

Abstract
Pulmonary arterial hypertension (PAH) is considered a disease of the pulmonary vasculature. Limited progress has been made in preventing or arresting progression of PAH despite extensive efforts. Our previous studies indicated that PAH could be considered a systemic disease since its pathology involves interplay of multiple organs. This, coupled with increasing implication of the gut and its microbiome in chronic diseases, led us to hypothesize that PAH patients exhibit a distinct gut microbiome that contributes to, and predicts, the disease. Fecal microbiome of 18 type 1 PAH patients (mean pulmonary arterial pressure [mPAP] 57.4, standard deviation 16.7 mmHg) and 13 reference subjects (REF) were compared by shotgun metagenomics to evaluate this hypothesis. Significant taxonomic and functional changes in microbial communities in the PAH cohort were observed. Pathways for the synthesis of arginine, proline and ornithine were increased in PAH cohort compared with REF cohort. Additionally, groups of bacterial communities associated with trimethylamine/ trimethylamine N-oxide and purine metabolism, were increased in PAH cohort. In contrast, butyrate-and propionate-producing bacteria such as Coprococcus, Butyrivibrio, Lachnospiraceae, Eubacterium, Akkermansia and Bacteroides were increased in REF cohort. A random forest model predicted PAH from the composition of the gut microbiome with 83% accuracy. Finally, virome analysis showed enrichment of Enterococcal and relative depletion of Lactococcal phages in the PAH cohort. In conclusion, PAH patients exhibit a unique microbiome profile that has high predictive potential for PAH. This highlights previously unknown roles of gut bacteria in this disease and could lead to new therapeutic, diagnostic or management paradigms for PAH.
Keywords: Pulmonary arterial hypertension, Gut microbiome, Virome, Metagenomics
Graphical Abstract
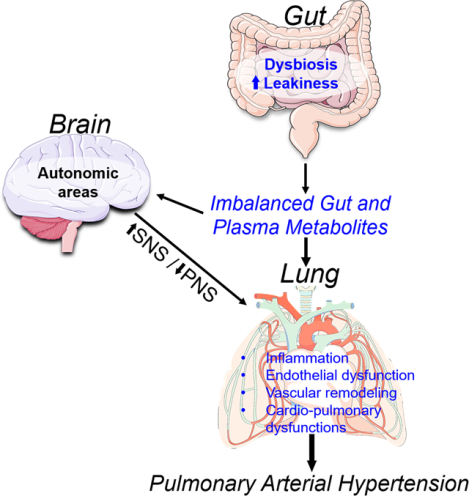
Summary
Despite evidence for the involvement of gut microbiota in many chronic diseases its contribution in PAH remains largely unknown. This study fills this gap in knowledge and demonstrates altered gut microbiota with prediction accuracy of 83% in PAH patients. The data suggest that uniquely altered microbiota may play a role pulmonary pathophysiology and thus presents novel target for both PAH diagnosis and therapy.
Introduction
Pulmonary arterial hypertension (PAH) is a chronic and progressive cardiopulmonary disease that leads to irreversible right heart failure, the primary cause of morbidity and mortality. Current therapeutic strategies target the pulmonary vasculature as vascular stiffening from endothelial dysfunction and fibrosis of pulmonary arteries are the major pathologies of PAH.1 Despite recent advances in using combination drug therapy and identification of new vascular signaling pathways as targets, treatment of PAH remains a significant challenge. Thus, it is imperative to consider innovative concepts for the discovery of new targets for a successful PAH regimen.2
Although the primary pathologic site is the pulmonary vasculature, PAH has characteristics of a systemic disease involving many other organs such as the central nervous system, immune system, heart, bone marrow and kidney in addition to lung.3 Another potentially important, but unexplored organ in PAH is the gut and its microbiome as evidenced in various cardiopulmonary diseases.4–6
There are remarkable similarities between lung and gut in relation to infection, immunity, epithelial barrier functions and microbiomes. For example; 1) both lung and gut are major organs directly in contact with the environment. Thus, risk factors for pulmonary disease such as smoking, toxins, infections, etc. may directly influence both organs. 2) Their epithelia serve as microbial barriers where mucin plays an important role. 3) Both have diverse microbiota. Although pulmonary bacterial populations are smaller in multitude than the gut’s, an imbalanced lung microbiome may contribute to respiratory infections and inflammation resulting in pulmonary diseases such as asthma, chronic obstructive pulmonary disease (COPD) or cystic fibrosis.7 However, there is a paucity of information about the potential role of gut microbial communities in pulmonary hypertension and nothing is known in PAH patients.
With the emerging role of the gut microbiome in health and disease, the gut-lung axis could offer an attractive alternative multi-organ pathway in the pathogenesis of PAH, as the gut and its microbiome play an important role in pulmonary vascular function. Thus, our objective in this study was to evaluate the hypothesis that patients with PAH have a unique gut microbiome profile that produces bacterial metabolites and molecules important in the pathogenesis of PAH.
Some results of these studies have been previously reported in abstract form.8
Methods
Data Availability Statement
Metadata and microbiome data (taxonomy and KO files) can be downloaded at https://drive.google.com/open?id=15NfMk7BAHo0Y_iyNlHnCNCE2Gg___v31
Patients
Eighteen type 1 PAH patients (mean pulmonary arterial pressure [mPAP] 57.4, standard deviation [SD] 16.7 mmHg) and 12 age- and sex-matched healthy reference (REF) subjects without a history of cardiopulmonary disease or PAH risk factors were recruited from Pulmonary Circulation Clinic, Clinicas Hospital of Porto Alegre, Brazil (Table 1). Their participation with informed consent was approved by the Institutional Scientific and Ethics Committee. Fecal samples were collected for comparison of gut microbial communities using shotgun metagenomics as described in the online supplement.
Table 1. Characteristics of patients with pulmonary arterial hypertension.
Sixteen subjects recruited for the reference (REF) cohort (13 women and 3 men) were aged 37 ± 11, BMI 22.7 ± 2.5 kg/m2 with no history of cardiopulmonary disease or PAH risk factors and with mPAP of 29. Age and BMI of the REF cohort were not different from those of the PAH cohort. Consecutive patients, with the appropriate clinical characteristics, were invited to participate in the studies. Among the recruited study subjects, four changed their mind and refused to enroll (all from the REF cohort), and one PAH subject did not return a sample after enrolling.
