

Abstract
Abdominal aortic aneurysm (AAA) is a major cause of sudden death in the elderly. While AAA has some overlapping genetic and environmental risk factors with atherosclerosis, there are substantial differences, and AAA-specific medication is lacking. A recent meta-analysis of genome-wide association studies has identified four novel single-nucleotide polymorphisms (SNPs) specifically associated with AAA. Here, we investigated the gene regulatory function for one of four non-coding SNPs associated with AAA, rs2836411, which is located in an intron of the ERG gene. Rs2836411 resides within a >70 kb super-enhancer that has high levels of H3K27ac and H3K4me1 in vascular endothelial and haematopoietic cell types. Enhancer luciferase assays in cell lines showed that the risk allele significantly alters enhancer activity. The risk allele also correlates with reduced ERG expression in aortic and other vascular tissues. To identify whether rs2836411 directly contacts the promoters of ERG and/or of genes further away, we performed allele-specific circular chromosome conformation capture sequencing. In vascular endothelial cells, which express ERG, the SNP region interacts highly within the super-enhancer, while in vascular smooth muscle cells, which do not express ERG, the interactions are distributed across a wider region that includes neighbouring genes. Furthermore, the risk allele has fewer interactions within the super-enhancer compared to the protective allele. In conclusion, our results indicate that rs2836411 likely affects ERG expression by altering enhancer activity and changing local chromatin interactions. ERG is involved in vascular development, angiogenesis, and inflammation in atherosclerosis; therefore mechanistically, rs2836411 could contribute to AAA by modulating ERG levels.
Introduction
Abdominal aortic aneurysm (AAA) is a common cause of death in the elderly Western population (1). It is characterized by an irreversible weakening and enlargement of the abdominal aorta, which can ultimately lead to rupture that is fatal in most cases. The risk of developing AAA is determined by a combination of environmental and genetic factors. Major risk factors include smoking, increasing age, male sex, white ethnicity and a family history of AAA (2,3). While some of the risk factors overlap with atherosclerotic diseases (such as increasing age and male sex), substantial differences exist (4). Smoking is a strong risk factor for AAA, while hypercholesterolemia is more weakly associated. Furthermore, diabetes is a risk factor for atherosclerosis but appears to be protective for AAA formation and progression (5). It is estimated that genetic components contribute to around 70% of the overall susceptibility to AAA (6), indicating that genetic factors play a major role in the aetiology.
The abdominal aortic wall consists of three layers: 1) the tunica intima, comprising a monolayer of endothelial cells and an internal elastic lamina, 2) the tunica media, comprising around 30 lamellar subunits consisting of elastic lamina, smooth muscle cells and collagen and 3) the tunica adventitia that is collagen rich and contains a network of vasa vasorum that supplies nutrients to the outer wall. AAA starts off with the formation of a lipid-rich plaque in the intima (atherosclerosis), which causes an inflammatory reaction that leads to smooth muscle cell apoptosis and degradation of the extracellular matrix (7). This process results in deep intima hypoxia, leading to neovascularization that extends into the tunica media, which further disrupts and weakens the aortic wall. Neovascularization allows for the infiltration of inflammatory cells (such as T-lymphocytes, macrophages and plasma cells) and causes a chronic inflammatory state within the outer aortic wall, characterized by the presence of small or large lymphoid aggregates (7). Although atherosclerosis underlies the formation of AAA, in other types of cardiovascular diseases (e.g. coronary artery disease), the inflammation is relatively confined to the tunica intima and causes an occlusive phenotype rather than a dilating phenotype.
Genome-wide association studies have identified 10 loci associated with AAA (8–10), which partially overlap with other cardiovascular diseases and risk factors, indicating there are both shared and independent genetic causal pathways. Four of the AAA-associated single-nucleotide polymorphisms (SNPs) appear to be specifically associated with AAA (9). All four are located in non-coding DNA, like the majority of disease-associated SNPs (11), and it is unclear how these SNPs contribute to AAA. Non-coding SNPs can alter the expression of genes by affecting the activity of regulatory elements, such as enhancers that can activate gene transcription (12,13). Disease-associated non-coding SNPs do not necessarily regulate the expression of the nearest gene and can alter the expression of distant genes by directly contacting them through chromatin looping (11,14). Therefore, identification of the gene(s) regulated by disease-associated non-coding SNPs requires identification of the chromatin interactions formed. Investigating the function of AAA-specific SNPs would help understand the molecular mechanisms involved in AAA pathogenesis.
Here, we studied the gene regulatory function of rs2836411, one of the SNPs that appears to have a AAA-specific association (9). Out of the four SNPs specifically associated with AAA, rs2836411 is located in a gene that could be directly relevant to AAA pathogenesis, which is why we focus on this SNP in this study. Rs2836411 is located in an intron of ERG (ETS related gene), over 50 kilobases (kb) away from the nearest ERG promoter. ERG is an erythroblast transformation-specific (ETS) domain containing transcription factor (TF) that is crucial for vascular development (15). It is also involved in angiogenesis and vascular endothelial cell inflammation. ERG has not been previously implicated in cardiovascular diseases, and, as such, it may be involved in AAA-specific pathobiological mechanism.
This study aims to identify if rs2836411 affects gene regulation and whether it affects the expression of ERG and/or of distant genes. We show that rs2846411 harbours enhancer activity in vascular endothelial cells and that the risk allele (T) lowers enhancer activity and correlates with reduced ERG expression in vascular tissues. To determine whether the SNP directly interacts with the promoters of ERG and/or of genes further away, we have performed allele-specific chromosome conformation capture sequencing (4C-seq) in human umbilical vein endothelial cells (HUVECs), which express ERG, and in human aortic smooth muscle cells (HASMCs), which do not express ERG.
Results
Rs2836411 is located in an enhancer in vascular endothelial cells
To investigate if the DNA region containing rs2836411 has gene regulatory activity, we explored epigenetic data in different cell types from the ENCODE and Roadmap Epigenomics projects (16,17). As rs2836411 is not in high linkage disequilibrium with any other SNP (Supplementary Material, Figure S1), in this study, we have focused on the function of rs2836411 alone. ENCODE regulatory tracks of the enhancer-marking histone modifications histone 3 lysine 27 acetylation (H3K27ac) and histone 3 lysine 4 monomethylation (H3K4me1) in seven commonly used cell lines show enrichment of H3K4me1 at rs2836411 in HUVECs, but not in other cell lines (Fig. 1). In primary aortic tissue from ascending and thoracic aorta, rs2836411 is marked by H3K4me1 and H3K27ac (Fig. 1). The ENCODE aortic tissue samples used are either homozygous for the protective allele (C/C) or heterozygous (C/T), and there are no observable differences in H3K4me1 or H3K27ac enrichment between the different genotypes (Fig. 1). We were unable to detect differences in enrichment between the C and T allele of heterozygous samples due to insufficient coverage at rs2836411 (<14 reads overlap rs2836411 directly). Rs2836411 is also located in DNase I hypersensitivity sites in HUVECs and other vascular endothelial cell types (Fig. 1). In addition to HUVECs and aortic tissue, HaploReg analysis showed that rs2836411 resides in an enhancer element based on chromatin segregation states by core 15-state chromHMM (chromatin hidden Markov model) in different blood cell types (e.g. haematopoietic stem cells, monocytes and neutrophils), mesenchymal stem cells, fetal muscle, thymus and the left ventricle (18).
