

Abstract
Macrocyclic ligands have been explored extensively as scaffolds for transition metal catalysts for oxygen and hydrogen atom transfer reactions. C-C reactions facilitated using earth abundant metals bound to macrocyclic ligands have not been well-understood but could be a green alternative to replacing the current expensive and toxic precious metal systems most commonly used for these processes. Therefore, the yields from direct Suzuki-Miyaura C-C coupling of phenylboronic acid and pyrrole to produce 2-phenylpyrrole facilitated by eight high-spin iron complexes ([Fe3+L1(Cl)2]+, [Fe3+L4(Cl)2]+, [Fe2+L5(Cl)]+, [Fe2+L6(Cl)2], [Fe3+L7(Cl)2]+, [Fe3+L8(Cl)2]+, [Fe2+L9(Cl)]+, and [Fe2+L10(Cl)]+) were compared to identify the effect of structural and electronic properties on catalytic efficiency. Specifically, catalyst complexes were compared to evaluate the effect of five properties on catalyst reaction yields: 1. the coordination requirements of the catalyst, 2. Redox half-potential of each complex, 3. topological constraint/rigidity, 4. N-atom modification(s) increasing oxidative stability of the complex, and 5. geometric parameters. The need for two labile cis-coordination sites was confirmed based on a 42% decrease in catalytic reaction yield observed when complexes containing penta-dentate ligands were used in place of complexes with tetra-dentate ligands. A strong correlation between iron(III/II) redox potential and catalytic reaction yields was also observed, with [Fe2+L6(Cl)2] providing the highest yield (81%, −405 mV). A Lorentzian fitting of redox potential versus yields predicts that these catalysts can undergo more fine tuning to further increase yields. Interestingly, the remaining properties explored did not show a direct, strong relationship to catalytic reaction yields. Altogether, these results show that modifications to the ligand scaffold using fundamental concepts of inorganic coordination chemistry can be used to control the catalytic activity of macrocyclic iron complexes by controlling redox chemistry of the iron center. Furthermore, the data provides direction for the design of improved catalysts for this reaction and strategies to understand the impact of a ligand scaffolds on catalytic activity of other reactions.
Keywords: C-C coupling, iron, catalysis, high-spin iron, cross-bridged cyclen, cross-bridged cyclam
Graphical Abstract
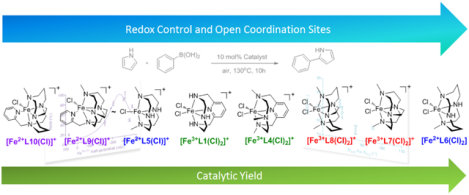
Introduction
Biological systems functionalize unreactive compounds such as alkanes and alkenes under mild conditions, but comparable laboratory transformations require multiple steps that include expensive and/or toxic materials such as palladium, protecting groups, and strong oxidants.1–9 Efforts to translate the chemistry of earth abundant metal systems such as iron-based metalloenzymes to the laboratory have resulted in many publications concentrating on the substrate scope of a particular metal salt or complex.10–13 These studies optimized directing groups, steric requirements, and electronic properties of the substrate using a single catalyst.
Although it is necessary to identify the limitations of the catalyst, it is also important to determine the properties of the metal center and the ligand scaffolds that allow catalytic processes to occur. For example, structural characterization of tetra-azamacrocycles in nature, specifically, hemoglobin, myoglobin, and cytochrome p450 revealed that the axial ligands tune the reactivity of the enzymes by changing the resting state and accessible oxidation states of the iron center, while ligand modification is also well-understood for tuning Fe-S clusters.14–15 The insight gained from characterization of natural systems has led to the development of biomimetic catalysts containing synthetic tetra-azamacrocycles to emulate the reactivity of natural systems.16–18 The reactivity of biomimetic catalysts has been altered by varying the ring size, rigidity, and amine functionalization of tetra-azamacrocycles bound to the metal center.11, 13, 19–22 The inorganic chemistry of metallo-biomimetic catalysts used for oxygen insertion or oxygen activation reactions has also been thoroughly investigated.23–27 For example, [Fe2+(L8)(OTf)2] catalyzes the epoxidation and cis-dihydroxylation of olefins,28 [Fe3+LN4Me2]+ (LN4Me2 = N,N’-dimethyl-2,11-diaza[3.3](2,6)pyridinophane) catalyzes cis-dihydroxylation of alkenes29 and intradiol-cleavage,30 and [Fe3+cyclen(Cl)2]+ (cyclen = 1,4,7,10-tetraazacyclododecane) catalyzes water oxidation.31 Mechanistic studies have shown that each oxidative reaction proceeds through a different catalytic pathway. The iron center is proposed to be redox active in both the cis-hydroxylation [iron (III/V)] and water oxidation [iron(III/IV)] mechanisms.29, 31 Conversely, the iron(III) catalyst was proposed to be redox inert in intradiol-cleavage reactions.30 Two labile cis-coordination sites are necessary for each of these catalytic reactions, despite the varied oxidation state of the iron catalyst.
Fewer details are understood for iron-based catalysts using tetra-azamacrocyclic scaffolds for C-C coupling reactions. Interestingly, Bedford and co-workers reported that iron catalysts derived from rigid tetra-azamacrocycles, such as L8, resulted in poor yields for the cross-coupling of pre-activated species such as 4-tolyl magnesium bromide with cyclohexylbromide.7 Conversely, iron salts in the presence or absence of organic chelates have also been reported to catalyze direct C-C coupling reactions.7, 12–13 Most studies have focused on the scope and versatility of specific ligand and metal salt mixtures, leaving the metal complex(es) unstudied. For example, Wen et al. reported that together, a 12-membered tetra-azamacrocycle (L1=1,4,7,10-tetra-aza-2,6-pyridinophane, Figure 1), oxygen, and an iron(II) salt are capable of catalyzing direct Suzuki-Miyaura type C-C coupling reactions. The direct coupling is highly advantageous by avoiding the need for reagent pre-modifications, such as aryl halides.32 Although a general catalytic mechanism was presented, the oxidation state of the iron catalyst was not assigned. Moreover, the authors claim to have mass spectrometry evidence for the catalytic species, but the proposed structure does not match the isotopic envelope reported (detailed in Figure S1). Recently, we reported that in the presence of oxygen, L1 and Fe(ClO4)2 form a high-spin iron(III) complex that is capable of arylation of pyrrole with phenylboronic acid to produce 2-phenylpyrrole, thus showing that the catalyst enters the catalytic cycle in the 3+ oxidation state.33 Iron(III) complexes of L2 and L3 (L2=1,4,7,10-tetra-aza-2,6-pyridinophane-14-ol; L3=1,4,7,10-tetra-aza-2,6-pyridinophane-13-ol]) were shown to be equally competent catalysts. Herein, a fundamental inorganic approach was used to analyze the properties of iron catalysts derived from a range of macrocyclic complexes that increase C-C efficiency for this reaction.
Figure 1.
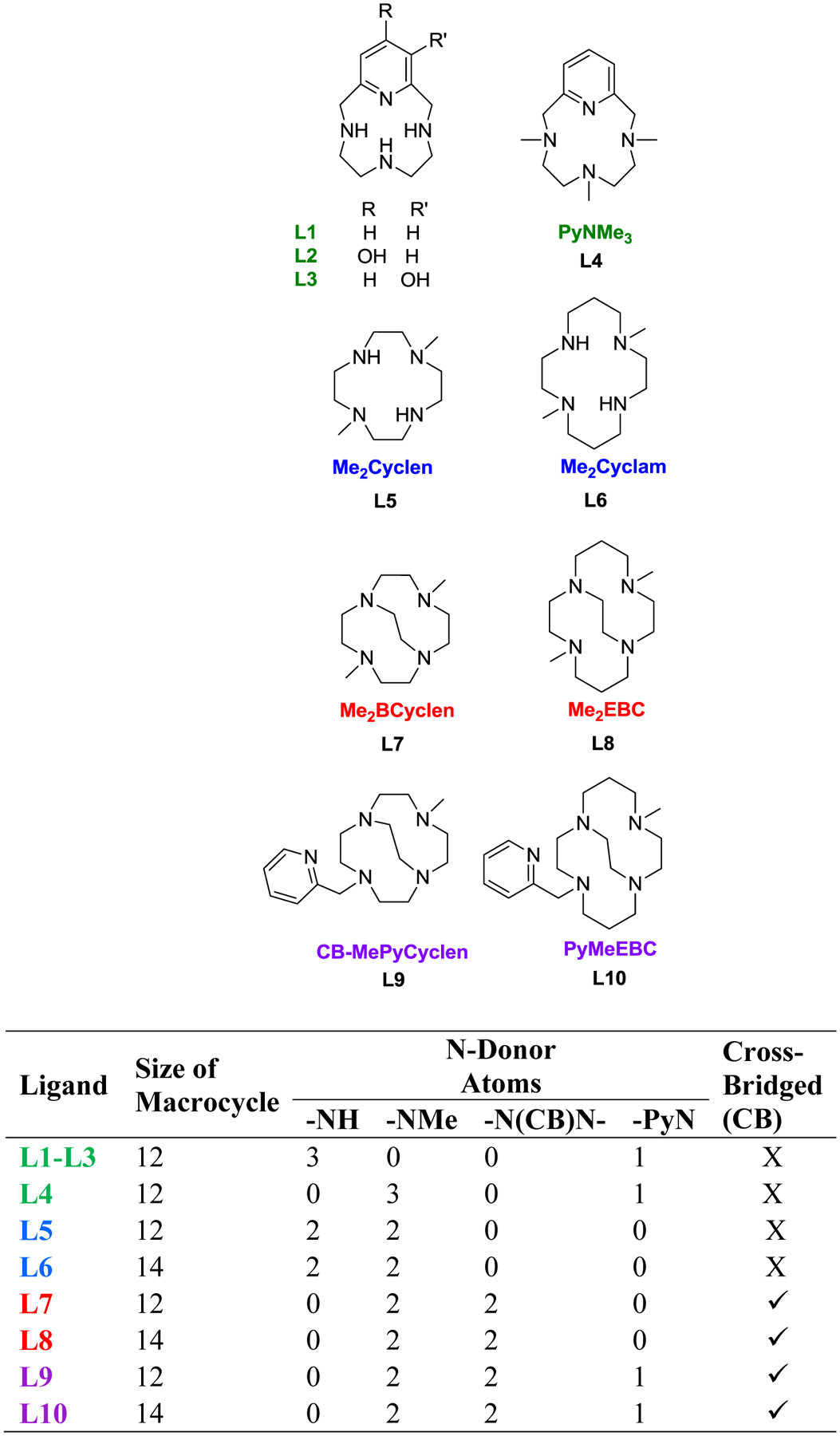
Tetra-azamacrocycles used to produce high-spin iron catalysts described herein and comparison of structures.33, 40, 32, 41, 42,43
Importance of the interplay of ligand properties.
This work seeks to evaluate the effect of five different properties (1. coordination number, 2. redox properties, 3. complex rigidity/topological constraint, 4. N-atom modification to increase resistance to oxidative degradation, and 5. complex geometric parameters) on the catalytic efficiency of a series of iron complexes. Our approach, below, will discuss each parameter separately. However, these parameters cannot typically be considered completely in isolation from one another, as they are related in a complex interplay that defines a given transition metal catalyst. Here, we would briefly like to point out some of the relationships between these parameters and provide an example of their interplay in a related catalytic system to provide more context for this current work.
Topological constraint (complexity of ligand donor atom interconnectedness) is intimately associated with complex stability through the chelate, macrocycle, and cryptate effects.34 In small ligand systems, such as those studied here, topological complexity also results in increased rigidity of the complex, as the ligand freedom of motion is restricted by the many rotation-restricted bonds holding its donor atoms in place. The “constraint” factors (increased topological complexity and increased rigidity) tend to make transition metal complexes much more kinetically stable towards solvolytic decomposition. The constraint factors have resulted in modern coordination complexes with stabilities well beyond the elementary matching of size, geometry, and electronic preferences of the ligand with the metal ion; these factors are known as “complementarity” factors.28, 35–39
Yet, these complementarity factors are still important and also help control the stability of a complex. The electronic match between the hard/soft acid/base properties of nitrogen donors and iron(III/II) ions control the redox potentials and reversibilities of the catalysts studied. Similarly, the geometric preference of an octahedral geometry for a d6 iron(II) cation must match the donor placement and cavity size of the azamacrocycle, and this match is evident in the bond angles and bond lengths of the Fe-N coordination sphere. Returning to the chelate effect, an additional chelated donor appended to the ligand is expected to increase complex stability via the chelate effect, but must also alter redox properties, geometric parameters, and coordination number in ways which may not be helpful to overall catalyst activity. Finally, changing secondary nitrogen donors to tertiary ones by simple methylation may be intended only to protect against oxidative degradation, yet will also increase slightly the rigidity of the ligand and will alter the redox potentials of the complex. Thus, the challenge in designing the “ideal ligand” is to “shim” these collected properties collectively by incrementally improving them one-by-one to give a collectively more stable, yet still reactive, catalyst.
