

SUMMARY
Long non-coding RNAs (lncRNAs) are critical regulators of numerous physiological processes and diseases, especially cancers. However, development of lncRNA-based therapies is limited because the mechanisms of many lncRNAs are obscure, and interactions with functional partners, including proteins, remain uncharacterized. The lncRNA SLNCR1 binds to and regulates the androgen receptor (AR) to mediate melanoma invasion and proliferation in an androgen-independent manner. Here, we use biochemical analyses coupled with selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) RNA structure probing to show that the N-terminal domain of AR binds a pyrimidine-rich motif in an unstructured region of SLNCR1. This motif is predictive of AR binding, as we identify an AR-binding motif in lncRNA HOXA11-AS-203. Oligonucleotides that bind either the AR N-terminal domain or the AR RNA motif block the SLNCR1-AR interaction and reduce SLNCR1-mediated melanoma invasion. Delivery of oligos that block SLNCR1-AR interaction thus represent a plausible therapeutic strategy.
In Brief
Androgen receptor (AR)-RNA complexes have been implicated in cancer, including melanoma. Schmidt et al. demonstrate that AR binds a single strand sequence in the long non-coding RNA (lncRNA) SLNCR. Point mutations or oligonucleotides that abrogate AR binding to SLNCR block melanoma invasion, suggesting that targeting lncRNA-protein complexes holds therapeutic promise.
Graphical Abstract
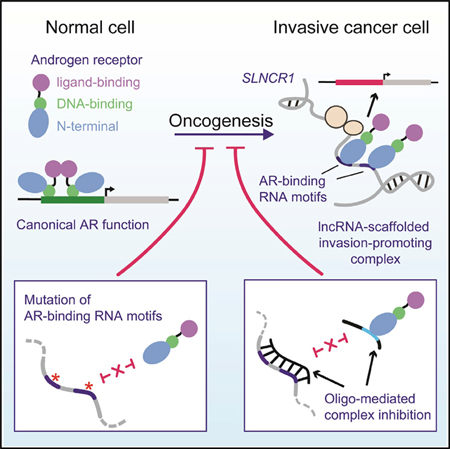
INTRODUCTION
Long non-coding RNAs (lncRNAs) are capped, usually polyadenylated, and often spliced transcripts of >200 nt. Many lncRNAs play critical roles in tissue physiology, disease processes, immune regulation, and cancer. Remarkably, despite their established importance, the fundamental mechanism of action of lncRNAs is poorly studied; only a few of the predicted 20,000–90,000 human lncRNAs (GENCODE version 7 [v.7] and NONCODE v.5, respectively) are well characterized (Fang et al., 2018; Harrow et al., 2012). It is believed that many lncRNAs function through direct interactions with protein partners, acting to scaffold the formation of ribonucleoprotein (RNP) complexes required for regulation of critical cellular processes (Ribeiro et al., 2018). Well-characterized examples of scaffolding lncRNAs include XIST, which regulates X chromosome inactivation through association with histone modification complexes, and NEAT1, a nuclear lncRNA that is essential for paraspeckle formation (Clemson et al., 2009).
It is currently difficult to predict lncRNA function from sequence analysis alone because so little is known about how lncRNA sequences and structural motifs interact with other macromolecules. The lack of understanding of the mechanisms of lncRNA activity hinders the development of therapeutic strategies that target lncRNAs involved in disease. Thus, there is a critical need for studies of the fundamental biology of lncRNAs and identification of functional lncRNA motifs. Functional characterization of lncRNA motifs and their interactions can begin to reveal the sequence and/or structural “rules” that govern lncRNA biology, advancing our ability to identify and characterize the function of other lncRNAs.
Many nuclear scaffolding lncRNAs regulate transcriptional activity through binding to transcription factors. For example, the lncRNAs SLNCR1 and HOTAIR, and possibly SRA1 and PCGEM1, bind to the androgen receptor (AR) protein and modulate its downstream transcriptional activity (Agoulnik and Weigel, 2009; Lanz et al., 1999; Prensner et al., 2014; Schmidt et al., 2016; Yang et al., 2013; Zhang et al., 2015). In contrast to canonical AR activation, in which an androgenic ligand, such as dihydrotestosterone (DHT), binds AR to induce nuclear localization and head-to-tail protein dimerization, lncRNA-mediated AR binding occurs in an androgen-independent manner (Schmidt et al., 2016; Yang et al., 2013; Zhang et al., 2015). RNA-driven AR function thus appears to fine-tune expression of a subset of genes, as opposed to global androgen-driven AR activation. In many cases, RNA-driven AR function promotes oncogenesis (Yang et al., 2013; Zhang et al., 2015). For example, our work has revealed that SLNCR1 increases AR occupancy at the promoter of the gene encoding the matrix metalloproteinase MMP9 to increase melanoma invasion, and regulates AR occupancy at the promoter of many growth-regulatory genes to increase cell proliferation (Schmidt et al., 2016, 2019). Although AR has been implicated in multiple cancers, including prostate, bladder, kidney, lung, liver, pancreatic, thyroid, and breast cancers, the fundamental mechanisms of AR activation in many of these cancers is not well-established (Chang et al., 2014; Kanda et al., 2014; Stanley et al., 2012). Thus, non-canonical AR activation, including RNA-mediated activation, is of clinical significance and understanding these pathways may explain many unresolved questions regarding AR’s oncogenic or tumor-suppressive functions.
In addition to AR, SLNCR1 binds to the transcription factor brain-specific homeobox protein 3a (Brn3a), and all three components are required for increased melanoma invasion (Schmidt et al., 2016). A highly conserved region of SLNCR1 (SLNCR1cons) spanning nucleotides 372–672 is necessary and sufficient to mediate increased melanoma invasion. The scaffolding model posits that lncRNAs bind multiple protein partners concurrently to coordinate their function. In agreement with this model, Brn3a and AR bind to adjacent sequences on SLNCR1cons, with Brn3a binding to SLNCR1462–572, while AR binds to SLNCR1568–637. It is an open question whether lncRNA scaffolds adopt complex structures that orient and enforce interactions between RNA-binding proteins or merely provide flexible short-distance linkers that bridge RNA-binding proteins together, likely through proximal sequence-specific RNA interactions. It is also currently unknown how ARs or Brn3a recognize their cognate binding regions in SLNCR1.
The predicted or confirmed AR-binding lncRNAs (SLNCR1, HOTAIR, SRA1, and PCGEM1) each contain a highly conserved sequence region of approximately 28 nt (Schmidt et al., 2016). This conserved sequence is located within the segment of SLNCR1, HOTAIR, and PCGEM1 required for AR-lncRNA association (SLNCRT568–637, HOTAIR1–360, and PCGEM 421–480), suggesting that AR interacts with RNA in a sequence-specific manner (Schmidt et al., 2016; Yang et al., 2013; Zhang et al., 2015). Moreover, HOTAIR and PCGEM1 mediate ligand-independent AR transcriptional activity, suggesting that these interactions may play an important role in AR-driven, androgen-independent cancers (Yang et al., 2013; Zhang et al., 2015). Given the clinical relevance of AR and its lncRNA binding partners, we chose to fully interrogate the AR-lncRNA interactions.
Here, we use a series of biochemical and phenotypic experiments to show that the N-terminal, regulatory domain (NTD) of AR (AR NTD) interacts with an unstructured, pyrimidine-rich RNA in a sequence-specific manner. Based on our definition of this binding site, we identify a novel AR-binding motif in HOXA11-AS-203 and confirm that this lncRNA interacts with AR in cells, suggesting that this lncRNA may function in a complex with AR. In-cell SHAPE and DMS probing reveals that the AR-binding motifs in SLNCR1 are found in a region of high structural flexibility, and that these motifs display in-cell protections consistent with protein binding. Mutational analysis confirms that the AR-binding motifs in SLNCR1 are required for SLNCR1- and AR-mediated melanoma invasion. Finally, we designed single-strand oligonucleotides to sterically block the interaction of AR and SLNCR1, and show that these oligonucleotides negate SLNCR1-mediated melanoma invasion. This work reveals how characterizing lncRNA structure and protein interactions enables identification of novel lncRNA function and supports design of unique lncRNA-targeting therapeutics.
RESULTS
AR NTD Binds to Unstructured RNA in a Sequence-Specific Manner
Previous RNA immunoprecipitation (RIP) assays identified an approximately 100-nt region of SLNCR1 required for interaction with AR in cells (nucelotides 568–673 [SLNCR1568–673]) (Schmidt et al., 2016). This sequence also encompasses the region of SLNCR1 (SLNCR1609–637) that is similar to other lncRNA sequences previously shown to bind AR in cells, including HOTAIR, SRA1, and PCGEM1, suggesting that AR binds to conserved nucleotides in these lncRNAs. Indeed, full-length AR protein bound an RNA probe containing nucleotides 597–637 of SLNCR1 (WT-41; Figure 1A) in a concentration-dependent manner, retarding the migration of the probe in an RNA electrophoretic mobility shift assay (REMSA; Figures 1B and S1A), and confirming that this RNA sequence is sufficient for AR binding. AR is composed of discreet protein domains, including a C-terminal ligand binding domain, a flexible hinge region, a DNA-binding domain, and an NTD (Figure 1A). The AR NTD is required for interactions with many protein and RNA cofactors, including the lncRNAs SRA1 and PCGEM1 (Lanz et al., 1999; Yang et al., 2013). A truncated version of AR (amino acids 2–566) spanning the AR NTD binds WT-41, indicating that the AR NTD is also sufficient to interact with SLNCR1 (Figures 1A and 1B). The AR NTD also binds shortened UC-rich SLNCR1 sequences WT-28 and WT-20 (Figure 1B). The multiple banding pattern of the shifted probe likely results from slight alterations in protein or RNA conformation that remain unresolved under native (i.e., non-denaturing) REMSA conditions. Addition of unlabeled WT-41 probes outcompetes the binding of labeled WT-41, WT-28, or WT-20 to either AR or AR NTD, consistent with the observed mobility shifts resulting from specific RNA binding (Figures S1B and S1C). The AR-RNA interaction was further examined using surface plasmon resonance (SPR) analysis, which enables quantification of bimolecular interactions in a label-free and real-time manner. The dissociation constant (KD) for the AR-WT-20 interaction, based on the ratio of the dissociation and association constants, was 270 ± 80 nM (Figure 1C). The combined data from RIP, REMSA, and SPR assays indicate that AR NTD binds with high affinity to short, pyrimidine-rich RNA sequences.
