastasis and further dampens the immune surveillance by recruiting immune-suppressive M2-like macrophages and neutrophils [35, 36].
b. Enzymes of the tricarboxylic acid cycle
The tricarboxylic acid (TCA) or the Kreb’s cycle operates in the mitochondrial matrix and is a central route for oxidative phosphorylation. Earlier, it was thought that the cancer cells bypass the TCA cycle entirely and operate only on the aerobic glycolytic pathway [37]. However, it is now clear that the TCA cycle not only contributes to energy production in cancer cells but also supports tumorigenesis. Moreover, the substrates of the TCA cycle also promote non-metabolic signaling involved in cancer progression. Major dysregulated enzymes in the TCA pathway are discussed below.
Pyruvate dehydrogenase complex/Pyruvate dehydrogenase kinase:
The pyruvate dehydrogenase (PDH) is comprised of three components, E1-E3. E1 contains thiamine pyrophosphate (TPP), which forms a carbanion and decarboxylates pyruvate [38]. PDH, in fact, bridges the glycolysis to the TCA cycle by facilitating the generation of acetyl CoA from pyruvate. High ATP-ADP ratio and elevated Acetyl CoA activate pyruvate dehydrogenase kinase (PDK), which phosphorylates and inhibits PDH at E1. In contrast, high ADP-ATP ratio and elevated pyruvate inhibit PDK and promote PDH activity [38]. Thus, PDK acts as a gatekeeper of altered glucose metabolism in cancer. The PDK family consists of 4 highly homologous members that phosphorylate 3 sites (Ser293, Ser300, and Ser 232) on E1-alpha of PDH. PDK overexpression is observed in several tumors and associated with chemoresistance, invasiveness, and metastasis [39]. It promotes tumor cell proliferation through the conservation of pyruvate for recycling of NAD+ via lactate dehydrogenase, production of Krebs cycle intermediates, and transamination through alanine aminotransferase [40]. Inhibition of PDKs is reported to increase PDH complex activity, mitochondrial activity, and production of reactive oxygen species (ROS) that slows tumor growth and promotes apoptosis [41]. Acetylation of PDH at Lys321 by acetyl-CoA acetyltransferase 1 (ACAT1) also leads to its inactivation via recruitment of PDK to its phosphorylation sites [42]. Deacetylation of that same lysine is catalyzed by SIRT3, and accordingly SIRT3 downregulation or ACAT1 upregulation is reported to enhance tumor growth [42]. Upregulation of HIF1α activates PDK transcriptionally, which inactivates PDH and allows increased recycling of NAD+ via lactate dehydrogenase under hypoxic or pseudohypoxic conditions [43]. PDK4 is paradoxically downregulated in hepatocellular carcinoma and lung cancer and suggested to have tumor suppressor functions [44, 45].
Isocitrate Dehydrogenase:
Isocitrate dehydrogenase (IDH) catalyzes the oxidation of isocitrate to alpha-ketoglutarate via a 2-step process that involves i) reduction of NAD+/NADP+ to NADH/NADPH to oxidize isocitrate to oxalosuccinate, and ii) decarboxylation of oxalosuccinate to alpha-ketoglutarate. IDH consists of 3 isozymes, IDH1-IDH3, of which the only IDH3 is found in the mitochondrial matrix [46]. Additionally, IDH3 uses NAD+ to oxidize isocitrate, while the other two use NADP+ [46]. Somatic mutations in IDH1 at R132 and in IDH2 at R172 and R140 result in a gain of new function i.e. catalyzing the conversion of alpha-ketoglutarate (α-KG) to D-2-hydroxyglutarate (D-2HG), which facilitates tumor proliferation [46, 47]. Mutant IDH1 also forms heterodimers with the wild-type IDH1 and inhibits its activity [48]. Knockdown of wildtype IDH1 in a glioma cell line resulted in an increased expression of HIF1α and VEGF. Similarly, forced expression of mutant IDH1 also inhibited wild type IDH1 by forming heterodimer and increased HIF1α [48]. These findings suggest that conversion of α-KG to D-2HG by mutant IDH weakens HIF1α degradation to promote tumorigenesis.
Succinate dehydrogenase:
The succinate dehydrogenase (SDH) complex proteins are encoded by four nuclear genes, SDHA, SDHB, SDHC, and SDHD, and their folding and assembly occur in mitochondria [49]. Tumor cells display aberrant promoter hypermethylation of SDH genes, and their expression is also repressed by certain oncogenic miRNAs (miR-210, miR-378, and miR-31) overexpressed in response to therapy, hypoxia and other stress conditions [49–51]. SDH complex proteins are also regulated in cancer through multiple post-translational modifications [49]. Mutations in any one of the subunits can disrupt the complex formation causing a loss of function. Additionally, mutations in SDHAF2/SDH5, an SDH complex assembly factor, also result in loss of function [52]. Diminished SDH expression or activity promotes a pseudohypoxic phenotype in cancer cells due to the accumulation of succinate, which inactivates prolyl hydroxylase (PHD), leading to HIF1α stabilization [53].
c. Enzymes in gluconeogenesis
Gluconeogenesis is a pathway that generates glucose from non-carbohydrate precursors, including lactate, pyruvate, glutamine, alanine, and glycerol. This process occurs in the liver, kidney, skeletal muscles and intestine to maintain blood glucose levels under fasting or after heavy exercise. Gluconeogenesis can antagonize aerobic glycolysis in cancer exclusively by three key gluconeogenesis enzymes, which are discussed below.
Phosphoenolpyruvate carboxykinase:
Phosphoenolpyruvate carboxykinase (PEPCK) exists as two isoforms, a cytosolic PCK1, and another mitochondrial, PCK2, and both catalyze the conversion of oxaloacetate to phosphoenolpyruvate. PEPCK is overexpressed in colon cancer cells and associated with decreased glucose and glutamine metabolism and diminished proliferation, clonogenic survival and tumorigenicity in a mouse model [54]. PCK2 mediates glycerol phosphate synthesis to support de novo synthesis of glycerophospholipid and its silencing reduces colony formation under the starved condition and in a subcutaneous mouse model of lung cancer [55]. Tumor-repopulating cells (TRCs) of melanoma in 3D culture reprogram glucose metabolism by hijacking the PCK1 without involving gluconeogenesis, but regulating glucose side-branch metabolism [56]. PCK1 silencing in TRCs drastically reduces glycine levels (a major metabolite of serine) and retards their growth in vitro and in vivo [56]. An overexpression of PCK2 is reported in pancreatic neuroendocrine tumors (pNET), where it regulates cellular metabolism by decreasing glycolysis but increasing mitochondrial oxidative phosphorylation [19, 57].
Fructose bisphosphatase:
Fructose-1,6-bisphosphatase (FBPase) is a rate-limiting enzyme that hydrolyzes fructose 1,6-bisphosphate to fructose 6-phosphate and inorganic phosphate (Pi) and has two isoforms liver FBPase (FBP1) and muscle FBPase (FBP2). FBP2 is overexpressed in metastatic breast cancer cells (MBCC) and supports their survival and proliferation by promoting gluconeogenesis and glutamine oxidation [58]. Several contradictory studies suggest a tumor-suppressive role of FBP2 and FBP as well. FBP1 is downregulated in various cancers, and low FBP1 levels are associated with poor overall survival [59]. Exogenous expression of FBP1 also decreases tumor cell growth in vitro and mouse models of multiple cancer types [60]. Under complete nutrient replenished conditions, FBP1 inhibits glycolysis in breast cancer, pancreatic cancer, lung cancer and HCC cells [59]. Overexpression of FBP1 in renal carcinoma cells (RCC) results in enhanced proliferation and migration, and reduction of glycolysis [61] and its role in EMT has also been suggested [60].