An example of a successfully realized catalytic system, manganese azamacrocyle oxidation catalysts, “shimmed” in this way may be useful to illustrate the challenges and the potential for this approach. Catalytic oxidation in water of “stain” molecules on clothing inspired the Mn(TMTACN) catalysts developed in the 1990’s (Figure 3).44–46 Yet, their lack of selectivity in oxidation reactivity contributed to their removal from consumer products because of their damage to cloth.46 Busch looked to the cross-bridged tetra-azamacrocycles of Weisman to increase the stability of the metal complexes by increasing the coordination number, as well as increasing the topological complexity and rigidity of the system.35, 47–65 However, the parent cross-bridged tetraazamacrocycles (like H2EBC, Figure 3) containing two secondary amines were ineffective oxidation catalysts under harsh aqueous conditions as they formed μ-oxo dimers that were coordinatively saturated and thermodynamic sinks.48 Methylation of the two secondary amines resulted in just enough steric bulk to prevent dimerization, decreased oxidative degradation of the complex, and resulted in monomeric catalysts stable enough to withstand harsh aqueous oxidation conditions. Selection of the cyclam-based ligand over the cyclen-based system was based on (1) greater kinetic stability toward solvolytic decomplexation of the larger ligand which could better engulf the metal ion and make stronger, more nearly octahedral bond angles; and (2) higher oxidation potentials (thus more oxidizing power) for the cyclam system due to a preference of the larger macrocycle for the larger manganese(II) ion. The resulting [Mn(L8)Cl2] oxidation catalyst has been extensively studied and patented over the last 20 years because it is the result of a combination of optimized parameters. Yet even this very successful complex may be “too reactive” for some purposes, and is not particularly selective for producing single specific oxidation products. A recent parameter modification was the appending of a pyridine pendant arm (L10), which increased the coordination number to five, reduced the oxidation potential(s), and made the catalyst more selective towards single major oxidation products.66 It is this type of iterative parameter examination that we explore here for the optimization of iron catalysts for C-C bond formation.
Figure 3.
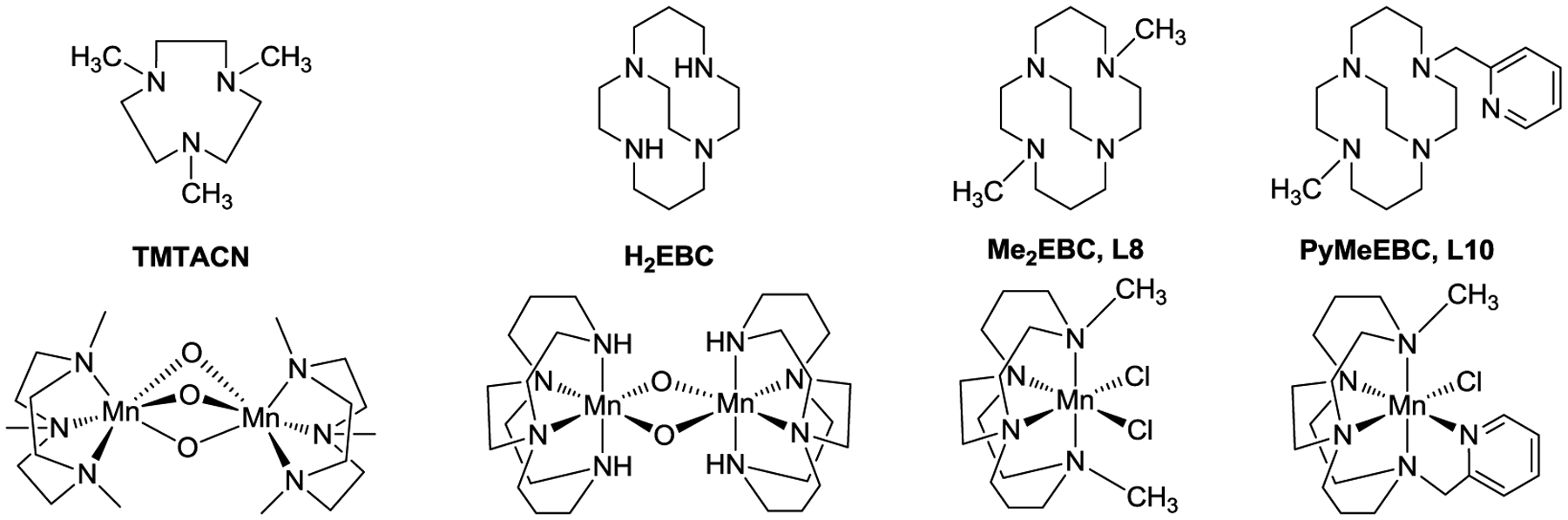
Ligands and catalytically active manganese complexes studied previously.44–46, 48, 51, 66–67
A comparison of the catalytic reaction yields of four high-spin iron(III) and four high-spin iron(II) complexes for the coupling of phenylboronic acid and pyrrole to form 2-phenylpyrrole was used to identify key properties of the iron catalyst responsible for providing enhanced yields. Specifically, the ligands in Figure 1 were chosen to vary five properties: the coordination requirements of the catalyst, increasing redox stability of the complex, topological constraint/rigidity, N-atom modification(s), and geometric parameters around the metal center of the complexes shown in Figure 2. Complexes, [Fe3+L7(Cl)2]PF6,47 [Fe3+L8(Cl)2]PF6,47 and [Fe2+L10(Cl)]PF666 were synthesized using previously reported procedures. However, complexes [Fe3+L4(Cl)2]PF6, [Fe2+L5(Cl)]Cl, [Fe2+L6(Cl)2], [Fe2+L9(Cl)]PF6, and [Fe3+L1(Cl)2]Cl were produced for the first time and are reported herein. As noted above, many of these ligands or similar systems have been used to support metal catalyzed oxidation, hydroxylation, or other reactions.29,35, 68–69,71 This study serves to show the versatility of macrocyclic systems and specific features that should be retained in these ligands to obtain optimized yields in the coupling C-C coupling of phenylboronic acid and pyrrole.
Figure 2.
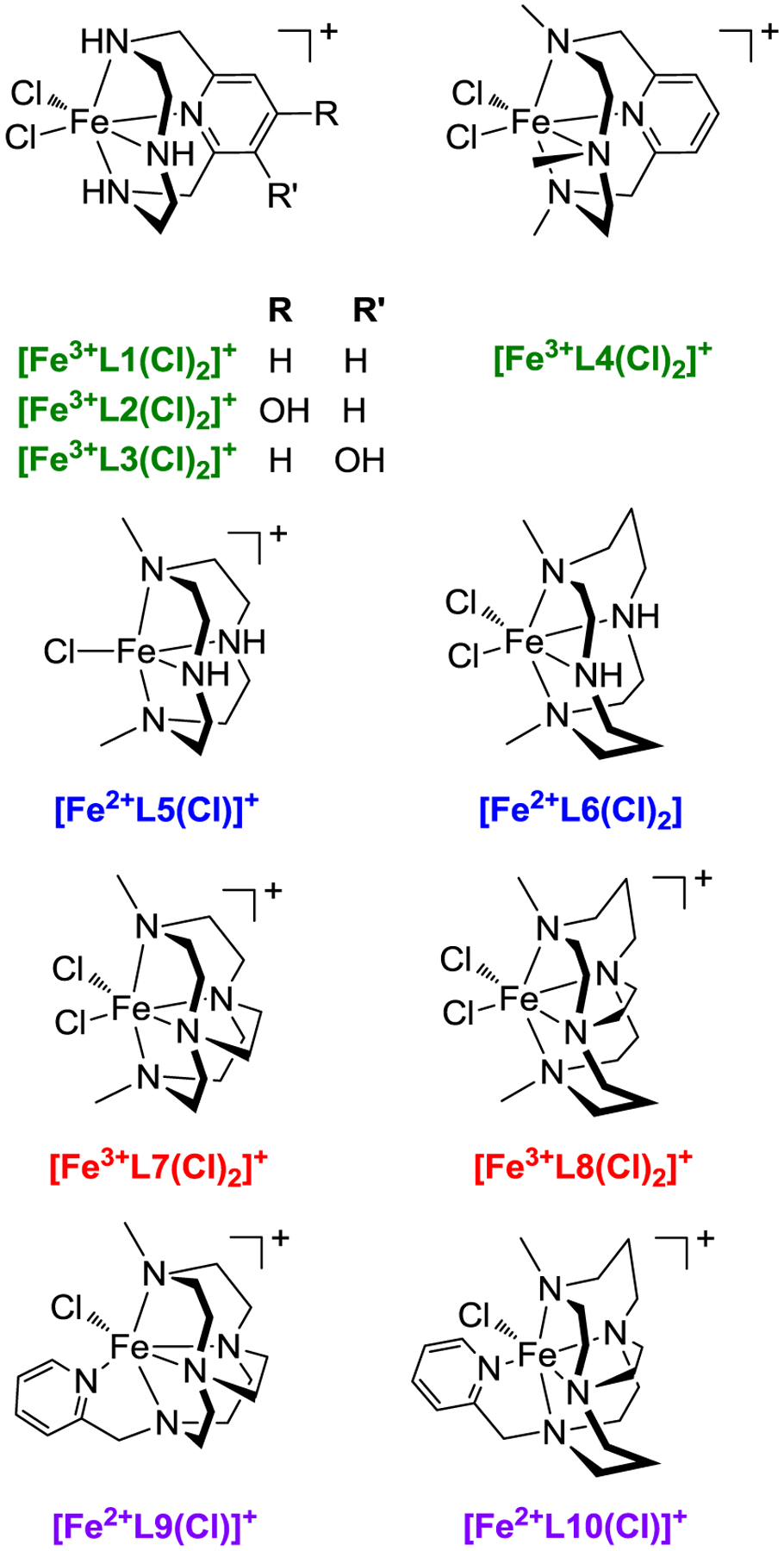
Iron catalysts derived from macrocyclic ligands discussed in this work.33, 47, 66
Experimental
General Methods.
Pyrrole was distilled under reduced pressure prior to use; all other reagents including anhydrous iron(II) chloride (Sigma Aldrich) used were obtained from commercial sources and used as received, unless noted otherwise. 1H-NMR spectra were obtained on a 300- or 400-MHz Bruker Avance spectrometer using deuterated solvents and spectra were referenced using the corresponding solvent resonance (in parts per million; CDCl3, δ = 7.26 ppm).70 For proper identification of the NMR signals the following abbreviations were used: s = singlet, d = doublet, t = triplet, m = multiplet. No NMR data were obtainable for [Fe3+L4(Cl)2]PF6 and [Fe3+L1(Cl)2]Cl. Elemental analysis was performed by Canadian Microanalytical Service Ltd. The following compounds were synthesized using previously published procedures: L1 – L3,71–72 PyNMe3 (L4),73 Me2Cyclen (L5;1,7-dimethyl-1,4,7,10-tetraazacyclododecane)74, Me2Cyclam (L6; 1,8-dimethyl-1,4,8,11-tetraazacyclotetradecane)75, Me2BCyclen (L7),42 Me2EBC (L8),35 CB-MePyCyclen (L9),43 PyMeEBC (L10),66 [Fe3+L7(Cl)2]PF6,47 [Fe3+L8(Cl)2]PF6,47 and [Fe2+L10]PF6.
Synthesis.
[Fe3+L4(Cl)2]PF6:
FeCl3·6H2O (109 mg, 0.404 mmol) was dissolved in 9 mL methanol and added drop-wise to L4 (124 mg, 0.500 mmol) dissolved in minimum volume of methanol. A large amount of precipitate formed when half of the iron solution was added, the reaction was allowed to stir overnight at which time the majority of precipitate re-dissolved. The remaining solid was removed by filtration through celite, and [(Bu)4N]PF6 (787 mg, 2.03 mmol) was added as a solid resulting in precipitate of a yellow powder. The product was isolated via filtration and washed with small amount of methanol. Yield: 78% (164 mg, 0.316 mmol). ESI-MS+ Calc (Found): 374.0727 (373.9824). Absorption spectrum (CH3CN), λmax, (ε, M−1·cm−1): 326 (5,000) and 408 (sh; 1,400) nm; ([Fe3+L4(Cl)2]+). Elemental Analysis: Calc. (Found) for C14H24Cl2FeN4PF6: C, 32.33(32.15); H, 4.65(4.90); N, 10.77(10.67).