Figure 1.
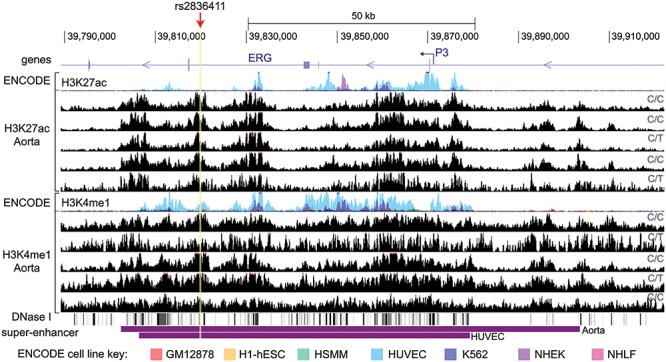
Rs2836411 is marked as an enhancer in HUVECs and aortic tissue. From top to bottom the following tracks are shown: UCSC genes with the ERG-P3 promoter indicated by a hooked arrow; overlaid ENCODE track of ChIP-seq signal for the histone modifications H3K4me1 and H3K27ac for seven different commonly used cell lines (see key for colours corresponding to each cell line); H3K27ac and H3K4me1 ChIP-seq tracks for five different thoracic and ascending aorta tissue samples (obtained from ENCODE); super-enhancer regions in aortic tissue and HUVECs obtained from dbSUPER. The genotypes of rs2836411 for the aortic tissue samples are indicated on the right side of each track. The location of rs2836411 is highlighted and marked by a red arrow. Tracks were obtained using the UCSC genome browser.
In both HUVECs and aortic tissue, rs2836411 is located in a large region (>70 kb) that contains high levels of H3K27ac and/or H3K4me1 starting from upstream of P3 to downstream of exon 6 (Fig. 1). This region is identified as a super-enhancer in both HUVECs and aortic tissue (19,20). Super-enhancers are clusters of multiple enhancers that drive the expression of cell type-specific genes that are important for cellular identity (21). The ERG super-enhancer was previously shown to drive ERG expression in myeloid leukemic cells (22,23). Rs2836411 could thus affect the activity of the ERG super-enhancer.
Aortic tissue consists mainly of ASMCs and has a small proportion of vascular endothelial cells that are present in the intima and vasa vasorum. To identify if the SNP region is active in ASMCs in addition to aortic endothelial cells, we gathered histone modification data (H3K4me2 and H3K27ac) in these two cell types from published literature (24,25). In human aortic endothelial cells, rs2836411 and the ERG super-enhancer have peaks of H3K4me2 and H3K27ac, while in ASMCs the peaks at rs2836411 are only present in one of three replicates, and fewer peaks are present at the super-enhancer region (Supplementary Material, Figure S2). This suggests that rs2836411 is located in an active enhancer in aortic endothelial cells.
The enhancer activity for the SNP region is consistent with the expression of ERG in the corresponding cell types. We performed quantitative polymerase chain reaction (qPCR) to measure ERG transcript levels in HUVEC and HASMCs and found that ERG is expressed in HUVECs and has minimal expression in HASMCs (Fig. 2A). We also investigated ERG expression in AAA tissue using in situ hybridization (RNAscope). ERG was expressed within endothelial cells lining the vasa vasorum in the inflamed tunica adventitia of AAA biopsies (Fig. 2B), but not in other cell types. This observation confirms that ERG is expressed primarily within vascular endothelial cells in AAA tissue.
Figure 2.
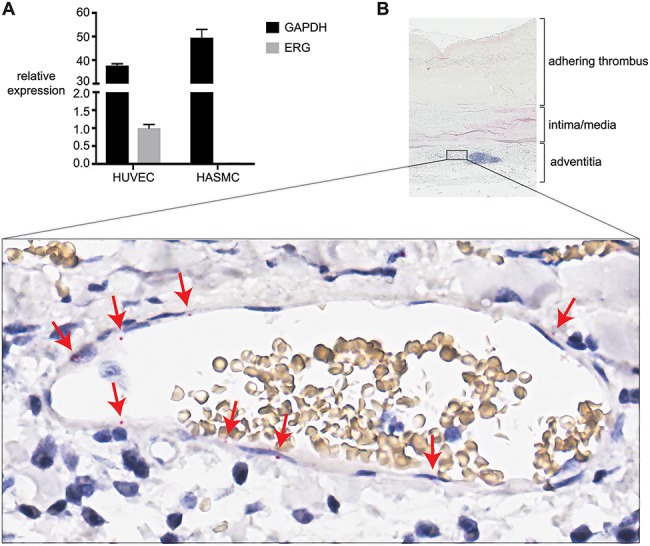
ERG is specifically expressed in vascular endothelial cells. (A) The expression of total ERG (all isoforms) and GAPDH was measured by qPCR in two different HUVEC lines and three different HASMC lines. ERG expression values were normalized to the GeoMean of the reference genes GAPDH and RPL13A and are expressed relative to ERG in HUVECs. The mean ± the standard error of the mean is shown. (B) RNAscope® was used to detect the presence of ERG transcripts in AAA tissue section. Part of the section is shown on top, with below it a zoomed-in view of a vasa vasorum located next to a lymphoid aggregate (purple cluster of cells in total view). Staining of ERG transcripts in endothelial cells lining the lumen (red dots) is indicated by red arrows.
The risk allele of rs2836411 reduces enhancer activity
To confirm that rs2836411 harbours enhancer activity and to assess whether there is a difference in enhancer activity between the risk and protective alleles, a luciferase reporter assay with constructs containing the protective (C) or risk (T) alleles was performed in HUVECs, HepG2 (human liver hepatocellular carcinoma) and human embryonic kidney (HEK293) cells. The construct containing the C allele harboured enhancer activity, which was significantly reduced for the construct containing the T allele in all three cell lines (Fig. 3A and Supplementary Material, Figure S3). These data confirm that rs2836411 harbours enhancer activity and that the enhancer activity is reduced by the risk allele.
