

ABSTRACT
Extracellular vesicles (EVs) are small membrane vesicles that serve as important intercellular signalling intermediaries in both malignant and non-malignant cells. For EVs formed by the plasma membrane, their biogenesis is characterized by an increase in intracellular calcium followed by successive membrane and cytoskeletal changes. EV-production is significantly higher in malignant cells relative to non-malignant cells and previous work suggests this is dependent on increased calcium mobilization and activity of calpain. However, calcium-signalling pathways involved in malignant and non-malignant EV biogenesis remain unexplored. Here we demonstrate; malignant cells have high basal production of plasma membrane EVs compared to non-malignant cells and this is driven by a calcium–calpain dependent pathway. Resting vesiculation in malignant cells occurs via mobilization of calcium from endoplasmic reticulum (ER) stores rather than from the activity of plasma membrane calcium channels. In the event of ER store depletion however, the store-operated calcium entry (SOCE) pathway is activated to restore ER calcium stores. Depleting both ER calcium stores and blocking SOCE, inhibits EV biogenesis. In contrast, calcium signalling pathways are not activated in resting non-malignant cells. Consequently, these cells are relatively low vesiculators in the resting state. Following cellular activation however, an increase in cytosolic calcium and activation of calpain increase in EV biogenesis. These findings contribute to furthering our understanding of extracellular vesicle biogenesis. As EVs are key mediators in the intercellular transfer of deleterious cancer traits such as cancer multidrug resistance (MDR), understanding the molecular mechanisms governing their biogenesis in cancer is the crucial first step in finding novel therapeutic targets that circumvent EV-mediated MDR.
KEYWORDS: Biogenesis, extracellular vesicles, microvesicles, calcium, endoplasmic reticulum, sarco/endoplasmic reticulum calcium ATPase, SERCA, store operated calcium entry, cancer, multidrug resistance
Introduction
Extracellular vesicles (EVs) are sub-micron sized membrane-enclosed vesicles released from all cell types [1,2]. EVs as signalling vectors and, regulate numerous physiological pathways ranging from inflammation, coagulation to immunity. EVs are also implicated in mediating many disease pathologies including but not limited to; autoimmune disease, infectious disease, cardiovascular disease and cancer. Of particular interest to our laboratory is their contribution to cancer survival via promotion of metastasis, and chemotherapeutic resistance. Tumour EVs mediate the transfer of functional resistance proteins and nucleic acids from drug-resistant to drug-sensitive cells resulting in intercellular conferral of multidrug resistance (MDR) [3–5]. We also showed through the study of EVs that MDR was complex and extended beyond drug – cell interactions. Specifically, EVs from MDR cells could confer increased metastatic capacity [6], active and passive drug sequestration [7], immune evasion [8] and altered tissue biomechanics [9].
The significance of EVs is not restricted to cellular signalling and they have emerged as novel drug-delivery vehicles, and important biomarkers [10,11].
Delineation of extracellular vesicle subtypes is typically based on size and biogenic origin [12,13]. The cellular machinery and signalling pathways involved in EV release also differ between EV subtypes. The process for exosome biogenesis has been extensively described by, Raposo and Stoorvogel [1], Colombo et al. [14] and Kalra et al. [15]. In contrast, the biogenesis of plasma membrane-derived EVs is less well defined, owing in part to early reports defining MVs as simply inert by-products of cellular stress and/or cell death [2,16–18].
Plasma membrane EV release can be initiated through cellular activation in response to stimuli such as exposure to serine proteases, thrombin, calcium ionophores, ADP, inflammatory cytokines, growth factors and shear inducers [19–23]. Cellular activation and a subsequent increase in intracellular Ca2+ affects both plasma membrane and cytosolic machinery leading to plasma membrane EV biogenesis. At steady state, phospholipids present in the plasma membrane are asymmetrically distributed. The anionic phospholipids phosphatidylserine (PS) and phosphatidylethanolamine (PE) localize to the inner leaflet whilst phosphatidylcholine (PC) and sphingomyelin (SM) localize to the outer [24]. A network of lipid translocases, modulated by calcium, maintains the asymmetry [25–27]. Increase in intracellular calcium results in the collapse of plasma membrane phospholipid asymmetry resulting in a destabilization of plasma membrane-cytoskeletal anchorage [28,29]. Calcium mobilization concurrently activates calpain, a cysteine protease, which in turn cleaves several cytoskeletal components including actin, ankyrin, protein 4.1 and spectrin [30,31]. Calpain-mediated cleavage of the cytoskeleton further disrupts attachment to the membrane and these localized regions bud outwards to form plasma membrane EVs [20,32,33].
An increase in intracellular calcium appears to be the initiating step in plasma membrane EV biogenesis [20,34–36]. The calcium ion (Ca2+) is a promiscuous signalling molecule, involved in diverse cellular processes ranging from neurological transmission to muscular contraction [37]. In non-excitable cells, cellular calcium activates signalling cascades through effects on Ca2+-regulated proteins and also via Ca2+ dependent transcription factors [38].
Cells maintain tight control of compartmental Ca2+ homoeostasis through a network of calcium channels, pumps and exchangers. This network facilitates simultaneous operation of calcium signalling pathways in cells. Dysregulation of Ca2+ signalling is a key feature of many diseases with calcium modulation forming a significant part of pharmacotherapy [39–42].
In the context of malignancy, dysregulation of Ca2+ homoeostasis significantly contributes to disease progression [43] as well as facilitating other cancer hallmarks [42,44]. In breast cancer cells, the calcium channel TRPC5 is important in MDR via extracellular vesicles [45]. For this reason, the therapeutic targeting of calcium signalling presents clinical opportunities [40,46]. Despite the known relationship between elevated intracellular calcium and plasma membrane vesiculation, the exact pathway(s) and channels involved in biogenesis remain to be fully defined.
We recently reported the presence of distinct vesiculation pathways in malignant and non-malignant cells at rest [47]. We demonstrated that a calcium–calpain dependent pathway regulated malignant cell biogenesis and an alternative pathway driving biogenesis was proposed for non-malignant cells at rest [47].
In this paper, we now investigate the Ca2+ signalling pathways involved in regulating plasma membrane EV biogenesis in malignant and non-malignant cells. Specifically, we propose a role for endoplasmic reticulum (ER) calcium stores and store-operated Ca2+ entry (SOCE) in EV biogenesis. The discovery of discrete pathways of biogenesis in malignant and non-malignant cells is promising in the search for new strategies to circumvent the acquisition and transfer of EV-mediated deleterious traits in cancer and in advancing knowledge in EV biology.
Methods
Cell culture
The drug-sensitive human breast adenocarcinoma cell line MCF-7 and its drug-resistant sub-line MCF-7/Dx (Dx for simplicity) were routinely cultured in RPMI-1640 (Sigma-Aldrich, NSW, Australia) supplemented with 10% FBS as previously described [48]. Incremental exposure of MCF-7 cells to doxorubicin was used to develop the Dx resistant sub-line as previously described [48–50]. The non-malignant human brain endothelial cell line (hCMEC-D3) was cultured in Endothelial Cell Basal Medium-2 (EBM-2; Lonza, MD, USA) as previously described [51]. All cell lines were maintained at 37°C in a humidified 5% CO2 incubator. All cell lines were routinely tested for mycoplasma infection.
Atomic force microscopy
The NanoWizard 4 BioScience Atomic Force Microscope (AFM; JPK Instruments, Germany) was used for cell surface topography studies. Cells were fixed in 4% paraformaldehyde (PFA) for 30 min, washed once with phosphate-buffered saline (PBS) and twice with distilled water. Cells were air-dried for ~1 h to allow the membrane-bound vesicles to transform to vesicle-derived pits as previously described [47]. Topographical AFM imaging was carried out in contact mode with sharpened silicon nitride cantilevers (L: 225 µm, W: 46 µm, T: 1.0 µm, SPM Probe Model: SHOCON; AppNano) with a tip radius of < 10 nm and spring constant of 0.6 N/m. Scans were performed in air at a rate of 1 Hz. Three areas were selected at random for scanning for each treatment condition and all experiments were performed in triplicate.