[Fe2+L5(Cl)]+:
In an inert atmosphere glovebox, 0.401 g (2.0 mmol) of L574 and 0.254 g (2.0 mmol) of anhydrous iron(II) chloride were added to a 20 mL reaction vial and 15 mL of anhydrous dimethylformamide was added with stirring, making a clear pale yellow solution. The reaction was stirred at room temperature for seven days. The solution was filtered into a 500 mL Erlenmeyer flask to remove any trace solids. Anhydrous ether (200 mL) was added and a precipitate formed immediately. Using a filter frit the solid was filtered off and washed with ether and left on the frit to dry in glovebox. A white solid powder product was obtained. Yield = 0.570g (87%). 1H NMR (CD3OH, 300 MHz): δ 0.837ppm (m, 4H); 1.129 (triplet, 6H); 1.332 (s, 2H); 1.237 (br singlet, 6H); 2.038 (s, 2H); 3.442 (m, 2H). ESI-MS+:m/z = 292 (FeLCl+). Absorption spectrum (CH3CN), λmax, (ε, M−1·cm−1): 276 (sh, 2,540) and 337 (2,480) nm. Elemental Analysis: Calc. (Found) for (Fe(C10H24N4)Cl2 ∙ 0.4 H2O): C 35.93(35.93), H 7.48(7.09), N 16.76(16.56). Blue X-ray quality crystals were obtained from ether diffusion into methanol solution, grown outside of the glovebox, and included an FeCl42- anion not present in the bulk white solid. It is possible trace unreacted FeCl2, or complex decomposition, led to formation of this anion during the crystal growth.
[Fe2+L6(Cl)2]:
In an inert atmosphere glovebox, 0.342 g (1.5 mmol) of L675 and 0.190 g (1.5 mmol) of anhydrous iron(II) chloride were added to a 20 mL reaction vial and 15 mL of anhydrous acetonitrile was added with stirring, making a clear, colorless solution. The reaction was stirred at room temperature for seven days. The solution was filtered into a 50 mL round bottom flask to remove any trace solids. The flask was fitted with a gas inlet valve, removed from the glovebox under nitrogen, and attached to a vacuum line to remove the solvent under vacuum without exposing the complex to air. A white solid powder product was obtained, which was pumped back into the glovebox without exposing to air. Yield = 0.347 g (65%). 1H NMR (CD3OH, 300 MHz): Paramagnetically broadened, only two peaks, free solvent, and metal-bound solvent observed; no peaks for the ligand observed. 3.240ppm (br singlet); 4.831ppm (br singlet) δ ESI-MS+: m/z = 319 (FeLCl+). Absorption spectrum (CH3CN), λmax, (ε, M−1·cm−1): 278 (2,520) and 330 (990) nm. Elemental Analysis: Calc. (Found) for ([Fe(C12H28N4)Cl2): C 40.59(40.65), H 7.95(8.29), N 15.78(15.62). Pale pink X-ray quality crystals were obtained from ether diffusion into a methanol solution, grown inside of the glovebox.
[Fe2+L9(Cl)]PF6:
(303 mg, 1.00 mmol) of L943 and (127 mg, 1.00 mmol) of anhydrous FeCl2 (Aldrich) were stirred together in 5 mL anhydrous acetonitrile in an inert atmosphere glovebox at room temperature for 48 h. Insoluble solids were removed and discarded. The orange-brown filtrate was allowed to evaporate to dryness (10 days) in the glovebox. The brown oil residue was dissolved in a minimum of anhydrous methanol. (805 mg, 5.00 mmol) of anhydrous NH4PF6 was dissolved in 5 mL anhydrous methanol and added dropwise with stirring to the complex solution causing a tan precipitate to form. This precipitate was filtered off, washed with minimal methanol, then ether, and allowed to dry in the glovebox to give the pure product (297 mg, 55%). 1H NMR (CD3CN, 300 MHz): δ 0.884 (m, 2H); 1.288 (s, 3H); 3.015, (br m, 10H); 3.304 (s, 4H); 3.550 (br m, 6H); 7.577 (m, 2H); 7.953 (m, 1H); 8.906 (m, 1H). ESI-MS+ = 394 (FeLCl+). Absorption spectrum (CH3CN), λmax, (ε, M−1·cm−1): 257(4,010) and 419(580). Elemental Analysis: Calc (Found) for FeC17H29N5ClPF6 (539.71 g/mol): C 37.83 (37.60); H 5.42 (5.22); N 12.98 (12.76).
[Fe3+L1(Cl)2]Cl:
Ligand (L1) (252 mg, 0.799 mmol) was dissolved in 3 mL DI water. Iron(III) chloride hexahydrate (222.3 mg, 0.823 mmol) was dissolved in 3 mL methanol, the metal was added dropwise to the ligand. Slow evaporation of methanol yielded yellow X-ray quality crystals. Yield: 12% (34 mg, 0.0932 mmol). Absorption spectrum (CH3CN), λmax, (ε, M−1·cm−1): 319 (4,500) and 390 (sh; 1,600) nm. Elemental analysis: Calc (Found) for C12H18Cl3FeN4·3H2O: C 35.85(35.29); H 4.92(4.95); N 15.20(14.92). ESI-MS+ was consistent with the ClO4− congener from previous reports.33
Catalytic Reactions:
Phenylboronic acid (24 mg, 0.20 mmol) and iron complex (0.02 mmol) were added to a 2–10 mL flask equipped with a stir bar. Pyrrole (1 mL) was added to flask, the mixture was heated to 130 °C for 10 hours.
Yield Determination:
Reactions were cooled to room temperature, and the pyrrole was removed in vacuo until no visible liquid was present. Care was taken to avoid product loss under high vacuum. The reaction was dissolved in a minimum amount of CDCl3 and 5 μL of dimethyldiphenyl silane was added. The solution was filtered through a 0.2 μm nylon filter and a known amount of sample was added to a pre-weighed NMR tube. Yield determinations were performed using three resonances 6.875, 6.532, and 6.307 ppm, corresponding to 2-phenylpyrrole and a resonance at 0.533 ppm corresponding to dimethyldiphenyl silane. The reported values are averages of all resonances; each reaction was run in triplicate.
Electrochemistry.
Cyclic voltammetry experiments utilized 2–3 mM complex and 100 mM tetrabutylammonium hexafluoroborate as the supporting electrolyte in DMF. The electrochemical cell was composed of a working glassy carbon electrode, a Pt auxiliary electrode, and a silver wire as the reference electrode. The potential values presented here have been normalized to the half-wave potential of the Fc+/Fc = 0.00 mV.
X-ray Crystallography.
(For [Fe3+L1(Cl)2]+) Crystal diffraction data for [Fe3+L1(Cl)2]+ were collected at 100 K on a Bruker D8 Quest Diffractometer. Data collection, frame integration, data reduction (multi-scan) were carried out using APEX3 software.76 The structure was solved via intrinsic phasing methods using ShelXT77 and refined with ShelXL78 within the Olex2 graphical user interface.79 All non-hydrogen atoms were refined using anisotropic thermal parameters, while the hydrogen atoms were treated as mixed. The ORTEP molecular plots (50 %) were produced using Platon.80 (For [Fe2+L5(Cl)]+) A small block-like crystal of [Fe2+L5(Cl)]2[FeCl4] having dimensions 0.13 × 0.07 × 0.05 mm3 was secured to a Mitegen micromount using Paratone oil and its single crystal reflection data was collected at 100 K using a Rigaku Oxford Diffraction (ROD) SuperNova X-ray diffractometer equipped with an Atlas S2 CCD detector and microfocused Cu Kα1 radiation (= 1.54184 Å). Data collection strategies to ensure completeness and redundancy were determined using CrysAlisPro.81 Data processing for all samples was done using CrysAlisPro and included a numerical absorption correction applied via face-indexing using the SCALE3 ABSPACK scaling algorithm.82 The structure was solved via intrinsic phasing methods using ShelXT77 and refined with ShelXL78 within the Olex2 graphical user interface.79 The space group was unambiguously verified by PLATON.83 The final structural refinement included anisotropic temperature factors on all constituent non-hydrogen atoms. Given the quality of the data collected, hydrogen atoms were located in the difference map and refined. (For [Fe2+L6(Cl)2]) Single crystal X-ray diffraction data were collected in series of x-scans using a Stoe IPSD2 image plate diffractometer utilizing monochromated Mo radiation (k = 0.71073 Å). Standard procedures were employed for the integration and processing of the data using X-RED.84 Samples were coated in a thin film of perfluoropolyether oil and mounted at the tip of a glass fiber located on a goniometer. Data were collected from crystals held at 150 K in an Oxford Cryosystems nitrogen gas cryostream. Crystal structures were solved using routine automatic direct methods implemented within SHELXS-97.85 Completion of structures was achieved by performing least squares refinement against all unique F2 values using SHELXL-97.85 All non-H atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed using a riding model. Where the location of hydrogen atoms was obvious from difference Fourier maps, C-H bond lengths were refined subject to chemically sensible restraints.
Results and Discussion
As previously shown, the iron(III) complexes [Fe3+L1(Cl)2]+, [Fe3+L2(Cl)2]+, and [Fe3+L3(Cl)2]+ catalyze the C-C coupling reaction between pyrrole and phenylboronic acid to yield 2-phenylpyrrole.33 This study established that the macrocyclic ligand, iron salt, and oxygen were all required for the reaction to proceed and that L1-L3 iron(III) complexes provided similar reaction yields. Encouraged by our preliminary results, the availability of a library of macrocyclic scaffolds, and the positive attributes of an iron catalyst to carry out direct C-C coupling reactions. Therefore, we set out to (1) exemplify/expand the versatility of macrocyclic ligands, previously shown to facilitate other catalytic transformations12,35, 68–69,71, to C-C chemistry to produce 2-phenylpyrrole for the first time and (2) identify properties of such an iron catalyst that result in increased catalytic yields. Modifications to tetra-azamacrocyclic ligands abound in the literature and are known to impact electronics and structural features of metal complexes.10, 12, 17, 29, 47, 86–92 For the study described herein, [Fe3+L1(Cl)2]+, [Fe3+L4(Cl)2]+, [Fe2+L5(Cl)]+, [Fe2+L6(Cl)2], [Fe3+L7(Cl)2]+, [Fe3+L8(Cl)2]+, [Fe2+L9(Cl)]+, and [Fe2+L10(Cl)]+ (Figure 2) at 10% loading resulted in a catalytic yields ranging from 19–81% (Table 1). Given that each catalyst was isolated and characterized prior to the catalytic studies, the inorganic properties of the iron catalysts could be compared for correlations to catalytic yield. Specifically, catalytic reaction yields, and the following properties were compared for each complex: (property 1) coordination requirements, (property 2) the reductive and oxidative properties of the complex, (property 3) the rigidity/topological constraint of the complex, (property 4) the resistance of the complex against oxidative degradation, and (property 5) geometric parameters about the iron center.
Table 1.
Half-potential and catalytic reaction yield of iron complexes used to couple pyrrole and phenylboronic acid to produce 2-phenylpyrrole using 10% catalyst loading in the presence of air.
 | ||
|---|---|---|
| Catalyst | Yield (%)a | E1/2 (mV)b |
| [Fe3+L1(Cl)2]+ | 57 ± 3 | −465 |
| [Fe3+L4(Cl)2]+ | 60 ± 1 | −285 |
| [Fe2+L5(Cl)]+ | 30 ± 2 | −442 |
| [Fe2+L6(Cl)2] | 81 ± 7 | −405 |
| [Fe3+L7(Cl)2]+ | 74 ± 3 | −391 |
| [Fe3+L8(Cl)2]+ | 68 ± 4 | −306 |
| [Fe2+L9(Cl)]+ | 32 ± 5 | −285 |
| [Fe2+L10(Cl)]+ | 19 ± 2 | 73 |
| L3 + FeC2O4 ·2H2O | 61 ± 5 | - |
Yields determined by NMR analysis.
Referenced vs. Fc/Fc+ 0.0 mV.
Rational for ligand/complex comparison.
As detailed in Figure 1, ligands L1-L8 have four N-atom donor atoms but vary from one another by ring size (12 versus 14), N-atom substitution (secondary versus tertiary amine), and the presence of the cross-bridge between N-atom donors. Ligands L9-L10 are 12- and 14-membered systems, respectively, and provide five N-atom donors. Therefore, the importance of coordination requirements (property 1) of the resulting iron catalyst was achieved by comparing complexes with tetra-dentate ligands ([Fe3+L7(Cl)2]+ and [Fe3+L8(Cl)2]+) to penta-dentate ligands ([Fe2+L9(Cl)]+ and [Fe2+L10(Cl)]+). The importance of the reductive and oxidative properties of the complexes (property 2) was determined by comparing the iron(III/II) half-wave potentials versus catalytic reaction yields of [Fe3+L1(Cl)2]+, [Fe3+L4(Cl)2]+, [Fe2+L5(Cl)]+, [Fe2+L6(Cl)2], [Fe3+L7(Cl)2]+, [Fe3+L8(Cl)2]+, [Fe2+L9(Cl)]+, and [Fe2+L10(Cl)]+. The effect of rigidity/topological ligand constraint (property 3) was investigated by comparing the most rigid metal complexes ([Fe3+L7(Cl)2]+ and [Fe3+L8(Cl)2]+) to the least rigid metal complexes ([Fe3+L1(Cl)2]+, [Fe2+L5(Cl)]+, and [Fe2+L6(Cl)2]), based on the factors described above. The presence of tertiary amines has been shown to increase the stability of complexes toward oxidative degradation; therefore, the impact of complex stability against oxidative degradation (property 4) on C-C catalytic reaction yields was evaluated directly by comparing the [Fe3+L1(Cl)2]+ (N-atom donors) with [Fe3+L4(Cl)2]+ (-NMe donor atoms).29 Finally, geometric parameters about the iron center (property 5) were probed by comparing geometric parameters of [Fe3+L1(Cl)2]+, [Fe2+L6(Cl)2], [Fen+L7(Cl)2]+, and [Fen+L8(Cl)2]+ determined through X-ray diffraction analysis.