Figure 1. REMSA Identifies Minimal Requirements for the AR-SLNCR1 Interaction.

(A) Top: schematic of AR’s protein domains. AR NTD used in these studies (amino acids 2–556) is denoted. Bottom: sequences of biotinylated probes used in Figure 1. DBD, DNA binding domain; LBD, ligand binding domain; NTD, N-terminal domain.
(B) The indicated probes (from A) were incubated with increasing concentrations of truncated protein corresponding to AR NTD, resolved on a 5% TBE gel and probed using Streptavidin-HRP following transfer to a negatively charged membrane. The lines indicate either an unbound labeled probe or the more slowly migrating RNA-protein complexes. The asterisk denotes a non-specific RNA band.
(C) Measurement of binding affinity by surface plasmon resonance (SPR) of the indicated concentrations of WT-20 to full-length recombinant AR. RU, resonance units.
See also Figure S1.
RNA-binding proteins may bind RNA in a sequence- and/or structure-specific manner. We next used a series of mutational analyses to probe the potential sequence requirements of the AR-RNA interaction, identifying a set of WT and mutated RNAs that retain the ability to bind AR NTD (Figures 2A–2E). REMSAs were performed using excess probing and a protein concentration (600 nM) roughly 2-fold greater than the calculated KD (270 ± 80 nM), and 6-fold greater than the protein concentration at which an interaction with WT-20 was still observed (Figure 1B, right panel; 0.1 μM). Thus, these conditions should detect interaction with a KD less than (higher affinity than) ~1.5 μM. We categorize sequences that fail to elicit a gel shift upon AR binding under these conditions as “non-binders.” Importantly, identification of non-binders confirms that REMSA can distinguish the sequence specificity of the AR-RNA interaction. Motif enrichment analysis (multiple EM for motif elicitation [MEME]) of this set of tested RNAs identified an enriched (E = 1.3 × 10−13) 9-nt motif CYUYUCCWS (Y = pyrimidine [C or U], W = weak [A or U], and S = strong [C or G]) required for AR-RNA binding (Figure 2A; Bailey et al., 2009). These results indicate that AR NTD binds to RNA in a sequence-specific manner.
Figure 2. AR Binds to Single-Strand RNA in a Sequence-Specific Manner.

(A) Consensus motif identified among AR-binding RNA sequences.
(B) Sequences ofbiotinylated probes used in these assays. Nucleotidesthat are mutated compared towild-type SLNCR1 are highlighted in red, bolded font. The predicted AR-binding motifs are highlighted in the gray boxes.
(C and D) REMSA, as in Figure 1B, of the indicated probes derived from WT-28 (C) or WT-20 (D) incubated with 600 nM of recombinant AR NTD.
(E) REMSA, as in (C) and (D), with the indicated WT or stem-loop probe. Left: secondary structure model (ΔG = −5.4, as predicted by RNAStructure, rna.urmc.rochester.edu/RNAstructureWeb) ofstem-loop probewith the non-endogenous nucleotides denoted by the red bar. Significance was calculated using the Student’s t test: *p < 0.05; **p < 0.005; ns, not significant.
See also Figure S2.
SLNCR1 contains two CYUYUCCWS motifs in close proximity to each other (nucleotides 612–620 and nucleotides 627–635, motif 1 and motif 2). However, several lines of evidence suggest that only one motif is required for interaction with AR (Figure 1B). First, mutations in either motif 1 (MUT L [left], containing three base changes) or motif 2 of WT-28 (MUT R [right], containing four base changes) do not abolish AR binding when the other motif is present (Figures 2B and 2C). The altered migration pattern of MUT L, however, suggests that AR engages each motif in a unique way. Second, combining MUT L and MUT R mutations (MUT full-length [MUT FL]) completely abolished AR binding at AR NTD concentrations up to 1.2 μM (Figures 2B, 2C, and S2A). In contrast, WT-20 and WT-17 contain intact single motifs and bind AR NTD (Figures 1B and 2E). Together, these data show that a single CYUYUCCWS motif is sufficient for specific recognition by AR NTD.
We next tested if AR binds other pyrimidine-rich sequences. AR does not bind 20-nt probes consisting of UC, UUUC, or CCCU repeats (REP-UC, REP-UUUC, or REP-CCCU, respectively) or to 18-nt probes containing two repeats of a UCUCCA sequence (Figure S2B). Although AR NTD can bind motifs as short as 11 nucleotides, AR NTD does not bind a 15-nt probe containing a near-consensus sequence (CUUCUCCAU) even at protein concentrations up to 1.2 μM (Figures S2C and S2D). Moreover, RIP assays confirm that AR does not bind this sequence in cells, as this near-consensus sequence is contained in SLNCR1 (nucleotides 547–555) but located outside of the region required for AR binding (SLNCR1568–637) (Schmidt et al., 2016). The inability of AR to bind the near-consensus sequence or tandem UCUCCA repeats suggests that AR does not tolerate a U in position 9 of the AR-binding motif. Mutations between motifs 1 and 2 that do not alter either CYUYUCCWS motif do not affect AR binding (MUT-I; Figures 2B and 2C). A probe containing seven pyrimidine transitions (U to C or C to U) across WT-28 (MUT-pyrimidine transitions [MUT PTs]) that disrupt both of the original motifs 1 and 2 but introduce a new CYUYUCCWS consensus motif (CCUUUCCUC) retains AR binding (Figures 2B and 2C). These data indicate that the CYUYUCCWS motif is necessary and sufficient for AR binding.
To test whether RNA structure interferes with AR binding, we compared AR RNA binding in the presence or absence of secondary structures (Figures 2E). Introduction of non-endogenous nucleotides at the 5’ end of WT-17 that are predicted to induce a stable stem-loop structure encompassing 6 nucleotides of motif 1 (ΔG = −5.4, as predicted by RNAStructure), abolished AR NTD binding (Figure 2E; Xu and Mathews, 2016). This finding agrees with results suggesting that AR NTD binds with higher affinity to WT-20 than WT-41, the latter of which is predicted to form a stable stem-loop structure (ΔG = −6.1) (Figures 1B and S2E). Although WT-41 contains both AR-binding motifs, only motif 2 is likely accessible for AR binding: motif 1 is occluded from AR binding by the stem-loop structure likely formed by the oligonucleotide (Figure S2E). Considering that REMSAs suggest AR has a higher affinity for WT-20 (containing only motif 1: CCUCUCCAC) compared to WT-41 (where only motif 2 is accessible: CUUCUCCUG), we postulate that AR has a higher affinity for motif 1 compared to motif 2 (Figure 1B). Together, these data reveal that AR NTD binds pyrimidine-rich RNA containing at least one accessible CYUYUCCWS motif.
HOXA11-AS-203 Contains a Novel AR-Binding Element
The ability to predict lncRNA-binding partners, and more generally function, from sequence analysis alone remains largely uninvestigated. We previously identified a region of sequence similarity between multiple lncRNAs predicted or shown to bind to AR, including HOTAIR, SRA1, and PCGEM1, that overlaps with the WT-28 SLNCR1 sequence (Schmidt et al., 2016). Of these lncRNAs, only SRA1 contains an intact potential AR-binding motif (CUUCUCCUG). Indeed, in REMSA, addition of AR results in a slight, but reproducible, shift of the SRA1 probe that is successfully competed away by unlabeled WT-41 probe (Figures 3A and S3A), suggesting that SRA1 directly binds AR. Further work is required to confirm this interaction.
Figure 3. AR Binds to a Sequence Contained in lncRNA HOXA11-AS-203.

(A) Sequences of probes corresponding to the regions of the indicated IncRNAs predicted to bind to AR.
(B) REMSA, as in Figure 1B, of the indicated probes incubated with and without 600 nM of recombinant AR NTD.
(C) Schematic of the genomic loci of HOXA11 gene (blue) and its cognate antisense RNAs (green). The HOXA11-AS transcript variant 203 contains the AR-binding sequence and is highlighted in red. Transcript variants are based on Ensembl genome assembly: GRCh38.p10 (GCA_000001405.25).
(D) Sequence of HOXA11-AS-203 variant, with the sequence corresponding to the probe used in (A) highlighted in yellow.
(E) RNA IP assays from HEK293T cells transfected with FLAG-tagged AR and the indicated HOXA11-AS-203-expressing plasmids using either α-FLAG or immunoglobulin G (IgG) nonspecific control. Top-left panel: schematic of the HOXA11-AS sequences expressed from pCDNA3.1, zoomed in on the region containing site-directed mutations within the AR binding motif. Bottom-left panel: western blot analysis of input (I), IgG-bound (IgG), or α-FLAG-bound (AR) proteins. Right panel: relative enrichment of the indicated transcripts from FLAG-AR IPs compared to the nonspecific IgG control. Error bars represent mean ± SD from 3 technical replicates. Significance was calculated using the Student’s t test: ***p < 0.0005.
See also Figure S3.
In contrast, probes corresponding to the conserved region of HOTAIR or PCGEM1, which do not contain a consensus CYUYUCCWS motif, do not bind detectably to AR NTD in REMSA assays, indicating that AR binds these sequences at best weakly (KD >1.5 μM; Figures 3A and 3B). REMSAs were performed in the absence of mammalian-specific post-translational modifications of AR or additional protein or RNA cofactors that may be required for mediating these interactions in vivo, as is required for the AR-PCGEM1 interaction (Yang et al., 2013). These results further confirm that, in a minimal in vitro system, AR requires the presence of at least one CYUYUCCWS motif for optimal RNA binding.