| Mean (SD) or Number (%) | |
|---|---|
| Age (years) | 39.5 (12.7) |
| Gender (male/female) | 3/16 |
| Race (white/other) | 18/1 |
| Weight (kg) | 65.2 (14.7) |
| Height (cm) | 160.7 (8.0) |
| Body mass index (BMI, kg/m2) | 25.3 (5.7) |
| PAH etiology (%) | |
| Idiopathic | 4 (21.1%) |
| Collagen vascular disease | 2 (10.5%) |
| HIV | 5 (26.3%) |
| Congenital heart disease with shunt | 7 (36.8%) |
| Portopulmonary hypertension | 1 (5.3%) |
| World Health Organization functional class (%) | |
| Class 1 | 9 (47.4%) |
| Class 2 | 5 (26.3%) |
| Class 3 | 3 (15.8%) |
| Class 4 | 2 (10.5%) |
| 6-minute walk distance (m) | 437 (89) |
| Echocardiographic data | |
| Transtricuspid gradient (mmHg) | 71 (27) |
| Systolic pulmonary arterial pressure (mmHg) | 76.4 (28.1) |
| Tricuspid annular plane systolic excursion (cm) | 17.5 (3.0) |
| Right ventricular diameter (M module, cm) | 3.4 (1.0) |
| Right heart catheterization | |
| Mean pulmonary arterial pressure (mmHg) | 57.4 (16.7) |
| Pulmonary artery occlusion pressure (mmHg) | 8.8 (4.3) |
| Cardiac output (L/minute) | 5.1 (2.0) |
| Pulmonary vascular resistance (Woods unit) | 11.5 (8.7) |
| Right Arterial Pressure (mmHg) | 8.8 (5.3) |
| Pulmonary arterial hypertension therapy | |
| None | 2 (10.5%) |
| Phosphodiesterese type 5 inhibitors | 16 (84.2%) |
| Endothelin antagonists | 7 (36.8%) |
| Prostanoids | 1 (5.3%) |
Taxonomic and Functional Profiling of Microbiome
Relative abundances of taxonomic profiles and functional pathways were multiplied by 1 million and formatted as described in Segata et al.9 CPM normalized KO data was converted to a QIIME formatted biom file to generate PLS-DA plots using the Bray-Curtis distance metric. CSS-normalized counts tables were separated into REF and PAH groups. These were aligned to Kyoto Encyclopedia of Gene and Genome (KEGG) pathways of interest using the R package, Pathview.10
Virus Analysis
Viral gene markers within filtered metagenome data were detected and annotated using the CLARK ((CLAssifier based on Reduced K-mers, version 1.2.3.1, http://clark.cs.ucr.edu/) annotation tool.11 Relative abundance estimates were formatted into a table and underwent LefSe and heatmap analysis within R using the “pheatmap” package. To affirm the accuracy of viral marker hits, the viromics tool Vipie was implemented for the assembly and annotation of viral genomes within the metagenome dataset.12
Full details of all experimental protocols are presented in Methods of the ‘Online Supplement’.
Results
PAH Patients Have Distinct Gut Microbiome Composition
Partial least squares discriminant analysis (PLS-DA) of the gut microbiome metagenomic data indicated differential taxonomic and functional gene (KEGG Orthology) profiles between PAH and REF cohorts (Figure 1A). Alpha diversity, the richness and evenness of bacteria were reduced in the gut microbiota of patients with PAH (Figure 1B–D). Differences in the bacterial taxa were detailed by linear discriminant analysis effective size (LEfSe), bar graphs and a bubble plot (Figure 1E, Figures S1 and S2). In addition, a network plot of the gut microbiota data showed that the bacterial genera within each cohort associated together but rarely with those in the other cohort resulting in two distinct clusters. The greater alpha diversity of the REF cohort was reflected by a cluster with a higher number of bacteria with more interactions than the PAH group. The differentially enriched bacterial taxa of each cohort are shown in Figure S3. Within the PAH cohort, there was enrichment of the Actinobacteria phylum, specifically the Bifidobacterium, a genus of acetate producers.13 More diverse taxa were enriched in the REF group, confirming the differences in alpha diversity. These included butyrate-producing bacteria such as Coprococcus, Butyrivibrio, Lachnospiraceae and Eubacterium13–15 and propionate-producing bacteria such as Akkermansia and Bacteroides16 (Figure 1E, 1F, Figure S3). These data demonstrate a distinct bacterial ecology in the gut microbiome of PAH patients compared to REF subjects.
Figure 1: Altered gut microbiota composition in type 1 pulmonary arterial hypertensive patients.
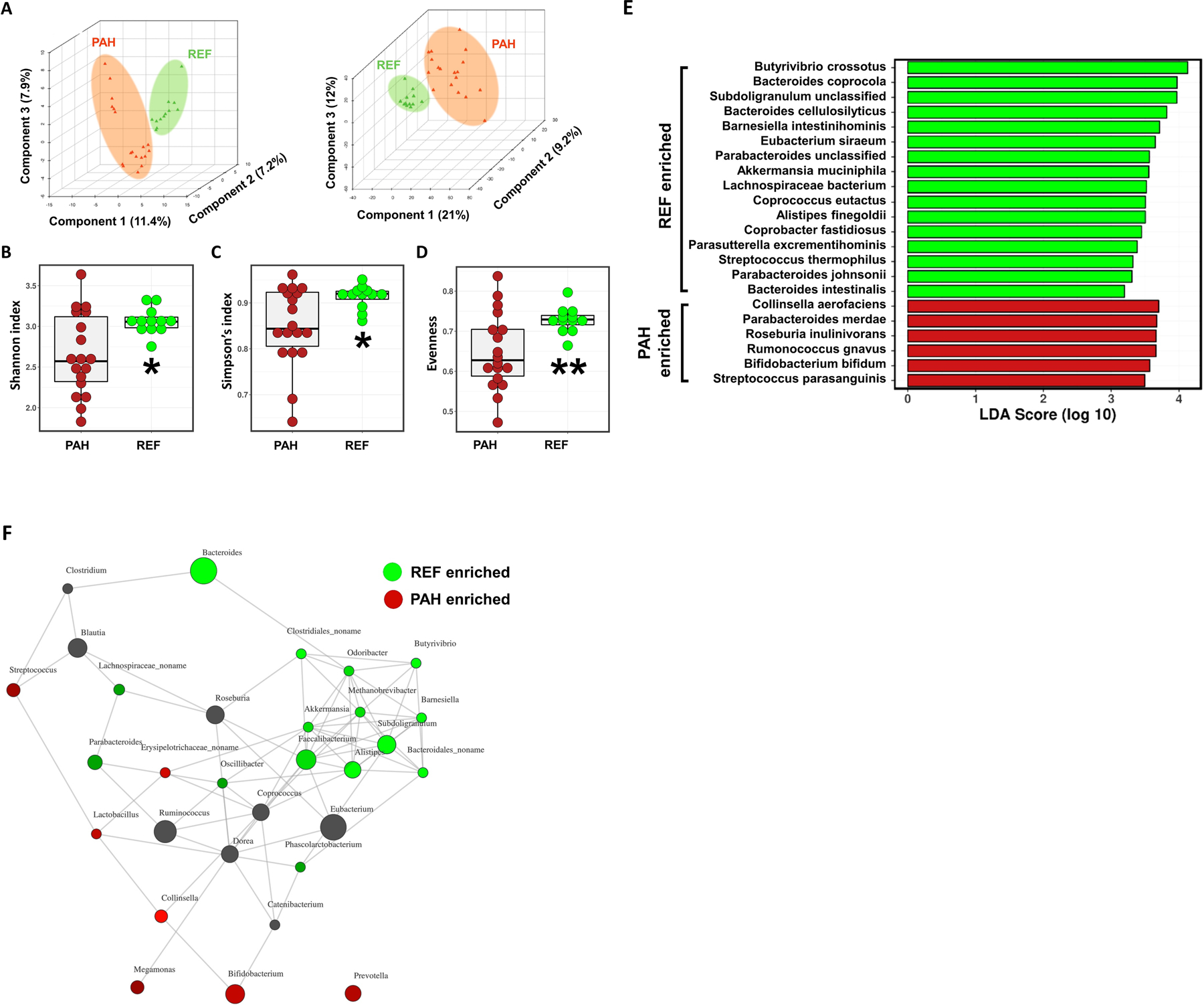
(A) Partial least squares discriminant analyses (PLS-DA) were conducted to compare the overall differences in taxonomy (left) and functional gene (KEGG Orthology) profiles (right) between the reference (REF) and pulmonary arterial hypertension (PAH) cohorts. (B-D) Alpha diversity measures showed significantly reduced Shannon and Simpson indices and evenness of fecal bacterial populations of PAH patients compared to age- and gender-matched healthy REF subjects. Student’s t-test was used to compare the means of the two groups. (E) Linear discriminant analysis effect size (LEfSe) of each cohort to visualize differences in bacterial species. (F) A network plot showing positive interactions (connected lines, distance determined from the Bray-Curtis method) between genera. Genera associated with the REF cohort were marked in green, genera enriched in PAH cohort in red and neutral genera in black in the nodes.