Figure 3.
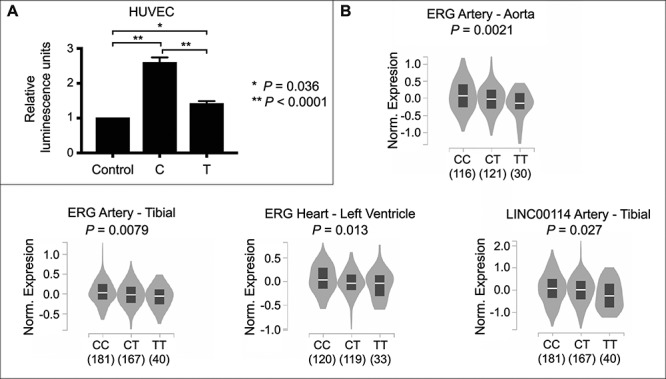
The risk allele (T) of rs2836411 reduces enhancer activity and correlates with lower ERG and LINC0014 expression. (A) Allele-specific enhancer activity of rs2836411 compared to the control pGL4.23 vector was assessed using a luciferase reporter assay in HUVECs. The C variant (protective allele) harboured enhancer activity that was significantly reduced for the T variant (risk allele). The average of five biological replicates ± the standard error of the mean is shown. Statistical significance was determined by a one-way ANOVA followed by a Sidak’s multiple comparisons test. (B) eQTL data from GTEx for rs2836411. Violin plots of normalized expression levels from individuals (number indicated below the genotypes) containing the rs2836411 CC, CT or TT alleles in the indicated vascular tissue types were obtained using the GTEx data portal (72). Only data that reached P < 0.05 in individual look-ups using the GTEx eQTL calculator for rs2836411 and the genes ERG, KCNJ15, LINC00114, ETS2, PSMG1 in vascular tissue types (aorta, tibial artery, left ventricle, coronary artery and arterial appendage) are shown here.
SNPs can alter enhancer activity by affecting the binding affinity of TFs (26,27). We investigated whether rs2836411 binds TFs and whether it might alter TF-binding motifs by 1) exploring experimentally identified TF-binding sites from published chromatin immunoprecipitation sequencing (ChIP-seq) studies and 2) prediction of TF-binding motif changes caused by rs2836411. Several in vivo TF-binding sites overlap rs2836411 at the flank of the binding sites, but not near the peak summits (Supplementary Material, Figure S4A). Haploreg (18) predicts that rs2836411 affects the FOXO3- and FOXO4-binding motifs and JASPAR (28) predicts the presence of a binding motif for FOXO3, but not for FOXO4 (Supplementary Material, Figure S4B). We did not find experimental evidence of any of the FOXO TFs binding to rs2836411 (Supplementary Material, Figure S4A). Based on in vivo TF binding and motif prediction changes, it is unclear through which TFs rs2836411 alters enhancer activity.
Rs2836411 is an expression quantitative trait loci with ERG and LINC0014 in vascular tissues
We next investigated whether rs2836411 is an expression quantitative trait loci (eQTL) with ERG and/or neighbouring genes in vascular tissues. Individual lookup of eQTL with ERG and the neighbouring genes KCNJ15, LINC00114, ETS2, PSMG1 in GTEx (genotype-tissue expression) showed that rs2836411 is an eQTL with ERG in aorta, tibial artery and left ventricle (P = 0.0021, 0.0079 and 0.013, respectively) and with LINC00114 (P = 0.027) in tibial artery (Fig. 3B and Supplementary Material, Table S1). The trend is that samples containing the T allele have lower ERG or LINC00114 expression than samples containing the C allele (Fig. 3B). Rs2836411 was also previously shown to be an eQTL with ERG in mammary artery using data from the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task study (9,29). The eQTL data indicate that rs2836411 mostly affects the expression of ERG.
Rs2836411 interacts within the super-enhancer in vascular endothelial cells
We then addressed whether the DNA region containing rs2836411 comes within physical proximity of ERG and/or of genes further away. To determine which genes are regulated by rs2836411, the chromatin interactions anchored at the rs2836411 DNA region were identified using 4C-seq in HUVECs heterozygous (C/T) for rs2836411 and in HASMCs homozygous for the protective allele (C/C). In HUVECs, ERG is expressed and the rs2836411 DNA region has enhancer activity, while in HASMCs, ERG is minimally expressed and the rs2836411 DNA region has minimal enhancer activity (Figs 1 and 2A). In HUVECs, ERG is transcribed from the P3 promoter, indicating that P1/P2 is inactive (Supplementary Material, Figure S5).
4C-seq profiles for the protective allele (C) were aligned with Hi-C data in HUVECs from Rao et al., 2014, so that the topologically associated domain (TAD) structure could be observed simultaneously. Hi-C data show that rs2836411 is located in a sub-domain spanning from a region upstream of P3 till the 3′ end of ERG, which is part of a larger TAD spanning from the 3′ end of ERG to SH3BGR (Fig. 4A). This sub-domain is not apparent in Hi-C data from cell lines that do not express ERG (Supplementary Material, Figure S6). Based on the sub-domain structure, statistically significant interactions for the 4C-seq data were called separately for the region within the ERG-P3 sub-domain compared to the total chromosome 21 and were overlapped between two replicates (see Materials and Methods and Supplementary Material, Figure S7). 4C-seq data showed that in HUVECs the majority of significant interactions were contained within the super-enhancer region, while significant interactions in HASMCs were distributed across the TAD and the downstream neighbouring TAD (Fig. 4A). This indicates that rs2836411-associated enhancer influences are mainly contained within the ERG-P3 sub-domain. In addition, both the interaction frequencies observed in the Hi-C data and the significant interactions observed in our 4C-seq data show that when ERG-P3 is transcriptionally active, the ERG-P3 sub-domain highly interacts within the sub-domain, while it does not when ERG-P3 is transcriptionally inactive.
Figure 4.