To test if our identified AR-binding motif is predictive of AR interaction, we used Nucleotide BLAST search (https://blast. ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch) to identify non-coding RNAs containing a consensus or nearconsensus AR-binding motif (corresponding to WT-28 or WT-20). Of the many coding and non-coding human transcripts containing a consensus or near-consensus AR-binding motif, we were particularly interested in a 1,169-nt transcript expressed divergently from the HOXA11 gene, lncRNA HOXA11-AS-203 (Ensembl transcript ENST00000520395, gene ENSG00000240990), which contains a region of high similarity to WT-20 (Figures 3C and 3D). Moreover, HOXA11-AS-203 and AR are co-expressed in several human tissues, including the prostate, uterus, and testis, suggesting that this interaction occurs in vivo (Figure S3B). A probe corresponding to 23 nucleotides of the HOXA11-AS-203 sequence is significantly shifted upon incubation with AR NTD, indicating that AR NTD directly binds to this sequence in vitro. We measured the interaction between ectopically expressed HOXA11-AS-203 and FLAG-tagged ARs in cells by RIP assay (Figure 3E). HOXA11-AS-203 was enriched (~4-fold) in RNAs immunoprecipitated with AR compared to an IgG control. HOXA11-AS-203 bearing a mutation in the AR-binding motif was not enriched in RNAs immunoprecipitated with ARs, confirming that the AR-binding motif is required for AR binding (Figure 3E). Identification of the AR-HOXA11-AS-203 interaction in cells confirms that REMSAs faithfully identified a bona fide AR-binding RNA motif, and that this motif may predict the ability of a lncRNA to bind to AR. While the functional implications of this interaction remain to be determined, these data imply that perturbing AR binding to lncRNAs can block AR-dependent functions of lncRNAs in multiple tissues.
AR- and Brn3a-Binding Sites Reside within an Unstructured Region of SLNCR1, In Vivo
Our in vitro data indicate that AR preferentially binds to unstructured, single-strand RNA in a sequence-specific manner. However, REMSA combined with sequence and structural prediction analyses do not reveal how an RNA sequence folds in the context of an entire transcript within a cell. We therefore sought to experimentally determine if the AR-binding motifs in SLNCR1 are (1) accessible to protein interaction and (2) bound by protein, in vivo.
We probed local RNA structure and nucleotide accessibility of SLNCR1 AR-binding motifs using SHAPE as analyzed by mutational profiling (MaP). SHAPE-MaP provides experimental evidence for RNA secondary structure formation at nucleotide resolution, including in living cells (Siegfried et al., 2014; Smola et al., 2015b). We applied SHAPE-MaP on the melanoma short-term culture (MSTC) WM1976 cell line, whose invasion potential is disrupted through siRNA-mediated depletion of SLNCR1 (Schmidt et al., 2016). MSTCs have undergone limited passages outside of the patient and therefore closely reflect the genetics and relative gene expression patterns of the tumor. We probed the structure of SLNCR1 using two cell-permeable reagents: the SHAPE reagent 5-nitroisatoic anhydride (5NIA), which reacts broadly with all four nucleotides, and the methylating agent dimethyl sulfate (DMS), which reacts most strongly with adenosine and cytidine (Busan et al., 2019; Smola and Weeks, 2018). We probed the nucleotide flexibilities of SLNCR1 both within WM1976 cells (in cell) and after gentle extraction of the RNA from WM1976 cells into a protein-free environment (cell free). SHAPE reactivities from the cell-free state can be used to model RNA structure in the absence of cellular factors, and in-cell SHAPE reactivities provide further evidence in support of structure probing-based models and can identify RNA sequences whose structure changes in the presence of proteins. Extracted RNA (cell free) was separated into nuclear and cytoplasmic fractions (the SLNCR1-AR complex regulates transcription in the nucleus). Sites of SHAPE and DMS modification within in-cell and cell-free RNAs were encoded as non-tem-plated sequence changes in the cDNA generated during MaP reverse transcription, recorded by massively parallel sequencing, and converted into per-nucleotide reactivity profiles (Homan et al., 2014; Siegfried et al., 2014; Smola et al., 2015b). We obtained single-nucleotide resolution structural information on nucleotides 403–780 of SLNCR1 (SLNCR1403–780) using a gene-specific reverse transcription and PCR primers. This region encompasses most of SLNCR1cons (nucleotides 372–672), including the Brn3a-binding region (SLNCR1462–572) and both AR-binding motifs, required for mediating melanoma invasion.
We observed high overall SHAPE reactivity across SLNCR1403–780 in the cell-free RNA (Figures 4A and 4C), suggesting this region is conformationally flexible and lacks a single, well-defined structure in the absence of cellular components. To calibrate the reactivity scale, we also collected SHAPE reactivities for the highly structured U1 small nuclear RNA (snRNA), extracted from WM1976 cells (Figures 5A and S4). SHAPE reactivities for SLNCR1 and U1 snRNA spanned a similar range of values; however, reactive nucleotides were more diffuse in SLNCR1403–780 than in the U1 snRNA, with fewer regions of well-defined, punctate lowly or highly reactive nucleotides (Figures 4A, 4C, 5A, 5B, and S4A). The distribution of SHAPE reactivities in SLNCR403–780 is strongly shifted to higher reactivities, relative to the U1 snRNA, indicating that this region of SLNCR1 is much less structured than the U1 snRNA (Figure 5B). We used cell-free SHAPE reactivities as restraints for RNA structure modeling (Smola et al., 2015b), generating base-pairing probabilities and consequent Shannon entropies for every nucleotide within SLNCR1403–780 (Figures 5C and S6A). As a control, we modeled the structure of the U1 snRNA extracted from WM1976 cells and recovered the accepted four-stem structure (Figures 5A and S4C). The global median Shannon entropy, a measure of the well determinedness and conformational diversity of an RNA, is much greater for SLNCR1403–780 (at 0.19) than that of the highly structured U1 snRNA (0.01) and suggests that the lncRNA region lacks a well-defined structure (Figures 5A and 5C). Indeed, standard approaches to identify stable RNA structure motifs based on SHAPE reactivity and Shannon entropies (Siegfried et al., 2014; Smola et al., 2015b) yielded no motifs with well-defined structure within SLNCR1403–780. We were able to model potential structural motifs using relaxed thresholds intended for large RNAs (Smola et al., 2016; Figure S6; see STAR Methods), but the relative high reactivity and entropy of these structures suggest that they do not persist stably in vivo.
Figure 4. AR and Brn3a Binding Sites Reside within an Unstructured Region of SLNCR1, In Vivo.

Reactivity profiles for nucleotides 403–780 of the SLNCR1 RNA from SHAPE-MaP of (A) cell-free nuclear RNA, (B) in-cell total cellular RNA, and (C) cell-free cytoplasmic RNA. SHAPE reactivities are colored bythe relativevalue (see scale) and SE is indicated. Significant in-cell protectionsfrom SHAPE (gray) and DMS (light blue) reactivity are indicated by vertical bars. The location of Brn3a and AR interaction sites are labeled with horizontal purple and dark blue lines, respectively. Nucleotides included in amplification primers and high-background positions (thus classified as no data) are indicated with gray rectangles below zero. See also Figures S4 and S5.
Figure 5. SHAPE-Directed RNA Structure Modeling Reveals SLNCR1403–780 is Largely Structured.

(A) Reactivity profile for the U1 small nuclear RNA(snRNA) from SHAPE-MaP of cell-free nuclear RNA. SHAPE reactivities are colored by relative value (see scale) and SE is indicated. The base-pairing probabilities of nucleotide pairs are represented by colored arcs linking the involved nucleotides. Entropy values (bottom) are local 50-nt windowed medians. A low entropy threshold consistent with limited conformational dynamics (equal to 0.03) is indicated with a dotted line. Nearly all nucleotides in U1 have windowed-median SHAPE and entropy values below thresholds suggestive of formation of well-defined RNA secondary structure (0.4 for SHAPE, 0.03 for entropy).
(B) Comparison of SHAPE reactivity distributions for SLNCR1 and U1 RNAs from nuclear extracts. 0nly nucleotides 403–780 of SLNCR1 were analyzed.
(C) Secondary structure model for nucleotides 403–780 of SLNCR1 lncRNA. SHAPE reactivities and Shannon entropies are plotted on the same scale as the U1 snRNA, shown in (A). Underlying SLNCR data are the same as shown in Figure 4A. See also Figure S6.
The lack of well-defined RNA structural motifs within SLNCR1403–780 supports the hypothesis that this region is accessible for interaction with single-stranded RNA-binding proteins in vivo. For example, unstructured regions in the lncRNA Xist, identified by SHAPE, are some of the most highly protein bound (Smola et al., 2016). The average SHAPE reactivity in SLNCR1 is highest across nucleotides 495–570 (Figure 5C), which overlaps the SLNCR1 region necessary for interaction with Brn3a (SLNCR1462−572) (Schmidt et al., 2016). SLNCR1 AR-binding motif 1 has moderate SHAPE reactivity (0.32) but high entropy (0.44), suggesting that the motif is partially base paired, but samples multiple base-pairing states and does not sample a single, stable conformation. AR-binding motif 2 shows moderate reactivity (0.32) and has low entropy (0.07), suggesting that this motif exists in a partially base-paired state with few alternative pairing partners. The SHAPE reactivity profiles of the SLNCR1 RNA, when extracted from the nucleus or the cytoplasm, are nearly identical, suggesting that events in either compartment do not induce stable changes to the structure of this region of SLNCR1 (Figures 4A and 4C). When SLNCR1 RNA structure was examined by SHAPE or DMS in cell, the reactivity profiles largely resembled the cell-free profiles (Figures 4B and S5). Together, these data suggest that the SLNCR1403–780 RNA region lacks extensive stable secondary structure, especially within the Brn3a-binding region (SLNCR1462–572) and that the cellular environment does not impose any wide-ranging structural shifts in the RNA. The SLNCR403–780 region thus appears to be broadly accessible to single-stranded RNA-binding proteins.