Prediction of PAH using Random Forest Modeling of Gut Microbiome
Having confirmed differential taxonomy and function of gut microbiota of the two cohorts, we attempted to identify bacteria most strongly correlated with PAH. A random forest machine-learning algorithm was applied to search for a highly fitting model discriminating the two cohorts (Figure 2). For each decision tree, a different training set was created with a random subset of samples and bacterial species, and its ability to subdivide the data into the cohorts evaluated. Please see the supplement for further description of the modeling. Our model contained 150 individual decision trees, ensembled by class designation. Thirty bacterial species with the highest Gini index predicted the presence or absence of PAH in subjects with 83% accuracy compared to an expected accuracy of ≤50% if there were no association of the gut microbiome with PAH. The relative abundance of the top 10 taxa was compared to their relative abundance in samples from patients with systemic hypertension.17 Most bacteria exhibited no similar differences in relative abundance between disease and no disease in these two conditions (Figure S4).
Figure 2: Random forest modeling identified a subset of taxa predictive of distinguishing REF (black) and PAH (red) cohorts.
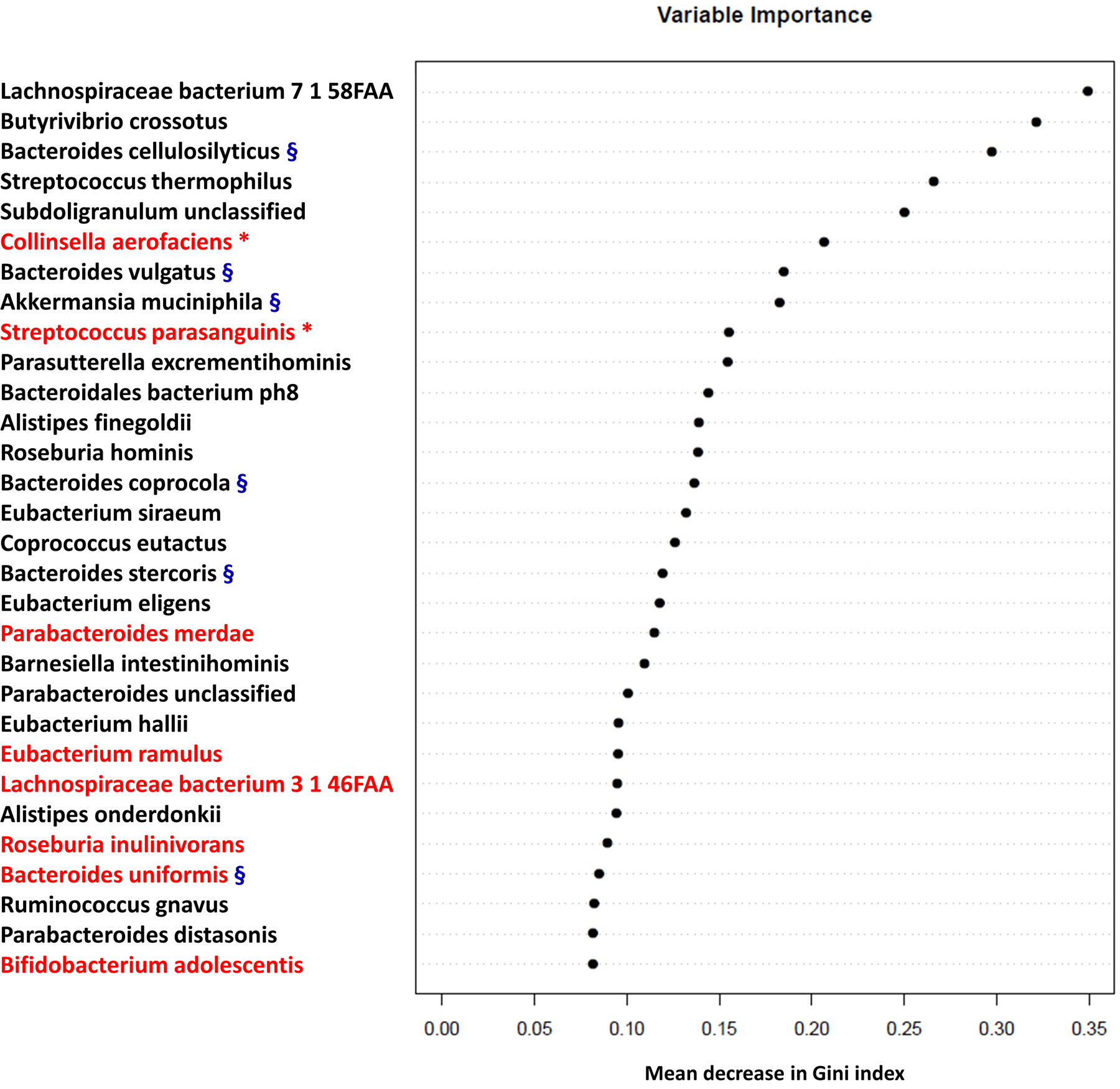
The 30 most important predictors of PAH vs. REF were ranked by Gini index determined from the random forest algorithm trained to distinguish the two cohorts. The taxa are ranked from top to bottom by decreasing Gini index scores. Since Gini index scores quantify the strength of each respective predictor, the best predictors of PAH are at the top of the plot. Bacteria with the red asterisk are positively correlated with TMA/TMAO production27. Bacteria with the blue § are negatively correlated with TMA/TMAO production28, 29.
PAH Gut Virome as a Potential Regulator of the Gut Ecosystem
We investigated the gut virome in the PAH population as viruses play an important regulatory role in the gut ecosystem.18 Viral gene marker profiles were different between PAH and REF cohorts by LEfSe analysis (Figure 3A). Multiple Lactococcal phages were enriched within the REF cohort, but Enterococcal phages were enriched within the PAH samples. The number of virus assemblies was also cohort-specific (Table S1). Phage infection of bacteria is highly specific,19 and is therefore essential in shaping bacterial population dynamics.20 We further confirmed that increased Lactococcal phages in the REF cohort were accompanied by increased Lactococcus lactis, a bacterium commonly used to test phage infection of Lactococcus (Figure 3B). Abundances of Lactococcal phages and Lactococcus within each subject were associated by both genus and species regardless of cohort (Figure 3C). Although the increase of Enterococcus in the PAH cohort was not statistically significant, enrichment of Enterococcus phage in PAH was associated with Enterococcus abundance (P=0.005), suggesting strong bacteria-phage interaction.
Figure 3: Significantly increased viral species and specific viruses in PAH (red) or REF (green) cohorts.