Property 1: Coordination Requirements
Mechanistic studies pertaining to catalytic oxygen-atom transfer and insertion reactions indicate that two exchangeable cis-sites are needed for oxygen activation during the catalytic cycle.28, 30 To date, the need for two cis-labile coordination sites has not been identified in iron catalyzed direct Suzuki-Miyaura C-C coupling reactions. Herein, the requirement for two labile cis-cites was determined by comparing catalysts with one exchangeable site ([Fe2+L9(Cl)]+, [Fe2+L10(Cl)]+) versus two cis-sites ([Fe3+L7(Cl)2]+, [Fe3+L8(Cl)2]+). The loss of an exchangeable cis-site led to a 42% reduction in catalyzing 2-phenylpyrrole formation using [Fe3+L7(Cl)2]+ (74 ± 3%) compared to [Fe2+L9(Cl)]+ (32 ± 5%). Similarly, a yield decrease of 49% was observed when comparing [Fe3+L8(Cl)2]+ (68 ± 4%) with [Fe2+L10(Cl)]+ (19 ± 2%). Next, catalytic yields obtained from a mixture of L3 and iron(II) oxalate (an iron(II) complex of L3 was not isolable) were compared to [Fe3+L1(Cl)2]+ (Table 1) to validate that it was not the iron(II) oxidation states of [Fe2+L9(Cl)]+ and [Fe2+L10(Cl)]+ that contribute to a decrease in catalytic yields versus the loss of a coordination site. As with all of the studies described herein, the conditions used mimic those reported previously.32 The mixture of L3 and iron(II) oxalate afforded 61 ± 5% of 2-phenylpyrrole, which is comparable to the 57 ± 3% obtained when [Fe3+L1(Cl)2]+ was used. Therefore, the use of an iron(II) complex as opposed to the iron(III) complex does not decrease the amount of 2-phenylpyrrole produced. This is further supported when comparing [Fe2+L5(Cl)]+ (30 ± 3%) and [Fe2+L6(Cl)2] (81 ± 7%), which provide the lowest and highest reaction yields, respectively, within the series of four-coordinate ligand based complexes explored. Furthermore, no significant differences are observed between catalytic yields of bona fide complexes with chloride or perchlorate counter ions versus conditions mixing iron oxalate and L3 (Table 1).32 This is in contrast to Wen et al., which reported that when mixtures of iron salts and L1 are used in the catalytic reaction, the oxalate salt afforded better yields than sulfate or chloride salts. The counter-ion effect previously reported may be attributed to solubility of the iron salts being prohibitive of catalyst formation in situ. Altogether, these results show that the addition of a 5th coordinating amine to the ligand scaffold significantly decreased catalytic yields, and therefore confirms the need for two exchangeable cis-sites.
Property 2: Reductive and Oxidative Properties of the Complexes
Ligand modifications are known to result in tuning the redox potential of the subsequent metal-ion complexes.93–95 In nature, the electrochemical potential of metalloenzymes is tuned by the ligand environment to produce different reactivities.15, 96–97 Therefore, the catalytic reaction yields of [Fe3+L1(Cl)2]+, [Fe3+L4(Cl)2]+, [Fe2+L5(Cl)]+, [Fe2+L6(Cl)2], [Fe3+L7(Cl)2]+, [Fe3+L8(Cl)2]+, [Fe2+L9(Cl)]+, and [Fe2+L10(Cl)]+ were compared to the iron(III/II) electrochemical potential of each catalyst to determine if the position of the iron redox potential effects the efficiency of 2-phenylpyrrole production (Table 1 and Figure 4–5). Cyclic voltammetry studies were carried out under identical conditions to one another and standardized to Fc/Fc+ (0.00 mV). As indicated in Figure 4, each scan was collected by beginning with the resting potential of each complex. The results indicate that the [Fe3+L1(Cl)2]+ complex affords the most negative iron(III/II) half potential (−465 mV), followed by [Fe2+L5(Cl)]+ (−442 mV), [Fe2+L6(Cl)2] (−405 mV), [Fe3+L7(Cl)2]+ (−391 mV), [Fe3+L8(Cl)2]+ (−306 mV), [Fe3+L4(Cl)2]+ (−285 mV), [Fe2+L9(Cl)]+ (−232 mV), and [Fe2+L10(Cl)]+ (73 mV). The more positive half-wave potentials derived from the pyridine pendant arm containing complexes, [Fe2+L9(Cl)]+ and [Fe2+L10(Cl)]+, could be related to having only one negatively charged chloride ion to stabilize the higher oxidation state of the iron center versus two in the remainder of the complexes. This observation is in accordance with previous reports of similar manganese complexes.66 The half potentials of the iron complexes, thereby, indicates that L1 is the most donating followed by L5 > L6 > L7 > L8 > L4 > L9 > L10. This order is in accordance to ring size and modifications of the N-atom donors from the macrocycle scaffold. Overall, the 14-membered complexes result in more positive iron(III/II) potentials compared to 12-membered congeners; e.g. a shift of +85 mV is observed for [Fe3+L7(Cl)2]+ versus [Fe3+L8(Cl)2]+. Conversion of –NH to –NMe donors also results in a shift to more positive potentials, consistent with the N-atoms becoming weaker donors upon methylation. For example, a difference of +180 mV is observed for [Fe3+L1(Cl)2]+ versus [Fe3+L4(Cl)2]+. Finally, the topological constraint obtain by substituting the -NH donor ligands (L5 and L6) with an ethylene cross-bridge (L7 and L8) results in a more positive iron(III/II) potential; [Fe3+L5(Cl)]+ versus [Fe2+L7(Cl)2]+ results in a shift of +51 mV.
Figure 4.
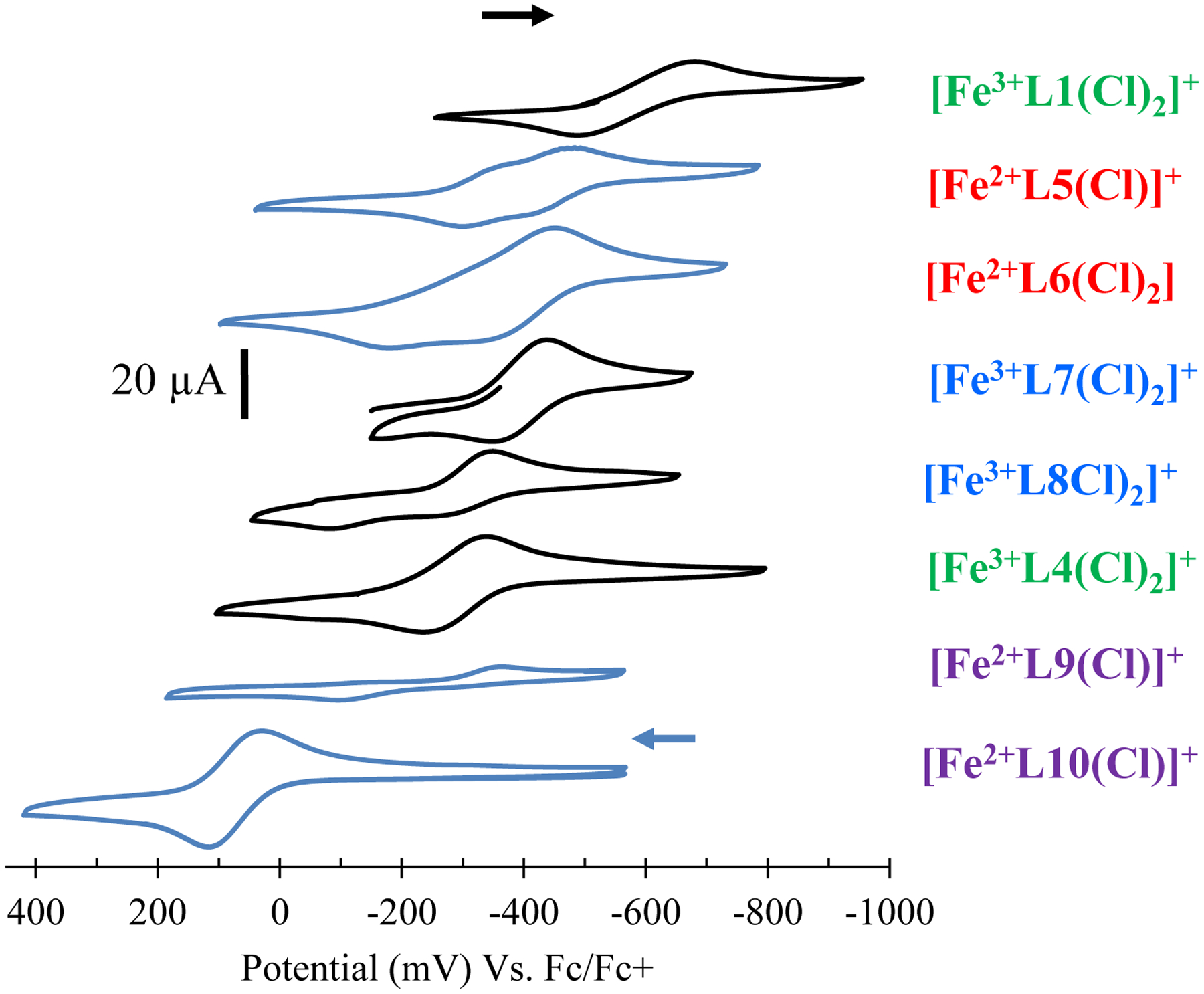
Cyclic voltammogram overlay of the iron(III/II) couple measured for [Fe3+L1(Cl)2]+, [Fe3+L4(Cl)2]+, [Fe2+L5(Cl)]+, [Fe2+L6(Cl)2], [Fe3+L7(Cl)2]+, [Fe3+L8(Cl)2]+, [Fe2+L9(Cl)]+, and [Fe2+L10(Cl)]+ in DMF containing 0.1 M [Bu4N][BF4] as electrolyte, Ag wire as the reference electrode, a glassy carbon working electrode, and a platinum auxiliary electrode at a scan rate of 100 mV/s. The trace color indicates the initial direction of each scan: Black trace (toward more positive potential), Blue trace (toward more negative potential).
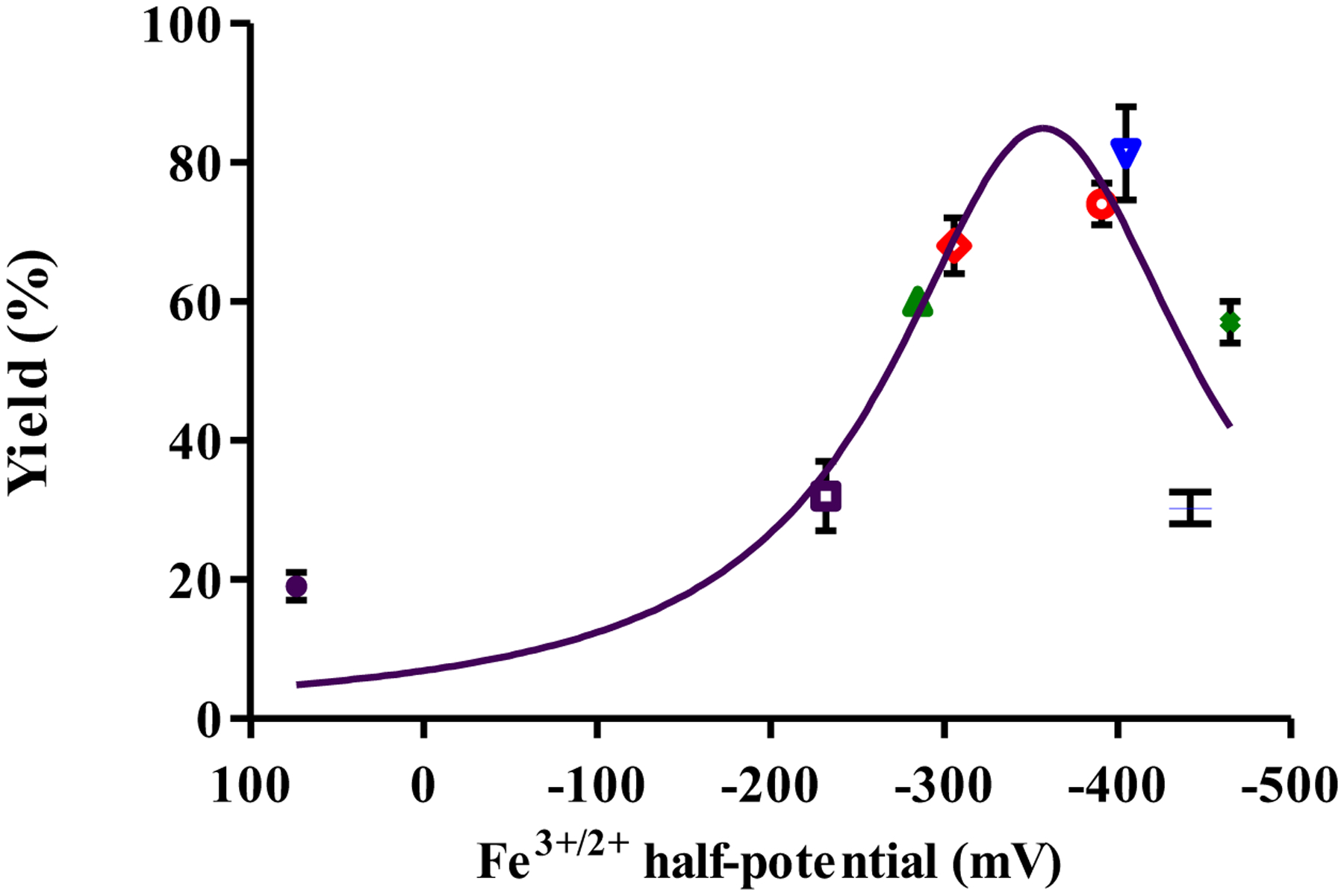
Figure 5.