The raw data from AFM height measurements were digitally filtered as previously described by Antonio et al. [52] and Root Mean Square Roughness (RRMS) was computed with the open source software Gwyddion [53]. The filtration procedure isolated features of interest (plasma membrane EV-derived pits) from features such as the overall cell shape. Given the size range of plasma membrane EVs, the specific filtration procedure applied in this case is separating features ranging from 100 to 1000 nm.
Each AFM image (30 µm2) was scanned with 512 points (scan step Δx = 58.6 nm) and a filtration procedure was applied with a cut-off normalized frequency (fc) of 0.07 to remove all features with steps λ longer than 1674.3 nm (λ = 2· Δx/fc). Previous work has demonstrated that plasma membrane vesiculation in occurs in perinuclear regions of the membrane [47]. Therefore, a “zoomed” 10 µm2 area was scanned within each 30 µm2 was also taken to determine roughness in these areas of surface. These scans consist of 512 points (scan step Δx = 19.5 nm) and are filtered to remove features larger than 975 nm (fc = 0.04; λ = 2· Δx/fc). The Root Mean Square Roughness (RRMS) was then computed as a quantitative measure of surface topography across different treatments.
Intracellular calcium imaging and quantification in MCF-7 cells using imageXpress
Cellular calcium was analysed using the ratiometric fluorescent indicator, Fura-2 AM as previously described [54,55]. Cells were loaded with Fura-2 AM for 30 min at 37°C in the dark with a dye-loading solution comprising 4 µM Fura-2 AM (Invitrogen) in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum with and without 50 μM 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM). Cells were allowed to incubate for a further 15 min in the dark at ambient temperature in physiological salt solution (PSS [nominal]; 10 mM HEPES, 5.9 mM KCl, 1.4 mM MgCl2, 1.2 mM NaH2PO4, 5 mM NaHCO3, 140 mM NaCl, 11.5 mM glucose, 1.8 mM CaCl2, pH 7.3). Loading-dye solution was removed and cells were washed twice with PSS Ca2+ (PSS supplemented with 1.8 mM CaCl2) and then twice with PSS no added extracellular (nominal) to ensure less than 100 µM Ca2+ was remaining in the test wells [56]. Fresh PSS containing 1.8 mM CaCl2 was added for intracellular calcium measurements.
MCF-7 cells loaded with Fura-2 AM were stimulated with the sarco-endoplasmic reticulum calcium ATPase (SERCA) inhibitor, thapsigargin (TG) (300 nM). Experiments were performed using the ImageXpressMICRO high content imaging system (Molecular Devices, Sunnyvale, CA, USA). Measurements were taken at an excitation wavelength of 340 and 387 nm with fluorescence emission detected using a 510–520 nm band pass filter. Two baseline measurements were taken at 0 and 10 s. TG was loaded into the reagent plate and added to test wells at 20 s. Fura-2 AM ratio data points were acquired every 5 s for a 300 s window. Relative changes in intracellular calcium [Ca2+]I were determined following background subtraction using the ratio of the two wavelengths, 340 and 387 nm for each time interval.
Cell viability following TG treatment
The effects of TG concentrations (1–300 nM) over a 10 min treatment window on cell viability were assessed by trypan blue dye exclusion at 24-h post treatment as described [20].
MV isolation and quantitative flow cytometric analysis of plasma membrane EVs
Plasma membrane EVs were isolated by a process of differential centrifugation as previously described [3,48]. Briefly, cell supernatants were collected from approximately 1.2 × 107 cells and centrifuged for 5 min at 500 × g to remove cells and large cellular debris. Smaller debris was removed by two further spins at 2000 × g for 1 min. The supernatant was collected and centrifuged at 18,000 × g for 30 min at 4°C to pellet EVs. Isolated EVs were resuspended in 200 µL PBS for analysis.
Flow cytometric analysis was conducted on the BD LSRFortessa™ X-20 flow cytometer (BD Biosciences) custom built with a 100 mW blue laser for small particle detection. Plasma membrane EV gates were established as previously described using latex sizing beads 0.3–1.1 µm in diameter (BD Biosciences). The threshold in side scatter was adjusted to 200 arbitrary units to avoid background noise [57]. The predefined gate was applied to all samples during analysis. The performance of lasers was validated before each experiment using BD FACSDiva™ CS&T Research Beads (BD, Australia/New Zealand).
EV samples resuspended in PBS were combined with TruCount™ beads and analysed according to the manufacturer’s instructions. The tube was gently vortexed and loaded onto the flow cytometer, after which the fluid stream was allowed to stabilize to a flow rate of 10 events/second for 30 s. To prevent cross-contamination of samples, the sample line was flushed with MilliQ water for at least 30 s between each run. The stop gate was set at a fixed number of 2000 of Trucount™ beads during data acquisition. A PBS blank buffer control was run to ensure baseline events were low (data not shown). The number of EVs per µL was calculated using the formula: N = (gated MV events/gated Trucount™ events) × total number of Trucount™ beads [11]. EV events were normalized to individual well cell counts to account for variation in cell numbers between samples as previously described [20]. FACSDiva version 8.0.1 (BD Biosciences) was used for analysis of flow cytometry data. Full description of methodologies was also submitted to EV-TRACK (ID: EV190073)
Statistics and data analysis
Data were analysed with a one-way analysis of variance (ANOVA) followed by a Dunnett’s multiple comparisons test using the GraphPad Prism version 7.02 for Windows (GraphPad Software, La Jolla, CA, USA). Data are presented as the mean or mean ± SD of 3 individual experiments with predictive results value of (****) P < 0.0001, (***) P < 0.0005, (**) P < 0.01 and (*) P < 0.05 were considered statistically significant.
Results
Mobilization of intracellular Ca2+ increases vesiculation
Using pharmacological modulators of Ca2+ release together with topographical AFM analysis we show that intracellular calcium mobilization is required for plasma membrane EV biogenesis. The Ca2+ ionophore, A23187 (1 µM) was used to study the effect of increasing intracellular Ca2+ on malignant (MCF-7 and Dx) cell and non-malignant (hCMEC-D3) cell vesiculation (Figure 1). Ca2+ ionophores mobilize Ca2+ through the translocation of Ca2+-ionophore complexes across the plasma membrane, activation of native plasma membrane Ca2+ channels, and via mobilization of Ca2+ from the ER [58].
Figure 1.
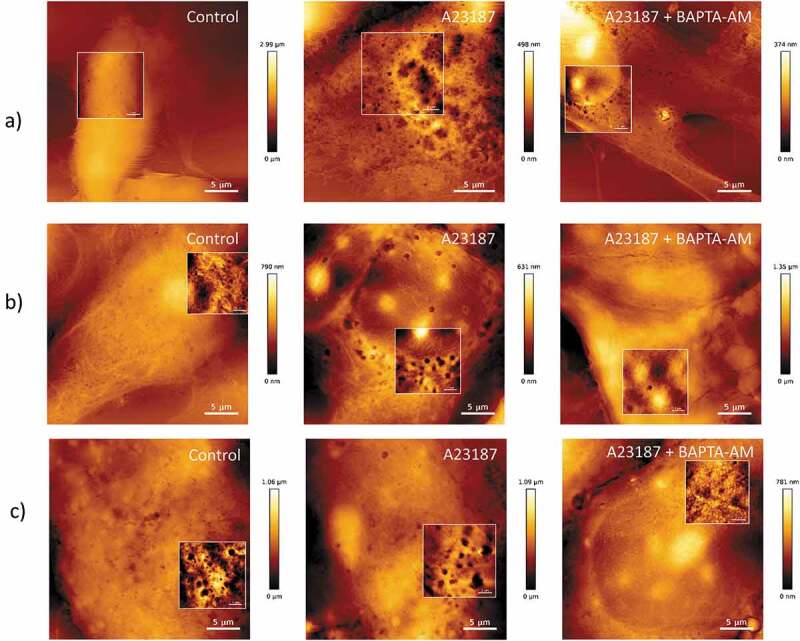
AFM topographical images of vesiculation following increased intracellular calcium in malignant and non-malignant cells.