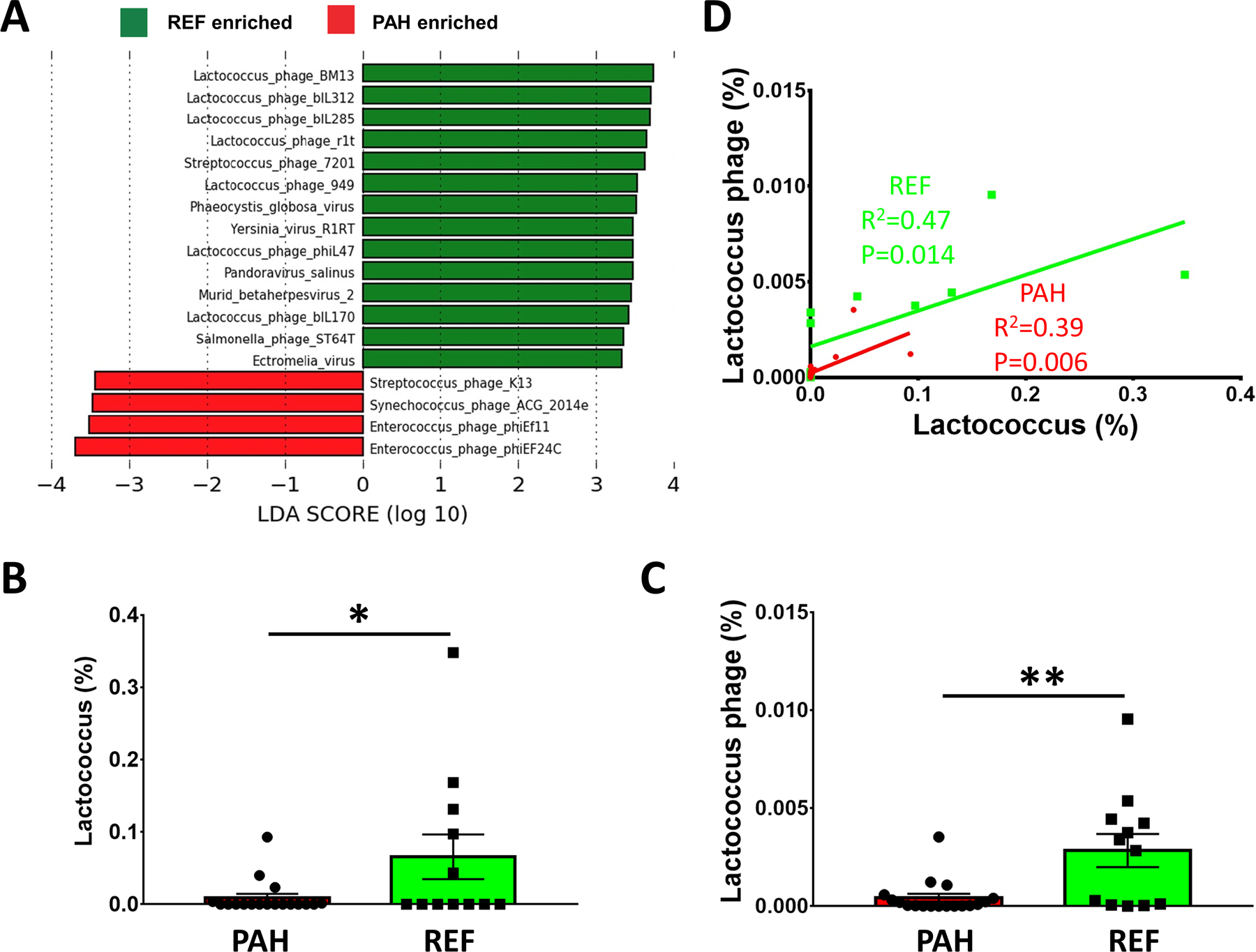
(A) LEfSe determined differential virus species in the gut microbiome. (B) Relative abundance of Lactococcus lactis in PAH and REF cohorts. (C) Relative abundance of Lactococcus phage. (D) A graph of Lactococcus phage and its bacterial host (Lactococcus) indicates interactions tend to occur among distinct groups of phages and bacteria. Out of 18 PAH samples, neither Lactococcus nor Lactococcus phage was detected in 4 samples (22% of total samples). Pearson correlation coefficient excluding those samples was comparable and it was statistically significant (R=0.59, P=0.033). Data are expressed as mean ± standard error of the mean (*p < 0.05; **p < 0.01).
Linkage of Functional Changes of Gut Microbiome with PAH
We investigated functional changes in the gut microbiomes by LEfSe analysis (Figure 4A). Pathways for the synthesis of several amino acids were enriched including arginine, proline, lysine, homoserine, methionine, ornithine and tryptophan. Elevated arginine catabolism by plasma arginase and a consequent decrease in arginine bioavailability was previously recognized as a hallmark of PAH pathogenesis.21–24 We found increased arginine, proline, ornithine biosynthesis and interconversion to be one of the main functional changes in the PAH microbiome (Figure 4B–D and Figure S5). Blautia and Bifidobacteria contributed most to arginine/ornithine biosynthesis in the PAH microbiome, while Collinsella was important for increased proline biosynthesis. The significant increase of ornithine transcarbamylase in PAH gut bacteria may have further contributed to decreased arginine bioavailability by metabolizing arginine to its major catabolic products, L-ornithine and L-citrulline. These data suggest the potential roles of enzymes and amino acids produced by the gut microbiota in PAH and cardiovascular disease risk (Figure 4F).4, 25
Figure 4: Functional changes of the gut microbiome and altered arginine metabolism in the PAH cohort.