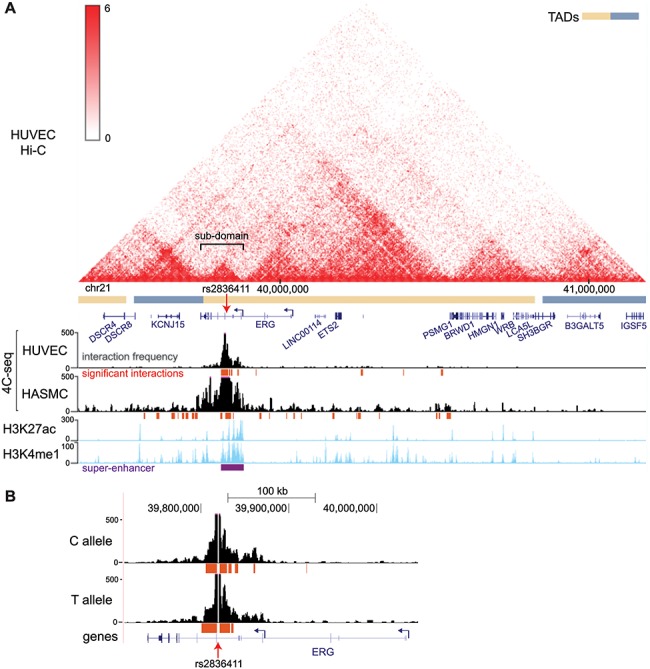
rs2836411 interact highly within the ERG super-enhancer in HUVECs and interactions are reduced for the T allele. (A) Hi-C contact map in HUVECs with TADs indicated are aligned with 4C-seq profiles using a restriction fragment containing rs2836411 as a bait in HUVECs and HASMCs harbouring the C allele of rs2836411. H3K4me1 and H3K27ac tracks (ENCODE data; light blue) and the super-enhancer region from dbSUPER (purple) in HUVECs are shown below the 4C-seq profiles. Hi-C data are from Rao et al., 2014 (61) and were obtained using the 3D genome browser (70). (B) 4C-seq profiles for the C (protective) and T (risk) allele of rs2836411 in HUVECs are shown. (A and B) 4C-seq frequency tracks (black) are the read per million normalized running mean of 21 successive MseI restriction fragments for the average of two replicates and are shown together with statistically significant interactions (the top fifth percentile of interactions with a FDR of < 0.01) that overlap between two replicates (red). UCSC reference genes are in blue and ERG P1/2 and P3 promoters are indicated by hooked arrows. Tracks were obtained using the UCSC genome browser.
The rs2836411 risk allele has fewer interactions with the ERG super-enhancer compared to the protective allele
We performed allele-specific 4C-seq in a HUVEC line that contained both the C and T alleles and compared the corresponding interaction profiles between alleles. The T allele had no significant interaction at the intron downstream of P3, while the C allele did (Fig. 4B). This suggests that the T allele may reduce interactions within the super-enhancer, which could potentially be a result of lower enhancer activity.
Discussion
We have investigated the gene regulatory function of rs2836411, a SNP that is independently associated with AAA. Rs2836411 is located in an intron of ERG, over 50 kb downstream of the P3 promoter that gives rise to the main isoform expressed in hematopoietic and vascular endothelial cells. The chromatin surrounding rs2836411 is marked as an enhancer in vascular endothelial cells and is part of a super-enhancer in this cell type. The risk allele (T) reduces enhancer activity, showing that rs2836411 affects gene regulation.
As the causal gene for disease is not necessarily the gene in which intronic SNPs lie (11,30,31), we sought to identify the gene(s) that are affected by rs2836411. Enhancers can connect with distant target genes through the formation of chromatin loops and these loops, as well as enhancer activity, are mostly contained within TADs (14). Rs2836411 and ERG are located in a TAD with several other genes, and we hypothesized that rs2836411 may influence the expression of any gene within the TAD, and each of these genes could be involved in AAA formation. For example, one of the other genes in the TAD is ETS2, another ETS domain containing TF that has been reported to co-regulate the expression of genes together with ERG (such as matrix metalloproteases 1 and 3) in adult human endothelial cells (32,33). ETS2 may thus be another potential candidate gene involved in AAA formation. To find out whether rs2836411 interacts with ERG and/or other genes, we performed 4C-seq in HUVECs, which express ERG and have enhancer activity at rs2836411, and in HASMCs, which do not express ERG and have no enhancer activity at rs2836411. In HUVECs, rs2836411 mainly interacts within an ERG-P3 subdomain containing the super-enhancer, while in HASMCs interactions are distributed across a larger region. These findings are consistent with Hi-C data in which the ERG-P3 sub-TAD is not apparent in cell types that do not express ERG. This suggests that rs2836411 mainly influences the expression of ERG and not of genes further away. This evidence is supported by the finding that the most significant eQTL for the SNP in vascular cell types is for ERG.
The 4C-seq data also shows that interactions within and outside of the ERG-P3 sub-domain are reduced for the risk (T) allele of rs2836411 compared to the protective allele (C). The risk allele also has reduced enhancer activity and is correlated to reduced expression of ERG. A recent study suggests that small changes in chromatin interactions caused by genetic variation could be a mechanism by which gene expression is regulated (34). Rs2836411 may regulate ERG expression by affecting enhancer activity, chromatin interactions or a combination of both. Based on the findings described in this study, a hypothetical model of the functional mechanism by which rs2836411 could act is shown in Figure 5.
Figure 5.
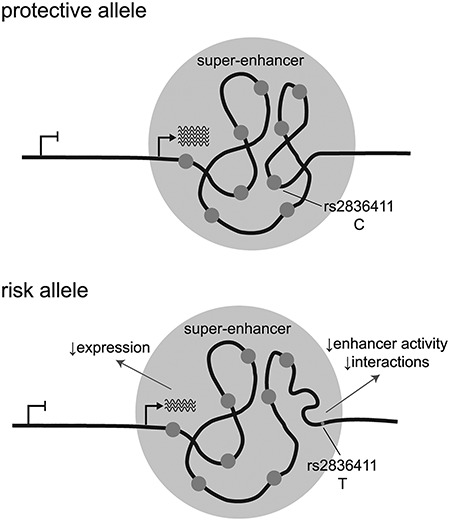
Hypothetical model of the functional mechanism by which rs2836411 could act. Rs2836411 is located in an enhancer element that is part of a super-enhancer downstream of ERG-P3. When ERG is expressed, the super-enhancer is active and forms a sub-domain structure. The risk allele (T) of rs2836411 reduces enhancer activity, correlates with reduced ERG expression and might alter interaction strength within the super-enhancer region.
In contrast to TADs, sub-TADs were shown to be more dynamic and tissue-specific (35,36). Furthermore, rs2836411 interacts mainly within the super-enhancer, which was shown to drive ERG expression previously (22,23). Super-enhancers are concatenated enhancers that drive the expression of key cell identity genes (21). Compared to regular enhancers, super-enhancers were found to mostly interact within their genomic location and to restrict super-enhancer activity to the genes within this region (36). The chromatin structure at rs2836411 and the presence of the super-enhancer are consistent with what has been reported for the majority of sub-domain and super-enhancer domains and further supports the finding that rs2836411 regulates ERG expression.