Plot of iron(III/II) half-potentials versus yield for complexes: [Fe2+L10(Cl)]+ ( );[Fe2+L9(Cl)]+ (
);[Fe2+L9(Cl)]+ ( ); [Fe3+L4(Cl)2]+ (
); [Fe3+L4(Cl)2]+ ( ); [Fe3+L8(Cl)2]+ (
); [Fe3+L8(Cl)2]+ ( ); [Fe3+L7(Cl)2]+ (
); [Fe3+L7(Cl)2]+ ( ); [Fe2+L6(Cl)2] (
); [Fe2+L6(Cl)2] ( ); [Fe2+L5(Cl)]+ (
); [Fe2+L5(Cl)]+ ( ); [Fe3+L1(Cl)2]+ (
); [Fe3+L1(Cl)2]+ ( ).
).
Figure 5 shows a plot of the iron(III/II) half-potentials versus catalytic reaction yields for each complex. Lorentzian fitting of the data predicts that a complex with iron(III/II) potentials in the range of −325.5 to −389.0 mV would be optimal for this C-C coupling reaction. The Lorentzian fitting parameters can be found in the supporting information (Table S1). The direct relationship between catalytic reaction yield and iron(III/II) redox potentials suggests that the reaction mechanism depends upon an iron based electron transfer, which can be modulated by ligand choice. Altogether, this comparison shows redox potentials of the iron center should be a factor to consider when tuning a catalyst. As detailed in the interplay of ligand properties description, this redox potential is clearly tied to the rigidity and modification to the donor and could serve as a more direct predictor for catalytic success. Finally, this result helps to explain why the C-C coupling reaction does not proceed without a ligand donor present33; the redox chemistry of the iron center needs to be tuned to match that of the key step(s) involved in the C-C coupling reaction cycle being carried out.
Property 3: Effect of Rigidity/Topological Constraint
The series of cross-bridge free ([Fe3+L1(Cl)2]+, [Fe2+L5(Cl)]+, and [Fe2+L6(Cl)2]) versus cross-bridged complexes ([Fe3+L7(Cl)2]+ and [Fe3+L8(Cl)2]+) provide a good comparison to evaluate the impact of rigidity on catalytic reaction yields as they vary by both ring size and presence of the rigidifying ethylene cross-bridge (CB). To the best of our knowledge, the incorporation of a cross-bridge into a ligand has not been explored as supports for direct C-C coupling reactions, particularly iron catalysts. For an example of a cross-bridged ligand supporting indirect C-C coupling (pre-activated substrates), see reference 7. The rigidity of a complex can be controlled by the presence of an ethylene cross-bridge and the size of the macrocyclic ligand (12 versus 14).48 Specifically, topological constraint34, a term that describes making a ligand’s donor atoms more interconnected, leads to more kinetically stable interactions between the metal ion and the ligand.36 The simple bridging of two nitrogen atoms of a cyclic ligand by a two-carbon chain dramatically changes how the ligand coordinates the metal ion, vastly increases the kinetic stability and ability to catalyze oxidation reactions.28, 35 In addition, smaller macrocyclic rings (12 versus 14 membered rings) also impart greater rigidity through ring strain that results when metals bind to the donor atoms. Based on these principles, the order of rigidity in the series studied is [Fe3+L7(Cl)2]+ (12 membered, CB) > [Fe3+L8(Cl)2]+ (14 membered, CB) > [Fe3+L1(Cl)2]+ (12 membered) ≥ [Fe2+L5(Cl)]+ (12 membered) > [Fe2+L6(Cl)2] (14 membered). The catalytic reaction yields do not parallel the rigidity/topological constraint: [Fe2+L6(Cl)2] (81 ± 7%) > [Fe3+L7(Cl)2]+ (74 ± 3%) > [Fe3+L8(Cl)2]+ (68 ± 4%) > [Fe3+L1(Cl)2]+ (57 ± 3%) > [Fe2+L5(Cl)]+ (30 ± 2%). The results indicate that cross-bridged complexes ([Fe3+L7(Cl)2]+ and [Fe3+L8(Cl)2]+) in general result in higher yields compared to 12-membered noncross-bridged complexes ([Fe3+L1(Cl)2]+ and [Fe2+L5(Cl)]+). Conversely, the absence of a cross-bridge in [Fe2+L6(Cl)2] compared to [Fe2+L8(Cl)2] results in higher (+13 %) catalytic reaction yields. This supported that the presence of a cross-bridge and ring strain (12- versus 14-membered rings) tunes the reactivity. However, the effect on reduction and oxidation properties was dominant compared to structural and stability considerations typically associated with cross-bridged systems alone.
Property 4: Stability of the Complex against Oxidative Degradation
The comparison of protected versus unmodified N-atoms in the ligands was prompted by Chow and co-workers, who reported that the use of iron catalysts for oxygen atom transfer reactions derived from azamacrocycle ligands with the N-atoms methylated (-NMe) afforded a 99% conversion of substrate. This is in contrast to the non-methylated congeners, which resulted in only 38% substrate conversion. The observed increase in conversion with N-atom protected complexes was attributed to the presence of tertiary versus secondary amine donors, that provided an increased stability by resistance to oxidative degradation of the catalyst.29 The effect of increased stability against oxidative degradation through protection of the N-atoms was evaluated for C-C coupling chemistry by comparison of [Fe3+L1(Cl)2]+ and [Fe3+L4(Cl)2]+, which utilize unprotected, secondary (-NH) amine donors versus tertiary (-NMe) donors, respectively (Figure 2). The [Fe3+L1(Cl)2]+ (57 ± 3%) and [Fe3+L4(Cl)2]+ (60 ± 1%) complexes afforded comparable yields under identical conditions. Therefore, the substitution of the N-atoms as protection from catalyst decomposition is not a property to be considered in catalyst design for this reaction. Furthermore, this, along with the need for oxygen, suggests that the catalytic cycle may not involve highly reactive oxidative intermediates or conditions.
Property 5: Effect of Geometric Parameters
A brief structural comparison between the novel catalysts presented here and related literature analogues may be helpful to orient the rationalization of catalytic performance with structural parameters. Figure 6 contains solid-state structures of each relevant complex and Table 2 includes useful geometric parameters for comparison. A full list of structural parameters related to previously unreported complexes [Fe3+L1(Cl)2]Cl, [Fe2+L5(Cl)]2[FeCl4], and [Fe2+L10(Cl)]PF6 are available in supplementary information and supporting files (Figure S2, Tables S2–S8).
Figure 6.
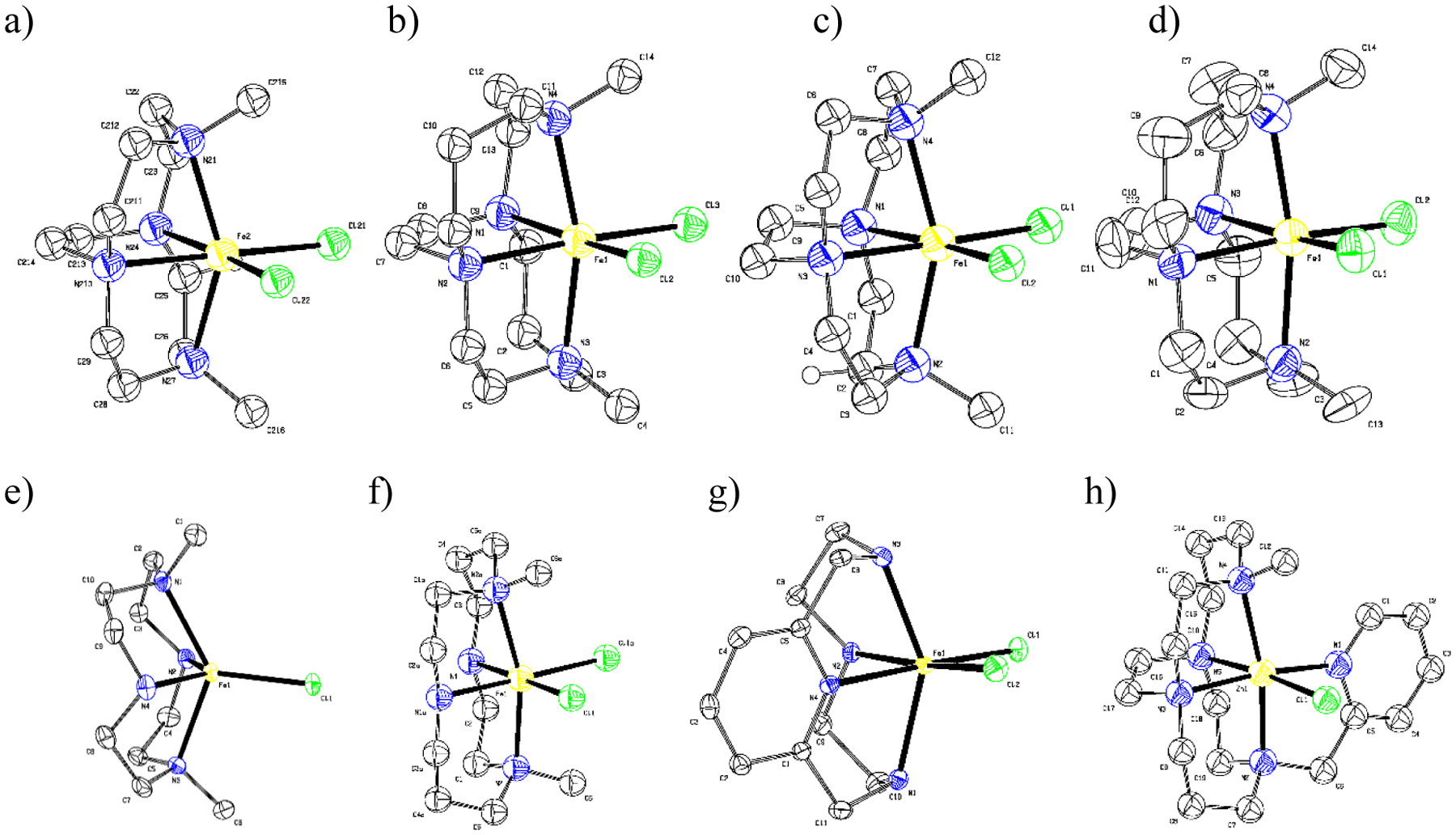
ORTEP (50% TELP) representation of a) [Fe2+L7(Cl)2] b) [Fe2+L8(Cl)2] c) [Fe3+L7(Cl)2]+ d) [Fe3+L8(Cl)2]+ e) [Fe2+(L5)Cl]+ f) [Fe2+(L6)Cl2] g) [Fe3+L1(Cl)2]+ h) [Fe2+L10(Cl)]+. Hydrogen atoms and counter-ions have been omitted for clarity.35, 98–99
Table 2.