Glucose 6 phosphatase:
Glucose-6-phosphatase (G6Pase, G6PC) is a key enzyme involved in gluconeogenesis and glycogenolysis. It hydrolyzes D-glucose 6-phosphate to D-glucose and orthophosphate. Overexpression of G6PC is reported in ovarian cancer and associated with poor overall survival [62]. Direct targeting of G6PC via the IL6-Stat3-miR-23a axis suppresses the gluconeogenesis in a mouse model of hepatocellular carcinoma and diminishes the serum glucose levels in tumor-bearing mice [63]. Low G6PC correlates with higher uptake of fluorodeoxyglucose (FDG) in poorly differentiated HCC as compared to that in moderately differentiated HCC, indicating a connection of G6PC with tumor-grade [64].
d. Enzymes of other cancer-promoting metabolic pathways
Apart from glucose metabolism, glutamine, and fatty acid metabolism also plays an active role in cancer cell growth and proliferation [1]. These pathways can directly contribute to the energy requirements of proliferating cells or support the generation of biomass through their integration with the glucose metabolic pathway. Further detail on the most frequently dysregulated enzymes from these pathways are below:
Glutaminase:
Glutamine is the most abundant amino acid in blood circulation and acts directly or indirectly as a carbon and nitrogen source for the synthesis of nucleic acids, fatty acids, and proteins. Glutaminase converts glutamine into glutamate and ammonia, and glutamate is further utilized in the amino acid biosynthetic pathway. Alternatively, it converted to α-KG, which enters into the TCA cycle for the production of energy and biosynthetic precursors for a wide range of metabolic pathways. Glutaminase is important for the production of glutathione, a key scavenger of reactive oxygen species (ROS) [65]. In humans, two isoforms of glutaminase are known, kidney-type (GLS) and liver-type (LGA/GLS2), of which the former has been studied extensively for its role in cancer pathogenesis [66]. Studies are also emerging that suggest the role of GLS in the microvesicle formation in cancer cells [67]. High expression of GLS is observed in several malignancies and positively correlated with disease progression, and the study demonstrated that c-myc-mediated GLS expression induced tumor cell growth and proliferation by stimulating glutamine catabolism [68]. In contrast, GLS2 is reported to have a dual role in tumorigenesis. It is downregulated in brain tumor cells where its forced expression reduces tumor cell growth in vitro and in vivo [69]. On the other hand, increased expression of GLS2 is crucial for the growth of lung, cervical and colon cancer cells [69, 70]. This functional disparity could be due to differential regulation of their activity. In the presence of phosphate, GLS has greater enzymatic acivity due to lower Km for glutamine as compared to GLS2 whereas the presence of ammonia increases GLS2 activity, but inhibits GLS activity [69, 71]. Small molecule inhibitors of both GLS and GLS2 have been developed of which Bis-2-(5-phenylacetamido-1, 2, 4-thiadiazol-2-yl) ethyl sulfide (BPTES) and dibenzophenanthridine-968 exhibit maximum potency [65].
Fatty acid synthase:
Fatty acid synthase (also referred to as FASN) is involved in lipogenesis and responsible for the production of long-chain fatty acids from acetyl-coenzyme A (CoA) and malonyl-CoA. Except for liver and adipose tissue, all the normal cells have low FASN expression and activity, whereas it is overexpressed in multiple cancer types [72]. A gene copy gain of FASN in prostate adenocarcinoma along with elevated protein levels has been demonstrated by FISH and IHC analyses [73]. Ubiquitin-specific protease 2a (USP2a), an androgen-regulated enzyme overexpressed in prostate cancer, promotes the expression of FASN to support cell survival [74]. FASN silencing or pharmacological inhibition is shown to suppress the tumor growth via apoptosis induction and increased survival of breast and prostate tumor-bearing mice [75, 76].
Metabolic reprogramming in stromal cells and associated mechanisms
Stromal cells are a pool of non-malignant, heterogeneous cell types residing in the tumor microenvironment (TME). They support tumor progression, metastasis, and therapy resistance via intercellular interactions with each other and the tumor cells. Like tumor cells, they also live in a continuously changing TME and thus make adaptive metabolic changes to survive. However, their adaptive metabolic reprogramming largely depends on either the tumor-derived factors (cytokines, growth factors, extracellular vesicles, etc.) or altered metabolic composition of the TME [77] (Figure 2). Below we describe changes in bioenergetics and associated mechanisms for three important stromal cell types.
Figure 2: Metabolic reprogramming in stromal cells.
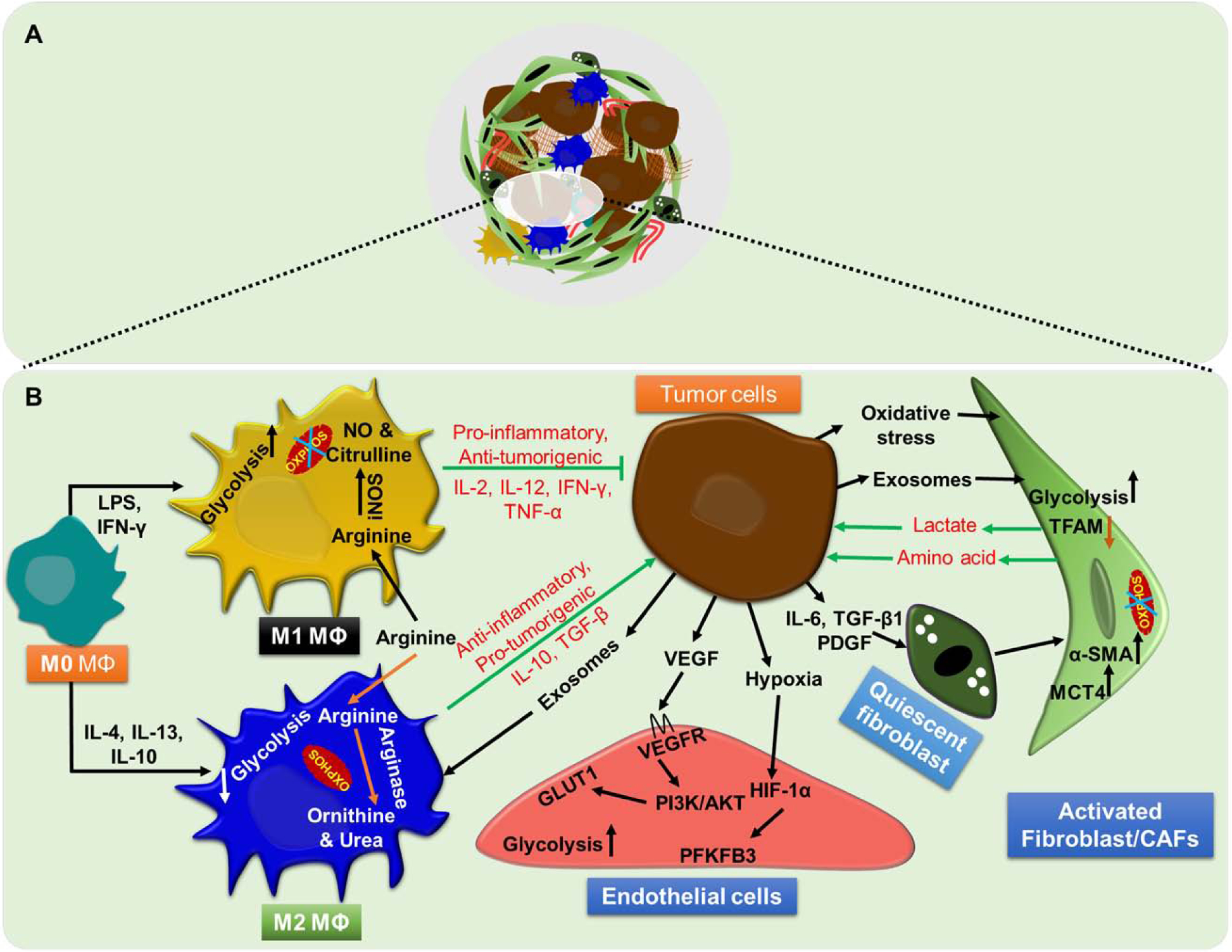
(A) Stromal cells, the pool of heterogeneous non-malignant cell types such as fibroblast, immune cells, and endothelial cells, are present within the tumor microenvironment. (B) Tumor cells metabolic reprogram surrounding stromal cells to meet their energy demand for sustaining growth, proliferation, aggressiveness and impairing host immune response.