ERG is a TF crucial for the development of the vascular system, angiogenesis and vascular inflammation (15). Differential expression of ERG could contribute to AAA pathogenesis by affecting any of these processes. ERG is involved in the development of both arteries and veins, including the aorta (37), and developmental processes could contribute to the risk of developing aneurysmal disease in adulthood (38). ERG has also been shown to repress the inflammatory response of endothelial cells in atherosclerosis or upon inflammatory stimuli and plays a critical role in maintaining the endothelium in an anti-inflammatory state (39–42). ERG knockdown also increases endothelial cell permeability (43–45), which allows for infiltration of inflammatory cells in atherosclerotic lesions. A lower expression of ERG in individuals carrying the risk allele of rs2836411 might increase the inflammatory response in AAA, which could accelerate AAA formation.
Another molecular mechanism by which ERG can influence AAA formation is through its role in angiogenesis. In AAA, neovascularization occurs to supply nutrients to the outer aortic wall, which is inherent to disease progression. ERG regulates crucial pathways involved in angiogenesis, including endothelial cell migration and the balance between endothelial cell apoptosis and survival (45–50). We show here that in AAA tissue, ERG is expressed in endothelial cells lining the vasa vasorum. Reduced ERG expression in individuals carrying the risk allele for rs2836411 could affect angiogenesis. Haematopoietic progenitor cells, which express ERG, were found to have elevated numbers in atherosclerotic and AAA tissue, where they can play a role in angiogenesis, inflammation and smooth muscle cell accumulation (51–53). Differential ERG expression in haematopoietic progenitor cells might also affect AAA progression; however, we have not investigated the function of rs2836411 in haematopoietic progenitor cells in this study.
The expansion of the vasa vasorum during AAA progression distinguishes AAA from occlusive atherosclerotic diseases where the inflammatory reaction is contained within the tunica intima and the medial and adventitial structures are unaffected. As rs2836411 is specifically associated with AAA, it might be more likely that the mechanism by which ERG contributes to AAA formation is through either influencing the inflammation in the tunica media and adventitia through endothelial cell function, or the expansion of vasa vasorum, or a combination of both. Further work would be required to test this hypothesis. The data presented here set the scene for further investigating the role of ERG in AAA formation and progression.
As ERG is specifically associated with AAA, it may provide a novel gene that can be targeted for the development of AAA-specific therapeutic targets. Currently, AAAs are treated with medication that is used to treat other cardiovascular diseases, such as antihypertensive and lipid-lowering drugs (statins). These therapies have not always proven effective for the treatment of AAA (54–57), although the latest reports show that statin treatment does attenuate AAA growth (58,59). Nevertheless, there is a need for the development of AAA-specific medication, and ERG may be a potential novel target gene.
Materials and Methods
Cell culture
Primary HUVECs were obtained from Lonza, Basel, Zwitserland (CC-2519) and ATCC, City of Manasses, Virginia, United States (PCS-100-010 and PCS-100-013) and were grown in EGM™-2 BulletKit™ (CC-3162, Lonza) according to the manufacturer’s instructions. Three different primary HASMCs were obtained from Lonza (CC-2571) and were grown in SmGM™-2 BulletKit™ (CC-3182, Lonza) according to the manufacturer’s instructions. For both HUVECs and HASMCs, growth media were renewed every 2 days and cells were sub-cultured using 0.05% Trypsin-EDTA (Invitrogen, Carlsbad, California, United States) when reaching a confluence of 70–80%.
HepG2 cells were grown in MEMα (12561056, Life Technologies, Carlsbad, California, United States) supplemented with 10% fetal bovine serum (Moregate, Hamilton, New Zealand). HEK293 cells were grown in DMEM (11995, Life Technologies) supplemented with 10% fetal bovine serum (Moregate, New Zealand). All cells were grown at 37°C, 5% CO2.
Cloning
A DNA fragment harbouring rs2836411 was amplified from the genomic DNA of an individual heterozygous for rs2836411 using Platinum® Taq DNA Polymerase (Life Technologies) and the primers listed in Supplementary Material, Table S2. Amplicons were cloned into pCR®8/GW/TOPO entry vector and recombined into pGL4.23-GW (Addgene, Cambridge, Massachusetts, United States) using the Gateway® system (LR Clonase® Enzyme mix, Invitrogen). The constructs obtained were Sanger-sequenced to identify constructs containing the C or T allele for rs2836411. Plasmids used for transfections were isolated using the NucleoBond Xtra Midi endotoxin-free kit (Machery-Nagel, Düren, Germany).
Luciferase enhancer assay
One day prior to transfection, HUVEC, HepG2 or HEK293 cells were seeded at 8 × 103, 2.4 × 104 or 8 × 103 cells per well, respectively, in a 96-well plate in 200 μl media. Transfections were carried out with Lipofectamine 3000 (Invitrogen) using 0.2 μl of Lipofectamine 3000 reagent per well. Per well, 100 ng of each pGL4.23 construct and 4 ng of pRL-TK (Renilla; Promega, Madison, Wisconsin, United States) for HepG2 and HEK293 cells, or 1 ng of pRL-CMV (Promega) for HUVECs, were transfected. Luminescence was measured 18 (HUVECs) or 48 (HepG2 and HEK293) hours post transfection, using the Dual-Glo® Luciferase Assay System (Promega), on a Perkin Elmer (Waltham, Massachusetts, United States) Victor X4 plate reader. Prior to adding Dual-Glo reagents, media were removed from wells and 40 μl of a 1:1 (HUVECs) or 1:3 (HepG2 and HEK293) dilution of Dual-Glo reagents was used. Firefly luminescence was normalized to that of Renilla, averaged for 3 (HepG2 and HEK293) or 8 (HUVECs) technical replicates, and expressed relative to pGL4.23 lacking a gateway cassette.
Quantitative PCR
Total RNA from HUVECs and HASMCs was extracted using the Nucleospin® RNA Plus Kit (Machery Nagel) and 500 ng of extracted RNA was used to synthesize cDNA (qScript™ cDNA SuperMix, Quanta Biosciences™, Beverly, Massachusetts, United States). SYBR® Premix Ex Taq™ (Tli RNase H Plus, Takara Bio, Kusatsu, Shiga Prefecture, Japan) was used to amplify cDNA in 10 μl reactions in a 384-well plate (LightCycler®480 Multiwell Plate 384, Roche Diagnostics, Almere, The Netherlands) on the LightCycler 384 (Roche Diagnostics). Cp values were determined by the second derivative method using Roche LightCycler® 480 software. PCR primers are listed in Supplementary Material, Table S2.