(a) hCMEC-D3, (b) MCF-7, and (c) Dx cells were imaged using AFM in contact mode (scan rate 1 Hz) in the presence and absence of treatment with the calcium ionophore A23187 (1 µM) ± BAPTA-AM (50 µM) for 18 h. Ca2+ activation with A23187 resulted in increased vesiculation in all cells; however, the effect is most prominent in non-malignant cells. The calcium chelator, BAPTA-AM inhibited the formation of EVs in all cells types. Zoomed images shown in focussed panels as shown in frame. Scale bar shown. Image representative of a typical field of view from three independent experiments.
As membrane-bound EVs are characteristically soft and deformable when undergoing AFM imaging, we used an indirect method for their examination whereby fixed cells are air-dried and membrane vesicles are transformed into vesicle-derived pits [47,59]. Prior to treatment with the ionophore, hCMEC-D3 cells displayed a smooth surface topography with little to no vesiculation at rest (Figure 1(a), left panel). Contrary to this, both malignant cell types, displayed an abundance of vesicle-derived pits on the cell surface at rest (Figure 1(b,c), left panels).
Increasing intracellular Ca2+ following A23187 treatment resulted in a pronounced and modest increase in vesiculation in non-malignant and malignant cells, respectively. Non-malignant cells underwent a pronounced change in topography with numerous vesicle-derived pits observed following treatment with A23187 (Figure 1(a), centre panel). In contrast, the increase in vesicle-derived pits observed in malignant cells treated with A23187 was less pronounced (Figure 1(b,c), centre panels). This is consistent with our previous findings [47]. Both cell types when treated with the calcium ionophore A23187 in combination with the calcium chelator, BAPTA-AM (µM), exhibited a smooth surface morphology devoid of vesicle-derived pits (Figure 1(a, b and c) right panels). Together these results evidence the involvement of intracellular Ca2+ mobilization in plasma membrane EV vesiculation in both malignant and non-malignant cells. Notably, the effects on vesiculation following Ca2+ activation were less in malignant cells compared with non-malignant cells and this is likely due to the already elevated baseline Ca2+ activation in MCF-7 cells at rest [60].
Increasing intracellular Ca2+ via store-operated Ca2+ entry (SOCE) activates EV vesiculation
The ER is the major intracellular store of calcium and is maintained at higher Ca2+ concentrations (100–800 µM) than that of the cytoplasm through the activity of Sarco/endoplasmic reticulum calcium ATPase (SERCA) [61,62]. We used Thapsigargin (TG) to selectively inhibit SERCA, deplete the ER Ca2+ stores and in turn activate SOCE [63]. Measurement of intracellular Ca2+ transients were carried out using the ImageXpressMICRO high content imaging system as described by [64]. As shown in Figure 2(a) treatment with TG resulted in a gradual increase in relative [Ca2+]i over a 5 min measurement interval. This corresponded to a significant increase in the F340/F387 ratio – indicative of an increase in intracellular [Ca2+]i (Figure 2(b)). The pre-treatment of cells with the intracellular Ca2+ chelator, BAPTA-AM, (50 µM) abolished the TG-induced increase in [Ca2+]i (Figure 2(a,b)).
Figure 2.
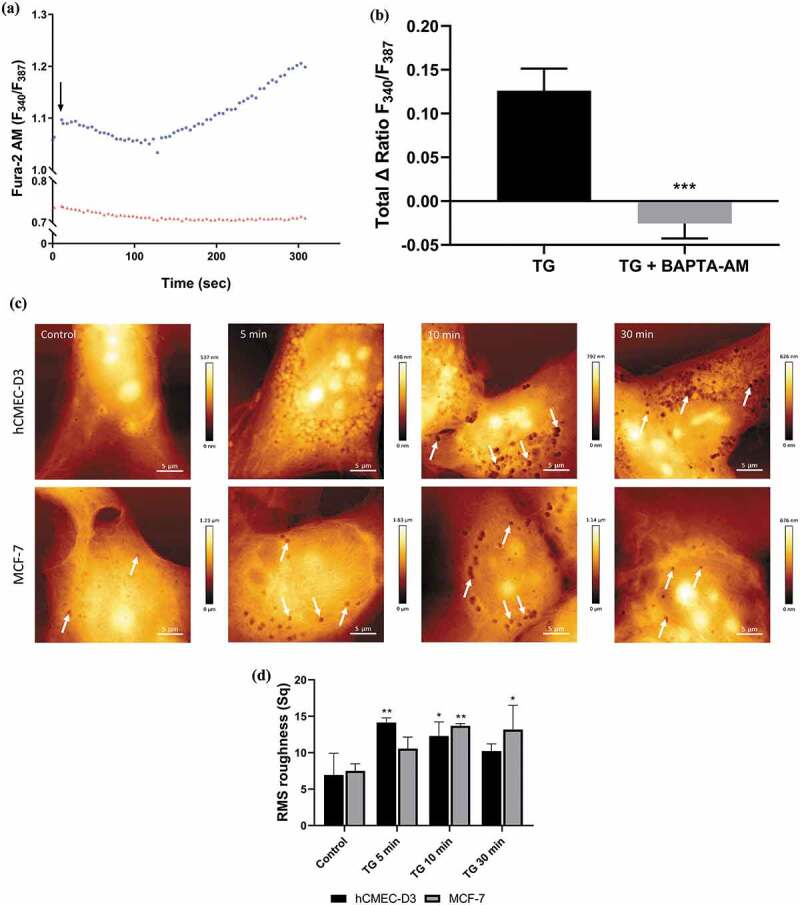
Increasing intracellular calcium with thapsigargin (TG) induces plasma membrane EV biogenesis in malignant MCF-7 cells.
MCF-7 cells were treated with TG (300 nM) for 5 min in the presence and absence of BAPTA-AM (50 uM). Changes to intracellular calcium was analysed using the ImageXpress high content imaging system and ratiometric calcium indicator Fura-2 AM. (a) Representative trace of change in relative fluorescence of Fura-2 AM following treatment with TG alone (●) and in the presence of BAPTA-AM (▴) in MCF-7 cells. The rise in fluorescence indicates increasing intracellular Ca2+ over 5 min. (b) Total TG-mediated increase in intracellular Ca2+ ± BAPTA-AM indicated by increase in relative fluorescence of Fura-2 AM (340/387) in MCF-7 cells. Data represents the mean ± SD of three experiments. ***p < 0.001 (paired t test). (c) Time course of TG-mediated increase in plasma membrane EV biogenesis. hCMEC-D3 (upper panel) and MCF-7 (lower panel) cells treated for 5, 10, and 30 min with TG and EV-derived pits visualized with AFM (arrows). (d) Analysis of hCMEC-D3 and MCF-7 cell topography illustrates there is an increase in surface roughness following thapsigargin treatment. Representative images shown from at least 3 experiments. Data represent the mean ± SD of at least 3 experiments. *p < 0.05, **p < 0.005 (one way ANOVA).
AFM was used to visualize the effects of TG induced [Ca2+]i increase on EV vesiculation in malignant and non-malignant cells. MCF-7 and hCMEC-D3 cells were treated with TG and the degree of vesiculation assessed for 30 min post treatment. As shown in Figure 2(c) (upper panels), non-malignant hCMEC-D3 cells display a smooth cellular topography supporting the presence of little to no vesiculation at rest. hCMEC-D3 cells treated for 5−30 min with TG displayed an uneven surface topography with a significant increase in Root Mean Squared (RMS) roughness (Figure 2(d)). This change in surface roughness was accompanied by the presence of vesicular pits, predominantly at perinuclear regions. hCMEC-D3 cells treated with TG for 10–30 min induced numerous vesicle-derived pits localized in the perinuclear region, consistent with the presence of active vesiculation (Figure 2).