Proteins Bind to Multiple Regions of SLNCR1cons, Including AR-Binding Motifs, In Vivo
Comparison of cell-free and in-cell reactivity profiles enables discovery of nucleotides that are protected in living cells, which is often indicative of protein binding in these regions of RNA (Smola et al., 2015a, 2016). For example, loops in the U1 snRNA engaged bythe 70K, U1A, and Sm proteins are highly reactive to SHAPE in the cell-free state but are unreactive in cell (Figure S4; Kondo et al., 2015; Pomeranz Krummel et al., 2009). We observed many in-cell protections from SHAPE and DMS reactivity throughout the SLNCR1403–780 region consistent with interactions with RNA-binding proteins (Figure 4). Statistically significant protections were observed at the 5′ end of the first AR-binding motif (Z factor = 0.59; Z score = 1.21) and adjacent to the 3′ end of the second AR-binding motif (Z factor = 0.60; Z score = 1.20), suggesting that these motifs are protein bound in vivo. Two large in-cell protections from SHAPE reactivity were also observed upstream of the AR-binding motifs within the Brn3a-interacting region (SLNCR1462–572). The two probing reagents show excellent agreement: all DMS protection sites are within five nucleotides of an in-cell SHAPE protection and in-cell DMS and SHAPE protections overlapped at the first AR-binding motif, further supporting that this motif is protein bound in cells (Figure 4). The stronger in-cell protection observed for motif 1 is consistent with our data showing that AR binds the SLNCR1 motif 1 with higher affinity than motif 2 (Figure 2G). Together, our REMSA and SHAPE data support a model wherein the SLNCR1cons region is unstructured and engages AR in vivo.
AR Binding to Its Cognate Motif Is Required for SLNCR1-Mediated Melanoma Invasion
To confirm the identity of the AR-binding motif and assess the consequence of AR binding to its cognate elements in vivo, we mutated the two AR-binding motifs in SLNCR1 and tested AR- and SLNCR1-mediated upregulation of MMP9 and subsequent melanoma invasions. We quantified cell invasion using the well-validated Matrigel invasion assay, capable of detecting physiologically relevant changes in cell invasion (Hall and Brooks, 2014). Transient overexpression of SLNCR1 significantly increased expression of MMP9 (~3-fold), and A375 melanoma invasion (~2-fold) (Figures 6 and S7) in agreement with our previous work (Schmidt et al., 2016). Mutation of individual AR-binding motifs 1 or 2 (SLNCR1AR MUT1 or SLNCR1AR MUT2, respectively) only partially attenuated SLNCR1-mediated invasion and MMP9 upregulation, suggesting that both motifs contribute to AR- and SLNCR1 -mediated melanoma invasions. Expression of SLNCR1 harboring mutant AR-binding motifs 1 and 2 (SLNCR1AR MUT 1+2) fully attenuated SLNCR1-mediated invasion and upregulation of MMP9 (Figures 6A, 6B, and S7). We note that all SLNCR1 mutants were expressed above the modest threshold (20-fold above endogenous SLNCR1 levels) necessary to increase invasion (Figures 7C and S7; data not shown). Thus, phenotypic analysis of SLNCR1 mutants confirms that AR-binding motifs 1 and 2, originally identified by REMSAs, mediate much of the AR-RNA interaction and is consistent with our model that SLNCR1 contains two AR-binding motifs.
Figure 6. Mutation of the AR-Binding Motif Blocks SLNCR1-Mediated Invasion.

(A) Schematic of the SLNCR1 sequences expressed in (B), zoomed in on the region containing site-directed mutations within the AR-binding motif.
(B) Matrigel invasion assays of A375 cells transfected with either an empty vector or a vector expressing the indicated SLNCR1 sequence. SLNCR1cons corresponds to SLNCR1 nucleotides 372–672. Invasion is calculated as the percent of invading cells compared to mobile cells as counted in eight fields of view. Top panels show representative images of the indicated invading and mobile cells. Quantification from three independent replicates, represented as mean ± SD, is shown at the bottom.
(C) RNA IP assaysfrom HEK293T cellstransfected with FLAG-tagged ARand the indicated SLNCR1-expressing plasmids using either α-FLAG orIgG nonspecific control. Left panel: western blot analysisofinput (I), IgG-bound (IgG), or α-FLAG bound (AR) proteins. Right panel: relative enrichment represented as mean ± SD of the indicated transcripts from FLAG-AR IPs compared to the nonspecific IgG control. Significance was calculated using the Student’s t test: *p < 0.05; **p < 0.005; ns, not significant.
See also Figure S7.
Figure 7. Sterically Blocking the AR-SLNCR1 Interaction Prevents SLNCRI-Mediated Melanoma Invasion.

(A) Sequences of FANA-modified steric-blocking oligonucleotides used in these assays.
(B) REMSA, as in Figure 1B, of the indicated probes incubated with and without 600 nM of recombinant AR NTD. Steric-blocking competitor oligonucleotides were added to a final concentration of 10 μM.
(C) Matrigel invasion assays of A375 cells transfected with either an empty vector or a vector expression SLNCR1. The indicated FANA-modified oligonucleotides were added 24 h post-transfection, at a final concentration of 500 nM. Invasion is calculated as the percent of invading cells compared to mobile cells as counted in eight fields of view. Top panels show representative images of the indicated invading and mobile cells. Quantification from three independent replicates, represented as mean ± SD, is shown at the bottom. Significance compared to the vector and scramble only control was calculated using the Student’s t test: p < 0.05; **p < 0.005; ***p < 0.0005; ns, not significant.
See also Figure S7.
To validate that both AR-binding motifs are important for protein interaction with SLNCR1, we repeated RIP assays using WT and the SLNCR1AR MUT1+2 mutant. Overexpressed WT SLNCR1 was enriched (~12-fold) in RNAs immunoprecipitated with ectopically expressed FLAG-tagged AR relative to a mock precipitation, consistent with previous results indicating that SLNCR1 and AR interact in cells (Figure 6C; Schmidt et al., 2016). In contrast, immunoprecipitation did not recover ectopically expressed SLNCR1AR MUT 1+2, instead enriching only background levels of endogenous SLNCR1 (i.e., levels immunoprecipitated from cells without SLNCR1 overexpression) (Figure 6C; data not shown). Collectively, these data confirm that AR binds RNA in a sequence-specific manner in vivo.
Inhibiting AR Binding Negates SLNCR1 -Mediated Melanoma Invasion
To assess the therapeutic potential of inhibiting the SLNCR1-AR interaction, we used short (21–28 nucleotides) chemically stabilized 2′-deoxy-2′-fluoro-D-arabinonucleic acid (2′-FANA) containing oligonucleotides that were either (1) antisense to the SLNCR1 AR-binding sequence, which then bound to SLNCR1 to generate double-strand RNAs incapable of AR binding (anti-senses 1 and 2), or (2) mimics of the SLNCR1 AR-binding sequence, which could bind directly to AR to inhibit its binding to SLNCR1 (mimics 1 and 2; Figure 7A; Ferrari et al., 2006). The antisense probes were designed to block both CY-UYUCCWS motifs present in SLNCR1. The 2′-FANA-modified oligonucleotides are incompatible with RNase H-mediated cleavage, thereby preventing downregulation of SLNCR1 or other potential off-target RNAs (Figure S7C; Schmidt et al., 2019). Consistent with a steric-blocking mechanism, these oligonucleotides decrease melanoma proliferation, another cancer phenotype attributed to the interaction of AR and SLNCR1, without decreasing SLNCR1 expression (Schmidt et al., 2019). Both SLNCR1-(antisense) and AR-binding (mimic) oligonucleotides sterically block the AR/SLNCR1 interaction in vitro (Figure 7B). Addition of antisense oligonucleotides upwardly shifts the biotinylated probe, confirming the formation of double-stranded RNA (dsRNA). Gymnotic delivery of these oligonucleotides (delivery of oligonucleotides independent of cell-transducing agents) to A375 melanoma cells reduced SLNCR1-mediated melanoma invasion, without impairing SLNCR1 overexpression (Figures 7C and S7C). Ectopic expression of SLNCR1 increased MMP9 mRNA levels, as expected, while addition of the steric-blocking oligonucleotides attenuated this increase (Figure S7D). The ability of the antisense oligonucleotides to attenuate SLNCR1-mediated proliferation, invasion, and upregulation of MMP9 is further evidence that AR binds single-stranded RNA. Collectively, these data are consistent with a model in which single-strand AR- or SLNCR1-targeting oligonucleotides block the AR and SLNCR1 interaction and attenuate SLNCR1-mediated melanoma invasion.
DISCUSSION
This work identifies a novel sequence-specific lncRNA motif that mediates protein binding and lncRNA function. We show that AR NTD binds with high affinity to unstructured, pyrimidine-rich RNA in a sequence-specific manner. Identifying and characterizing protein-lncRNA interactions enables prediction of novel lncRNA function, as demonstrated by the prediction and confirmation of a bona fide AR-binding sequence in the HOXA11-AS-203 lncRNA (Figure 3). Similarly, identifying RNA motifs bound by specific proteins may be crucial for the development of oligonucleotide-based therapeutics that target protein-lncRNA interactions.
Previous results suggested that lncRNA-mediated AR activity occurs independently of canonical androgen-mediated AR dimerization and activation (Schmidt et al., 2016; Yang et al., 2013; Zhang et al., 2015). Our current data indicate that AR likely binds to SLNCR1 as a monomer because (1) AR binds RNA independently of androgens in vitro (Figure S1A); (2) AR NTD, incapable of head-to-tail dimerization, binds RNA in vitro (Figure 2B); and (3) only one AR-binding motif is required for AR-RNA interaction (Figures 2 and 3). Interestingly, multiple AR-driven cancers show limited response to anti-androgen therapies, consistent with androgen-independent mechanisms for oncogenic AR activation (Morvillo et al., 1995, 2002; Rose et al., 1985). Increasing our understanding of novel RNA-mediated and androgen-independent AR function may help explain the failure of anti-androgens in the treatment of these tumors.