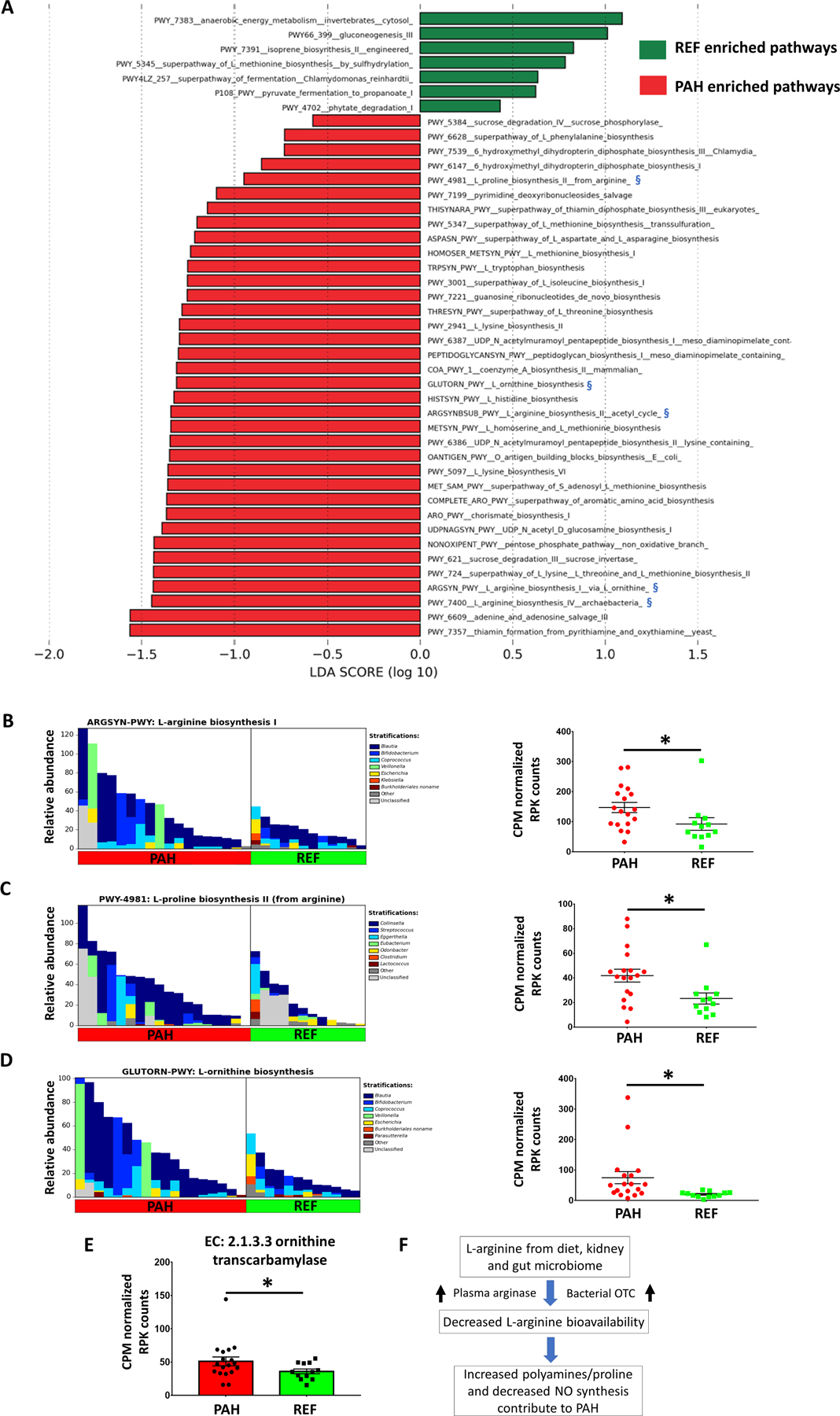
(A) LEfSe of altered functional pathways of the microbiomes in PAH and REF cohorts. Biosynthesis of several amino acids was increased in PAH such as arginine, lysine, homoserine, methionine, ornithine and tryptophan. Arginine/proline/ornithine biosynthesis was increased in PAH microbiome and marked with blue symbols (§). (B) Genera contributing to L-arginine biosynthesis (left) and CPM normalized RPK counts for the respective pathway by MetaCyc analysis (right). (C) Genera contributing to L-proline biosynthesis (left) and CPM normalized RPK counts for the respective pathway (right). (D) Genera contributing to L-ornithine biosynthesis (left) and CPM normalized RPK counts for the respective pathway (right). (E) Ornithine transcarbamylase (OTC), which converts L-ornithine to L-citrulline, was significantly increased in PAH microbiome. (F) Schematic diagram of potential bacterial contribution to PAH pathogenesis. Along with increased plasma arginase, bacterial OTC may contribute to decreased arginine bioavailability in PAH47.
Another significantly increased groups of bacteria in the PAH cohort was Coriobacteriales and its subpopulations Collinsella and Collinsella aerofaciens (Figure 1E, S3, 5A and 5B). Since these bacteria strongly correlate with production of the microbiome dependent metabolites, trimethylamine (TMA) and trimethylamine N-oxide (TMAO),26 we compared the sum of TMA/TMAO-associated bacteria and observed a significant increase in PAH patients (Figure 5C).27 In contrast, bacterial taxa that are negatively correlated with TMA/TMAO, such as Bacteroides were enriched in the REF cohort (Figure 2).28, 29
Figure 5: Increase in TMA/TMAO-associated bacteria and purine metabolism- related enzymes in PAH microbiome.
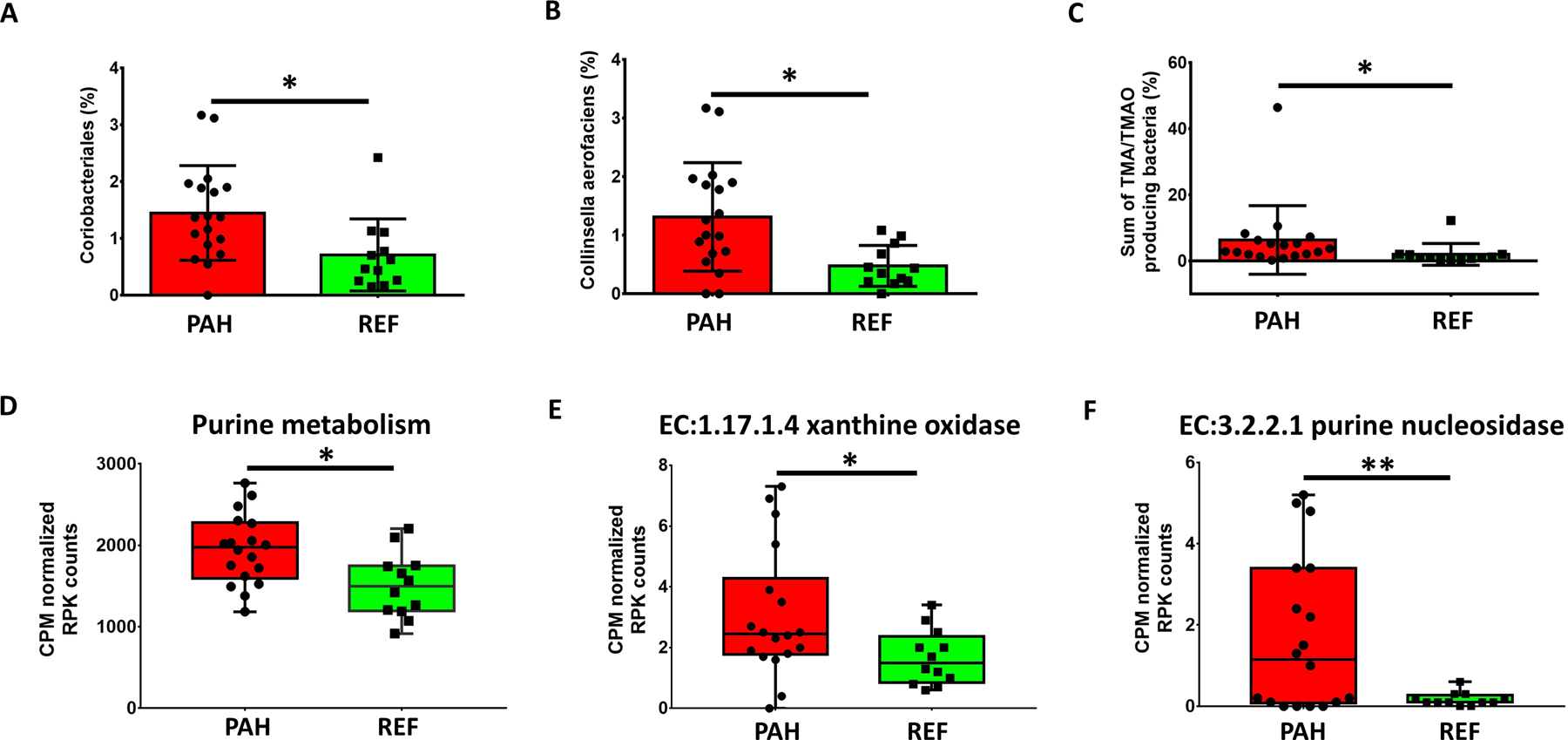
(A, B) Relative abundance of Coriobacteriales and Collinsella aerofaciens, major TMATMAO producers, were significantly increased in PAH microbiota. (C) Sum of TMA/TMAO producing bacteria in each cohort. (D) Comparison of enzymes of purine metabolism in gut microbiome of PAH and REF cohorts. Summed CPM normalized RPK counts of KEGG Orthology in the purine metabolism pathway were analyzed using Pathview in R studio (E, F). Xanthine oxidase/dehydrogenase and purine nucleosidase were significantly increased in the PAH microbiome. A full heatmap of purine metabolism (KEGG) comparing REF and PAH enzyme abundances can be found in Figure S5. Bar graph data are expressed as mean ± standard deviation with p values (*p<0.05; **p<0.01). The boxes of the box-whisker plots extend from the 25th to 75th percentiles, medians are shown in the boxes as a line. Whiskers encompass the maximum and minimum values and individual values are superimposed on the graph.