4C-seq library preparation
4C-seq libraries were prepared as described previously (60,61) with modifications. The following libraries were prepared: two replicates and one control per cell line for HUVECs (Lonza CC-2519) heterozygous (CT) for rs2836411 and for HASMCs homozygous (CC) for rs2836411. For each cell line, genotypes for rs2836411 were identified by amplifying the DNA fragment from genomic DNA and Sanger sequencing.
Per library, 2.5 million cells were harvested by trypsinization to obtain a single-cell suspension and then cross-linked in 2% formaldehyde, 5% FBS and 1x PBS for 10 min at RT while rotating. Formaldehyde was quenched with a final concentration of 125 mM glycine for 5 min on ice while inverting several times. Cells were lysed (10 mM Tris pH 8.0, 10 mM NaCl, 0.2% NP-40, protease inhibitors) by resuspending in 2.5 ml ice-cold lysis buffer and incubating for 10 min on ice. Cell pellets were spun down, snap-frozen and stored at −80°C. A second lysis was performed in 10 ml ice-cold lysis buffer for 30 min on ice, followed by 10x strokes of a Dounce homogenizer. Nuclei were then resuspended in ice-cold PBS and transferred to Eppendorf tubes. Pellets were resuspended in 50 μl 0.05% SDS in MQ and incubated at 60°C for 10 min. To quench the SDS, 153.3 μl MQ and 16.7 μl of 15% Triton X-100 (final concentration of 1.14%) were added and the suspension was incubated at 37°C for 20 min, while breaking up aggregates every 5 min by resuspending. Nuclei were digested horizontally in a total volume of 600 μl with 400 units of MseI (R0525M, New England Biolabs [NEB], Ipswich, Massachusetts, United States) by incubating at 37°C overnight. While incubating, samples were mixed regularly by gently inverting the tube. MseI was inactivated by incubating at 65°C for 20 min. Ligation was performed (omitted for the controls) in a total volume of 2.5 ml using 1675 units T4 DNA ligase (M0202M, NEB) by incubating at 16°C overnight at 30 revolutions per minute (rpm). Samples were proteinase K (Invitrogen) treated and reverse-crosslinked overnight at 65°C. Samples were then treated with RNase A (Thermo Scientific, Waltham, Massachusetts, United States) at 37°C for 30 min. DNA was purified by 2× phenol/chloroform extraction and ethanol precipitated.
A second digestion was performed with 25 units CviQI (R0639L, NEB) by incubating at 25°C overnight at 30 rpm. The restriction enzyme was removed via phenol/chloroform extraction followed by ethanol precipitation. A second ligation was performed in the same way as the first ligation, in a total volume of 3.5 ml using 1675 units of ligase. DNA was ethanol precipitated with 70 μg glycogen.
Digestion and ligation efficiencies were checked after each step by running aliquots on an agarose gel. DNA concentrations were measured using a Qubit® 3.0 Fluorometer (Life Technologies) and Qubit® double-stranded DNA (dsDNA) Broad Range or High-Sensitivity Assay kits (Life Technologies).
For each replicate, a total of 1 μg in 5 × 25 μl reactions was amplified by PCR using Q5 Hot Start High-Fidelity 2X Master Mix (NEB). Bait primer sequences are listed in Supplementary Material, Table S2. PCR products were purified twice using the QIAquick PCR Purification kit (QIAGEN, Hilden, Germany) and Agencourt AMPure XP beads (Beckman-Coulter, Brea, California, United States). DNA concentrations were measured using a Qubit® 3.0 Fluorometer and were run on a 2100 Bioanalyser (Agilent Technologies, Santa Clara, California, United States) using a High Sensitivity DNA Kit (Agilent Technologies). Amplicons were mixed equally based on the concentration and ratio of demultiplexed 4C baits obtained from an in-house MiSeq run. Libraries were prepared using the Thruplex Kit (Takara Bio) with 10 ng of input DNA according to the manufacturer’s instructions, using 7 cycles of adapter amplification and 30 s of extension time. Libraries were mixed in equal concentrations and sequenced as 2 × 150 bp (paired-end) reads on a HiSeq X by Admera Health (South Plainfield, New Jersey, United States).
4C-seq data analysis
4C-seq data was analysed in command-line and the R statistical environment (http://www.R-project.org/) (62), and visualized using the University of California Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu/) using assembly hg19 (63,64). Baits were demultiplexed based on bait primer sequences up to and including the digestion site using grep, allowing 0 mismatches. Only read pairs that had the forward and reverse bait sequences in the correct orientation were selected. Adapter sequences and bases with a Phred quality score under 20 were then trimmed from the reads using fastq-mcf (65). Bait sequences up to, but excluding, the digestion site were trimmed from the reads using seqtk (https://github.com/lh3/seqtk). Quality of reads was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) (66).
Reads were mapped to the hg19 reference genome using Burrows-Wheeler Aligner (MEM algorithm) (67). Mapped reads were assigned to MseI digestion fragments using fourSig (68). In the replicate samples, bait and neighbouring fragments for which the read count was high in the control (unligated) libraries were removed (includes self-ligated and uncut reads). The running mean of windows of 21 successive fragments was calculated from the read per million-normalized read counts, which was obtained using fourSig (68).
Significant interaction calling was performed using fourSig with the following settings: window size of 3, 1000 iterations, false discovery rate (FDR) of 0.01, fdr.prob of 0.05 (which selects the top fifth percentile of interactions with a FDR < 0.01) and only included mappable fragments (68). Significant interactions were called for two regions: 1) the whole of chr21 and 2) within the ERG-P3 sub-domain at chr21:39730000-39 911 000 (see Supplementary Material, Figure S7). Significant interactions in both replicates were overlapped using the bedIntersect tool from UCSC (63). The significant interactions per replicate and the overlap between the two replicates are shown in Supplementary Material, Figure S7.
In the fourSig package, significant interactions can be categorized into three categories: 1) interactions that are significant after the reads from the fragment with the highest read count is removed, 2) interactions that are significant when the fragment with the highest read count is averaged to the read counts of the neighbouring fragments and 3) interactions that are significant only when all fragment read counts are included (68). For this study, only category 1 and 2 interactions that overlap between both replicates were included, as they are more likely to represent true interactions (because they span multiple fragments) and were previously shown to be more reproducible between replicates than single-fragment interactions (68).