Comparison of the catalytic reaction yields and useful geometric parameters, derived from solid-state structural data. Values in () indicate yields based on congener with complementary oxidation state to the species used for catalysis.
| Fig. 7 | Complex (Coord Geometry) | Catalytic Yield |
Mn+ | ar (pm) | Nax-Fe-Nax Angle (°) | Neq-Fe-Neq Angle (°) | Cl-Fe-Cl Angle (°) | Cl-Cl Bond Dist (Å) | M-N Bond Dist (Å) | M-Cl Bond Dist (Å) | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a |
[Fe2+L7(Cl)2]+ (Octahedral) |
(74) | Fe2+ | 92 | 145.78 | 77.31 | 93.42 | 3.511 | 2.276 | 2.416 | 35 |
| b |
[Fe2+L8(Cl)2] (Octahedral) |
(68) | Fe2+ | 92 | 161.88 | 78.36 | 95.53 | 3.593 | 2.275 | 2.426 | 35 |
| c |
[Fe3+L7(Cl)2]+ (Octahedral) |
74 | Fe3+ | 79 | 153.20 | 77.81 | 95.10 | 3.377 | 2.179 | 2.2853 | 98 |
| d |
[Fe3+L8(Cl)2]+ (Octahedral) |
68 | Fe3+ | 79 | 166.8 | 79.8 | 95.70 | 3.412 | 2.188 | 2.278 | 99 |
| e |
[Fe2+L5Cl]+ (Square Pyramidal (τ) = 0.098b) |
30 | Fe2+ | 92 | 131.14 | 125.27 | - | - | 2.296 | 2.304 | This work |
| f |
[Fe2+L6(Cl)2] (Octahedral) |
81 | Fe2+ | 92 | 162.4 | 91.5 | 94.4 | 3.58 | 2.27 | 2.438 | This work |
| g |
[Fe3+L1(Cl)2]+ (Octahedral) |
57 | Fe3+ | 79 | 146.68 | 86.62 | 95.78 | 3.388 | 2.181 | 2.2862 | This work |
| h |
[Fe2+L10(Cl)]+ (Octahedral) |
19 | Fe2+ | 92 | 168.27 | 80.82 | - | - | 2.204 | 2.3805 | 66 |
Shannon, R. D. Acta Crystallogr. Sect. A 1976, 32, 751–767.
Addison, A. W. et al. J Chem Soc Dalton 1984, 7, 1349–1356.
The X-ray derived crystal structures of iron(II) and (III) dimethyl cross-bridged cyclen and cyclam complexes are found in Figure 6a–d and all have distorted octahedral geometries with the macrocyclic nitrogen atoms at the axial and two cis equatorial positions, leaving the remaining cis equatorial positions filled by chloro- ligands. The iron(II) complexes (Figure 6a–b,h) are presented for comparison with the other iron(II) structures, since we were unable to obtain iron(III) structures of [Fe2+L5(Cl)]+, [Fe2+(L6)Cl2], and [Fe2+L10(Cl)]+. For our purposes, these structures mainly differ by the extent to which the metal ion extends from the macrocyclic cavity to be available for catalytic reactivity. The iron ion extends further from the cavity in both cyclen analogues (L5, L7) than the larger cyclam analogues (L6, L8), which can more fully engulf the metal ion and thereby more closely approach octahedral geometry. Finally, the larger iron(II) cations are extended further away from the macrocycle cavity than the smaller iron(III) cations.
The unbridged analogues ([Fe2+L5(Cl)]+ and [Fe2+(L6)Cl2]) of these just-discussed iron(II) complexes are found in Figure 6e–f and demonstrate the enhanced flexibility available without the ethylene cross-bridges. A direct comparison can be made between both octahedral cross-bridged cyclam complexes, which have Neq-Fe-Neq bond angles of 79.8° and 78.36°, respectively, to the Neq-Fe-Neq bond angle of 91.5° in the likewise octahedral [Fe2+L6(Cl)2] (Figure 6f). The Fe-N and Fe-Cl bond distances change less than 0.02 Å between the cross-bridged and unbridged iron(II) complexes even as the bond angles are adjusted. The unbridged ligand has more nearly octahedral bond angles and likely better orbital overlap for bonding, while the cross-bridged complex has gained topological constraint that is most readily demonstrated as higher kinetic stability.40 The [Fe2+L5(Cl)]+ (Figure 6e) complex, on the other hand, has a unique 5-coordinate geometry that is best described as distorted square pyramidal (τ = 0.098).100 The flexible cyclen macrocycle unfolds from its tight cis-configuration in the two cross-bridged cyclen octahedral structures in Figure 6a,c which have Neq-Fe-Neq bond angles of 77.81° and 77.3°, respectively, to the analogous smallest Neq-Fe-Neq bond angle of 125.27° in the square pyramidal structure of [Fe2+L5(Cl)]+. This expansion of the macrocycle around the “equator” allows steric space for only one chloro- ligand, which is more tightly held, as would be expected, in this 5-coordinate complex (Fe2+-Cl = 2.304 Å) than in the octahedral iron(II) bridged cyclen complex (Fe2+-Cl = 2.407 Å and 2.416 Å). Presumably, the unbridged ligand could have folded more tightly to accommodate an octahedral structure with a second chloro- ligand, but at least in the solid state, the 5-coordinate mono chloro- complex was favored.
The [Fe3+L1(Cl)2]+ complex (Figure 6g) is pseudo-octahedral, folded like the cross-bridged ligands, with two cis chloro- ligands and the pyridine donor located equatorially. This complex has a Neq-Fe-Neq bond angle of 86.62°, which is larger than the 77.81° of the constrained complex [Fe3+L7(Cl)2]+, but much smaller than the highly flexible 125.27° of [Fe2+L5(Cl)]+. Interestingly, the Nax-Fe-Nax bond angle of 146.68° for [Fe3+L1(Cl)2]+ is also intermediate between the 153.20° of [Fe3+L7(Cl)2]+ and the 131.14° of [Fe2+L5(Cl)]+. It appears that inclusion of a pyridine in the macrocyclic ring is an effective strategy to select these intermediate bond angles between those of the ethylene cross-bridged and unbridged cyclen analogues and could be used for tuning complexes in future ligand design.
Finally, the octahedral [Fe2+L10(Cl)]+ complex (Figure 6h) has metric parameters fairly similar to its L8 analogue (Figure 6b), with the trend of slightly larger N-Fe-N bond angles, as the pyridine pendant arm helps pull the iron(II) ion more fully into the ligand cavity.66 However, this ligand is pentadentate donor, and its pendant pyridine may interfere with the catalytic process rather than enhance it, as the catalytic yield of [Fe2+L10(Cl)]+ is the lowest studied. Presumably, the [Fe2+L9(Cl)]+ complex, though no crystal structure was obtained, would also have a pentadentate macrocyclic ligand and was likewise not a very active catalyst.
In summary, comparison of the N-Fe-N angles versus reaction yields indicate that equatorial space about the exchangeable cis-sites and/or iron accessibility does not correlate to increased catalytic reaction yield; the Neq-Fe-Neq and Nax-Fe-Nax were 91.5° and 162.4° for [Fe2+L6(Cl)2], 86.62° and 146.68° for [Fe3+L1(Cl)2]+, 79.83° and 166.8° for [Fe3+L8(Cl)2]+, and 79.62° and 153.20° for [Fe3+L7(Cl)2]+. Likewise, the differences in the corresponding Cl-Fe-Cl(°) and Cl-Cl(Å) geometric parameters for the cis-chloride ions are derived from the chelate composition and were analyzed as well. Small variations between [Fe3+L1(Cl)2]+ (95.78°, 3.388 Å), [Fe3+L7(Cl)2]+ (95.10°, 3.377 Å), and [Fe3+L8(Cl)2]+ (95.70°, 3.412 Å) are noted, but, like N-Fe-N angles, none correlate to the observed trends in catalytic yields. These studies help to support that the redox chemistry of the iron center plays the key role in controlling the catalytic reaction yields. Although, the accessibility to the iron center is a factor to consider, as shown by the need for two labile cis sites (property 1), the small changes in the axial and equatorial iron angles within the series studied does not correlate to catalytic reaction yields. This encourages the use of the range of tetra azamacrocycles available to synthetic chemists as considerations for tuning redox potentials to optimal ranges for this reaction.
Conclusion
Catalytic reaction yields of a library of high-spin iron complexes [Fe3+L1(Cl)2]+, [Fe3+L4(Cl)2]+, [Fe2+L5(Cl)]+, [Fe2+L6(Cl)2], [Fe3+L7(Cl)2]+, [Fe3+L8(Cl)2]+, [Fe2+L9(Cl)]+, and [Fe2+L10(Cl)]+ has been determined for the C-C coupling reaction that yields 2-phenylpyrrole. Complex [Fe2+L6(Cl)2] afforded the highest yield (81%). The systematic evaluation of the coordination requirements, the reductive and oxidative properties of the complex, rigidity/topological constraint of the complex, the stability of the complex toward oxidative degradation by N-atom modification, and geometric parameters about the iron center revealed that optimized iron redox potential are responsible for the increase in catalytic reaction yield. The need for two labile cis-sites was probed and confirmed as a 42% decrease in yield was observed when only one labile labile-site was present. Altogether the fundamental approach to this series of catalysts for C-C coupling reactions shows some similarities to iron catalysts used in oxidation catalytic reactions, but there are differences that should be taken in to consideration when carrying out these reactions. The studies presented herein suggest that electronic control of the iron center via rationalized modification of the macrocycle ring size, modification to the N-atoms (methylation or cross-bridging) and/or further modification to the pyridine for electronic tuning could be used to improved yields obtained to date. Such a new class of molecules is the focus of our future studies. Altogether, this work provides an inorganic/coordination chemistry focused approach to optimizing catalytic reactions and expands the applications in which macrocycles and cross-bridged macrocycles are used.
Supplementary Material
Synopsis.
The systematic evaluation of a library of macrocyclic based iron complexes revealed that two cis-sites and optimized iron redox potential are responsible for the increase in catalytic reaction yield in the direct C-C coupling of phenylboronic acid with pyrrole.
ACKNOWLEDGMENT
The authors are grateful for generous financial support from TCU Andrews Institute of Mathematics & Science Education (to KG), TCU Research and Creativity Activity Grant (to KG), TCU Invests in Scholarship, and INFOR Moncrief Foundation Support (to KG). TJH acknowledges Southwestern Oklahoma State University for internal funding through a Proposal Development Award. TJH acknowledges the Donors of the American Chemical Society Petroleum Research Fund; Health Research award for project number HR13-157, from the Oklahoma Center for the Advancement of Science and Technology; and Grant Number P20RR016478 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) for partial support of this research. TJH also acknowledges the Henry Dreyfus Teacher-Scholar Awards Program for support of this work.
Footnotes
Supporting Information
This material is available free of charge via the Internet at pubs.acs.com Exact mass analysis of the mass spectrometry assignments made by Wen. et al., Lorentzian fitting parameters used to produce Figure 5, X-ray crystallographic data for [Fe3+L1(Cl)2]Cl, [Fe2+L5(Cl)]2[FeCl4], and [Fe2+L10(Cl)]PF6.
Conflict of Interest: The authors declare no competing financial interest.