Fibroblasts:
Fibroblasts have unique phenotypic plasticity. Cancer-associated fibroblasts (CAFs) acquire myofibroblasts-like phenotypic characteristics and are the most abundant component of TME. They are also a significant source of extracellular matrix (ECM) proteins. They undergo metabolic reprogramming, from oxidative phosphorylation to aerobic glycolysis, to support their proliferative growth [77]. It is shown that breast cancer cells induce oxidative stress in fibroblasts by secreting hydrogen peroxide, which inhibits their mitochondrial activity and increases glycolysis. This metabolic change also generates high energy intermediates like lactate and pyruvate, which are taken up by the breast cancer cells to meet their high energy demand [78]. Breast cancer cells express high levels of integrin beta 4 (ITB4), which is packed into exosomes and transferred to the stromal cells (CAFs). This exosomal transfer induces glycolysis by causing BNIP3L-dependent mitophagy in CAFs [79]. Microvesicles derived from the oral squamous cell carcinoma are also shown to change normal fibroblasts into CAFs and induce metabolic switching to aerobic glycolysis. In return, CAFs provide high energy metabolite, lactate, to the cancer cells by monocarboxylate transporter 1 and 4 [80]. Primary and metastatic tumor cell-derived exosomes can activate quiescent fibroblasts and give rise to a functionally distinct subset of CAFs by upregulating glycogen metabolism, amino acid biosynthesis, and glycolysis [80]. Breast tumor cells increase glucose and glutamine metabolism in CAFs in an MYC-dependent manner by transferring miR-105 through exosomes. In exchange, reprogrammed CAFs produce metabolic intermediates to support tumor cell growth [81]. Similarly, in prostate cancer, metabolically reprogrammed CAFs secret high amount of lactate that is taken up by prostate cancer cells and induces SIRT/PGC-1α-dependent mitochondrial activity and prostate cancer aggressiveness [82].
Immune cells:
Tumor-associated macrophages (TAMs) are the most abundant immune component of TME. Increased TAM infiltration usually correlates with poor clinical outcomes in multiple cancer types [83]. TAMs are reprogrammed macrophages that not only support tumor progression and metastasis but also cause further immune suppression. Cytokines and growth factors derived from TAMs also support tumorigenesis by promoting neoangiogenesis [83]. In addition, TAMs provide nutrient availability to the tumor cells in TME. For example, TAMs secrete a high amount of iron in TME, which is taken up by the tumor cells and induces their growth and proliferation [84]. Macrophages can polarize to M1-like (tumor-suppressive) or M2-like (tumor supportive) phenotypes depending on the extracellular stimuli [85]. Data suggest that M1 macrophages rely more on glycolysis, and glycolysis inhibition renders their tumor-suppressive activity due to reduced phagocytic ability, production of pro-inflammatory cytokines and reactive oxygen species (ROS). In contrast, M2 macrophages preferentially utilize oxidative phosphorylation for their activity [85].
Glutamine metabolism is one of the indispensable pathways for macrophage activation and immune responses, and α-ketoglutarate derivative of glutamine induces the macrophage polarization to the M2 phenotype. Another amino acid, arginine, is equally important. It is catabolized by two enzymes, inducible nitric oxide synthase (iNOS) and arginase (two isozymes, arginase I, and II). iNOS facilitates the conversion of arginine into nitric oxide (NO), while arginase hydrolyzes the arginine into ornithine and urea [85]. M1 macrophages utilize iNOS for arginine metabolism and generate reactive nitrogen species (RNS) from NO that has a microbicidal or tumoricidal function. On the contrary, tumor-supportive M2 macrophages catabolize arginine into ornithine and urea by arginase II [85].
Enzyme activity profile in normal human macrophages and TAMs from colorectal tumors have suggested different metabolic profiles among M1-like and M2-like macrophages. Further, infiltrating M2-like macrophages have reduced GAPDH activity compared to M1-like [86]. Adenosine, a purine ribonucleoside in TME, is involved in immune evasion by eliciting inhibitory signaling in natural killer (NK) cells through adenosine receptor (A2AR) [87]. Glycolytic melanoma cells produce a high amount of lactic acid, which potentially inhibits the activity of NK- and T-cells by downregulating the nuclear factor of activated T cells (NFAT) signaling [34]. Data also suggest that a low level of triglycerides anabolic enzyme, abhydrolase domain containing 5, in tumor-associated macrophages (TAMs) promotes tumor metastasis via NF-κB-dependent matrix metalloproteinases activation [88]. The anti-diabetic drug, metformin, rewires glucose metabolism in CD11b+ myeloid cells (TAMs) and induces their repolarization to the M1 phenotype leading to the suppression of the growth of osteosarcoma cells in vivo [89].
Tumor endothelial cells:
Endothelial cells (ECs) exhibit metabolic plasticity and preferentially adopt a glycolytic phenotype despite having high oxygen in their proximity. Glycolysis in these cells is attributed majorly to increased glucose transporter1 (GLUT1), hexokinase 2 (HK2), and phosphofructokinase 1 (PFK1). Tumor-associated endothelial cells (TECs) exhibit even more glycolysis as compared to normal ECs and glycolysis inhibition in TECs reduces their proliferation, survival, and migration [90]. Data suggest that vascular endothelial growth factor (VEGF) secreted by cancer cells can increase glycolysis in TECs by upregulating glucose transporter, GLUT-1 [91]. Other TME associated factors, including hypoxia, cytokines, and growth factors, are shown to increase the expression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) in TECs that controls PFK1 activity in glycolytic pathway.
Targeting metabolic abnormalities in cancer for therapeutic exploitation
In light of the essential roles of adaptive metabolic changes in the growth of cancer, their targeting is considered a novel avenue for cancer therapy (Table 1). Some of the examples are described below:
TABLE 1:
List of metabolic enzymes, their inhibitors, and underlying mechanism(s) of action.