Conversely, the malignant MCF-7 cells are observed to have numerous vesicle derived pits compared to their non-malignant counterparts (Figure 2(c) (lower panels)). This supports these cells to be actively vesiculating at rest, consistent with earlier studies by us [20,47]. Again, there was a modest change in the surface topography following treatment with TG over 30 min in MCF-7 cells following TG treatment (Figure 2(c,d)). These studies demonstrate an increase in vesiculation following TG induced intracellular Ca2+ mobilization, particularly pronounced in the non-malignant cells.
Pre-treatment with the intracellular calcium chelator, BAPTA-AM effectively inhibited TG-induced vesiculation in both malignant and non-malignant cells (Figure 3(a)). Together, these results establish a role for [Ca2+]i in vesiculation in malignant and non-malignant cells.
Figure 3.
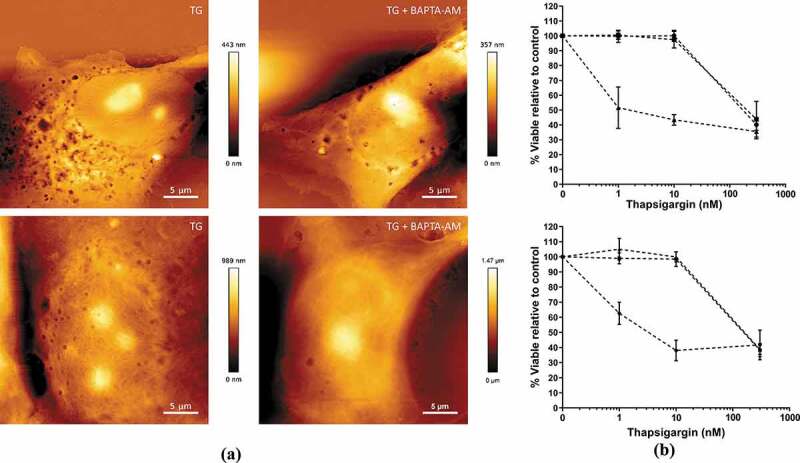
Effects of Thapsigargin (TG)-induced microvesicle (MV) biogenesis on cell viability over 24 h.
hCMEC-D3 and MCF-7 cells were treated with TG (1–300 nM) for up to 10 min. Cells were washed and then incubated for a further 24 h and analysed for viability and plasma membrane EV biogenesis. (a) AFM topographical images following TG (10 nM) treatment for 5 min ± BAPTA-AM (50 µM). hCMEC-D3 (upper panels) and MCF-7 (lower panels). Images representative of three independent experiments. (b) hCMEC-D3 (upper panel) and MCF-7 (lower panel) cell viability was assessed using a Trypan Blue dye Exclusion assay. Cells were treated with 1, 10 and 300 nM TG for 1 min (▴), 5 min (∎), and 10 min (●), washed and analysed 24 h post treatment. Data represent the mean ± SD of three experiments.
The high potency and irreversible binding of TG to SERCA means that it can impact cell viability [65,66]. The effects of TG on the viability of hCMEC-D3 and MCF-7 cells were assessed over 24 h using a Trypan Blue Exclusion Assay to ensure viability under experimental conditions. As shown in Figure 3(a,b), 10 nM TG treatment for 5 min resulted in an observable increase in vesiculation, whilst not affecting cell viability. This concentration was chosen for further investigations.
Flow cytometry was used to interrogate the Ca2+ signalling pathways regulating plasma membrane EV biogenesis. The gating parameters for the EV region were defined using 0.3 µm latex beads (R1: the lowest possible limit on forward scatter for BD LSRII) and 1.1 µm beads (R2: represents the upper limit for quantitation of the EV population; Figure 4(a)), as previously described [11]. We observed a significant, 1.5 fold increase in EV biogenesis in hCMEC-D3 cells following 300 nM TG treatment (Figure 4(b)). Resting hCMEC-D3 cells treated with the calpain inhibitor, ALLM (10 µM), displayed a 1.14 fold increase in EV production (Figure 4(b)) consistent with our previous findings [47]. This suggests to us that at rest vesiculation is driven by a calpain-independent pathway in non-malignant HCEM-D3 cells. Pre-treatment of hCMEC-D3 cells with ALLM inhibited the TG-induced plasma membrane EV production consistent with the involvement of a calpain-dependent pathway driving vesiculation following TG treatment. Conversely, the addition of the calpain inhibitor, ALLM to resting MCF-7 cells significantly reduced EV production by 1.4 fold, consistent with the involvement of calpain in EV vesiculation of malignant MCF-7 cells at rest. ALLM also significantly inhibited vesiculation by 2 fold in TG-treated MCF-7 cells, with the effect on EV vesiculation being greater than ALLM alone (Figure 4(b)).
Figure 4.
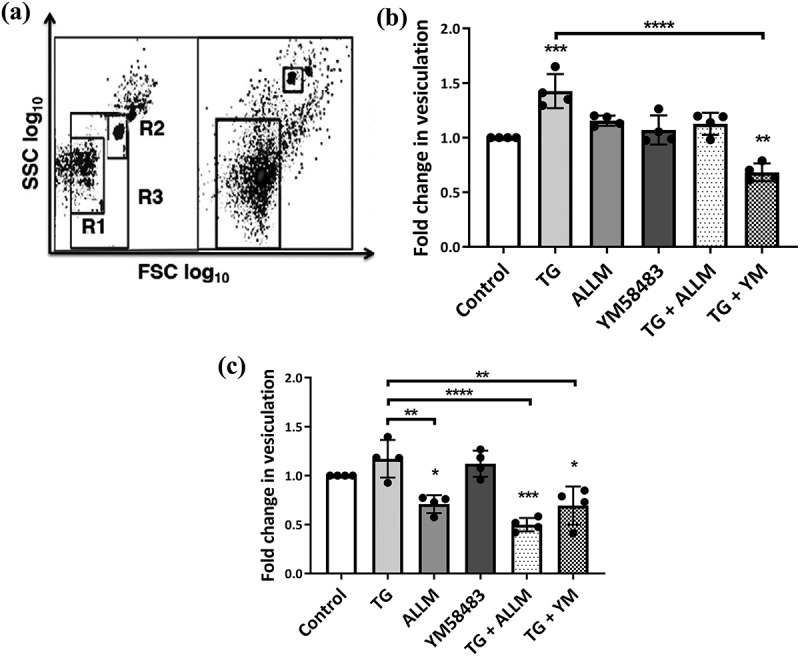
Vesiculation in malignant and non-malignant cells following manipulation of the calcium-calpain plasma membrane EV biogenic pathway.
hCMEC-D3 and MCF-7 cells were treated with modulators of vesiculation and Ca2+ for 24 h EV release was quantified by flow cytometry. (a) EV size gating strategy. Latex sizing beads of (0.3–1.1 um diameter) were used to define the gate for EVs. R1 represents the lower (0.3 µm) and R2 represents the upper (1.1 µm) limits set by the beads. R3 defines the region that was used to quantitate EVs and was applied to all samples. The total number of acquired events when the stop gate was set at 5000 TruCount bead counts is shown on the right. Data are expressed as fold change of EV release relative to vehicle control. (b) Non-malignant hCMEC-D3 cells displayed a significant increase in vesiculation following thapsigargin (TG) treatment and a significant reduction in vesiculation when treated with TG and YM58483. While treatments produced a consistent, small increase in vesiculation, it did not reach statistical significance. (c) Treatment with TG only resulted in a modest increase in vesiculation in malignant MCF-7 cells compared to vehicle control. There were significant reductions in malignant cell vesiculation following treatment with calpain inhibitor II (ALLM), TG + ALLM and TG + YM58483. Data represents the mean ± SD of at least 3 experiments. *p < 0.05, **p < 0.005 ***p < 0.001 (one way ANOVA).