Although monomeric AR can bind SLNCR1, it is intriguing that the lncRNA contains two AR-binding motifs in close proximity (spanning 24 nucleotides). AR appears to interact independently and uniquely with each site, as evidenced by altered REMSA migration patterns and phenotypic differences upon mutation of either motif 1 or motif 2 (Figures 2C and S7B). In-cell protections and phenotypic data suggest that motif 1 is more potent for AR binding (Figures 4A, 6B, and S7B), possibly due to differences in sequence affinity or accessibility between motif 1 and motif 2. The high-affinity AR-binding motif 1, which exists at the junction of the highly flexible Brn3a-binding domain and a lower SHAPE and entropy 3’ region, may be more accessible than AR-binding motif 2, which resides within the lower SHAPE and entropy region (Figure 5C). Moreover, mutation of AR-binding motif 2 does not abolish transcriptional upregulation of MMP9 and increased invasion, indicating that motif 1 is sufficient for activity (Figures 6 and S7). The second, lower-affinity binding site may accommodate or anchor AR dimers following canonical activation, or independently bind a second AR monomer, forcing proximal dimerization and possibly initiating cooperative activity. We note that high concentrations of full-length AR, but not AR NTD, resulted in a secondary sub-shift of the WT-41 probe, suggesting a change in protein:RNA stoichiometry possibly resulting from AR dimerization (Figures S1A and 1B). Further work is required to determine if dimeric AR binds SLNCR1, and what the functional implications of this interaction might be.
AR NTD directly binds SLNCR1 and HOXA11-AS, but not PCGEM1 or HOTAIR, in vitro, despite the presence of a near-consensus AR-binding motif (Schmidt et al., 2016). It is possible that AR directly binds these lncRNAs with lower affinity than SLNCR1. Mutating only a few nucleotides within the AR-binding motif abolished lncRNA binding even at AR concentrations up to 1.2 μM (which should capture interactions with affinities up to ~7 μM), suggesting that the AR-RNA interaction is exquisitely sequence specific (Figures S2A and S2C). It is also possible that certain AR-interacting lncRNAs have evolved from a direct to an indirect interaction with AR (reflected as a near-consensus AR-binding motif), or may require additional cofactors or post-translational modifications of AR to facilitate the interaction. Indirect interaction or interactions that require additional cofactors or modifications would not be captured in our minimal in vitro system. Indeed, the AR-PCGEM1 interaction requires binding of the lncRNA PRNCR1 to the carboxyl-terminus of AR, recruitment of DOT1L, and methylation at AR K349 (Yang et al., 2013). It is unknown if other cofactors or post-translational modifications are required to mediate the interaction of AR with PCGEM1 or HOTAIR.
Our results also suggest that AR NTD binds SRA1 (Figure S3A), agreeing with prior work indicating that SRA1 is bound by full-length, but not N-terminally truncated, AR (Lanz et al., 1999). The interaction of AR with either SRA1 or HOXA11-AS appears weaker and/or more transient compared to the AR-SLNCR1 interaction (Figure 3E versus Figure 6C; see also Figure S3A), possibly due to the presence of only one CYUYUCCWS motif found within a non-pyrimidine-rich sequence compared to the two motifs found in the highly pyrimidine-rich sequence of SLNCR1. A combination of chemical and enzymatic probing in vitro suggests the AR motif of SRA1 lies in a structurally dynamic domain, which has characteristics of well-defined structure or flexibility depending on the probing technique employed and that the motif itself is single stranded in the absence of additional proteins (Novikova et al., 2012). REMSA conditions and the sequence of the SRA1 probe used here should be optimized further to verify the interaction of SRA1 and AR, and cell-based experiments may be necessary to discern whether other factors are necessary for the interaction in vivo.
Our results indicate that AR binds RNA independently of its DNA-binding domain. While this work focuses on interactions between AR and free RNA functioning in trans, it is tempting to postulate that AR also binds actively transcribed lncRNAs, recruiting AR in cis to particular genomic regions independently of DNA-encoded AR response elements. The proximal recruitment of AR to these regions may be sufficient to elicit AR-mediated transcriptional regulation of the lncRNA or other neighboring genes. In this way, AR may regulate expression of many non-ARE-containing genes. Indeed, many lncRNAs are expressed divergently or antisense to protein-coding genes, and expressions of many of these lncRNAs have been linked to transcriptional changes in expression of cognate mRNA. In addition to AR, many other transcription factors also bind RNA independently of canonical DNA interactions (Long et al., 2017). Identification of RNA-binding transcription factors and elucidation of their RNA sequence or structure requirements is critical to fully appreciate the complexities of regulated gene expression.
Many lncRNAs are likely composed of varying combinations of discrete domains that impart functional specificity through assembly of associated proteins, RNA and DNA (Guttman and Rinn, 2012). These lncRNA domains might impart function through a variety of means, for example, by (1) acting as allosteric regulators, assuming complex secondary and tertiary structures that present a unique surface for interaction and impart new or enhanced activities on associated macromolecules (Shamovsky et al., 2006; Wang et al., 2008); (2) acting as sponges that compete for binding to factors and thus inhibit their effects (Lee et al., 2016); or (3) providing protein-specific landing platforms to scaffold the formation of higher-order complexes, thereby coordinating the activities of multiple associated proteins through induced proximity (Cerase et al., 2015). SLNCR1403–780 behaves most like category 3, serving as a flexible scaffold for AR and Brn3 to coordinate their transcriptional activities (Schmidt et al., 2016). In other cell lines, SLNCR1 interacts with epigenetic regulators EZH2, DNMT1, and LSD1, although it is unknown if these interactions occur in the melanomas used in this study (Ba et al., 2017; Huang et al., 2017; Ma et al., 2017; Shi et al., 2016). Given that the most significant SHAPE protection in cell within SLNCR1403–780 lies adjacent to the Brn3a and AR-interacting domains (nucleotides 672–678), it is likely that additional factors associate with SLNCR1 and guide ribonucleoprotein complex assembly.
Given their critical role in the regulation of complex gene expression patterns and their implication in many human cancers, lncRNAs are rapidly emerging as an attractive class of novel pharmacological targets. Despite several hurdles early in their development, several oligonucleotide-based therapeutics have been approved by the Food and Drug Administration (FDA), including Onpattro (Patisiran), for the treatment of hereditary transthyretin amyloidosis; Eteplirsen (Exondys 51), for the treatment of Duchenne muscular dystrophy; and Nusinersen (Spinraza), for the treatment of spinal muscular atrophy (Adams et al., 2018; Juliano, 2016; Lundin et al., 2015; Ottesen, 2017; Stein and Castanotto, 2017). Here, we show that chemically stable oligonucleotides that block the AR-SLNCR1 interaction inhibit SLNCR1-mediated melanoma invasion, suggesting that disrupting this interaction has potential therapeutic value. Thus, characterizing lncRNA-protein interactions presents a novel avenue for the design and implementation of lncRNA-targeting therapies. In addition to oligonucleotide-based approaches, small molecules could also be used to selectively inhibit the AR-SLNCR1 interaction. Importantly, the SLNCR1 structural information generated here is critical to enabling identification of small molecules that bind to SLNCR1 and selectively inhibit AR binding (Connelly et al., 2016).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Carl Novina (Carl_Novina@dfci.harvard.edu). Reagents generated in this study will be made available on request but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
A375 (female) and HEK293T (female) cells were purchased from ATCC. WM1976 cells were from the collection of the Wistar Institute (Philadelphia, PA). Cells were cultured as adherent cells in DMEM (Dulbecco’s modified eagle medium, Invitrogen) without glutamine supplemented with 10% fetal bovine serum (FBS). Cells were maintained at 37°C, 95% relative humidity, and 5% CO2. The A375 cell line was authenticated via short tandem repeat profiling at the American Tissue Culture Repository on May 19, 2016. WM1976 and HEK293T cells were not subject to additional authentication.
METHOD DETAILS
Reagents and Plasmid Construction
Biotinylated and unmodified RNA probes were purchased from Integrated DNA Technologies. Full-length, recombinant AR protein containing an N terminus DDDKtag was purchased from Abcam (ab82609). N-terminal recombinant AR protein containing an N-terminal 6x HIS tag was purchased from RayBiotech (RB-14–0003P). Steric-blocking 2′-FANA modified oligonucleotides were gifts from AUM LifeTech. The plasmid expressing full-length SLNCR1 was previously described (Schmidt et al., 2016). Site-directed mutagenesis was used to obtain all SLNCR1 mutant plasmids described here, using oligo sequences listed in Table S1. Wild-type and mutant HOXA11-AS-203 were ordered as gblocks from IDT, and cloned into pcDNA3.1 (1) using flanking EcoRI and HindIII sites. The FLAG-tagged AR expression plasmid (pFLAG-EGFP-C1-AR) was generated by cloning a FLAG tag and 3x glycine linker flanked by NheI and AgeI sites (sequence of duplexed oligonucleotides found in Table S1) into pEGFP-C1-AR, a gift from Michael Mancini (Addgene plasmid #28235).
RNA Electrophoretic Mobility Shift Assays
REMSAs were performed using Thermo Fisher Scientific LightShift Chemiluminescent RNA EMSA (REMSA) Kit, according to manufacturer’s instructions. Briefly, 20 μl binding reactions were assembled in low-adhesion tubes in 1X binding buffer (10 mM HEPESpH 7.3, 20 mM KCl, 1 mM MgCl2,1 mM DTT), with 2 μg of yeast tRNA, the indicated amount of either recombinant AR or AR NTD, 10 μM of unlabeled competitor RNA (if applicable), and 0.5 nM final concentration of the biotinylated RNA probe (added last). Reactions were incubated at room temperature for 20 minutes, at which time 5 μl of loading dye was added, and 20 μl was electrophoresed on Bio-Rad’s 5% Mini-PROTEAN® TBE Gel, 10 well, 30 ml (Cat number #4565013). RNA and protein/RNA complexes were transferred to GE Healthcare Amersham Hybond -N+ Membrane in 0.5x TBE at 400 mA for 30 minutes in 0.5X TBE on Bio-Rad’s Trans Blot Turbo Transfer System. Detection was performed as described, using Bio-Rad’s ChemiDoc XRS+ System. At least 2 replicate EMSAs were performed for each RNA probe; one representative image is shown. MEME analysis (http://meme-suite.org/) was performed using the normal motif discovery mode, searching for any number of motif repetitions using 0-order background model of sequences within the input sequences WT-41, WT-28, WT-20, WT-17, MUT R, MUT L, MUT PT, MUT I, HOXA11-AS.