Purine metabolism was the third functional change we observed in PAH microbiome. Previous studies have shown a strong correlation of urate with PAH severity and mortality.23, 30, 31 We observed significant increases of overall purine metabolism, and key enzymes for metabolizing purine and producing urate such as xanthine oxidase and purine nucleosidase in the gut microbiota of PAH patients (Figure 5D–F and Figure S6). This suggests that testing the hypothesis that gut microbiome- derived purine and urate contribute to PAH pathogenesis would be valuable.
Discussion
The most significant observation of this study is that it demonstrates a unique profile of gut microbial communities in patients with PAH. Genome wide sequencing of these patients’ gut microbiota offers insight into the functions of the altered bacterial communities and their contribution to PAH pathophysiology. Finally, our data suggest that some of the previously identified potential biomarkers of PAH could be derived from altered bacterial functions in PAH patients. There is a distinct gut microbiome composition in the PAH patients (Figure 1). Many of the REF cohort enriched (and PAH cohort depleted) bacteria are associated with processes conferring gut health, such as polysaccharide fermentation and short chain fatty acid production (B. crossotus, B. cellulosilyticus, E. siraeum, B. vulgatus, A. muciniphila),13, 16 immune regulation (B. intestinihominis)32, and gut barrier fortification (A. muciniphila).33 In contrast, some of the enriched taxa of the PAH cohort have been directly linked to lung infections such as acute exacerbation of COPD (S. parasanguinis)34 and lung/intestine inflammation (R. gnavus, C. aerofaciens)35–38 (Figure 1, 2). This microbiome composition may be human PAH specific, as important bacterial predictors for PAH were not similarly changed in human systemic hypertension (Figure S4)17 nor an animal model of pulmonary hypertension.39 In addition, our data indicate the gut virome may be an interesting regulatory mechanism of the gut microbiome ecosystem and a novel therapeutic target (Figure 3).
Our results showing taxonomic and functional changes in gut microbiome of PAH patients supply several clues about the unique roles of the gut microbiome in the disease process. First, decreased beneficial short-chain fatty acid (SCFA) producing bacteria in the PAH cohort, such as Coprococcus, Butyrivibrio, Lachnospiraceae, Eubacterium and Clostridia may epigenetically or otherwise modify gut epithelial cells resulting in increased gut permeability.40 Reduced butyrate, or butanoate, weakens gut barrier function and favors oxidative metabolism, known to increase the potential for gut inflammation and leakiness. This is supported by evidence of increased gut permeability8 and depletion of anaerobic energy metabolizing bacterial communities (Figure 4A) in PAH cohort. Bacterial genes generating another SCFA derived from bacteria, propionate, or propanoate, were significantly reduced in the PAH microbiome (Figure 4A). These alterations in the PAH microbiome potentially enable microbial metabolites to leak into the circulation, disrupt metabolic and immune homeostasis, and influence the pulmonary vasculature. Although the exact role of SCFAs in PAH needs to be further validated, our data accentuate potential roles of the gut environment in PAH pathogenesis. Finally, it is interesting to point out that although distinct gut microbial communities are present in PAH and hypertension patients they both show increased gut permeability.8, 17, 41 This could be related to decreases in butyrate-producing bacteria in both diseases.17 Butyrate and related gut microbial species have been intimately implicated in gut leakiness in diverse diseases.42
Another possible mechanism is via increased Collinsella in the PAH cohort. This may contribute to disease pathogenesis by increasing gut permeability. Collinsella downregulates gut tight junction proteins and increases epithelial interleukin (IL)-17A production. These processes are generally accepted to result in gut barrier dysfunction and inflammation. Our data of elevated circulating markers of gut permeability and inflammation support this.8 Secondly, Collinsella contributed most of the increased genes for proline biosynthesis (Figure 4C). Thirdly, it suggests the important role of TMA/TMAO metabolism in PAH. Collinsella and its parent population Coriobacteriales have a strong positive correlation with TMA/TMAO production in atherosclerosis.26 TMA/TMAO producing taxa (Clostridium, Desulfovibrio, Enterobacter, Escherichia, Klebsiella, Pseudomonas, Rothia, Prevotella, Clostridium, Staphylococcus, Streptococcus, Citrobacter, Collinsella) were significantly elevated in the PAH microbiome (Figure 5). This coincided with decreases of bacterial taxa negatively associated with TMA/TMAO production such as B. cellulosilyticus, B. vulgatus, A. muciniphila, B. coprocola, B.stercoris (Figure 2).28, 29 Furthermore, a study in apolipoprotein E null mice fed with a Paigen diet (high fat and high choline) resulted in development of PAH with pulmonary vascular abnormalities.43 Another study showed gut microbiomes with high TMAO production increase thrombotic potential,44 an important risk factor for certain types of PAH.45 These provide experimental evidence supporting the hypothesis that diet and gut microbiome exert a significant influence on PAH pathogenesis.46
Urate and arginine bioavailability are known PAH risk factors correlating with the severity and mortality of PAH.30, 31, 47 Diet and endogenous synthesis were previously suggested as their origin, but our results indicate additional contributions from the gut microbiome. Indeed, xanthine oxidase/dehydrogenase and purine nucleosidase, essential enzymes for urate formation and overall purine metabolism were significantly elevated in the PAH gut microbiome. Similarly, a component of the altered L-arginine metabolism in PAH may arise in the gut microbiota. Key bacteria, such as Blautia likely increase L-arginine and L-ornithine biosynthesis in the PAH microbiome (Figure 1, 4) that are subsequently metabolized by increased ornithine transcarbamylase, an enzyme of the urea cycle, in the PAH microbiome (Figure 4E). Combined with elevated plasma arginase from hemolysis,48 this may contribute to the reduction in L-arginine bioavailability and PAH pathogenesis.47, 49
We observed increases in Streptococcus, Coprococcus and bacterial tryptophan biosynthesis, which may also have major mechanistic implications (Figures 1, S3 and S5). Firstly, the gut microbiome plays the key role in the synthesis of bioactive tryptophan metabolites, regulating serotonin (5-hydroxytryptamine) in the gut, and thereby, circulating serotonin.50 Serotonin has been implicated in PAH pathogenesis.51 Furthermore, inhibition of serotonin synthesis prevented experimental PAH in rats.52 In fact, inhibitors of tryptophan hydroxylase 1, the rate-limiting enzyme in serotonin synthesis in the gut, decrease gut serotonin by 90%, and attenuate pulmonary vascular pathology.50 Finally, SCFAs such as butyrate, are important stimulants to the synthesis of serotonin by the enterochromaffin cells of the gut.53 Thus, the decrease in butyrate-producing bacteria in PAH may be a brain-gut axis-mediated attempt to reduce serotonin synthesis and thereby ameliorate serotonin-associated PAH pathology.