| Enzymes | Inhibitors | Mechanism of action | Refs |
|---|---|---|---|
| HK | 3-Bromopyruvate (3BP) | Suppresses the production of ATP by alkylating HK-II | [92, 94] |
| PFK1 | 3PO | Indirect targets PFK-1 by inhibiting its allosteric activator, PFKFB3 | [96–98] |
| PKM2 | Tanshione IIA | miR122-mediated PKM2 downregulation | [101] |
| PDC/PDK | CPI-613 | Inhibits PDC via activation of PDK | [103, 104] |
| LDH | FX-11 | Non-specific/competitive inhibition at NADH binding site | [106, 112] |
| PEPCK | 3-Mercaptopicolinic | Competitive inhibition at the substrate-binding site and allosteric inhibition | [113] |
| G6Pase | Chlorogenic acid | Inhibits glucose-6-phosphatase transporter | [115] |
| Glutaminase | CB-839, BPTES, and trans-CBTBP | Inhibition through allosteric binding at GLS1 pocket | [116] |
| IDH | Ivosidenib (AG-120) | Rapid-equilibrium inhibitor of mutated IDH1 | [117] |
Glucose analog, 2-deoxy-D-glucose (2-DG), inhibits glucose metabolism with high efficacy. Hexokinase phosphorylates 2-DG, which cannot be metabolized any further by the cells, leading to its intracellular accumulation and inhibition of hexokinase activity [92]. Upregulation of hexokinase II is reported during the metabolic shift from oxidative phosphorylation to glycolysis, and its inhibition is shown to sensitize the cancer cells to oxidative stress therapy [93]. 3-bromopyruvate (3BP), a brominated derivative of pyruvic acid, is structurally similar to pyruvic and lactic acid. It inhibits HK II and possesses alkylating properties towards biomacromolecules [88, 93]. Moreover, 3BP also targets other glycolytic enzymes such as glyceraldehyde 3-phosphate dehydrogenase, lactate dehydrogenase, pyruvate carboxylase, and pyruvate dehydrogenase [94]. The ATP depleting effect of 3BP has been promising for the treatment of several cancers, and in some cases, it restored chemo-sensitivity [94]. Lonidamine (LND), a derivative of indazole-3 carboxylic acid, has been used to inhibit aerobic glycolysis in cancer cells. LND inhibits the HK II and other enzymes of pentose shunt- and glycolytic-pathways, possibly via cytosolic acidification. Moreover, it is also shown to inhibit mitochondrial respiration by abolishing the pyruvate import via mitochondrial pyruvate carrier, and mitochondrial electron transport complex II or complex I or both in case of ubiquinone reduction [95]. Due to its ability to inhibit the energy metabolism, LND, in combination with other anti-cancer agents, has been tested for the treatment of glioblastoma multiforme, breast cancer, lung, and ovarian cancers and is currently in a clinical trial [95].
A study has suggested that PFK-1 activity is regulated through allosteric inhibition by ATP and citrate. Small-molecule inhibitor, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3-PO), indirectly targets PFK-1 by inhibiting its allosteric activator, 6-phosphofructose-2-kinase, and thus reduces glucose metabolism and synthesis of the ATP, NAD+, NADH, and fructose 2,6 bisphosphate. 3-PO has been shown to diminish the growth of many human malignant hematopoietic and adenocarcinoma cell lines in vitro and in vivo [96–98].
Among the four isozymes, PKM2 is considered vital due to its elevated expression in cancer [21]. The knockdown of PKM2 sensitizes pancreatic cancer cells to gemcitabine therapy and colorectal cancer cells to oxaliplatin treatment [99, 100]. Tanshione IIA, a natural compound isolated from Chinese herb Salvia miltiorrhiza, inhibits human esophageal cancer cell growth via miR-122-mediated downregulation of PKM2 [101]. Moreover, Shikonin, alkannin, and their structural analogs also inhibit the activity of PKM2 and reduce cancer cell proliferation and survival. In a clinical trial, shikonin mixture reduced the tumor size (25% in diameter) and increased the survival rate (36.9%) in lung cancer patients [102].
Another study has suggested that CPI-613, a lipoid analog, strongly disrupts the mitochondrial metabolism by phosphorylating pyruvate dehydrogenase (PDH). It also targets alpha-ketoglutarate dehydrogenase through redox modification [103]. Dichloroacetate (DCA) inhibits the pyruvate dehydrogenase kinase (PDK) and decreases the glycolytic flux towards the lactate; however, the clinical trial data suggest that it is effective in some cancers only. 2-chloroproprtionate, an allosteric inhibitor of PDK, also works similar to DCA [104]. Gossypol, a polyphenolic binaphthyl disesquiterpene compound, targets lactate dehydrogenase by competitively inhibiting the interconversion of NADH and NAD+ [105]. A small-molecule inhibitor, FX11, inhibits LDH-A-dependent tumor growth by reducing ATP levels and increasing oxidative stress. Quinoline 3-sulphonamide also exerts LDH inhibitory activity and impairs survival in numerous cancer cell lines [106]. Oxamic acid, a structural isostere of pyruvic acid, inhibits the LDH activity, and induces ROS in cancer cell lines [88]. Additionally, inhibition of HIF-1α could be a practical approach to reduce tumor growth by altering glucose metabolism. For example, FDA-approved drug (Topotecan) for the treatment of ovarian cancer, inhibits HIF-1α leading to a reduced expression of GLUT-1 in patients with advanced solid cancers [107].
Pharmacological inhibition of gluconeogenesis enzyme, PEPCK (PEPCKi), reduces phosphoenolpyruvate (PEP) in colon cancer cells leading to an increase in PEPCK substrate, oxaloacetate, and growth inhibition [108]. Another PEPCK inhibitor, 3-mercaptopicolinic, induces apoptosis in lung cancer cells by depleting glucose. Interestingly, dexamethasone treatment induces gluconeogenesis by upregulating PEPCK in low PEPCK-expressing hepatocellular carcinoma cells and inhibits their growth and angiogenesis. Forced expression of PEPCK1 in T cells induces metabolic reprogramming bolstering their tumoricidal activity and thus increasing the survival of tumor-bearing mice [88]. HDAC inhibitors (sodium butyrate, vorinostat, and panobinostat) upregulate FBP1 expression and inhibit glycolysis in multiple cancers [59]. In another report, FBP1-derived small peptide inhibitor, FBP1-E4, is shown to enhance gemcitabine toxicity in pancreatic tumor cells [59]. Competitive inhibitor of G6PT (catalytic subunit of G6PC), chlorogenic acid, inhibits migration of glioblastoma cells by blocking matrix metalloproteinase (MMP)-2 secretion. Human brain tumor-initiating cells (BTIC) require G6PC to counteract the effect of 2-DG, and its silencing reduces their survival and malignant potential under glycolysis inhibited condition [109].
Since tumor and stromal cells, especially immune cells, have overlapping metabolic needs, targeting of metabolic reprogramming for effective anti-tumor therapies should be very specific. For example, 2-DG inhibits tumor growth by inhibiting glycolysis, but it has an unwanted effect on T cell metabolism. Although in another case, inhibition of lactic acid secretion by lenalidomide reduces tumor growth and enhances T cell activation. In colorectal cancer, inhibition of lactate production is shown to reduce tumor cell growth and improve the therapeutic response of 5-fluorouracil [47].
CONCLUSION AND FUTURE PERSPECTIVES
Metabolic adaptation of tumor and stromal cells via altered expression of metabolic enzymes is now well established. It is also proven that metabolic reprogramming plays an essential role in cancer development. Altered metabolism not only helps cancer cells survive and grow but confers resistance to therapeutic interventions as well. Identifying the dependence of tumor cells on certain metabolic enzymes in preclinical and clinical studies has opened up new avenues for therapy development or the improvement of existing therapies. However, there are challenges along the way. Certain available drugs that can target cancer metabolism may not be clinically useful due to their lack of specificity and broad effects on multiple cell types. In some instances, metabolic alterations appear to play contrasting roles in different cancers, suggesting their context-dependent actions. Thus, efforts are required to improve understanding of associated mechanisms in parallel investigations. Regardless, existing data and current thrust in cancer metabolic research give us hope that novel therapies will soon emerge to provide additional options to oncologists to better manage lethal malignancies.
Highlights:
To maintain a proliferative state and survive under hostile environments, cancer cells make adaptive changes in cellular metabolism.
Transformed cellular metabolism is associated with dysregulation of metabolic enzymes.
High metabolic demand of cancer cells affects the tumor microenvironment due to unusual uptake of certain nutrients and release of metabolic derivatives and associated factors in the extracellular milieu.