Surface Plasmon Resonance
SPR experiments were performed on a Biacore 3000. Full length AR protein (Abcam) was dialyzed against 10 mM NaOAc pH 5 and subsequently coupled to a CM5 chip (GE) using EDC/NHS coupling. The system was equilibrated to running buffer (10mM HEPESpH 7.3,20 mM KCl, 1 mM MgCl2,1 mM DTT) and RNA(IDT) was subsequently injected (30uL/minflow rate, one minute injection followed by 5–15 minutes dissociation). Measurements for each RNA concentration were collected across three independent experiments, and fit to a 1:1 Langmuir model. All measurements made are within the physical limits of the instrument.
Cell-Based Assays
Lipofectamine® 2000 (Life Technologies) was used for all plasmid transfections. RIP assays were performed as previously published, using HEK293T cells transfected with 10 μg of FLAG-AR expressing plasmid, and 10 μg of lncRNA-expressing plasmid (Schmidt et al., 2016). Immunoprecipitation of FLAG-tagged AR was performed as previously published (Schmidt et al., 2017). For invasion assays, A375 cells were seeded at 25 × 104 cells in a 6-well plate and transfected with 2,500ng of the indicated plasmid 24 hours later. For assays using FANA-modified oligos, media was changed 7–8 hours post-transfection to media containing 1 μM final concentration of the indicated oligo. Approximately 28 hours post-transfection, 2.5 × 104 cells in serum-free media were plated in either BD BioCoat matrigel inserts or uncoated control inserts (Corning), placed into DMEM with 30% FBS (fetal bovine serum), and incubated for 16 hours. Cells that did not migrate or invade were removed using a cotton tipped swap, chambers were rinsed twice with PBS, and stained using Fisher HealthCare PROTOCOL Hema 3 Fixative and Solutions. Cells were imaged on 20x magnification in 8 fields of view for 3 independent replicates.
RNA Extraction and Quantification
RNA was isolated using QIAGEN RNeasy® Mini Kit and treated with DNase. cDNA was generated using Bio-Rad’s iScript™ cDNA synthesis kit and quantified as previously described (Schmidt et al., 2016). For tissue specific expression analysis, cDNA was generated from Human Total RNA Master Panel II (Clontech). AR mRNA expression was taken from the Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS (GTEX Consortium, 2015). The GTEx data used for the analyses described in this manuscript were obtained from The Human Protein Atlas available at www.proteinatlas.org (Uhlén et al., 2015).
Denatured Control RNA SHAPE Probing
WM1976 cells were grown in 6-well plates (2 wells per experiment) to ~80% confluency. Media was aspirated, cells were washed once in 1 mL PBS, and total RNA was extracted using 1 mL TRIzol reagent (Thermo Fisher) per well. Resulting total RNA precipitate was resuspended in 30 μL of nuclease-free water. Total RNA (15 μL) was combined with 5 μL of 10x denaturing buffer [500 mM HEPES pH 8,40 mM EDTA] and 25 μL of formamide, and the mixture was incubated at 95°Cfor 1 minute. 5 mL of 10x SHAPE reagent [250 mM 5-nitroisatoic anhydride (5NIA)] was added to the denatured RNA and incubation at 95°C continued for 1 min. Denatured modified RNA was purified using a 1.8x ratio of Agencourt RNA Clean XP beads (Beckman Coulter) and eluted in 88 μL of nuclease-free water.
In-Cell SHAPE Probing
WM1976 cells were grown in 6-well plates (4 wells per experiment) to ~80% confluency. Cells were washed once in PBS before adding to each well 900 μL of PBS. To two wells, 100 μL of 10x SHAPE reagent [250 mM 5-nitroisatoic anhydride (5NIA)] was added while 100 μL of neat DMSO were added to each of the remaining two wells. Plates were mixed concurrently with reagent addition and then incubated at 37°C for 10 minutes. Media was aspirated, cells were washed once in 1 mL PBS per well, and total RNA was extracted using 1 mL TRIzol reagent (Thermo Fisher) per well. Resulting pellets were resuspended in 44 μL of water and like wells were combined into 88 μL volumes.
In-Cell DMS Probing
WM1976 cells were grown in 6-well plates (4 wells per experiment) to ~80% confluency. Cells were washed once in PBS before adding to each well 900 μL of DMS Folding Buffer [300 mM Bicine pH 8,100 mM NaCl, 5 mM MgCl2]. To two wells, 100 μL of 10x DMS reagent [1:5 dimethyl sulfate (DMS): ethanol] was added while 100 μL of neat ethanol were added to each of the remaining two wells. Plates were mixed concurrently with reagent addition and then incubated at 37°C for 6 minutes. DMS was quenched with 200 μL of beta-mercaptoethanol and cells were placed on ice for 5 minutes. Excess buffer was removed, cells were washed once in 1 mL PBS per well, and total RNA was extracted using 1 mL TRIzol reagent (Thermo Fisher) per well. Resulting pellets were resuspended in 44 μL of water and like wells were combined into 88 μL volumes.
Cell-Free RNA Extraction and Protein Digestion
WM1976 cells were grown in two 10 cm dishes to ~80% confluency. Both plates were washed once in 5 mL of PBS before scraping and lysis in 2.5 mL of ice-cold cytoplasmic lysis buffer [40 mM Tris pH 8,175 mM NaCI, 6 mM MgCl2,1 mM CaCl2, 256 mM Sucrose, 0.5% Triton X-100, 0.5 Units/μL RNasin (Promega), 0.45 Units/μl DNase I (Roche)]. Cells were lysed for 5 minutes on ice with intermittent mixing. Cell nuclei were pelleted at 3000 xg for 5 minutes at 4°C (nuclear fraction), and the resulting supernatant (cytoplasmic fraction) was transferred to a new tube. A volume of 2.5 mL of proteinase K buffer [40 mM Tris pH 8, 200 mM NaCl, 1.5% Sodium Dodecyl Sulfate, and 0.5 mg/mL Proteinase K] was added to the nuclear pellets. The supernatant (cytoplasmic fraction) volume was increased to 5 mL to include a final concentration of 200 mM NaCl, 1.5% Sodium Dodecyl Sulfate, and 0.5 mg/mL Proteinase K (Thermo Fisher). Proteins were digested for 45 minutes at 23°C with intermittent mixing.
RNA Separation and Buffer Exchange
Nucleic acids were extracted twice with 1 volume of Phenol Chloroform Isoamyl Alcohol (25:24:1) that was pre-equilibrated with either 1.1x SHAPE Folding Buffer [110 mM HEPES pH 8, 110 mM NaCl, 5.5 mM MgCl2] or 1.1x DMS Folding Buffer [330 mM Bicine pH 8,110 mM NaCl, 5.5 mM MgCl2]. Excess Phenol was removed through two subsequent 1 volume chloroform extractions. The final aqueous layer was buffer exchanged into 1.1x SHAPE Folding Buffer or 1.1x DMS Folding Buffer using PD-10 Desalting Columns (GE Healthcare Life Sciences). The resulting RNA solution (3.5 mL nuclear/7 mL cytoplasmic) was incubated at 37°C for 20 minutes before being split into two equal volumes for control and probe treatment samples.
5NIA SHAPE Probing of Extracted RNA
A 1/10th volume of 10x SHAPE reagent [250 mM 5-nitroisatoic anhydride (5NIA)] in DMSO was added to one sample of RNA while neat DMSO was added to the other. Samples were incubated at 37°C for 10 minutes.
DMS Probing of Extracted RNA
A 1/10 volume of 10x DMS Solution [1:5 Dimethyl Sulfate (DMS):Ethanol] was added to one sample of RNA while neat ethanol was added to the other. Samples were incubated at 37°C for 6 minutes. DMS was quenched with 1/5 volume of beta-mercaptoethanol and immediately placed on ice for 5 minutes.
Precipitation of Probe-Reacted RNA
Cell-free RNA was precipitated with 1/10 volume of 2 M NH4OAc and 1 volume of isopropanol. RNA was pelleted at 10,000 x g for 10 minutes at 4°C. After 1 wash in 75% ethanol, the resulting pellet was dried and then resuspended in 88 μL of water.
DNase Treatment of Cell-Derived RNAs
To 88 μL of in-cell or cell-free RNA samples, 10 μL of 10x TURBO DNase buffer and 4 Units of TURBO DNase (Thermo Fisher) were added and the mixture was incubated at 37°C for 1 hour. Following this incubation, 2 more Units of TURBO DNase were added and incubation continued at 37°C for another 1 hour. RNA was purified using a 1.8x ratio of Agencourt RNAClean XP magnetic beads (Beckman Coulter) and eluted into 20–30 μL of nuclease-free water.
SLNCR MaP
From each sample of RNA from WM1976 in-cell and cell-free probing experiments, 1–3 μg were subjected to mutational profiling (MaP) reverse transcription (Siegfried et al., 2014; Smola et al., 2015b) using a primer specific to SLNCR. The cDNA generated was buffer exchanged over Illustra microspin G-50 columns (GE Healthcare). Output cDNA (5 μL) was used as a template for 50 μL PCR reactions (Q5 Hot-start polymerase, NEB) with primers made to amplify nt 403–780 of SLNCR1 and add adaptor sequences [1xQ5 reaction buffer, 250 nM each primer, 200 μM dNTPs, 3% DMSO, 0.02 Units/μLQ5 Hot-start polymerase]. PCR proceeded in a touchdown format: 98°C for 2 minutes, 20 cycles of [98°C for 10 s, 68°C (decreasing by 1°C each cycle until reaching 63°C) for 30 s, 72°C 30 s], and 72°C for 2 min. Step 1 PCR products were purified using a 0.8x ratio of Agencourt AMPure XP beads (Beckman Coulter) and eluted in 20 μL of nuclease-free water. Purified PCR products (3 ng) were used as a template in 50 μL of PCR meant to add multiplex indices and remaining sequence necessary for Illumina sequencing [1xQ5 reaction buffer, 500 nM each index primer, 200 μM dNTPs, 3% DMSO, 0.02 Units/μLQ5 Hot-start polymerase]. Step 2 PCR proceeded as follows: 98°C for 2 minutes, 10 cycles of [98°Cfor 10 s, 66°C for 30 s, 72°C 20 s], and 72°Cfor 2 min. Step 2 PCR products were purified using a 0.8x ratio of Agencourt AMPure XP beads (Beckman Coulter) and eluted in 20 μL of nuclease-free water.