RNAscope® in situ hybridization
Tissue from an 83-year-old male with an 8 cm AAA was obtained from an aneurysm repair surgery and was formalin-fixed O/N, paraffin-embedded and cut into 4 μm sections. The RNAscope® 2.5 HD Reagent Kit-RED kit (Advanced Cell Diagnostics, Newark, California, United States) was used according to the manufacturer’s instructions. Probes used were RNAscope® Probe-Hs-ERG, Negative Control Probe-DapB and Positive Control Probe-Hs-PPIB. Staining patterns were confirmed in two additional AAA samples from different individuals. Stained sections were scanned using the Aperio Scanscope CS digital pathology slide scanner (Leica Biosystems, Wetzlar, Germany).
ChIP-seq, DNase I, Hi-C, TF binding and eQTL data
Histone modification ChIP-seq, DNase I hypersensitivity and 15-state ChromHMM data was obtained from the ENCODE (69) and Roadmap Epigenomics (17) projects through the UCSC genome browser (https://genome.ucsc.edu/) using assembly hg19 (63,64) or through HaploReg (18). ENCODE donor identification numbers of the aortic biosamples used and the corresponding genotypes for rs2836411 are ENCDO845WKR (C/C), ENCDO451RUA (C/C), ENCDO793LXB (C/T), ENCDO271OUW (C/C) and ENCDO424HVB (C/T). Genotypes were identified from the vcf files of each donor. The genotype for 2 836 411 of the HUVECs used by ENCODE is unknown. Previously published Hi-C data in HUVEC, GM12878, HMEC, NHEK and IMR90 cells were obtained using the 3D genome browser (http://www.3dgenome.org) (61,70). TF-binding data were obtained using ReMap (71). EQTL data were obtained from GTEx (72).
TF-binding motif prediction changes
Haploreg was used to predict alterations in TF-binding affinities between the two alleles of rs2836411 (18). Findings were confirmed using the scan tool from JASPAR (http://jaspar.genereg.net/) (28).
Supplementary Material
Acknowledgements
HUVECs, HepG2 and HEK293 cells were kindly provided by Dr Rajesh Katare and Dr Martin Fronius, Dr Adele Woolley and Dr Heather Cunliffe (University of Otago, Dunedin, New Zealand), respectively.
The 4C-seq dataset is accessible through GEO accession number GSE137288.
Conflict of interest statement
The authors declare no conflict of interest.
Funding
Heart Foundation of New Zealand (post-doctoral research fellowship [1691] and small project grant [1804]); the Dunedin School of Medicine Dean’s Bequest fund; Health Research Council of New Zealand Grants [14-155, 17-402].
References
- 1. Stather P.W., Sidloff D.A., Rhema I.A., Choke E., Bown M.J. and Sayers R.D. (2014) A review of current reporting of abdominal aortic aneurysm mortality and prevalence in the literature. Eur. J. Vasc. Endovasc. Surg., 47, 240–242. [DOI] [PubMed] [Google Scholar]
- 2. Kent K.C., Zwolak R.M., Egorova N.N., Riles T.S., Manganaro A., Moskowitz A.J., Gelijns A.C. and Greco G. (2010) Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J. Vasc. Surg., 52, 539–548. [DOI] [PubMed] [Google Scholar]
- 3. Singh K., Bonaa K.H., Jacobsen B.K., Bjork L. and Solberg S. (2001) Prevalence of and risk factors for abdominal aortic aneurysms in a population-based study: The Tromso Study. Am. J. Epidemiol., 154, 236–244. [DOI] [PubMed] [Google Scholar]
- 4. Toghill B.J., Saratzis A. and Bown M.J. (2017) Abdominal aortic aneurysm-an independent disease to atherosclerosis? Cardiovasc. Pathol., 27, 71–75. [DOI] [PubMed] [Google Scholar]
- 5. Lederle F.A. (2012) The strange relationship between diabetes and abdominal aortic aneurysm. Eur. J. Vasc. Endovasc. Surg., 43, 254–256. [DOI] [PubMed] [Google Scholar]
- 6. Wahlgren C.M., Larsson E., Magnusson P.K., Hultgren R. and Swedenborg J. (2010) Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J. Vasc. Surg., 51, 3–7discussion 7. [DOI] [PubMed] [Google Scholar]
- 7. Jones G.T. (2011) The pathohistology of abdominal aortic aneurysm. In Grundmann R. (ed), Diagnosis, Screening and Treatment of Abdominal, Thoracoabdominal and Thoracic Aortic Aneurysms. InTech, Europe, Rijeka, Croatia, pp. 53–74. [Google Scholar]
- 8. Bown M.J., Jones G.T., Harrison S.C., Wright B.J., Bumpstead S., Baas A.F., Gretarsdottir S., Badger S.A., Bradley D.T., Burnand K. et al. (2011) Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am. J. Hum. Genet., 89, 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones G.T., Tromp G., Kuivaniemi H., Gretarsdottir S., Baas A.F., Giusti B., Strauss E., Van't Hof F.N., Webb T.R., Erdman R. et al. (2017) Meta-analysis of genome-wide association studies for abdominal aortic aneurysm identifies four new disease-specific risk loci. Circ. Res., 120, 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pinard A., Jones G.T. and Milewicz D.M. (2019) Genetics of thoracic and abdominal aortic diseases. Circ. Res., 124, 588–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spitz F. (2016) Gene regulation at a distance: from remote enhancers to 3D regulatory ensembles. Semin. Cell Dev. Biol., 57, 57–67. [DOI] [PubMed] [Google Scholar]
- 13. Schaub M.A., Boyle A.P., Kundaje A., Batzoglou S. and Snyder M. (2012) Linking disease associations with regulatory information in the human genome. Genome Res., 22, 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krijger P.H. and de Laat W. (2016) Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol., 17, 771-782. [DOI] [PubMed] [Google Scholar]
- 15. Shah A.V., Birdsey G.M. and Randi A.M. (2016) Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul. Pharmacol., 86, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M. et al. (2015) The UCSC Genome Browser database: 2015 update. Nucleic Acids Res., 43, D670–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roadmap Epigenomics C., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ward L.D. and Kellis M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khan A. and Zhang X. (2016) dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res., 44, D164–D171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A. and Young R.A. (2013) Super-enhancers in the control of cell identity and disease. Cell, 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I. and Young R.A. (2013) Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell, 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamamoto R., Kawahara M., Ito S., Satoh J., Tatsumi G., Hishizawa M., Suzuki T. and Andoh A. (2018) Selective dissociation between LSD1 and GFI1B by a LSD1 inhibitor NCD38 induces the activation of ERG super-enhancer in erythroleukemia cells. Oncotarget, 9, 21007–21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugino N., Kawahara M., Tatsumi G., Kanai A., Matsui H., Yamamoto R., Nagai Y., Fujii S., Shimazu Y., Hishizawa M. et al. (2017) A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia, 31, 2303–2314. [DOI] [PubMed] [Google Scholar]