YM58483 (N-[4-[3,5-Bis(trifluoromethyl)-1 H-pyrazol-1-yl]phenyl]-4-methyl-1,2,3-thiadiazole-5-carboxamide) is a selective inhibitor of SOCE and acts by blocking plasma membrane Ca2+ channels which are activated upon ER store depletion [67]. We observed no significant difference in vesiculation in hCMEC-D3 cells treated with 1 µM YM58483 alone compared to untreated control (Figure 4). Inhibition of TG activated SOCE with YM58483 resulted in a 1.5 fold decrease in vesiculation in hCMEC-D3 cells compared to cells treated with TG. This effect was also seen in malignant MCF-7 cells. Collectively, this data suggests that depletion of internal calcium stores by TG induced MVs in both hCMEC-D3 and MCF-7 cells and demonstrates that ER-mediated calcium regulation primarily drives vesiculation in malignant and non-malignant cells. Consistent with this, the lack of reduction in vesiculation of MCF-7 cells by the SOCE blocker YM58483 alone, suggests that elevated basal EVs in MCF-7 cells occurs through a pathway independent of SOCE.
Taken together, these results establish a role for ER Ca2+ as well as SOCE in the biogenesis of plasma membrane EVs in malignant and non-malignant cells. Non-malignant hCMEC-D3 cells are low vesiculators at rest and display a large increase in vesiculation following ER Ca2+ store depletion and activation of SOCE. Contrary to this, malignant MCF-7 cells are actively vesiculating at rest, and although SOCE can contribute to MVs, SOCE is not the driver of increased EVs in resting MCF-7 cells.
Discussion
Elevated intracellular Ca2+ is required in the biogenesis of microvesicles [35–37,47]. Despite the central role that Ca2+ plays in plasma membrane EV biogenesis, the source of Ca2+ mobilization – intra- or extracellular – driving plasma membrane vesiculation has remained undefined. We have previously shown that distinct EV biogenic pathways exist between malignant and non-malignant cells, the differences of which appear related to intracellular calcium levels at rest [47]. Uncovering the mechanistic differences between malignant and non-malignant plasma membrane EV biogenesis is important as it could point to novel and selective treatment strategies for the circumvention of MV-mediated acquisition and dissemination of MDR and other deleterious traits in cancer cell populations. This study sought to identify the calcium signalling pathways involved in the vesiculation of malignant MCF-7 and non-malignant hCMEC-D3 cells. We evidence the involvement of the ER and SOCE pathway in EV biogenesis. Malignant cells are high vesiculators at rest compared to non-malignant cells, however, observed differences do not appear to be related to mobilization of Ca2+ following SOCE activation. SOCE does play a role in plasma membrane EV biogenesis; however, it serves as a failsafe mechanism – driving vesiculation in the event ER stores are depleted of Ca2+.
At rest, non-malignant hCMEC-D3 cells are low vesiculators and increase in intracellular Ca2+ result in the induction of plasma membrane EV production. Consistent with our previous work, high-resolution AFM images show hCMEC-D3 cells have a smooth topography with very few vesicle-derived pits observable in their resting state (Figure 1(a)). Following cellular activation with the Ca2+ ionophore, A23187, plasma membrane vesicle biogenesis is efficiently “turned on” in non-malignant cells. A23187 has been shown previously to elevate [Ca2+]i and induce plasma membrane EV formation in a number of cell types [58] and these results are consistent with this. Non-malignant cells stimulated with A23187 displayed a dramatically altered surface morphology consistent with cells undergoing active membrane vesiculation.
During plasma membrane EV biogenesis, the increase in intracellular Ca2+ triggers a number of cytoplasmic and membrane alterations including: externalization of PS through the activation of lipid translocases, loss of membrane asymmetry, activation of calpain and the remodelling of the cytoskeleton. Ultimately, anchorage of the plasma membrane to the cytoskeleton is destabilized, localized regions of the membrane form membrane blebs and are released as EVs. These are observed in morphological AFM studies as uneven cellular surfaces with the presence of vesicle derived pits (Figures 1–3).
Co-treatment of hCMEC-D3 cells with A23187 and the intracellular Ca2+ chelator, BAPTA-AM, inhibited vesiculation, again evidencing the essential role of Ca2+ mobilization in MV biogenesis (Figure 1(a)). Interestingly, inhibiting calpain with ALLM in resting hCMEC-D3 cells resulted in a modest increase in vesiculation compared to untreated control (Figures 1(a), 4(b)). Calpains are highly conserved cysteine proteases that, when activated by Ca2+ cleave a number of substrates including kinases and phosphatases, membrane receptors, and cytoskeletal proteins and associated proteins [68]. Some calpain substrates are responsible for alternative MV biogenesis pathways. For example, calpain directly inhibits the small GTPase, RhoA that is an upstream activator of ROCK1-mediated MV biogenesis [21]. Thus, its inhibition can activate the RhoA mediated vesiculation pathway. Given chelation of intracellular Ca2+ with BAPTA-AM prevents calpain activation, one would expect to see a similar rise in MV production as with ALLM. However, treatment with BAPTA-AM completely blocks vesiculation in resting non-malignant cells. This result suggests the alternative plasma membrane EV biogenic pathway may also be Ca2+ dependent.
Conversely, resting malignant cells produce significantly more plasma membrane vesicles than their non-malignant counterparts [20] (Figure 1). Our results show that the higher resting level of vesiculation in malignant cells is dependent on both intracellular calcium and the activity of calpain, as production can be inhibited by both calcium modulators and the calpain inhibitor ALLM (Figures 1 and 4). Resting malignant cells display a surface topography consistent with actively vesiculating cells: with an abundance of vesicle-derived pits (Figure 1(b,c)). MCF-7 cells treated with the calcium chelator, BAPTA-AM were devoid of vesicle-derived pits and displayed a smooth surface morphology (Figure 1(b,c)). Quantitative flow cytometry verified these observations and demonstrated a significant 1.4-fold reduction in vesiculation in MCF-7 cells treated with ALLM (Figure 4(c)). Analysis of surface topography with AFM on fixed cells is somewhat cumbersome as image acquisition is slow. This disadvantage makes this approach unsuited the to analysis of broader changes in vesiculation. AFM is however, capable of resolving fine details on the cellular surface such as vesicle-derived, pits down to 100 nm in diameter. It is also important to note that while analysis of surface roughness provides a measure of topographical surface changes in different cells types and treatment conditions, only the formation of plasma membrane EV-derived pits have been correlated to production of plasma membrane EVs previously.
These results evidence our proposal that resting malignant cell plasma membrane EV biogenesis is dependent on both Ca2+ and calpain and supports the presence of discrete biogenic pathways in malignant and non-malignant cells at rest [47].
A network of pumps, channels and exchangers maintain tight regulatory control of intracellular Ca2+ homoeostasis [69]. This enables a multitude of signalling modalities to be simultaneously operational in cells [70]. Ca2+ is a highly promiscuous intracellular signalling molecule and cellular effects are dependent on the size, kinetics and subcellular localization of incoming Ca2+ signals [71]. An increase in intracellular Ca2+ is the initiating stimulus for both the collapse of membrane phospholipid asymmetry, and for the activation of calpain and cytoskeletal remodelling required for plasma membrane EV biogenesis. What is intriguing is the propensity of malignant cells to shed spontaneous EVs at rest. This is contrary to non-malignant cells, which require cellular activation. The process in malignant cells is Ca2+-dependent as chelation of intracellular Ca2+ attenuates biogenesis (Figure 1). In adriamycin-resistant breast cancer cells, TRPC5 calcium channels promote the formation of MVs [24]. While that study demonstrated the importance of calcium signalling in vesiculation, the differences in the biogenic pathways between malignant and non-malignant cells were not examined.
The ER is the primary intracellular store of Ca2+, which can be mobilized in response to numerous stimuli [61,72]. It provides the ideal starting point for the interrogation of the specific Ca2+ signalling pathways involved in plasma membrane EV biogenesis as it is well documented to be involved in many cellular processes. TG is commonly employed as an experimental tool to interrogate Ca2+ signalling pathways and it has high selectivity and potency as an irreversible inhibitor of SERCA [73]. SERCA is responsible for the maintenance of high Ca2+ concentration in the ER (≈100–800 µM) and its inhibition ultimately results in an influx of Ca2+ into the cytosol. (Figure 5).