This study has several limitations. First, the small sample size of study subjects from a limited geographical area limits its broad applicability and global conclusions. Unfortunately, the high cost of whole genome sequencing (WGS) restricted this to a “proof of concept” study. However, WGS of human gut microbiome should be the preferred strategy since it provides unlimited information not only on taxa, species etc. but also on functional aspects of the entire microbiome (bacteria, viruses and fungi etc.) for future data mining. Second, PAH is associated with multiple etiologies although all patients had one common feature, increased mPAP. No attempt was made to determine contribution of each etiology on microbiota because of small sample size. Third, the random forest model-based ROC curve used in this analysis is appropriate for a small sample size in predicting PAH with high probability. However, this needs to be validated by an appropriately powered clinical trial with an independent test cohort. If confirmed, it would present an alternative diagnostic tool for PAH compared to the costly and risky right heart catheterization presently in use. Fourth, questions regarding the origin of the altered microbial community, how gut microbiome derived signals affect remote organs such as lung, and the causal relationship between these changes and PAH remains unknown. Nonetheless, it is tempting to suggest that diverse pro- pulmonary hypertensive signals (environmental, genetic, etc.) induce gut dysbiosis and leakiness. This would create an environment where gut bacteria, viruses and their metabolites could distribute in the systemic circulation and lung, which may initiate pulmonary inflammation, and infection, leading to vascular remodeling and PAH.
Analysis of gut bacteria and viruses in PAH suggest they have previously unknown roles in PAH. PAH-associated taxa such as Blautia, Bifidobacteria, Collinsella contributed genes for carnitine metabolism or biosynthesis of arginine, ornithine, proline, providing evidence that taxonomic changes are closely associated with altered gut microbiome function in the PAH cohort. Therefore, identification and characterization of PAH specific bacteria and metabolites in both gut and lung hold promise for the development of innovative strategies for the control and treatment of PAH by modification of diet, prebiotics, microbiota transplantation and pro/antibiotics.
Supplementary Material
Perspectives:
Significant evidence exists for the involvement of gut microbiota in a variety of pulmonary diseases but not for the role and contribution of the gut and its microbiome in the pathogenesis of pulmonary hypertension. The present study begins to fill this gap in our knowledge. It compares functional and taxonomic gut microbiome profiles of patients with PAH with control subjects to imply a role in the pathogenesis of pulmonary hypertension. We demonstrate a taxonomically and functionally distinct microbiome in PAH patients with a prediction accuracy of 83%. Some of these distinct bacterial make metabolites known to be altered in the circulation in PAH patients. Therefore, our data suggest that uniquely altered gut microbiome may play an important role in pulmonary pathology and thus present novel targets for both PAH diagnosis and therapy.
Novelty and Significance.
What Is New?
First study to show alterations in gut microbiota in patients with pulmonary arterial hypertension (PAH).
Taxonomically and functionally distinct microbiota in PAH patients with a prediction accuracy of 83%. Some of these bacterial communities produce metabolites known to be altered in circulation of PAH patients.
What Is Relevant?
PAH is considered a disease of pulmonary vasculature. Our study demonstrates involvement of the gut and its microbiota and supports the hypothesis of a dysfunctional gut-lung axis in PAH.
Gut microbiota holds potential for PAH therapy.
Source of funding
This work was supported by National Institute of Health (NIH) grants (R01 HL102033 to M.K. Raizada; R01 HL146158, R01 HL 132448, R01 HL091005, U01 HL064924, U01 HL087366, UM1 HL087366; UM1 HL087366, UM1 HL087364 to C.J. Pepine), NIH/National Center for Research Resources grant (UL1 TR000064 to C.J. Pepine), and US Department of Defense grants (W81XWH1720030, W81XWH-17-2-0030 to C.J. Pepine)
Footnotes
Disclosure
None.
References
- 1.Fallah F. Recent strategies in treatment of pulmonary arterial hypertension, a review. Glob J Health Sci. 2015;7(4):307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurtu V, Michelakis ED. A Paradigm Shift Is Needed in the Field of Pulmonary Arterial Hypertension for Its Entrance Into the Precision Medicine Era. Circ Res.119(12):1276–1279. [DOI] [PubMed] [Google Scholar]
- 3.West J. Pulmonary arterial hypertension as a systemic disease. Advances in Pulmonary Hypertension. 2012;11(3):130–131. [Google Scholar]
- 4.Tang WH, Hazen SL. The Gut Microbiome and Its Role in Cardiovascular Diseases. Circulation. 2017;135(11):1008–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marsland BJ, Trompette A, Gollwitzer ES. The Gut-Lung Axis in Respiratory Disease. Ann Am Thorac Soc. 2015;12 Suppl 2:S150–156. [DOI] [PubMed] [Google Scholar]
- 6.Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;7(3):245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moffatt MF, Cookson WO. The lung microbiome in health and disease. Clin Med (Lond). 2017;17(6):525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, M. WJ, Rigatto, Jordan R, Shapiro B, Pepine CJ, Richards EM, Raizada MK. Altered Gut Microbiome Contributes to Metabolite Biomarkers in Patients With Pulmonary Hypertension. Hypertension. 2018;72(Suppl 1):https://www.ahajournals.org/doi/10.1161/hyp.1172.suppl_1161.P1167. [Google Scholar]
- 9.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo W, Pant G, Bhavnasi YK, Blanchard SG, Jr., Brouwer C. Pathview Web: user friendly pathway visualization and data integration. Nucleic Acids Res. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ounit R, Wanamaker S, Close TJ, Lonardi S. CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics. 2015;16:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin J, Kramna L, Autio R, Hyoty H, Nykter M, Cinek O. Vipie: web pipeline for parallel characterization of viral populations from multiple NGS samples. BMC Genomics. 2017;18(1):378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riviere A, Selak M, Lantin D, Leroy F, De Vuyst L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front Microbiol. 2016;7:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio. 2014;5(2):e00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett. 2002;217(2):133–139. [DOI] [PubMed] [Google Scholar]
- 16.Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41. [DOI] [PubMed] [Google Scholar]