Metabolic rewiring is also observed in stromal cells likely resulting from their adaptation to the remodeled tumor microenvironment and facilitated by tumor-derived factors.
Tumor-stromal metabolic crosstalk is vital for progressive growth of tumors providing opportunities for therapeutic exploitation.
ACKNOWLEDGMENT
The authors would like to acknowledge the funding from NIH/NCI [R01CA175772, R01CA224306, U01CA185490 (to APS) and R01CA204801, R01CA231925 (to SS)] and USA MCI. The authors would like to acknowledge Mr. Josiah Perry for his assistance during the manuscript preparation.
ABBREVIATIONS:
- LDH
Lactate dehydrogenase
- HK
Hexokinase
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- EGFR
Epidermal growth factor receptor
- NFκB
Nuclear factor kappa light chain enhancer of activated B cells
- STAT3
Signal transducer and activator of transcription 3
- TCA
Tricarboxylic acid cycle
- CAFs
Cancer-associated fibroblasts
- TAMs
Tumor-associated macrophages
- 2-DG
2-deoxy-D-glucose
- ATP
Adenosine triphosphate
- ROS
Reactive oxygen species
- TME
Tumor microenvironment
- HIF1α
Hypoxia-inducible factor 1-alpha
- PDC
Pyruvate dehydrogenase complex
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that there are no conflict of interests.
REFERENCES
- [1].Sreedhar A, Zhao Y, Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells, Biomed Rep, 8 (2018) 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, Jones LW, Ko YH, Le A, Lea MA, Locasale JW, Longo VD, Lyssiotis CA, McDonnell E, Mehrmohamadi M, Michelotti G, Muralidhar V, Murphy MP, Pedersen PL, Poore B, Raffaghello L, Rathmell JC, Sivanand S, Vander Heiden MG, Wellen KE, Target Validation T, Dysregulated metabolism contributes to oncogenesis, Semin Cancer Biol, 35 Suppl (2015) S129–S150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Warburg O, On the origin of cancer cells, Science, 123 (1956) 309–314. [DOI] [PubMed] [Google Scholar]
- [4].DeBerardinis RJ, Chandel NS, Fundamentals of cancer metabolism, Sci Adv, 2 (2016) e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Weyandt JD, Thompson CB, Giaccia AJ, Rathmell WK, Metabolic Alterations in Cancer and Their Potential as Therapeutic Targets, Am Soc Clin Oncol Educ Book, 37 (2017) 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schnittert J, Heinrich MA, Kuninty PR, Storm G, Prakash J, Reprogramming tumor stroma using an endogenous lipid lipoxin A4 to treat pancreatic cancer, Cancer Lett, 420 (2018) 247–258. [DOI] [PubMed] [Google Scholar]
- [7].Ward PS, Thompson CB, Metabolic reprogramming: a cancer hallmark even warburg did not anticipate, Cancer Cell, 21 (2012) 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Marbaniang C, Kma L, Dysregulation of Glucose Metabolism by Oncogenes and Tumor Suppressors in Cancer Cells, Asian Pac J Cancer Prev, 19 (2018) 2377–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wilson JE, Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function, J Exp Biol, 206 (2003) 2049–2057. [DOI] [PubMed] [Google Scholar]
- [10].Londhe P, Yu PY, Ijiri Y, Ladner KJ, Fenger JM, London C, Houghton PJ, Guttridge DC, Classical NF-kappaB Metabolically Reprograms Sarcoma Cells Through Regulation of Hexokinase 2, Front Oncol, 8 (2018) 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yeung SJ, Pan J, Lee MH, Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer, Cell Mol Life Sci, 65 (2008) 3981–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Goel A, Mathupala SP, Pedersen PL, Glucose metabolism in cancer. Evidence that demethylation events play a role in activating type II hexokinase gene expression, J Biol Chem, 278 (2003) 15333–15340. [DOI] [PubMed] [Google Scholar]
- [13].Pastorino JG, Hoek JB, Regulation of hexokinase binding to VDAC, J Bioenerg Biomembr, 40 (2008) 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jiang WG, Raz A, Douglas-Jones A, Mansel RE, Expression of autocrine motility factor (AMF) and its receptor, AMFR, in human breast cancer, J Histochem Cytochem, 54 (2006) 231–241. [DOI] [PubMed] [Google Scholar]
- [15].Kho DH, Nangia-Makker P, Balan V, Hogan V, Tait L, Wang Y, Raz A, Autocrine motility factor promotes HER2 cleavage and signaling in breast cancer cells, Cancer Res, 73 (2013) 1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mor I, Cheung EC, Vousden KH, Control of glycolysis through regulation of PFK1: old friends and recent additions, Cold Spring Harb Symp Quant Biol, 76 (2011) 211–216. [DOI] [PubMed] [Google Scholar]
- [17].Obach M, Navarro-Sabate A, Caro J, Kong X, Duran J, Gomez M, Perales JC, Ventura F, Rosa JL, Bartrons R, 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia, J Biol Chem, 279 (2004) 53562–53570. [DOI] [PubMed] [Google Scholar]
- [18].Li X, Zheng Y, Lu Z, PGK1 is a new member of the protein kinome, Cell Cycle, 15 (2016) 1803–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Qian X, Li X, Cai Q, Zhang C, Yu Q, Jiang Y, Lee JH, Hawke D, Wang Y, Xia Y, Zheng Y, Jiang BH, Liu DX, Jiang T, Lu Z, Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy, Mol Cell, 65 (2017) 917–931 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zubair H, Azim S, Srivastava SK, Ahmad A, Bhardwaj A, Khan MA, Patel GK, Arora S, Carter JE, Singh S, Singh AP, Glucose Metabolism Reprogrammed by Overexpression of IKKepsilon Promotes Pancreatic Tumor Growth, Cancer Res, 76 (2016) 7254–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Israelsen WJ, Vander Heiden MG, Pyruvate kinase: Function, regulation and role in cancer, Semin Cell Dev Biol, 43 (2015) 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, Burga LN, Xie J, Jurczak MJ, DePinho RA, Clish CB, Jacks T, Kibbey RG, Wulf GM, Di Vizio D, Mills GB, Cantley LC, Vander Heiden MG, PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells, Cell, 155 (2013) 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gao X, Wang H, Yang JJ, Liu X, Liu ZR, Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase, Mol Cell, 45 (2012) 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC, Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses, Science, 334 (2011) 1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Eigenbrodt E, Reinacher M, Scheefers-Borchel U, Scheefers H, Friis R, Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells, Crit Rev Oncog, 3 (1992) 91–115. [PubMed] [Google Scholar]
- [26].Hsu MC, Hung WC, Pyruvate kinase M2 fuels multiple aspects of cancer cells: from cellular metabolism, transcriptional regulation to extracellular signaling, Mol Cancer, 17 (2018) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Feng Y, Xiong Y, Qiao T, Li X, Jia L, Han Y, Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy, Cancer Med, 7 (2018) 6124–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Azim S, Zubair H, Srivastava SK, Bhardwaj A, Zubair A, Ahmad A, Singh S, Khushman M, Singh AP, Deep sequencing and in silico analyses identify MYB-regulated gene networks and signaling pathways in pancreatic cancer, Sci Rep, 6 (2016) 28446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bhardwaj A, Srivastava SK, Singh S, Tyagi N, Arora S, Carter JE, Khushman M, Singh AP, MYB Promotes Desmoplasia in Pancreatic Cancer through Direct Transcriptional Up-regulation and Cooperative Action of Sonic Hedgehog and Adrenomedullin, J Biol Chem, 291 (2016) 16263–16270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Khan MA, Srivastava SK, Bhardwaj A, Singh S, Arora S, Zubair H, Carter JE, Singh AP, Gemcitabine triggers angiogenesis-promoting molecular signals in pancreatic cancer cells: Therapeutic implications, Oncotarget, 6 (2015) 39140–39150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li Y, Luo S, Ma R, Liu J, Xu P, Zhang H, Tang K, Ma J, Zhang Y, Liang X, Sun Y, Ji T, Wang N, Huang B, Upregulation of cytosolic phosphoenolpyruvate carboxykinase is a critical metabolic event in melanoma cells that repopulate tumors, Cancer Res, 75 (2015) 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen H, Gao S, Cheng C, MiR-323a-3p suppressed the glycolysis of osteosarcoma via targeting LDHA, Hum Cell, 31 (2018) 300–309. [DOI] [PubMed] [Google Scholar]