Figure 5.
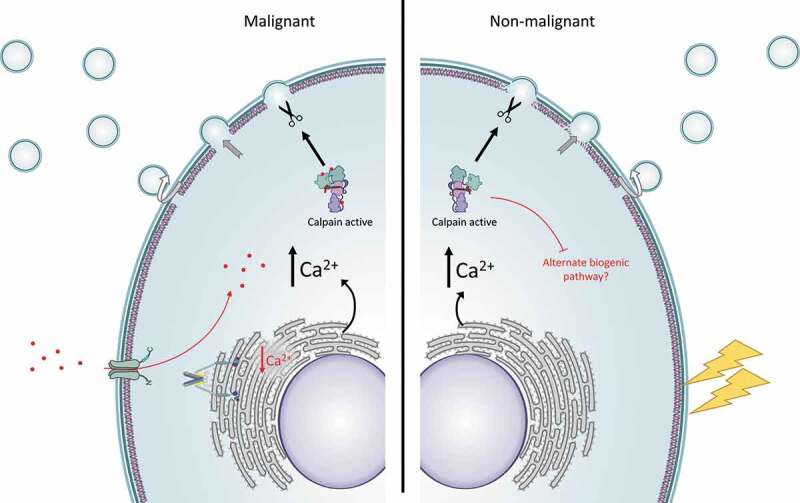
Proposed model for the role of the endoplasmic reticulum and SOCE in MV biogenesis.
Malignant cells have high basal production of plasma membrane EVs compared to non-malignant cells which is driven by a calcium–calpain dependent pathway. Resting vesiculation in MCF-7 cells is dependent on mobilization of calcium from stores within the endoplasmic reticulum rather than from activity of plasma membrane channels. In the event of ER store-depletion – as observed when SERCA is inhibited with thapsigargin – the store-operated calcium entry (SOCE) pathway is activated to restore ER calcium stores. Depleting ER calcium stores and blocking the activity of Orai1 channels with a selective inhibitor of SOCE, YM58483, prevents intracellular calcium increases and inhibits the production of EVs. Calcium signalling pathways are not activated in resting non-malignant hCMEC-D3 cells. Consequently, these cells are relatively low vesiculators in the resting state. Following cellular activation (lightning bolt) or activation of SOCE with thapsigargin, there is an increase in cytosolic calcium and activation of calpain. Calpain mediates remodelling of cytoskeletal proteins, disrupts anchorage of the plasma membrane to the cytoskeleton and results in an increase in plasma membrane EV biogenesis.
SERCA inhibitors such as TG are favoured in the Ca2+ signalling experimental toolkit, as direct depletion of Ca2+ from the ER bypasses other, interfering biochemical signals. As TG has also been employed in the study of numerous cellular processes ranging from cytokine release regulation in lymphocytes [74] to the dysregulation of Ca2+ homoeostasis in cancer [40,54,71], it was chosen to interrogate Ca2+ signalling and plasma membrane EV biogenesis in our cell lines. We demonstrate that TG induces an increase in [Ca2+]i within 5 min in MCF-7 cells (Figure 2(a,b)). As expected, pre-treatment with BAPTA-AM inhibited TG-mediated [Ca2+]i increases (Figure 2(a,b)). Importantly, corresponding to Ca2+ increases, we observed a time-dependent change in surface morphology in both MCF-7 and hCMEC-D3 cells indicative of an increase in vesiculation (Figure 2(c,d)). Demonstrating for the first time that TG-mediated activation of SOCE can generate production of plasma membrane EVs.
Having established TG as a suitable molecule for activation of plasma membrane EV biogenesis and a concentration that does not compromise cell viability (Figure 3(b)), we interrogated the role of ER and SOCE pathway in MV biogenesis.
We demonstrate that SOCE is involved in plasma membrane EV biogenesis in both malignant MCF-7 and non-malignant hCMEC-D3 cells. Upon depletion of intracellular stores of Ca2+ with TG, a significant increase in vesicle biogenesis is observed in hCMEC-D3 cells. While statistical significance was not reached by flow cytometry, there is a consistent increase in MCF-7 vesiculation observable in these and in our AFM studies (Figures 1–2). Some malignant cells lines have been reported to maintain higher basal Ca2+ concentrations than normal cell lines [60], which could explain why there is such a stark difference in vesiculation before stimulation between hCMEC-D3 and MCF-7 cells. Roseblade et al. [20] proposed that higher basal Ca2+ and activity of calpain contribute to higher levels of basal vesiculation in malignant cells. Indeed, ALLM pre-treatment of non-malignant cells reduced TG-mediated vesicle production to levels comparable to control (Figure 4(b)). In malignant cells, ALLM significantly reduces TG-mediated vesiculation by 0.5 fold to levels well below control (Figure 4(c)). This further supports a calcium–calpain pathway driving malignant cell plasma membrane vesiculation at rest and demonstrates also a role for SOCE in the pathway.
Dysregulation of Ca2+ homoeostasis and signalling is complex and highly varied between cancer types [45]. Malignant cells often have remodelled Ca2+ signalling pathways that provide them with a survival advantage [43]. In the present study, differences between ER Ca2+ signalling pathways and, in particular, the SOCE pathway in breast cancer cells was assessed in terms of plasma membrane EV production with the selective SOCE inhibitor, YM58483. The pyrazole analogue YM58483 potently inhibits TG-mediated intracellular Ca2+ increases in non-excitable cells [67], effectively inhibits SOCE in breast cancer cells [75], as well as inhibiting T-cell activation mediated hypersensitivity reactions in mice [76]. At rest, both malignant and non-malignant vesiculation were unaffected when treated with YM58483 alone (Figure 4(a,b)). This demonstrates that basal vesiculation in both cell types is not dependent on SOCE specifically, hCMEC-D3 cells remain low vesiculators and MCF-7 cells remain high vesiculators.
We show that treatment of non-malignant hCMEC-D3 cells with YM58483 induced a significant reduction in TG-promoted vesiculation compare to control (Figure 4(b)). In this case, TG inhibits SERCA and results in Ca2+ depletion from the ER, however SOCE is blocked and so less extracellular Ca2+ enters cells. This renders the plasma membrane EV biogenic machinery inactive and less vesicles are released. These results demonstrate that SOCE is involved in the biogenesis of plasma membrane EVs from both MCF-7 and hCMEC-D3 cells. However, it appears that SOCE acts as a secondary pathway of Ca2+ entry that is activated down stream of ER store depletion.
While this does not completely explain differences in vesiculation at rest between MCF-7 and hCMEC-D3 cells, it does demonstrate differences in EV production between these malignant and non-malignant cells could be related to altered ER Ca2+ mobilization pathways (Figure 5). Importantly, results reported here mainly focus on larger (≈1000 nm) EV populations only as particle analysis with flow cytometry is limited in the detection of particles below a few hundred nanometres. This particle size detection limitation prevents analysis on small EVs and therefore changes in their production were not considered.
These results also provide interesting avenues for future work. Malignant cells have higher basal Ca2+ activity and some reports suggest this could be related to impaired intracellular Ca2+ storage [77]. On the other hand, malignant cells have also been reported to have increased expression and activity of ER Ca2+ channels – resulting in more frequent mobilization from intracellular stores. In each case, the ability of malignant cells to maintain high Ca2+ concentrations within intracellular Ca2+ store is impaired and is therefore more susceptible to SERCA inhibition and sensitive to store depletion. Future work aims to determine if malignant cell vesiculation is vulnerable to store depletion as this could prove to be useful in endeavours to mitigate EV-mediated cancer progression with novel therapeutics.