- 17.Kim S, Goel R, Kumar A, Qi Y, Lobaton G, Hosaka K, Mohammed M, Handberg EM, Richards EM, Pepine CJ, Raizada MK. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci (Lond). 2018;132(6):701–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almand EA, Moore MD, Jaykus LA. Virus-Bacteria Interactions: An Emerging Topic in Human Infection. Viruses. 2017;9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chibani-Chennoufi S, Bruttin A, Dillmann ML, Brussow H. Phage-host interaction: an ecological perspective. J Bacteriol. 2004;186(12):3677–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koskella B, Meaden S. Understanding bacteriophage specificity in natural microbial communities. Viruses. 2013;5(3):806–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Peng J, Lu C, Hsin M, Mura M, Wu L, Chu L, Zamel R, Machuca T, Waddell T, Liu M, Keshavjee S, Granton J, de Perrot M. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS One. 2014;9(2):e88727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao YD, Chu L, Lin K, Granton E, Yin L, Peng J, Hsin M, Wu L, Yu A, Waddell T, Keshavjee S, Granton J, de Perrot M. A Biochemical Approach to Understand the Pathogenesis of Advanced Pulmonary Arterial Hypertension: Metabolomic Profiles of Arginine, Sphingosine-1-Phosphate, and Heme of Human Lung. PLoS One. 2015;10(8):e0134958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhodes CJ, Ghataorhe P, Wharton J, Rue-Albrecht KC, Hadinnapola C, Watson G, Bleda M, Haimel M, Coghlan G, Corris PA, Howard LS, Kiely DG, Peacock AJ, Pepke-Zaba J, Toshner MR, Wort SJ, Gibbs JS, Lawrie A, Graf S, Morrell NW, Wilkins MR. Plasma Metabolomics Implicates Modified Transfer RNAs and Altered Bioenergetics in the Outcomes of Pulmonary Arterial Hypertension. Circulation. 2017;135(5):460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kao CC, Wedes SH, Hsu JW, Bohren KM, Comhair SA, Jahoor F, Erzurum SC. Arginine metabolic endotypes in pulmonary arterial hypertension. Pulm Circ. 2015;5(1):124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang WH, Wang Z, Cho L, Brennan DM, Hazen SL. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J Am Coll Cardiol. 2009;53(22):2061–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ, Hazen SL. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163(7):1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Obaide MAI, Singh R, Datta P, Rewers-Felkins KA, Salguero MV, Al-Obaidi I, Kottapalli KR, Vasylyeva TL. Gut Microbiota-Dependent Trimethylamine-N-oxide and Serum Biomarkers in Patients with T2DM and Advanced CKD. J Clin Med. 2017;6(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen ML, Yi L, Zhang Y, Zhou X, Ran L, Yang J, Zhu JD, Zhang QY, Mi MT. Resveratrol Attenuates Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. MBio. 2016;7(2):e02210–02215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagaya N, Uematsu M, Satoh T, Kyotani S, Sakamaki F, Nakanishi N, Yamagishi M, Kunieda T, Miyatake K. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med. 1999;160(2):487–492. [DOI] [PubMed] [Google Scholar]
- 31.Zhang CY, Ma LL, Wang LX. Relationship between serum uric acid levels and ventricular function in patients with idiopathic pulmonary hypertension. Exp Clin Cardiol. 2013;18(1):e37–39. [PMC free article] [PubMed] [Google Scholar]
- 32.Daillere R, Vetizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, Duong CPM, Flament C, Lepage P, Roberti MP, Routy B, Jacquelot N, Apetoh L, Becharef S, Rusakiewicz S, Langella P, Sokol H, Kroemer G, Enot D, Roux A, Eggermont A, Tartour E, Johannes L, Woerther PL, Chachaty E, Soria JC, Golden E, Formenti S, Plebanski M, Madondo M, Rosenstiel P, Raoult D, Cattoir V, Boneca IG, Chamaillard M, Zitvogel L. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity. 2016;45(4):931–943. [DOI] [PubMed] [Google Scholar]
- 33.Chelakkot C, Choi Y, Kim DK, Park HT, Ghim J, Kwon Y, Jeon J, Kim MS, Jee YK, Gho YS, Park HS, Kim YK, Ryu SH. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp Mol Med. 2018;50(2):e450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan KG, Ng KT, Pang YK, Chong TM, Kamarulzaman A, Yin WF, Tee KK. Genome Anatomy of Streptococcus parasanguinis Strain C1A, Isolated from a Patient with Acute Exacerbation of Chronic Obstructive Pulmonary Disease, Reveals Unusual Genomic Features. Genome Announc. 2015;3(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chua HH, Chou HC, Tung YL, Chiang BL, Liao CC, Liu HH, Ni YH. Intestinal Dysbiosis Featuring Abundance of Ruminococcus gnavus Associates With Allergic Diseases in Infants. Gastroenterology. 2018;154(1):154–167. [DOI] [PubMed] [Google Scholar]
- 36.Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, Lagoudas GK, Vatanen T, Fornelos N, Wilson R, Bertha M, Cohen M, Garber J, Khalili H, Gevers D, Ananthakrishnan AN, Kugathasan S, Lander ES, Blainey P, Vlamakis H, Xavier RJ, Huttenhower C. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017;9(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makivuokko H, Tiihonen K, Tynkkynen S, Paulin L, Rautonen N. The effect of age and non-steroidal anti-inflammatory drugs on human intestinal microbiota composition. Br J Nutr. 2010;103(2):227–234. [DOI] [PubMed] [Google Scholar]
- 38.Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, Nelson H, Matteson EL, Taneja V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016;8(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callejo M, Mondejar-Parreno G, Barreira B, Izquierdo-Garcia JL, Morales-Cano D, Esquivel-Ruiz S, Moreno L, Cogolludo A, Duarte J, Perez-Vizcaino F. Pulmonary Arterial Hypertension Affects the Rat Gut Microbiome. Sci Rep. 2018;8(1):9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015;17(5):662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goel R, Kim S, Rigatto K, Richards E, Pepine C, Raizada M. Increased Gut Dysbiosis and Leakiness in Patients With Pulmonary Arterial Hypertension. Circulation. 2017;136(Supplement 1):A20620. [Google Scholar]
- 42.Hiippala K, Jouhten H, Ronkainen A, Hartikainen A, Kainulainen V, Jalanka J, Satokari R. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients.10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawrie A, Hameed AG, Chamberlain J, Arnold N, Kennerley A, Hopkinson K, Pickworth J, Kiely DG, Crossman DC, Francis SE. Paigen diet-fed apolipoprotein E knockout mice develop severe pulmonary hypertension in an interleukin-1-dependent manner. Am J Pathol. 2011;179(4):1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, Sartor RB, McIntyre TM, Silverstein RL, Tang WHW, DiDonato JA, Brown JM, Lusis AJ, Hazen SL. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell. 2016;165(1):111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolf M, Boyer-Neumann C, Parent F, Eschwege V, Jaillet H, Meyer D, Simonneau G. Thrombotic risk factors in pulmonary hypertension. Eur Respir J. 2000;15(2):395–399. [DOI] [PubMed] [Google Scholar]
- 46.Kinlay S, Michel T, Leopold JA. The Future of Vascular Biology and Medicine. Circulation. 2016;133(25):2603–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris CR, Gladwin MT, Kato GJ. Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders. Curr Mol Med. 2008;8(7):620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM Jr., Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rafikova O, Meadows ML, Kinchen JM, Mohney RP, Maltepe E, Desai AA, Yuan JX, Garcia JG, Fineman JR, Rafikov R, Black SM. Metabolic Changes Precede the Development of Pulmonary Hypertension in the Monocrotaline Exposed Rat Lung. PloS one. 2016;11(3):e0150480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacLean MR. Pulmonary hypertension and the serotonin hypothesis: where are we now? Int J Clin Pract Suppl. 2007(156):27–31. [DOI] [PubMed] [Google Scholar]
- 51.MacLean MMR. The serotonin hypothesis in pulmonary hypertension revisited: targets for novel therapies (2017 Grover Conference Series). Pulm Circ. 2018;8(2):2045894018759125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun XQ, Peters E, Schalij I, Andersen S, Da Silva Goncalves Bos D, Vonk-Noordegraaf A, De Man FS, Van Der Laarse W, Bogaard HJ. The effect of Monoamine oxidase A inhibition on experimentally induced pulmonary arterial hypertension. European Respiratory Journal. 2018;52:https://erj.ersjournals.com/content/52/suppl_62/PA3072. [Google Scholar]
- 53.Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O’Donnell TA, Brierley SM, Ingraham HA, Julius D. Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell. 2017;170(1):185–198 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metadata and microbiome data (taxonomy and KO files) can be downloaded at https://drive.google.com/open?id=15NfMk7BAHo0Y_iyNlHnCNCE2Gg___v31