- [33].Lei W, Kang W, Nan Y, Lei Z, Zhongdong L, Demin L, Lei S, Hairong H, The Downregulation of miR-200c Promotes Lactate Dehydrogenase A Expression and Non-Small Cell Lung Cancer Progression, Oncol Res, 26 (2018) 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, Kastenberger M, Bogdan C, Schleicher U, Mackensen A, Ullrich E, Fichtner-Feigl S, Kesselring R, Mack M, Ritter U, Schmid M, Blank C, Dettmer K, Oefner PJ, Hoffmann P, Walenta S, Geissler EK, Pouyssegur J, Villunger A, Steven A, Seliger B, Schreml S, Haferkamp S, Kohl E, Karrer S, Berneburg M, Herr W, Mueller-Klieser W, Renner K, Kreutz M, LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells, Cell Metab, 24 (2016) 657–671. [DOI] [PubMed] [Google Scholar]
- [35].Shime H, Yabu M, Akazawa T, Kodama K, Matsumoto M, Seya T, Inoue N, Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway, J Immunol, 180 (2008) 7175–7183. [DOI] [PubMed] [Google Scholar]
- [36].Nie W, Yu T, Sang Y, Gao X, Tumor-promoting effect of IL-23 in mammary cancer mediated by infiltration of M2 macrophages and neutrophils in tumor microenvironment, Biochem Biophys Res Commun, 482 (2017) 1400–1406. [DOI] [PubMed] [Google Scholar]
- [37].Anderson NM, Mucka P, Kern JG, Feng H, The emerging role and targetability of the TCA cycle in cancer metabolism, Protein Cell, 9 (2018) 216–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Patel MS, Nemeria NS, Furey W, Jordan F, The pyruvate dehydrogenase complexes: structure-based function and regulation, J Biol Chem, 289 (2014) 16615–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Saunier E, Benelli C, Bortoli S, The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents, Int J Cancer, 138 (2016) 809–817. [DOI] [PubMed] [Google Scholar]
- [40].Stacpoole PW, Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer, J Natl Cancer Inst, 109 (2017). [DOI] [PubMed] [Google Scholar]
- [41].Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth, Cancer Cell, 11 (2007) 37–51. [DOI] [PubMed] [Google Scholar]
- [42].Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, Gu TL, Aguiar M, Lonning S, Chen H, Mohammadi M, Britton LM, Garcia BA, Aleckovic M, Kang Y, Kaluz S, Devi N, Van Meir EG, Hitosugi T, Seo JH, Lonial S, Gaddh M, Arellano M, Khoury HJ, Khuri FR, Boggon TJ, Kang S, Chen J, Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex, Mol Cell, 53 (2014) 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC, HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption, Cell Metab, 3 (2006) 187–197. [DOI] [PubMed] [Google Scholar]
- [44].Li M, Jin R, Wang W, Zhang T, Sang J, Li N, Han Q, Zhao W, Li C, Liu Z, STAT3 regulates glycolysis via targeting hexokinase 2 in hepatocellular carcinoma cells, Oncotarget, 8 (2017) 24777–24784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Choiniere J, Wu J, Wang L, Pyruvate Dehydrogenase Kinase 4 Deficiency Results in Expedited Cellular Proliferation through E2F1-Mediated Increase of Cyclins, Mol Pharmacol, 91 (2017) 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Al-Khallaf H, Isocitrate dehydrogenases in physiology and cancer: biochemical and molecular insight, Cell Biosci, 7 (2017) 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li X, Zhao H, Zhou X, Song L, Inhibition of lactate dehydrogenase A by microRNA-34a resensitizes colon cancer cells to 5-fluorouracil, Mol Med Rep, 11 (2015) 577–582. [DOI] [PubMed] [Google Scholar]
- [48].Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y, Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha, Science, 324 (2009) 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rutter J, Winge DR, Schiffman JD, Succinate dehydrogenase - Assembly, regulation and role in human disease, Mitochondrion, 10 (2010) 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shi SS, Wang YF, Bao W, Ye SB, Wu N, Wang X, Xia QY, Li R, Shen Q, Zhou XJ, Genetic and epigenetic alterations of SDH genes in patients with sporadic succinate dehydrogenase-deficient gastrointestinal stromal tumors, Pathol Int, 69 (2019) 350–359. [DOI] [PubMed] [Google Scholar]
- [51].Lee MR, Mantel C, Lee SA, Moon SH, Broxmeyer HE, MiR-31/SDHA Axis Regulates Reprogramming Efficiency through Mitochondrial Metabolism, Stem Cell Reports, 7 (2016) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, Rutter J, SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma, Science, 325 (2009) 1139–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E, Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells, Mol Cell Biol, 27 (2007) 3282–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Montal ED, Dewi R, Bhalla K, Ou L, Hwang BJ, Ropell AE, Gordon C, Liu WJ, DeBerardinis RJ, Sudderth J, Twaddel W, Boros LG, Shroyer KR, Duraisamy S, Drapkin R, Powers RS, Rohde JM, Boxer MB, Wong KK, Girnun GD, PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth, Mol Cell, 60 (2015) 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Leithner K, Triebl A, Trotzmuller M, Hinteregger B, Leko P, Wieser BI, Grasmann G, Bertsch AL, Zullig T, Stacher E, Valli A, Prassl R, Olschewski A, Harris AL, Kofeler HC, Olschewski H, Hrzenjak A, The glycerol backbone of phospholipids derives from noncarbohydrate precursors in starved lung cancer cells, Proc Natl Acad Sci U S A, 115 (2018) 6225–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sun S, Liang X, Zhang X, Liu T, Shi Q, Song Y, Jiang Y, Wu H, Jiang Y, Lu X, Pang D, Phosphoglycerate kinase-1 is a predictor of poor survival and a novel prognostic biomarker of chemoresistance to paclitaxel treatment in breast cancer, Br J Cancer, 112 (2015) 1332–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chu PY, Jiang SS, Shan YS, Hung WC, Chen MH, Lin HY, Chen YL, Tsai HJ, Chen LT, Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) regulates the cell metabolism of pancreatic neuroendocrine tumors (pNET) and de-sensitizes pNET to mTOR inhibitors, Oncotarget, 8 (2017) 103613–103625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, Wang Y, He J, Bollu LR, Gao G, Su F, Briggs J, Liu X, Melman T, Asara JM, Fidler IJ, Cantley LC, Locasale JW, Weihua Z, Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain, Cancer Res, 75 (2015) 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu GM, Zhang YM, Targeting FBPase is an emerging novel approach for cancer therapy, Cancer Cell Int, 18 (2018) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liu GM, Li Q, Zhang PF, Shen SL, Xie WX, Chen B, Wu J, Hu WJ, Huang XY, Peng BG, Restoration of FBP1 suppressed Snail-induced epithelial to mesenchymal transition in hepatocellular carcinoma, Cell Death Dis, 9 (2018) 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, Mancuso A, Gade TP, Keith B, Nissim I, Simon MC, Fructose-1,6-bisphosphatase opposes renal carcinoma progression, Nature, 513 (2014) 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Guo T, Chen T, Gu C, Li B, Xu C, Genetic and molecular analyses reveal G6PC as a key element connecting glucose metabolism and cell cycle control in ovarian cancer, Tumour Biol, 36 (2015) 7649–7658. [DOI] [PubMed] [Google Scholar]