Conclusion
Cancer EVs are capable of transferring functional proteins and nucleic acids to recipient cells, bestowing upon them a complex MDR phenotype [3,7,9,78,7,9]. In the effort to develop novel strategies for the circumvention of EV-mediated cancer progression, resistance and survival it is imperative to understand the biogenic mechanisms that govern vesiculation. Here, we have demonstrated that malignant MCF-7 cells are high vesiculators at rest compared to non-malignant hCMEC-D3 cells and this is dependent on Ca2+ mobilization and activation of calpain. We found that the ER is the primary source of Ca2+ for plasma membrane EV biogenesis and that SOCE acts as a backup in the event of store depletion. Further, we report that differences in malignant and non-malignant vesiculation are related to altered mobilization of Ca2+ from ER stores. Pharmacological manipulation of ER Ca2+ stores has been a focus for many researchers attempting target Ca2+ signalling in the treatment of cancer. It now appears Ca2+ signalling may also be a novel avenue to pursue in the effort to reduce EV-mediated MDR and other deleterious cancer traits.
Declaration
GRM has had projects with UniQuest the commercialisation company of the University of Queensland.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- [1].Raposo G, Stoorvogel W.. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wolf P. Nature and significance of platelet products in human plasma. Br J Haematol. 1967;13:269-&. [DOI] [PubMed] [Google Scholar]
- [3].Bebawy M, Combes V, Lee E, et al. Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia. 2009;23:1643–1649. [DOI] [PubMed] [Google Scholar]
- [4].Lu JF, Luk F, Gong J, et al. Microparticles mediate MRP1 intercellular transfer and the re-templating of intrinsic resistance pathways. Pharmacol Res. 2013;76:77–83. [DOI] [PubMed] [Google Scholar]
- [5].Lu JF, Pokharel D, Padula MP, et al. A novel method to detect translation of membrane proteins following microvesicle intercellular transfer of nucleic acids. J Biochem. 2016;160:281–289. [DOI] [PubMed] [Google Scholar]
- [6].Gong J, Luk F, Jaiswal R, et al. Microparticles mediate the intercellular regulation of microRNA-503 and proline-rich tyrosine Kinase 2 to alter the migration and invasion capacity of breast cancer cells. Front Oncol. 2014;4:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gong J, Luk F, Jaiswal R, et al. Microparticle drug sequestration provides a parallel pathway in the acquisition of cancer drug resistance. Eur J Pharmacol. 2013;721:116–125. [DOI] [PubMed] [Google Scholar]
- [8].Jaiswal R, Johnson MS, Pokharel D, et al. Microparticles shed from multidrug resistant breast cancer cells provide a parallel survival pathway through immune evasion. Bmc Cancer. 2017;17:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pokharel D, Wijesinghe P, Oenarto V, et al. Deciphering cell-to-cell communication in acquisition of cancer traits: extracellular membrane vesicles are regulators of tissue biomechanics. OMICS. 2016;20:462–469. [DOI] [PubMed] [Google Scholar]
- [10].De Rubis G, Krishnan SR, Bebawy M. Liquid biopsies in cancer diagnosis, monitoring, and prognosis. Trends Pharmacol Sci. 2019;40:172–186. [DOI] [PubMed] [Google Scholar]
- [11].Krishnan SR, Luk F, Brown RD, et al. Isolation of human CD138(+) microparticles from the plasma of patients with multiple myeloma. Neoplasia. 2016;18:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lötvall J, Hill AF, Hochberg F, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the international society for extracellular vesicles. J Extracell Vesicles. 2014;3:26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Witwer KW, Buzás EI, Bemis LT, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2:20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles In: Schekman R, Lehmann R, editors. Annual review of cell and developmental biology. Vol. 30 Palo Alto: Annual Reviews; 2014:255–289. [DOI] [PubMed] [Google Scholar]
- [15].Kalra H, Drummen GPC, Mathivanan S. Focus on extracellular vesicles: introducing the next small big thing. Int J Mol Sci. 2016;17:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chargaff E, West R. The biological significance of the thromboplastic protein of blood. J Biol Chem. 1946;166:189–197. [PubMed] [Google Scholar]
- [17].Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. [DOI] [PubMed] [Google Scholar]
- [18].Warren BA, Vales O. Release of vesicles from platelets following adhesion to vessel walls in-vitro. Br J Exp Pathol. 1972;53:206-&. [PMC free article] [PubMed] [Google Scholar]
- [19].Freyssinet JM, Toti F. Formation of procoagulant microparticles and properties. Thromb Res. 2010;125:S46–S48. [DOI] [PubMed] [Google Scholar]
- [20].Roseblade A, LUK F, UNG A, et al. Targeting microparticle biogenesis: a novel approach to the circumvention of cancer multidrug resistance. Curr Cancer Drug Targets. 2015;15:205–214. [DOI] [PubMed] [Google Scholar]
- [21].Sapet C, Simoncini S, Loriod B, et al. Thrombin-induced endothelial microparticle generation: identification of a novel pathway involving ROCK-II activation by caspase-2. Blood. 2006;108:1868–1876. [DOI] [PubMed] [Google Scholar]
- [22].Vanwijk MJ, Vanbavel E, Sturk A, et al. Microparticles in cardiovascular diseases. Cardiovasc Res. 2003;59:277–287. [DOI] [PubMed] [Google Scholar]
- [23].Zwaal RFA, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bretscher MS. Asymmetrical lipid bilayer structure for biological-membranes. Nat New Biol. 1972;236:11-+. [DOI] [PubMed] [Google Scholar]
- [25].Daleke DL. Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res. 2003;44:233–242. [DOI] [PubMed] [Google Scholar]
- [26].Daleke DL, Lyles JV. Identification and purification of aminophospholipid flippases. Biochim Biophys Acta Mol Cell Biol Lipids. 2000;1486:108–127. [DOI] [PubMed] [Google Scholar]
- [27].Daleke DL, Lyles JV, Nemergut E, et al. Purification and substrate specificity of the human erythrocyte aminophospholipid transporter In: Trafficking of intracellular membranes: from molecular sorting to membrane fusion. Vol. 91 1995. p. 49–59. Heidelberger: Springer-Verlag Berlin. [Google Scholar]
- [28].Kunzelmann-marche C, Freyssinet JM, Martinez MC. Regulation of phosphatidylserine transbilayer redistribution by store-operated Ca2+ entry - role of actin cytoskeleton. J Biol Chem. 2001;276:5134–5139. [DOI] [PubMed] [Google Scholar]
- [29].Martin SJ, Reutelingsperger CPM, Mcgahon AJ, et al. Early redistribution of plasma-membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus - inhibition by overexpression of BCL-2 and ABL. J Exp Med. 1995;182:1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Perrin BJ, Amann KJ, Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell. 2006;17:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Storr SJ, Carragher NO, Frame MC, et al. The calpain system and cancer. Nat Rev Cancer. 2011;11:364–374. [DOI] [PubMed] [Google Scholar]
- [32].Basse F, Gaffet P, Bienvenue A. Correlation between inhibition of cytoskeleton proteolysis and anti-vesiculation effect of calpeptin during A23187-induced activation of human platelets - are vesicles shed by filopod fragmentation. Biochim Biophys Acta, Biomembr. 1994;1190:217–224. [DOI] [PubMed] [Google Scholar]
- [33].Fox JEB, Austin CD, Reynolds CC, et al. Evidence that agonist-induced activation of calpain causes the shedding of procoagulant-containing microvesicles from the membrane of aggregating platelets. J Biol Chem. 1991;266:13289–13295. [PubMed] [Google Scholar]
- [34].Distler JHW, Huber LC, Hueber AJ, et al. The release of microparticles by apoptotic cells and their effects on macrophages. Apoptosis. 2005;10:731–741. [DOI] [PubMed] [Google Scholar]