REFERENCES
- 1.Beck EM; Gaunt MJ, C-H Activation. In Top. Curr. Chem, C-H Activation ed.; Yu JQ; Shi Z, Eds. Springer: 2010; Vol. 292. [PubMed] [Google Scholar]
- 2.Doerwald FZ; Dorwald FZ, Electrophilic Arylation of Arenes In Side Reactions in Organic Synthesis Ii: Aromatic Substitutions, Wiley-VCH Verlag GmbH & Co. KGaA: 2014; pp 61–84. [Google Scholar]
- 3.Bedford RB, Eight irons hit the right spin. Nature Chemistry 2016, 8, 904–905. [DOI] [PubMed] [Google Scholar]
- 4.Daifuku SL; Al-Afyouni MH; Snyder BER; Kneebone JL; Neidig ML, A Combined Mössbauer, Magnetic Circular Dichroism, and Density Functional Theory Approach for Iron Cross-Coupling Catalysis: Electronic Structure, In Situ Formation, and Reactivity of Iron-Mesityl-Bisphosphines. J Am Chem Soc 2014, 136 (25), 9132–9143. [DOI] [PubMed] [Google Scholar]
- 5.Daifuku SL; Kneebone JL; Snyder BER; Neidig ML, Iron(II) Active Species in Iron–Bisphosphine Catalyzed Kumada and Suzuki–Miyaura Cross-Couplings of Phenyl Nucleophiles and Secondary Alkyl Halides. J Am Chem Soc 2015, 137 (35), 11432–11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agata R; Iwamoto T; Nakagawa N; Isozaki K; Hatakeyama T; Takaya H; Nakamura M, Iron Fluoride/N-Heterocyclic Carbene Catalyzed Cross Coupling between Deactivated Aryl Chlorides and Alkyl Grignard Reagents with or without β-Hydrogens. Synthesis 2015, 47 (12), 1733–1740. [Google Scholar]
- 7.Bedford RB; Brenner PB; Elorriaga D; Harvey JN; Nunn J, The influence of the ligand chelate effect on iron-amine-catalysed Kumada cross-coupling. Dalton T 2016, 45 (40), 15811–15817. [DOI] [PubMed] [Google Scholar]
- 8.Bedford RB; Brenner PB, In Iron Catalysis II. Topics in Organometallic Chemistry, vol 50, Bauer E, Ed. Springer: Cham, 2015; Vol. 50, pp 19–46. [Google Scholar]
- 9.Bedford RB, How Low Does Iron Go? Chasing the Active Species in Fe-Catalyzed Cross-Coupling Reactions. Accounts Chem Res 2015, 48 (5), 1485–1493. [DOI] [PubMed] [Google Scholar]
- 10.Chen MS; White CW, A predictably selective aliphatic C–H oxidation reaction for complex molecule synthesis. Science 2007, 318, 783–787. [DOI] [PubMed] [Google Scholar]
- 11.Kist LT; Trujillo MJF; Szpoganicz B; Manez MA; Basallote MG, Kinetics of reaction of the FeII-cyclam complex with H2O2 in acetonitrile and the mechanism of catalyzed epoxidation of cyclohexene. Polyhedron 1997, 16, 3827–3833. [Google Scholar]
- 12.Bauer I; Knölker H-J, Iron Catalysis in Organic Synthesis. Chem Rev 2015, 115 (9), 3170–3387. [DOI] [PubMed] [Google Scholar]
- 13.Marek I; Rappoport Z; Liebman JF; Patai S, The Chemistry of Organoiron Compounds: Fe. Wiley: West Sussex, England, 2014. [Google Scholar]
- 14.Lippard SJ; Berg JM, Principles of Bioinorgainic Chemistry. University Science Books: United State of America, 1994. [Google Scholar]
- 15.Concepts and Models in Bioinorganic Chemistry. Wiley-VCH: Weinheim, 2006. [Google Scholar]
- 16.Talsi EP; Bryliakov KP, Chemo- and stereoselective CH oxidations and epoxidations/cis-dihydroxylations with H2O2, catalyzed by non-heme iron and manganese complexes. Coordin Chem Rev 2012, 256 (13), 1418–1434. [Google Scholar]
- 17.Tseberlidis G; Intrieri D; Caselli A, Catalytic Applications of Pyridine-Containing Macrocyclic Complexes. Eur J Inorg Chem 2017, 2017 (30), 3589–3603. [Google Scholar]
- 18.Ray K; Pfaff FF; Wang B; Nam W, Status of Reactive Non-Heme Metal–Oxygen Intermediates in Chemical and Enzymatic Reactions. J Am Chem Soc 2014, 136 (40), 13942–13958. [DOI] [PubMed] [Google Scholar]
- 19.Nam W; Kim HJ; Kim SH; Ho RYN; Valentine JS, Metal Complex-Catalyzed Epoxidation of Olefins by Dioxygen with Co-Oxidation of Aldehydes. A Mechanistic Study. Inorganic Chemistry (Washington, DC, United States) 1996, 35, 1045–1049. [DOI] [PubMed] [Google Scholar]
- 20.Nam W; Ho R; Valentine JS, Iron-cyclam complexes as catalysts for the epoxidation of olefins by 30% aqueous hydrogen peroxide in acetonitrile and methanol. J. Am. Chem. Soc 1991, 113, 7052–7054. [Google Scholar]
- 21.Hong S; Lee YM; Cho KB; Seo MS; Song D; Yoon J; Garcia-Serres R; Clemancey M; Ogura T; Shin W; Latour JM; Nam W, Conversion of high-spin iron(III)-alkylperoxo to iron(IV)-oxo species via O-O bond homolysis in nonheme iron models. Chem Sci 2014, 5 (1), 156–162. [Google Scholar]
- 22.Watkins DD Jr.; Riley DP; Stone JA; Busch DH, Iron (II) complexes with unsubstituted saturated tetraaza macrocyclic ligands of varying ring size. Inorganic Chemistry (Washington, DC, United States) 1976, 15, 387–393. [Google Scholar]
- 23.Serrano-Plana J; Aguinaco A; Belda R; García-España E; Basallote MG; Company A; Costas M, Exceedingly Fast Oxygen Atom Transfer to Olefins via a Catalytically Competent Nonheme Iron Species. Angewandte Chemie International Edition 2016, 55 (21), 6310–6314. [DOI] [PubMed] [Google Scholar]
- 24.Decker A; Clay MD; Solomon EI, Spectroscopy and electronic structures of mono- and binuclear high-valent non-heme iron–oxo systems. J Inorg Biochem 2006, 100 (4), 697–706. [DOI] [PubMed] [Google Scholar]
- 25.Shaik S; Hirao H; Kumar D, Reactivity of High-Valent Iron–Oxo Species in Enzymes and Synthetic Reagents: A Tale of Many States. Accounts Chem Res 2007, 40 (7), 532–542. [DOI] [PubMed] [Google Scholar]
- 26.Bigelow JO; England J; Klein JEMN; Farquhar ER; Frisch JR; Martinho M; Mandal D; Münck E; Shaik S; Que L, Oxoiron(IV) Tetramethylcyclam Complexes with Axial Carboxylate Ligands: Effect of Tethering the Carboxylate on Reactivity. Inorg Chem 2017, 56 (6), 3287–3301. [DOI] [PubMed] [Google Scholar]
- 27.Singh KK; Tiwari M. k.; Ghosh M; Panda C; Weitz A; Hendrich MP; Dhar BB; Vanka K; Sen Gupta S, Tuning the Reactivity of FeV(O) toward C–H Bonds at Room Temperature: Effect of Water. Inorg Chem 2015, 54 (4), 1535–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng Y; England J; Que L, Iron-Catalyzed Olefin Epoxidation and cis-Dihydroxylation by Tetraalkylcyclam Complexes: the Importance of cis-Labile Sites. ACS Catalysis 2011, 1 (9), 1035–1042. [Google Scholar]
- 29.Chow TW-S; Wong EL-M; Guo Z; Liu Y; Huang J-S; Che C-M, cis-Dihydroxylation of Alkenes with Oxone Catalyzed by Iron Complexes of a Macrocyclic Tetraaza Ligand and Reaction Mechanism by ESI-MS Spectrometry and DFT Calculations. J Am Chem Soc 2010, 132 (38), 13229–13239. [DOI] [PubMed] [Google Scholar]
- 30.Koch WO; Krüger H-J, A Highly Reactive and Catalytically Active Model System for Intradiol-Cleaving Catechol Dioxygenases: Structure and Reactivity of Iron(III) Catecholate Complexes of N,N′-Dimethyl-2,11-diaza[3.3](2,6)pyridinophane. Angewandte Chemie International Edition in English 1996, 34 (23–24), 2671–2674. [Google Scholar]
- 31.Wang Z-Q; Wang Z-C; Zhan S; Ye J-S, A water-soluble iron electrocatalyst for water oxidation with high TOF. Applied Catalysis A: General 2015, 490, 128–132. [Google Scholar]
- 32.Wen J; Qin S; Ma LF; Dong LA; Zhang J; Liu SS; Duan YS; Chen SY; Hu CW; Yu XQ, Iron-Mediated Direct Suzuki-Miyaura Reaction: A New Method for the ortho-Arylation of Pyrrole and Pyridine. Org Lett 2010, 12 (12), 2694–2697. [DOI] [PubMed] [Google Scholar]
- 33.Brewer SM; Palacios PM; Johnston HM; Pierce BS; Green KN, Isolation and Identification of the Pre-Catalyst in Iron-Catalyzed Direct Arylation of Pyrrole with Phenylboronic Acid. Inorg Chim Acta 2018, 478, 139–147. [Google Scholar]
- 34.Busch DH, Distinctive Coordination Chemistry and Biological Significance of Complexes with Macrocyclic Ligands. Accounts Chem Res 1978, 11 (10), 392–400. [Google Scholar]
- 35.Hubin TJ; McCormick JM; Collinson SR; Buchalova M; Perkins CM; Alcock NW; Kahol PK; Raghunathan A; Busch DH, New iron(II) and manganese(II) complexes of two ultra-rigid, cross-bridged tetraazamacrocycles for catalysis and biomimicry. J Am Chem Soc 2000, 122 (11), 2512–2522. [Google Scholar]
- 36.Busch DH, Metals and Enzymes - Multiple Juxtopositional Fixedness. Chem. Eng. News 1970, p 9. [Google Scholar]
- 37.S. D; R. D. P, Nonpeptidyl mimetics of superoxide dismutase in clinical therapies for diseases. Cell Mol Life Sci. 2000. 57, 1489–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taktak S; Ye W; Herrera AM; Rybak-Akimova EV, Synthesis and Catalytic Properties in Olefin Epoxidation of Novel Iron(II) Complexes with Pyridine-Containing Macrocycles Bearing an Aminopropyl Pendant Arm. Inorg Chem 2007, 46 (7), 2929–2942. [DOI] [PubMed] [Google Scholar]
- 39.Ye W; Ho DM; Friedle S; Palluccio TD; Rybak-Akimova EV, Role of Fe(IV)-oxo intermediates in stoichiometric and catalytic oxidations mediated by iron pyridine-azamacrocycles. Inorg Chem 2012, 51 (9), 5006–21. [DOI] [PubMed] [Google Scholar]
- 40.Lincoln KM; Arroyo-Currás N; Johnston HM; Hayden TD; Pierce BS; S. B; Green KN, Chemical characteristics of the products of the complexation reaction between copper(II) and a tetra-aza macrocycle in the presence of chloride ions. Journal of Coordination Chemistry 2015, 68 (16), 2810–2826. [Google Scholar]
- 41.Weisman GR; Rogers ME; Wong EH; Jasinski JP; Paight ES, Cross-bridged cyclam. Protonation and lithium cation (Li+) complexation in a diamond-lattice cleft. J Am Chem Soc 1990, 112 (23), 8604–8605. [Google Scholar]
- 42.Bazzicalupi C; Bencini A; Bianchi A; Ciampolini M; Fusi V; Micheloni M; Nardi N; Paoli P; Valtancoli B, Two macrocycles of different molecular topology obtained by the same synthetic procedure. Their crystal structures and ligational properties. Supramolecular Chemistry 1994, 3 (4), 279–290. [Google Scholar]
- 43.Wilson KR; Cannon-Smith DJ; Burke BP; Birdsong OC; Archibald SJ; Hubin TJ, Synthesis and structural studies of two pyridine-armed reinforced cyclen chelators and their transition metal complexes. Polyhedron 2016, 114, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brinksma J; La Crois R; Feringa BL; Donnoli MI; Rosini C, New ligands for manganese catalysed selective oxidation of sulfides to sulfoxides with hydrogen peroxide. Tetrahedron Lett 2001, 42 (24), 4049–4052. [Google Scholar]
- 45.Shul’pin GB; Süss-Fink G; Shul’pina LS, Oxidations by the system “hydrogen peroxide–manganese(IV) complex–carboxylic acid”: Part 3. Oxygenation of ethane, higher alkanes, alcohols, olefins and sulfides. Journal of Molecular Catalysis A: Chemical 2001, 170 (1), 17–34. [Google Scholar]
- 46.Lindsay Smith JR; Gilbert BC; Mairata i Payeras A; Murray J; Lowdon TR; Oakes J; Pons i Prats R; Walton PH, Manganese 1,4,7-trimethyl-1,4,7-triazacyclononane complexes: Versatile catalysts for the oxidation of organic compounds with hydrogen peroxide. Journal of Molecular Catalysis A: Chemical 2006, 251 (1), 114–122. [Google Scholar]
- 47.Hubin TJ; McCormick JM; Alcock NW; Busch DH, Topologically Constrained Manganese(III) and Iron(III) Complexes of Two Cross-Bridged Tetraazamacrocycles. Inorganic Chemistry 2001, 40 (3), 435–444. [DOI] [PubMed] [Google Scholar]
- 48.Hubin TJ; McCormick JM; Collinson SR; Alcock NW; Clase HJ; Busch DH, Synthesis and X-ray crystal structures of iron(II) and manganese(II) complexes of unsubstituted and benzyl substituted cross-bridged tetraazamacrocycles. Inorganica Chimica Acta 2003, 346, 76–86. [Google Scholar]
- 49.Hubin TJ, Synthesis and coordination chemistry of topologically constrained azamacrocycles. Coordin Chem Rev 2003, 241 (1–2), 27–46. [Google Scholar]
- 50.Yin GC; Buchalova M; Danby AM; Perkins CM; Kitko D; Carter JD; Scheper WM; Busch DH, Olefin oxygenation by the hydroperoxide adduct of a nonheme manganese(IV) complex: Epoxidations by a metallo-peracid produces gentle selective oxidations. J Am Chem Soc 2005, 127 (49), 17170–17171. [DOI] [PubMed] [Google Scholar]