- [63].Wang B, Hsu SH, Frankel W, Ghoshal K, Jacob ST, Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha, Hepatology, 56 (2012) 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Izuishi K, Yamamoto Y, Mori H, Kameyama R, Fujihara S, Masaki T, Suzuki Y, Molecular mechanisms of [18F]fluorodeoxyglucose accumulation in liver cancer, Oncol Rep, 31 (2014) 701–706. [DOI] [PubMed] [Google Scholar]
- [65].Masisi BK, El Ansari R, Alfarsi L, Rakha EA, Green AR, Craze ML, The Role of Glutaminase in Cancer, Histopathology, (2019). [DOI] [PubMed] [Google Scholar]
- [66].Katt WP, Lukey MJ, Cerione RA, A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis, Future Med Chem, 9 (2017) 223–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Santana SM, Antonyak MA, Cerione RA, Kirby BJ, Cancerous epithelial cell lines shed extracellular vesicles with a bimodal size distribution that is sensitive to glutamine inhibition, Phys Biol, 11 (2014) 065001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM, Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC, Proc Natl Acad Sci U S A, 109 (2012) 8983–8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cluntun AA, Lukey MJ, Cerione RA, Locasale JW, Glutamine Metabolism in Cancer: Understanding the Heterogeneity, Trends Cancer, 3 (2017) 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Xiang L, Xie G, Liu C, Zhou J, Chen J, Yu S, Li J, Pang X, Shi H, Liang H, Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation, Biochim Biophys Acta, 1833 (2013) 2996–3005. [DOI] [PubMed] [Google Scholar]
- [71].Botman D, Tigchelaar W, Van Noorden CJ, Determination of phosphate-activated glutaminase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry), J Histochem Cytochem, 62 (2014) 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Menendez JA, Lupu R, Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis, Nat Rev Cancer, 7 (2007) 763–777. [DOI] [PubMed] [Google Scholar]
- [73].Shah US, Dhir R, Gollin SM, Chandran UR, Lewis D, Acquafondata M, Pflug BR, Fatty acid synthase gene overexpression and copy number gain in prostate adenocarcinoma, Hum Pathol, 37 (2006) 401–409. [DOI] [PubMed] [Google Scholar]
- [74].Graner E, Tang D, Rossi S, Baron A, Migita T, Weinstein LJ, Lechpammer M, Huesken D, Zimmermann J, Signoretti S, Loda M, The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer, Cancer Cell, 5 (2004) 253–261. [DOI] [PubMed] [Google Scholar]
- [75].Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW, Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity, Cancer Res, 64 (2004) 2070–2075. [DOI] [PubMed] [Google Scholar]
- [76].Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP, Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts, Cancer Res, 60 (2000) 213–218. [PubMed] [Google Scholar]
- [77].Xing Y, Zhao S, Zhou BP, Mi J, Metabolic reprogramming of the tumour microenvironment, FEBS J, 282 (2015) 3892–3898. [DOI] [PubMed] [Google Scholar]
- [78].Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F, Lisanti MP, Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors, Cell Cycle, 10 (2011) 2504–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sung JS, Kang CW, Kang S, Jang Y, Chae YC, Kim BG, Cho NH, ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts, Oncogene, (2019). [DOI] [PubMed] [Google Scholar]
- [80].Jiang E, Xu Z, Wang M, Yan T, Huang C, Zhou X, Liu Q, Wang L, Chen Y, Wang H, Liu K, Shao Z, Shang Z, Tumoral microvesicle-activated glycometabolic reprogramming in fibroblasts promotes the progression of oral squamous cell carcinoma, FASEB J, 33 (2019) 5690–5703. [DOI] [PubMed] [Google Scholar]
- [81].Yan W, Wu X, Zhou W, Fong MY, Cao M, Liu J, Liu X, Chen CH, Fadare O, Pizzo DP, Wu J, Liu L, Liu X, Chin AR, Ren X, Chen Y, Locasale JW, Wang SE, Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells, Nat Cell Biol, 20 (2018) 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ippolito L, Morandi A, Taddei ML, Parri M, Comito G, Iscaro A, Raspollini MR, Magherini F, Rapizzi E, Masquelier J, Muccioli GG, Sonveaux P, Chiarugi P, Giannoni E, Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer, Oncogene, 38 (2019) 5339–5355. [DOI] [PubMed] [Google Scholar]
- [83].Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, Soong DYH, Cotechini T, Anur P, Lin EY, Fidanza A, Lopez-Yrigoyen M, Millar MR, Urman A, Ai Z, Spellman PT, Hwang ES, Dixon JM, Wiechmann L, Coussens LM, Smith HO, Pollard JW, Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets, Cancer Cell, 35 (2019) 588–602 e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mertens C, Mora J, Oren B, Grein S, Winslow S, Scholich K, Weigert A, Malmstrom P, Forsare C, Ferno M, Schmid T, Brune B, Jung M, Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment, Oncoimmunology, 7 (2018) e1408751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Netea-Maier RT, Smit JWA, Netea MG, Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship, Cancer Lett, 413 (2018) 102–109. [DOI] [PubMed] [Google Scholar]
- [86].Miller A, Nagy C, Knapp B, Laengle J, Ponweiser E, Groeger M, Starkl P, Bergmann M, Wagner O, Haschemi A, Exploring Metabolic Configurations of Single Cells within Complex Tissue Microenvironments, Cell Metab, 26 (2017) 788–800 e786. [DOI] [PubMed] [Google Scholar]
- [87].Chambers AM, Lupo KB, Matosevic S, Tumor Microenvironment-Induced Immunometabolic Reprogramming of Natural Killer Cells, Front Immunol, 9 (2018) 2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Shang S, Ji X, Zhang L, Chen J, Li C, Shi R, Xiang W, Kang X, Zhang D, Yang F, Dai R, Chen P, Chen S, Chen Y, Li Y, Miao H, Macrophage ABHD5 suppresses NF-kappaB-dependent matrix metalloproteinase expression and cancer metastasis, Cancer Res, (2019). [DOI] [PubMed] [Google Scholar]
- [89].Uehara T, Eikawa S, Nishida M, Kunisada Y, Yoshida A, Fujiwara T, Kunisada T, Ozaki T, Udono H, Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: implications for metabolic reprogramming of myeloid cells and anti-tumor effects, Int Immunol, 31 (2019) 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Fitzgerald G, Soro-Arnaiz I, De Bock K, The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer, Front Cell Dev Biol, 6 (2018) 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Yeh WL, Lin CJ, Fu WM, Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia, Mol Pharmacol, 73 (2008) 170–177. [DOI] [PubMed] [Google Scholar]