- [35].Morrell AE, Brown GN, Robinson ST, et al. Mechanically induced Ca2+ oscillations in osteocytes release extracellular vesicles and enhance bone formation. Bone Res. 2018;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pasquet JM, Dacharyprigent J, Nurden AT. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur J Biochem. 1996;239:647–654. [DOI] [PubMed] [Google Scholar]
- [37].Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. [DOI] [PubMed] [Google Scholar]
- [38].Berridge MJ. Calcium signalling remodelling and disease. Biochem Soc Trans. 2012;40:297–309. [DOI] [PubMed] [Google Scholar]
- [39].Azimi I, Roberts-Thomson SJ, Monteith GR. Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. Br J Pharmacol. 2014;171:945–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev. 2009;89:1341–1378. [DOI] [PubMed] [Google Scholar]
- [41].Missiaen L, Robberecht W, Van Den Bosch L, et al. Abnormal intracellular Ca2+ homeostasis and disease. Cell Calcium. 2000;28:1–21. [DOI] [PubMed] [Google Scholar]
- [42].Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem. 2012;287:31666–31673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Monteith GR, Mcandrew D, Faddy HM, et al. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer. 2007;7:519–530. [DOI] [PubMed] [Google Scholar]
- [44].Stewart TA, Yapa K, Monteith GR. Altered calcium signaling in cancer cells. Biochim Biophys Acta, Biomembr. 2015;1848:2502–2511. [DOI] [PubMed] [Google Scholar]
- [45].Ma X, Chen Z, Hua D, et al. Essential role for TrpC5-containing extracellular vesicles in breast cancer with chemotherapeutic resistance. Proc Natl Acad Sci U S A. 2014;111:6389–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pera E, Kaemmerer E, Milevskiy MJG, et al. The voltage gated Ca2+-channel Ca(v)3.2 and therapeutic responses in breast cancer. Cancer Cell Int. 2016;16:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Taylor J, Jaiswal R, Bebawy M. Calcium-calpain dependent pathways regulate vesiculation in malignant breast cells. Curr Cancer Drug Targets. 2017;17:486–494. [DOI] [PubMed] [Google Scholar]
- [48].Jaiswal R, Gong J, Sambasivam S, et al. Microparticle-associated nucleic acids mediate trait dominance in cancer. Faseb J. 2012a;26:420–429. [DOI] [PubMed] [Google Scholar]
- [49].Donmez Y, Akhmetova L, Iseri OD, et al. Effect of MDR modulators verapamil and promethazine on gene expression levels of MDR1 and MRP1 in doxorubicin-resistant MCF-7 cells. Cancer Chemother Pharmacol. 2011;67:823–828. [DOI] [PubMed] [Google Scholar]
- [50].Kars MD, Iseri OD, Gunduz U, et al. Development of rational in vitro models for drug resistance in breast cancer and modulation of MDR by selected compounds. Anticancer Res. 2006;26:4559–4568. [PubMed] [Google Scholar]
- [51].Jaiswal R, Luk F, Dalla PV, et al. Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. Plos One. 2013;8:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Antonio PD, Lasalvia M, Perna G, et al. Scale-independent roughness value of cell membranes studied by means of AFM technique. Biochim Biophys Acta, Biomembr. 2012;1818:3141–3148. [DOI] [PubMed] [Google Scholar]
- [53].Necas D, Klapetek P. Gwyddion: an open-source software for SPM data analysis. Cent Eur J Phys. 2012;10:181–188. [Google Scholar]
- [54].Feng MY, Grice DM, Faddy HM, et al. Store-independent activation of orai1 by SPCA2 in Mammary Tumors. Cell. 2010;143:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Robinson JA, Jenkins NS, Holman NA, et al. Ratiometric and nonratiometric Ca2+ indicators for the assessment of intracellular free Ca2+ in a breast cancer cell line using a fluorescence microplate reader. J Biochem Biophys Methods. 2004;58:227–237. [DOI] [PubMed] [Google Scholar]
- [56].Azimi I, Flanagan JU, Stevenson RJ, et al. Evaluation of known and novel inhibitors of orai1-mediated store operated Ca2+ entry in MDA-MB-231 breast cancer cells using a fluorescence imaging plate reader assay. Bioorg Med Chem. 2017;25:440–449. [DOI] [PubMed] [Google Scholar]
- [57].Lacroix R, Robert S, Poncelet P, et al. Overcoming limitations of microparticle measurement by flow cytometry. Semin Thromb Hemost. 2010;36:807–818. [DOI] [PubMed] [Google Scholar]
- [58].Dedkova EN, Sigova AA, Zinchenko VP. Mechanism of action of calcium ionophores on intact cells: ionophore-resistant cells. Membr cell biol. 2000;13:357–368. [PubMed] [Google Scholar]
- [59].Zhang X, Chen Y, Chen Y. An AFM-based pit-measuring method for indirect measurements of cell-surface membrane vesicles. Biochem Biophys Res Commun. 2014;446:375–379. [DOI] [PubMed] [Google Scholar]
- [60].Pottle J, Sun C, Gray L, et al. Exploiting MCF-7 Cells¡¯ calcium dependence with interlaced therapy. J Cancer Ther. 2013;04(7):9. [Google Scholar]
- [61].Marchi S, Patergnani S, Missiroli S, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72. [DOI] [PubMed] [Google Scholar]
- [62].Pinton P, Giorgi C, Siviero R, et al. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarcoendoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19:131–135. [DOI] [PubMed] [Google Scholar]
- [64].Peters AA, Simpson PT, Bassett JJ, et al. Calcium channel TRPV6 as a potential therapeutic target in estrogen receptor-negative breast cancer. Mol Cancer Ther. 2012;11:2158–2168. [DOI] [PubMed] [Google Scholar]
- [65].Sagara Y, Wade JB, Inesi G. A conformational mechanism for formation of a dead-end complex by the sarcoplasmic-reticulum atpase with thapsigargin. J Biol Chem. 1992;267:1286–1292. [PubMed] [Google Scholar]
- [66].Sehgal P, Szalai P, Olesen C, et al. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J Biol Chem. 2017;292:19656–19673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ishikawa J, Ohga K, Yoshino T, et al. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J Iimmunol. 2003;170:4441–4449. [DOI] [PubMed] [Google Scholar]
- [68].Chan SL, Mattson MP. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res. 1999;58:167–190. [PubMed] [Google Scholar]
- [69].Carafoli E. Calcium signaling: a tale for all seasons. Proc Natl Acad Sci U S A. 2002;99:1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lipskaia L, Lompre AM. Alteration in temporal kinetics of Ca2+ signaling and control of growth and proliferation. Biol Cell. 2004;96:55–68. [DOI] [PubMed] [Google Scholar]
- [71].Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The calcium-cancer signalling nexus. Nat Rev Cancer. 2017;17:367–380. [DOI] [PubMed] [Google Scholar]
- [72].Rizzuto R, Pinton P, Ferrari D, et al. Calcium and apoptosis: facts and hypotheses. Oncogene. 2003;22:8619–8627. [DOI] [PubMed] [Google Scholar]
- [73].Putney JW. Pharmacology of store-operated calcium channels. Mol Interv. 2010;10:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Takezawa R, Cheng H, Beck A, et al. A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol Pharmacol. 2006;69:1413–1420. [DOI] [PubMed] [Google Scholar]
- [75].Azimi I, Bong AH, Poo GXH, et al. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: consequences on calcium signalling, cell migration and proliferation. Cell Mol Life Sci. 2018;75:4525–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ohga K, Takezawa R, Arakida Y, et al. Characterization of YM-58483/BTP2, a novel store-operated Ca(2+) entry blocker, on T cell-mediated immune responses in vivo. Int Immunopharmacol. 2008;8:1787–1792. [DOI] [PubMed] [Google Scholar]
- [77].Pinton P, Ferrari D, Magalhaes P, et al. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J Cell Biol. 2000;148:857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jaiswal R, Luk F, Gong J, et al. Microparticle conferred microRNA profiles - implications in the transfer and dominance of cancer traits. Mol Cancer. 2012b;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lu JF, Pokharel D, Bebawy M. A novel mechanism governing the transcriptional regulation of ABC transporters in MDR cancer cells. Drug Deliv Transl Res. 2017;7:276–285. [DOI] [PubMed] [Google Scholar]