- 51.Yin GH; Danby AM; Kitko D; Carter JD; Scheper WM; Busch DH, Olefin epoxidation by alkyl hydroperoxide with a novel cross-bridged cyclam manganese complex: Demonstration of oxygenation by two distinct reactive intermediates. Inorg Chem 2007, 46 (6), 2173–2180. [DOI] [PubMed] [Google Scholar]
- 52.Yin GC; Danby AM; Kitko D; Carter JD; Scheper WM; Busch DH, Oxidative Reactivity Difference among the Metal Oxo and Metal Hydroxo Moieties: pH Dependent Hydrogen Abstraction by a Manganese(IV) Complex Having Two Hydroxide Ligands. J Am Chem Soc 2008, 130 (48), 16245–16253. [DOI] [PubMed] [Google Scholar]
- 53.Chattopadhyay S; Geiger RA; Yin GC; Busch DH; Jackson TA, Oxo- and Hydroxomanganese(IV) Adducts: A Comparative Spectroscopic and Computational Study. Inorg Chem 2010, 49 (16), 7530–7535. [DOI] [PubMed] [Google Scholar]
- 54.Shi S; Wang YJ; Xu AH; Wang HJ; Zhu DJ; Roy SB; Jackson TA; Busch DH; Yin GC, Distinct Reactivity Differences of Metal Oxo and Its Corresponding Hydroxo Moieties in Oxidations: Implications from a Manganese(IV) Complex Having Dihydroxide Ligand. Angew Chem Int Edit 2011, 50 (32), 7321–7324. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y; Shi S; Wang H; Zhu D; Yin G, Kinetics of hydrogen abstraction by active metal hydroxo and oxo intermediates: revealing their unexpected similarities in the transition state. Chem Commun 2012, 48 (63), 7832–7834. [DOI] [PubMed] [Google Scholar]
- 56.Wang YJ; Sheng JY; Shi S; Zhu DJ; Yin GC, Influence of the Net Charge on the Reactivity of a Manganese(IV) Species: Leading to the Correlation of Its Physicochemical Properties with Reactivity. J Phys Chem C 2012, 116 (24), 13231–13239. [Google Scholar]
- 57.Wang YJ; Shi S; Zhu DJ; Yin GC, The oxidative properties of a manganese(IV) hydroperoxide moiety and its relationships with the corresponding manganese(IV) oxo and hydroxo moieties. Dalton T 2012, 41 (9), 2612–2619. [DOI] [PubMed] [Google Scholar]
- 58.Dong L; Wang Y; Lv Y; Chen Z; Mei F; Xiong H; Yin G, Lewis-Acid-Promoted Stoichiometric and Catalytic Oxidations by Manganese Complexes Having Cross-Bridged Cyclam Ligand: A Comprehensive Study. Inorg Chem 2013, 52 (9), 5418–5427. [DOI] [PubMed] [Google Scholar]
- 59.Yin G, Understanding the Oxidative Relationships of the Metal Oxo, Hydroxo, and Hydroperoxide Intermediates with Manganese(IV) Complexes Having Bridged Cyclams: Correlation of the Physicochemical Properties with Reactivity. Accounts Chem Res 2013, 46 (2), 483–492. [DOI] [PubMed] [Google Scholar]
- 60.Weisman GR; Rogers ME; Wong EH; Jasinski JP; Paight ES, Cross-Bridged Cyclam - Protonation and Li+ Complexation in a Diamond-Lattice Cleft. J Am Chem Soc 1990, 112 (23), 8604–8605. [Google Scholar]
- 61.Busch DHC, S. R.; Hubin TJ; Perkins CM; Labeque R; Williams BK; Johnston JP; Kitko DJ; Berkett-St. Laurent JCTR Bleach Compositions. . April 17, 2001, 2001. [Google Scholar]
- 62.Busch DHC, S. R.; Hubin TJ; Perkins CM; Labeque R; Williams BK; Johnston JP; Kitko DJ; Berkett-St. Laurent JCTR; Burns, M. E. Bleach Compositions May 14, 2002, 2002. [Google Scholar]
- 63.Busch DHC, S. R.; Hubin TJ; Perkins CM; Labeque R; Williams BK; John-ston JP; Kitko DJ; Berkett-St. Laurent JCTR; Burns, M. E. Bleach Compositions . August 19, 2003, 2003. [Google Scholar]
- 64.Busch DHC, S. R.; Hubin TJ Catalysts and methods for catalytic oxidation. June 14, 2005, 2005. [Google Scholar]
- 65.Busch DHC, S. R.; Hubin TJ; Perkins CM; Labeque R; Williams BK; Johnston JP; Kitko DJ; Berkett-St. Laurent JCTR Bleach Compositions. October 24, 2006, 2006. [Google Scholar]
- 66.Jones DG; Wilson KR; Cannon-Smith DJ; Shircliff AD; Zhang Z; Chen Z; Prior TJ; Yin G; Hubin TJ, Synthesis, Structural Studies, and Oxidation Catalysis of the Late-First-Row-Transition-Metal Complexes of a 2-Pyridylmethyl Pendant-Armed Ethylene Cross-Bridged Cyclam. Inorg Chem 2015, 54 (5), 2221–2234. [DOI] [PubMed] [Google Scholar]
- 67.Yin GC; McCormick JM; Buchalova M; Danby AM; Rodgers K; Day VW; Smith K; Perkins CM; Kitko D; Carter JD; Scheper WM; Busch DH, Synthesis, characterization, and solution properties of a novel cross-bridged cyclam manganese(IV) complex having two terminal hydroxo ligands. Inorg Chem 2006, 45 (20), 8052–8061. [DOI] [PubMed] [Google Scholar]
- 68.Goedken VL; Busch DH, Facile Promotion of Oxidative Dehydrogenation by Iron Ions and Synthesis of New Complexes of Iron with Highly Unsaturated Tetraaza Macrocycles. J Am Chem Soc 1972, 94 (21), 7355–7363. [Google Scholar]
- 69.Yin GC, Active transition metal oxo and hydroxo moieties in nature’s redox, enzymes and their synthetic models: Structure and reactivity relationships. Coordin Chem Rev 2010, 254 (15–16), 1826–1842. [Google Scholar]
- 70.Fulmer GR; Miller AJM; Sherden NH; Gottlieb HE; Nudelman A; Stoltz BM; Bercaw JE; Goldberg KI, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29 (9), 2176–2179. [Google Scholar]
- 71.Lincoln KM; Gonzalez P; Richardson TE; Julovich DA; Saunders R; Simpkins JW; Green KN, A potent antioxidant small molecule aimed at targeting metal-based oxidative stress in neurodegenerative disorders. Chem. Commun 2013, 49, 2712–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lincoln KM; Offutt ME; Hayden TD; Saunders RE; Green KN, Structural, Spectral, and Electrochemical Properties of Nickel(II), Copper(II), and Zinc(II) Complexes Containing 12-Membered Pyridine- and Pyridol-Based Tetra-aza Macrocycles. Inorg Chem 2014, 53 (3), 1406–1416. [DOI] [PubMed] [Google Scholar]
- 73.Shin B; Sutherlin KD; Ohta T; Ogura T; Solomon EI; Cho J, Reactivity of a Cobalt(III)–Hydroperoxo Complex in Electrophilic Reactions. Inorg Chem 2016, 55 (23), 12391–12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amoyaw PNA; Pham K; Cain AN; McClain JM; Hubin TJ; Khan MOF, Synthesis of Novel Tetraazamacrocyclic Bisquinoline Derivatives as Potential Antimalarial Agents. Curr Org Synth 2014, 11 (6), 916–921. [Google Scholar]
- 75.Royal G; Dahaoui-Gindrey V; Dahaoui S; Tabard A; Guilard R; Pullumbi P; Lecomte C, New synthesis of trans-disubstituted cyclam macrocycles - Elucidation of the disubstitution mechanism on the basis of X-ray data and molecular modeling. Eur J Org Chem 1998, (9), 1971–1975. [Google Scholar]
- 76.Solutions., B. A. X.-R.; APEX2, (Version 2014.9–0), Bruker AXS Inc., Madison, WI: (2007). [Google Scholar]
- 77.Sheldrick GM, Crystal structure refinement with SHELXL. Acta Crystallographica Section A-Structural Chemistry 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sheldrick GM, Crystal structure refinement with SHELXL. Acta Crystallogr C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dolomanov OVB, L.J.; Gildea RJ; Howard JAK; Puschmann H, OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst 2009, 42, 339–341. [Google Scholar]
- 80.Spek A, Structure validation in chemical crystallography. Acta Crystallographica Section D 2009, 65 (2), 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.CrysAlisPro, Rigaku Oxford Diffraction, version 171.38.42a; 2015.
- 82.SCALE3 ABSPACK – A Rigaku Oxford Diffraction program for Absorption Correc-tions, Rigaku Oxford Diffraction, 2017.
- 83.Spek AL, Structure validation in chemical crystallography. Acta Crystallogr 2009, D65, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.X-AREA v 1.64, Darmstadt: STOE & Cie GmbH, 2012, 2012.
- 85.Sheldrick GM, A short history of SHELX. Acta Crystallogr A 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- 86.Lyakin OY; Ottenbacher RV; Bryliakov KP; Talsi EP, Asymmetric Epoxidations with H2O2 on Fe and Mn Aminopyridine Catalysts: Probing the Nature of Active Species by Combined Electron Paramagnetic Resonance and Enantioselectivity Study. Acs Catal 2012, 2 (6), 1196–1202. [Google Scholar]
- 87.Raffard N; Carina R; Simaan AJ; Sainton J; Riviere E; Tchertanov L; Bourcier S; Bouchoux G; Delroisse M; Banse F; Girerd JJ, Biomimetic catalysis of catechol cleavage by O-2 in organic solvents - Role of accessibility of O-2 to Fe-III in 2,11-diaza[3,3](2,6)pyridinophane-type catalysts. Eur J Inorg Chem 2001, (9), 2249–2254. [Google Scholar]
- 88.Sink RM; Buster DC; Sherry AD, Synthesis and Characterization of a Series of Macrocyclic Chelates Containing O-Donors and N-Donors - Prospects for Use as NMR Shift Agents for Alkali-Metal Cations. Inorg Chem 1990, 29 (19), 3645–3649. [Google Scholar]
- 89.Koch WO; Schünemann V; Gerdan M; Trautwein AX; Krüger H, Evidence for an unusual thermally induced low-spin (S=1/2)reversible arrow intermediate-spin (S=3/2) transition in a six-coordinate iron(III) complex: Structure and electronic properties of a (1,2-benzenedithiolato)iron(III) complex containing N,N ‘-dimethyl-2,11-diaza[3.3](2,6)pyridinophane as ligand. Chem. Eur. J 1998, 4, 686–691. [Google Scholar]
- 90.Hua WS; Ajiboye SI; Haining G; Mcghee L; Peacock RD; Peattie G; Siddique RM; Winfield JM, Coordination Chemistry of Iodine(I) with Tetraazamacrocycles or Monodentate Ligands - Comparisons with Bromine(I) and with Some D-Block Metals. J Chem Soc Dalton 1995, (23), 3837–3841. [Google Scholar]
- 91.Collinson S; Alcock NW; Raghunathan A; Kahol PK; Busch DH, Synthesis and Properties of Iron(II) and Manganese(II) Complexes Derived from a Topologically Constrained Pentadentate Ligand. Inorganic Chemistry 2000, 39 (4), 757–764. [DOI] [PubMed] [Google Scholar]
- 92.Shannon RD, Revised Effective Ionic-Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr A 1976, 32 (Sep1), 751–767. [Google Scholar]
- 93.Pulukkody R; Kyran SJ; Drummond MJ; Hseigh C-H; Darensbourg DJ; Darensbourg MY, Hammett correlations as a test of mechanism of CO-induced disulfide elimination from dinitrosyl iron complexes. Chem Sci 2014, 5, 3795–3802. [Google Scholar]
- 94.Green KN; Jeffery SP; Reibenspies JH; Darensbourg MY, A nickel tripeptide as a metallodithiolate ligand anchor for resin-bound organometallics. J Am Chem Soc 2006, 128 (19), 6493–6498. [DOI] [PubMed] [Google Scholar]
- 95.Green KN; Brothers SM; Thomas CW; Darensbourg MY, Macrocylization, sulfenation, and computation explorations of the thiolate carboxyamido NiN2S2 complex Ni(ema)2-. Abstr Pap Am Chem S 2007, 233, 698–698. [Google Scholar]
- 96.Voet D; Voet JG; Pratt CW, Fundamentals of biochemistry : life at the molecular level. 2nd ed.; Wiley: Hoboken, N.J., 2006. [Google Scholar]
- 97.Crabtree RH, Principles of Bioinorganic Chemistry - Lippard,Sj, Berg,Jm. Science 1994, 266 (5190), 1591–1592. [DOI] [PubMed] [Google Scholar]
- 98.McClain Ii JM; Maples DL; Maples RD; Matz DL; Harris SM; Nelson ADL; Silversides JD; Archibald SJ; Hubin TJ, Dichloro-(4,10-dimethyl-1,4,7,10-tetra-azabicyclo-[5.5.2]tetra-decane)-iron(III) hexa-fluoro-phosphate. Acta Crystallographica Section C 2006, 62 (11), m553–m555. [DOI] [PubMed] [Google Scholar]
- 99.Funwie NL; Cain AN; Fanning BZ; Hageman SA; Mullens M; Roberts TK; Turner DJ; Valdez CN; Vaughan RW; Ermias HG; Silversides JD; Archibald SJ; Hubin TJ; Prior TJ, Crystal structure of dichlorido(4,11-dimethyl-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane)iron(III) hexafluoridophosphate. Acta Crystallographica Section E 2015, 71 (9), 1073–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Addison AW; Rao TN; Reedijk J; Vanrijn J; Verschoor GC, Synthesis, Structure, and Spectroscopic Properties of Copper(Ii) Compounds Containing Nitrogen Sulfur Donor Ligands - the Crystal and Molecular-Structure of Aqua[1,7-Bis(N-Methylbenzimidazol-2’-Yl)-2,6-Dithiaheptane]Copper(Ii) Perchlorate. J Chem Soc Dalton 1984, (7), 1349–1356. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.