U1 snRNA MaP
The procedure for U1 snRNA MaP RT was similar to that of SLNCR MaP with minor modifications: (1) U1-specific primers were used for RT and step 1 PCR, (2) During step 1 PCR, the touchdown protocol began with an annealing temperature of 72°C and only decreased to 64°C, and (3) Step 1 PCR included 500 nM of each primer, no DMSO, and only 25 μL of total volume.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism version 7.01 for Windows (GraphPad Software, La Jolla California USA). For invasion assays, error bars represent the mean ± SD of 3 technical replicates, imaging 8 fields of view per replicate. RT-qPCR data is represented as the fold change compared to scramble control, normalized to GAPDH. Error bars represent standard deviations calculated from 3 reactions. Significance for RNA quantification were calculated using the two-tailed Student’s t test.
Sequencing and Profile Generation
Purified SLNCR amplicons were pooled and sequenced on an Illumina Miseq instrument, outputting 2 × 300 paired end datasets. For U1 libraries, 2× 150 paired end datasets were generated instead. Reverse transcription of SLNCR yielded three products, corresponding to 3 alternatively spliced forms of SLNCR (SLNCR1: 2257 nt, SLNCR2: 2522 nt, newly observed isoform SLNCR4: 2160 nt). Data from these isoforms were separated before alignment by fragment lengths after merging paired reads. SLNCR1-specific merged reads were then aligned to the SLNCR1/LINC00673 sequence (NCBI Reference NR_036488.1) using the ShapeMapper2 software (Busan and Weeks, 2017; Smola et al., 2015b) with the command shapemapper-target [TARGET].fa-modified-U [MERGED-TREATED-READS].fastq-untreated-U [MERGED-UNTREATED-READS].fastq-denatured-U [MERGED-DENATURED-READS].fastq. When aligning DMS reads, the denatured control was excluded. U1 reads were similarly aligned to the U1 snRNA sequence (NCBI Reference NR_004430.2). SHAPE reactivities are first calculated as a combination of multiple measured mutation rates [(SHAPE treated - untreated)/denatured], and these values are then divided by a normalization factor. Normalization factors are derived via the boxplot method (Hajdin et al., 2013), where reactivities above the larger of the 90th percentile and 1.5 x the interquartile range beyond the 75th percentile are first removed as outliers and the top 10% of remaining reactivities are averaged (the normalization factor). The error of each mutation rate measurement is estimated as , and then propagated through the combination of rates and normalization to assign a standard error to a SHAPE reactivity.
Discovery of In-Cell SHAPE Protections with ΔSHAPE
In-cell SHAPE and DMS protection sites were identified through comparison of in-cell and cell-free reactivities using the ΔSHAPE program with default settings (Smola et al., 2015a, 2016). In-cell data were scaled such that the 95th percentile reactivity matched the average of cell-free 95th percentile reactivities. Significant nt protections were identified in both nuclear and cytoplasmic fractions with the criteria of being centered in window of 5 nt that contains 3 nt with positive smoothed (average of 3 nt window) ΔSHAPE values (cell-free minus in-cell reactivities), positive Z-factors, and standard scores above 1. Additional regions with smoothed ΔSHAPE values above 1 and Z-factors above 0.5 were also considered to account for skewing of Z-score by highly reactive sites. To achieve positive Z-factors, the absolute difference of smoothed in-cell and cell-free reactivities must be greater that 2 times the sum of their smoothed errors (smoothed in quadrature), while a standard score of 1 is achieved when a ΔSHAPE value is more than 1 standard deviation from the mean.
RNA Structure Modeling with SuperFold
RNA partition functions, informed by SHAPE reactivities, were calculated to generate base-pairing probabilities and Shannon entropy (the negative sum of all probabilities multiplied by the natural log of those probabilities) using the SuperFold analysis pipeline (Smola et al., 2015b). For Figure 5, SLNCR1403–780 data were scaled to the U1 normalization factor. The original SLNCR1 data were divided by this U1 normalization factor to obtain U1-scaled reactivity profiles. Low SHAPE, low entropy regions were identified by two methods: [1] as nts where the 50 nt window-median SHAPE and entropy values fall below 0.4 and 0.03, respectively (Figure 5), and [2] nts where the 50 nt window-median SHAPE and entropy values fall below the global medians (Figure S6). Regions separated by 5 or fewer nts were combined and only regions spanning at least 30 nts were retained before expanding to include overlapping base-pairs. RNA secondary structure representations of SLNCR1 were generated from ShapeMapper and SuperFold outputs using VARNA (Darty et al., 2009).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat# F3165, RRID:AB_259529 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| recombinant DDDK-AR protein | abcam | Cat#ab82609 |
| recombinant N-terminal AR-6xHIS protein | RayBiotech | Cat#RB-14-0003P |
| 5-nitroisatoic anhydride | Millipore Sigma | Cat#S428396 CAS#4693-02-1 |
| Dimethyl Sulfate | Millipore Sigma | Cat#D186309 CAS#77-78-1 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596026 |
| Recombinant RNasin Ribonuclease Inhibitor | Promega | Cat#N2511 |
| Recombinant DNase I, RNase-free | Roche | Cat#04716728001 |
| TURBO DNase | Thermo Fisher Scientific | Cat#AM2238 |
| SuperScript II Reverse Transcriptase | Thermo Fisher Scientific | Cat#18064014 |
| Q5 Hot-start high Fidelty DNA Polymerase | New England Biolabs | Cat#M0493 |
| Proteinase K, Lyophilized | Millipore Sigma | Cat#70663 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat#11668019 |
| DMEM, high glucose, no glutamine | Invitrogen | cat# 11960044 |
| GIBCO FBS | Thermo Fisher Scientific | cat# 26140 |
| Critical Commercial Assays | ||
| PD-10 Desalting Columns | GE Healthcare Life Sciences | Cat#17-0851-01 |
| Agencourt RNA Clean XP kit | Beckman Coulter | Cat#A63987 |
| Agencourt AMPure XP kit | Beckman Coulter | Cat#A63881 |
| Illustra Micro-Spin G50 Columns | GE Healthcare Life Sciences | Cat#27533001 |
| LightShift Chemiluminescent RNA EMSA (REMSA) Kit | Thermo Fisher Scientific | Cat#20158 |
| iScript™ cDNA synthesis kit | BioRad | Cat#1708890 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| Human Total RNA Master Panel II | Clontech | Cat#636643 |
| Fisher HealthCare PROTOCOL Hema 3 Fixative and Solutions | Fisher Scientific | Cat#23-123869 |
| Deposited Data | ||
| The Human Protein Atlas | Uhlén etal., 2015 | www.proteinatlas.org |
| Sequencing Reads and MaP Profiles | Generated in this study | GEO Accession#GSE140048 |
| Experimental Models: Cell Lines | ||
| A375 | American Type Culture Collection | cat# ATCC CRL-1619; RRID:CVCL_0132 |
| WM1976 human melanoma cell line | Wistar Institute | WM1976 |
| HEK293T/17 | American Type Culture Collection | ATCC Cat# CRL-11268, RRID:CVCL_1926 |
| Oligonucleotides | ||
| All Oligonucleotides | See Table S1, this study | N/A |
| Recombinant DNA | ||
| pEGFP-C1-AR | Stenoien et al., 1999; Addgene | Cat#28235 |
| pCDNA3.1(−)+SLNCR1 | Schmidt et al., 2016; Addgene | Cat#86816 |
| Software and Algorithms | ||
| ShapeMapper2 | RNA. 2018 Feb;24(2):143–148. https://doi.org/10.1261/rna.061945.117 | https://github.com/Weeks-UNC/shapemapper2 |
| ΔSHAPE | Biochemistry. 2015 Nov 24;54(46):6867 75. https://doi.org/10.1021/acs.biochem.5b00977 | http://weeks.chem.unc.edu/software.html |
| SuperFold | Nat Protoc. 2015 Nov;10(11):1643–69. https://doi.org/10.1038/nprot.2015.103 | http://weeks.chem.unc.edu/software.html |
| MEME Suite | Bailey et al., 2009 | http://meme-suite.org |
| GraphPad Prism version 7.01 for Windows | GraphPad Software | https://www.graphpad.com/ |
| Other | ||
| 5% Mini-PROTEAN® TBE Gel, 10 well, 30 μl | BioRad | Cat#456501S |
| Series S Sensor Chip CM5, pack of 1 | GE Healthcare | Cat#29104988 |
| Corning BioCoat Matrigel Invasion Chamber with Corning Matrigel Matrix | Fisher Scientific | Cat#S54480 |
Highlights.
The N-terminal region of AR binds unstructured RNA in a sequence-specific manner
An unstructured lncRNA region scaffolds an invasion-promoting transcription complex
An identified AR RNA-binding motif is predictive of other lncRNA-AR interactions
Sterically blocking the SLNCR-AR interaction blocks melanoma invasion
ACKNOWLEDGMENTS
We gratefully acknowledge the members of our laboratories for technical advice and critical discussions. We thank Veenu Aishwarya and AUM Technologies for providing 2′-FANA modified oligonucleotides, and Meenhard Herlyn for MSTCs. This work was supported by funding from NIH R01 CA140986 and R01 CA185151 and a Claudia Adams Barr Award (to C.D.N.); by NIH R35 GM122532 (to K.M.W.); and by the Intramural Research Program of the NIH, Center for Cancer Research, the National Cancer Institute (NCI), NIH 1 ZIA BC011585 (to J.S.S.). K.S. was supported by funding from T32 AI007386; C.A.W. was supported by funding from T32 CA009156 through June 2017 and is currently a Postdoctoral Fellow of the American Cancer Society (130845-RSG-17–114-01-RMC).