- [92].Wang H, Wang L, Zhang Y, Wang J, Deng Y, Lin D, Erratum to: Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth, Cancer Cell Int, 16 (2016) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].El Sayed SM, Abou El-Magd RM, Shishido Y, Chung SP, Sakai T, Watanabe H, Kagami S, Fukui K, D-amino acid oxidase gene therapy sensitizes glioma cells to the antiglycolytic effect of 3-bromopyruvate, Cancer Gene Ther, 19 (2012) 1–18. [DOI] [PubMed] [Google Scholar]
- [94].Fan T, Sun G, Sun X, Zhao L, Zhong R, Peng Y, Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment, Cancers (Basel), 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Guo L, Shestov AA, Worth AJ, Nath K, Nelson DS, Leeper DB, Glickson JD, Blair IA, Inhibition of Mitochondrial Complex II by the Anticancer Agent Lonidamine, J Biol Chem, 291 (2016) 42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, Lane A, Trent JO, Chesney J, Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth, Mol Cancer Ther, 7 (2008) 110–120. [DOI] [PubMed] [Google Scholar]
- [97].Lea MA, Altayyar M, desBordes C, Inhibition of Growth of Bladder Cancer Cells by 3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one in Combination with Other Compounds Affecting Glucose Metabolism, Anticancer Res, 35 (2015) 5889–5899. [PubMed] [Google Scholar]
- [98].Rao TN, Hansen N, Hilfiker J, Rai S, Majewska JM, Lekovic D, Gezer D, Andina N, Galli S, Cassel T, Geier F, Delezie J, Nienhold R, Hao-Shen H, Beisel C, Di Palma S, Dimeloe S, Trebicka J, Wolf D, Gassmann M, Fan TW, Lane AN, Handschin C, Dirnhofer S, Kroger N, Hess C, Radimerski T, Koschmieder S, Cokic VP, Skoda RC, JAK2-mutant hematopoietic cells display metabolic alterations that can be targeted to treat myeloproliferative neoplasms, Blood, 134 (2019) 1832–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Gines A, Bystrup S, Ruiz de Porras V, Guardia C, Musulen E, Martinez-Cardus A, Manzano JL, Layos L, Abad A, Martinez-Balibrea E, PKM2 Subcellular Localization Is Involved in Oxaliplatin Resistance Acquisition in HT29 Human Colorectal Cancer Cell Lines, PLoS One, 10 (2015) e0123830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kim DJ, Park YS, Kang MG, You YM, Jung Y, Koo H, Kim JA, Kim MJ, Hong SM, Lee KB, Jang JJ, Park KC, Yeom YI, Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells, Exp Cell Res, 336 (2015) 119–129. [DOI] [PubMed] [Google Scholar]
- [101].Zhang HS, Zhang FJ, Li H, Liu Y, Du GY, Huang YH, Tanshinone A inhibits human esophageal cancer cell growth through miR-122-mediated PKM2 down-regulation, Arch Biochem Biophys, 598 (2016) 50–56. [DOI] [PubMed] [Google Scholar]
- [102].Guo XP, Zhang XY, Zhang SD, [Clinical trial on the effects of shikonin mixture on later stage lung cancer], Zhong Xi Yi Jie He Za Zhi, 11 (1991) 598–599, 580. [PubMed] [Google Scholar]
- [103].Stuart SD, Schauble A, Gupta S, Kennedy AD, Keppler BR, Bingham PM, Zachar Z, A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process, Cancer Metab, 2 (2014) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Whitehouse S, Cooper RH, Randle PJ, Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids, Biochem J, 141 (1974) 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Granchi C, Bertini S, Macchia M, Minutolo F, Inhibitors of lactate dehydrogenase isoforms and their therapeutic potentials, Curr Med Chem, 17 (2010) 672–697. [DOI] [PubMed] [Google Scholar]
- [106].Billiard J, Dennison JB, Briand J, Annan RS, Chai D, Colon M, Dodson CS, Gilbert SA, Greshock J, Jing J, Lu H, McSurdy-Freed JE, Orband-Miller LA, Mills GB, Quinn CJ, Schneck JL, Scott GF, Shaw AN, Waitt GM, Wooster RF, Duffy KJ, Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells, Cancer Metab, 1 (2013) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kummar S, Raffeld M, Juwara L, Horneffer Y, Strassberger A, Allen D, Steinberg SM, Rapisarda A, Spencer SD, Figg WD, Chen X, Turkbey IB, Choyke P, Murgo AJ, Doroshow JH, Melillo G, Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor-1alpha in advanced solid tumors, Clin Cancer Res, 17 (2011) 5123–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Montal ED, Bhalla K, Dewi RE, Ruiz CF, Haley JA, Ropell AE, Gordon C, Haley JD, Girnun GD, Inhibition of phosphoenolpyruvate carboxykinase blocks lactate utilization and impairs tumor growth in colorectal cancer, Cancer Metab, 7 (2019) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Abbadi S, Rodarte JJ, Abutaleb A, Lavell E, Smith CL, Ruff W, Schiller J, Olivi A, Levchenko A, Guerrero-Cazares H, Quinones-Hinojosa A, Glucose-6-phosphatase is a key metabolic regulator of glioblastoma invasion, Mol Cancer Res, 12 (2014) 1547–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Thamrongwaranggoon U, Seubwai W, Phoomak C, Sangkhamanon S, Cha’on U, Boonmars T, Wongkham S, Targeting hexokinase II as a possible therapy for cholangiocarcinoma, Biochem Biophys Res Commun, 484 (2017) 409–415. [DOI] [PubMed] [Google Scholar]
- [111].Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X, Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2, Oncogene, 30 (2011) 4297–4306. [DOI] [PubMed] [Google Scholar]
- [112].Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV, Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression, Proc Natl Acad Sci U S A, 107 (2010) 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Balan MD, McLeod MJ, Lotosky WR, Ghaly M, Holyoak T, Inhibition and Allosteric Regulation of Monomeric Phosphoenolpyruvate Carboxykinase by 3-Mercaptopicolinic Acid, Biochemistry, 54 (2015) 5878–5887. [DOI] [PubMed] [Google Scholar]
- [114].Ma R, Zhang W, Tang K, Zhang H, Zhang Y, Li D, Li Y, Xu P, Luo S, Cai W, Ji T, Katirai F, Ye D, Huang B, Switch of glycolysis to gluconeogenesis by dexamethasone for treatment of hepatocarcinoma, Nat Commun, 4 (2013) 2508. [DOI] [PubMed] [Google Scholar]
- [115].Arion WJ, Canfield WK, Ramos FC, Schindler PW, Burger HJ, Hemmerle H, Schubert G, Below P, Herling AW, Chlorogenic acid and hydroxynitrobenzaldehyde: new inhibitors of hepatic glucose 6-phosphatase, Arch Biochem Biophys, 339 (1997) 315–322. [DOI] [PubMed] [Google Scholar]
- [116].Ramachandran S, Pan CQ, Zimmermann SC, Duvall B, Tsukamoto T, Low BC, Sivaraman J, Structural basis for exploring the allosteric inhibition of human kidney type glutaminase, Oncotarget, 7 (2016) 57943–57954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, Cianchetta G, Cai Z, Zhou D, Cui D, Chen P, Straley K, Tobin E, Wang F, David MD, Penard-Lacronique V, Quivoron C, Saada V, de Botton S, Gross S, Dang L, Yang H, Utley L, Chen Y, Kim H, Jin S, Gu Z, Yao G, Luo Z, Lv X, Fang C, Yan L, Olaharski A, Silverman L, Biller S, Su SM, Yen K, Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers, ACS Med Chem Lett, 9 (2018) 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Yen K, Travins J, Wang F, David MD, Artin E, Straley K, Padyana A, Gross S, DeLaBarre B, Tobin E, Chen Y, Nagaraja R, Choe S, Jin L, Konteatis Z, Cianchetta G, Saunders JO, Salituro FG, Quivoron C, Opolon P, Bawa O, Saada V, Paci A, Broutin S, Bernard OA, de Botton S, Marteyn BS, Pilichowska M, Xu Y, Fang C, Jiang F, Wei W, Jin S, Silverman L, Liu W, Yang H, Dang L, Dorsch M, Penard-Lacronique V, Biller SA, Su SM, AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations, Cancer Discov, 7 (2017) 478–493. [DOI] [PubMed] [Google Scholar]