Footnotes
DATA AND CODE AVAILABILITY
ShapeMapper 2, ΔSHAPE, and SuperFold software is freely available on the Weeks laboratory website (weeks.chem.unc.edu/soft-ware.html). The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE140048.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.011.
DECLARATION OF INTERESTS
K.S. and E.Y. are current employees of Alnylam Pharmaceuticals. K.S. and C.D.N. are inventors of two patents based on work presented in this manuscript (PCT/US2016/041343 and PCT/US2018/050597). K.M.W. is an advisor to and holds equity in Ribometrix, to which mutational profiling (MaP) technologies have been licensed.
REFERENCES
- Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, et al. (2018). Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 379, 11–21. [DOI] [PubMed] [Google Scholar]
- Agoulnik IU, and Weigel NL (2009). Coactivator selective regulation of androgen receptor activity. Steroids 74, 669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba MC, Long H, Cui SZ, Gong YF, Yan ZF, Wu YB, and Tu YN (2017). Long noncoding RNA LINC00673 epigenetically suppresses KLF4 by interacting with EZH2 and DNMT1 in gastric cancer. Oncotarget 8, 95542–95553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busan S, and Weeks KM (2017). Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with ShapeMapper 2. RNA 24, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busan S, Weidmann CA, Sengupta A, and Weeks KM (2019). Guidelines for SHAPE Reagent Choice and Detection Strategy for RNA Structure Probing Studies. Biochemistry 58, 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerase A, Pintacuda G, Tattermusch A, and Avner P (2015). Xist localization and function: new insights from multiple levels. Genome Biol. 16, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Lee SO, Yeh S, and Chang TM (2014). Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene 33, 3225–3234. [DOI] [PubMed] [Google Scholar]
- Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, and Lawrence JB (2009). An architectural role for a nuclear non-coding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 33, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly CM, Moon MH, and Schneekloth JS Jr. (2016). The Emerging Role of RNA as a Therapeutic Target for Small Molecules. Cell Chem. Biol. 23, 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K, Denise A, and Ponty Y (2009). VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 25, 1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Zhang L, Guo J, Niu Y, Wu Y, Li H, Zhao L, Li X, Teng X, Sun X, et al. (2018). NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 46 (D1), D308–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari N, Bergeron D, Tedeschi AL, Mangos MM, Paquet L, Renzi PM, and Damha MJ (2006). Characterization of antisense oligonucleotides comprising 2′-deoxy-2′-fluoro-beta-D-arabinonucleicacid (FANA): specificity, potency, and duration of activity. Ann. N Y Acad. Sci. 1082, 91–102. [DOI] [PubMed] [Google Scholar]
- GTEx Consortium (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, and Rinn JL (2012). Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, and Weeks KM (2013). Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl. Acad. Sci. USA 110, 5498–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DM, and Brooks SA (2014). In vitro invasion assay using matrigel™: a reconstituted basement membrane preparation. Methods Mol. Biol. 1070, 1–11. [DOI] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homan PJ, Favorov OV, Lavender CA, Kursun O, Ge X, Busan S, Dokholyan NV, and Weeks KM (2014). Single-molecule correlated chemical probing of RNA. Proc. Natl. Acad. Sci. USA 111, 13858–13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Hou J, Wang Y, Xie M, Wei C, Nie F, Wang Z, and Sun M (2017). Long Noncoding RNA LINC00673 Is Activated by SP1 and Exerts Oncogenic Properties by Interacting with LSD1 and EZH2 in Gastric Cancer. Mol.Ther. 25, 1014–1026. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Juliano RL (2016). The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44, 6518–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Jiang X, and Yokosuka O (2014). Androgen receptor signaling in hepatocellular carcinoma and pancreatic cancers. World J. Gastroenterol. 20, 9229–9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Oubridge C, van Roon AM, and Nagai K (2015). Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. eLife 4, e04986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, and O’Malley BW (1999). A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 97, 17–27. [DOI] [PubMed] [Google Scholar]
- Lee S, Kopp F, Chang TC, Sataluri A, Chen B, Sivakumar S, Yu H, Xie Y, and Mendell JT (2016). Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 164, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y, Wang X, Youmans DT, and Cech TR (2017). How do lncRNAs regulate transcription? Sci. Adv. 3, eaao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin KE, Gissberg O, and Smith CI (2015). Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther. 26, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Wu G, Zhu Q, Liu H, Yao Y, Yuan D, Liu Y, Lv T, and Song Y (2017). Long intergenic noncoding RNA 00673 promotes non-small-cell lung cancer metastasis by binding with EZH2 and causing epigenetic silencing of HOXA5. Oncotarget 8, 32696–32705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morvillo V, Luthy IA, Bravo AI, Capurro MI, Donaldson M, Quintans C, Calandra RS, and Mordoh J (1995). Atypical androgen receptor in the human melanoma cell line IIB-MEL-J. Pigment Cell Res. 8, 135–141. [DOI] [PubMed] [Google Scholar]
- Morvillo V, Lüthy IA, Bravo AI, Capurro MI, Portela P, Calandra RS, and Mordoh J (2002). Androgen receptors in human melanoma cell lines IIB-MEL-LES and IIB-MEL-IAN and in human melanoma metastases. Melanoma Res. 12, 529–538. [DOI] [PubMed] [Google Scholar]
- Novikova IV, Hennelly SP, and Sanbonmatsu KY (2012). Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 40, 5034–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottesen EW (2017). ISS-N1 makes the First FDA-approved Drug for Spinal Muscular Atrophy. Transl. Neurosci. 8, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeranz Krummel DA, Oubridge C, Leung AK, Li J, and Nagai K (2009). Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature 458, 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner JR, Sahu A, Iyer MK, Malik R, Chandler B, Asangani IA, Poliakov A, Vergara IA, Alshalalfa M, Jenkins RB, et al. (2014). The IncRNAs PCGEM1 and PRNCR1 are not implicated in castration resistant prostate cancer. Oncotarget 5, 1434–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro DM, Zanzoni A, Cipriano A, Delli Ponti R, Spinelli L, Ballarino M, Bozzoni I, Tartaglia GG, and Brun C (2018). Protein complex scaf-folding predicted as a prevalent function of long non-coding RNAs. Nucleic Acids Res. 46,917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C, Pedersen L, and Mouridsen HT (1985). Endocrine treatment with antiestrogen, anti-androgen or progestagen of advanced malignant melanoma: three consecutive phase II trials. Eur. J. Cancer Clin. Oncol. 21, 1171–1174. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Joyce CE, Buquicchio F, Brown A, Ritz J, Distel RJ, Yoon CH, and Novina CD (2016). The lncRNA SLNCR1 Mediates Melanoma Invasion through a Conserved SRA1-like Region. Cell Rep. 15, 2025–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Buquicchio F, Carroll JS, Distel RJ, and Novina CD (2017). RATA: A method for high-throughput identification of RNA bound transcription factors. J. Biol. Methods 4, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Carroll JS, Yee E, Thomas DD, Wert-Lamas L, Neier SC, Sheynkman G, Ritz J, and Novina CD (2019). The lncRNA SLNCR Recruits the Androgen Receptor to EGR1-Bound Genes in Melanoma and Inhibits Expression of Tumor Suppressor p21. Cell Rep. 27, 2493–2507.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamovsky I, Ivannikov M, Kandel ES, Gershon D, and Nudler E (2006). RNA-mediated response to heat shock in mammalian cells. Nature 440, 556–560. [DOI] [PubMed] [Google Scholar]
- Shi X, Ma C, Zhu Q, Yuan D, Sun M, Gu X, Wu G, Lv T, and Song Y (2016). Upregulation of long intergenic noncoding RNA 00673 promotes tumor proliferation via LSD1 interaction and repression of NCALD in non-small-cell lung cancer. Oncotarget 7, 25558–25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried NA, Busan S, Rice GM, Nelson JA, and Weeks KM (2014). RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 11, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, and Weeks KM (2018). In-cell RNA structure probing with SHAPE-MaP. Nat. Protoc. 13, 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Calabrese JM, and Weeks KM (2015a). Detection of RNA-Protein Interactions in Living Cells with SHAPE. Biochemistry 54, 6867–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Rice GM, Busan S, Siegfried NA, and Weeks KM (2015b). Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 10, 1643–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Christy TW, Inoue K, Nicholson CO, Friedersdorf M, Keene JD, Lee DM, Calabrese JM, and Weeks KM (2016). SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc. Natl. Acad. Sci. USA 113, 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley JA, Aruldhas MM, Chandrasekaran M, Neelamohan R, Suthagar E, Annapoorna K, Sharmila S, Jayakumar J, Jayaraman G, Srinivasan N, and Banu SK (2012). Androgen receptor expression in human thyroid cancer tissues: a potential mechanism underlying the gender bias in the incidence of thyroid cancers. J. Steroid Biochem. Mol. Biol. 130, 105–124. [DOI] [PubMed] [Google Scholar]
- Stein CA, and Castanotto D (2017). FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 25, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, De-Martino GN, Marcelli M, Weigel NL, and Mancini MA (1999). Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum. Mol. Genet. 5, 731–741. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, and Kurokawa R (2008). Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZZ, and Mathews DH (2016). Secondary Structure Prediction of Single Sequences Using RNAstructure. Methods Mol. Biol. 1490, 15–34. [DOI] [PubMed] [Google Scholar]
- Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, Merkurjev D, Ohgi KA, Meng D, Zhang J, et al. (2013). lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 500, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Zhao JC, Kim J, Fong KW, Yang YA, Chakravarti D, Mo YY, and Yu J (2015). LncRNA HOTAIR Enhances the Androgen-Receptor-Mediated Transcriptional Program and Drives Castration-Resistant Prostate Cancer. Cell Rep. 13, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.