- 24. Hogan N.T., Whalen M.B., Stolze L.K., Hadeli N.K., Lam M.T., Springstead J.R., Glass C.K. and Romanoski C.E. (2017) Transcriptional networks specifying homeostatic and inflammatory programs of gene expression in human aortic endothelial cells. Elife, 6, e22536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller C.L., Pjanic M., Wang T., Nguyen T., Cohain A., Lee J.D., Perisic L., Hedin U., Kundu R.K., Majmudar D. et al. (2016) Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun., 7, 12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cowper-Sal lari R., Zhang X., Wright J.B., Bailey S.D., Cole M.D., Eeckhoute J., Moore J.H. and Lupien M. (2012) Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet., 44, 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deplancke B., Alpern D. and Gardeux V. (2016) The genetics of transcription factor DNA binding variation. Cell, 166, 538–554. [DOI] [PubMed] [Google Scholar]
- 28. Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J.A., van der Lee R., Bessy A., Cheneby J., Kulkarni S.R., Tan G. et al. (2018) JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res., 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bjorkegren J.L.M., Kovacic J.C., Dudley J.T. and Schadt E.E. (2015) Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J. Am. Coll. Cardiol., 65, 830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smemo S., Tena J.J., Kim K.H., Gamazon E.R., Sakabe N.J., Gomez-Marin C., Aneas I., Credidio F.L., Sobreira D.R., Wasserman N.F. et al. (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature, 507, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gupta R.M., Hadaya J., Trehan A., Zekavat S.M., Roselli C., Klarin D., Emdin C.A., Hilvering C.R.E., Bianchi V., Mueller C. et al. (2017) A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell, 170, e515, 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carrere S., Verger A., Flourens A., Stehelin D. and Duterque-Coquillaud M. (1998) Erg proteins, transcription factors of the Ets family, form homo, heterodimers and ternary complexes via two distinct domains. Oncogene, 16, 3261–3268. [DOI] [PubMed] [Google Scholar]
- 33. Buttice G., Duterque-Coquillaud M., Basuyaux J.P., Carrere S., Kurkinen M. and Stehelin D. (1996) Erg, an Ets-family member, differentially regulates human collagenase1 (MMP1) and stromelysin1 (MMP3) gene expression by physically interacting with the Fos/Jun complex. Oncogene, 13, 2297–2306. [PubMed] [Google Scholar]
- 34. Greenwald W.W., Li H., Benaglio P., Jakubosky D., Matsui H., Schmitt A., Selvaraj S., D'Antonio M., D'Antonio-Chronowska A., Smith E.N. et al. (2019) Subtle changes in chromatin loop contact propensity are associated with differential gene regulation and expression. Nat. Commun., 10, 1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., Ong C.T., Hookway T.A., Guo C., Sun Y. et al. (2013) Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell, 153, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schujiers J., Lee T.I., Zhao K. et al. (2014) Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell, 159, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wythe J.D., Dang L.T., Devine W.P., Boudreau E., Artap S.T., He D., Schachterle W., Stainier D.Y., Oettgen P., Black B.L. et al. (2013) ETS factors regulate Vegf-dependent arterial specification. Dev. Cell, 26, 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Norman P.E. and Powell J.T. (2010) Site specificity of aneurysmal disease. Circulation, 121, 560–568. [DOI] [PubMed] [Google Scholar]
- 39. Sperone A., Dryden N.H., Birdsey G.M., Madden L., Johns M., Evans P.C., Mason J.C., Haskard D.O., Boyle J.J., Paleolog E.M. et al. (2011) The transcription factor Erg inhibits vascular inflammation by repressing NF-kappaB activation and proinflammatory gene expression in endothelial cells. Arterioscler. Thromb. Vasc. Biol., 31, 142–150. [DOI] [PubMed] [Google Scholar]
- 40. McLaughlin F., Ludbrook V.J., Kola I., Campbell C.J. and Randi A.M. (1999) Characterisation of the tumour necrosis factor (TNF)-(alpha) response elements in the human ICAM-2 promoter. J. Cell Sci., 112(Pt 24), 4695–4703. [DOI] [PubMed] [Google Scholar]
- 41. Yuan L., Nikolova-Krstevski V., Zhan Y., Kondo M., Bhasin M., Varghese L., Yano K., Carman C.V., Aird W.C. and Oettgen P. (2009) Antiinflammatory effects of the ETS factor ERG in endothelial cells are mediated through transcriptional repression of the interleukin-8 gene. Circ. Res., 104, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dryden N.H., Sperone A., Martin-Almedina S., Hannah R.L., Birdsey G.M., Khan S.T., Layhadi J.A., Mason J.C., Haskard D.O., Gottgens B. et al. (2012) The transcription factor Erg controls endothelial cell quiescence by repressing activity of nuclear factor (NF)-kappaB p65. J. Biol. Chem., 287, 12331–12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yuan L., Le Bras A., Sacharidou A., Itagaki K., Zhan Y., Kondo M., Carman C.V., Davis G.E., Aird W.C. and Oettgen P. (2012) ETS-related gene (ERG) controls endothelial cell permeability via transcriptional regulation of the claudin 5 (CLDN5) gene. J. Biol. Chem., 287, 6582–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shah A.V., Birdsey G.M., Peghaire C., Pitulescu M.E., Dufton N.P., Yang Y., Weinberg I., Osuna Almagro L., Payne L., Mason J.C. et al. (2017) The endothelial transcription factor ERG mediates Angiopoietin-1-dependent control of Notch signalling and vascular stability. Nat. Commun., 8, 16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Birdsey G.M., Shah A.V., Dufton N., Reynolds L.E., Osuna Almagro L., Yang Y., Aspalter I.M., Khan S.T., Mason J.C., Dejana E. et al. (2015) The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/beta-catenin signaling. Dev. Cell, 32, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Birdsey G.M., Dryden N.H., Shah A.V., Hannah R., Hall M.D., Haskard D.O., Parsons M., Mason J.C., Zvelebil M., Gottgens B. et al. (2012) The transcription factor erg regulates expression of histone deacetylase 6 and multiple pathways involved in endothelial cell migration and angiogenesis. Blood, 119, 894–903. [DOI] [PubMed] [Google Scholar]
- 47. Birdsey G.M., Dryden N.H., Amsellem V., Gebhardt F., Sahnan K., Haskard D.O., Dejana E., Mason J.C. and Randi A.M. (2008) Transcription factor erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood, 111, 3498–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baltzinger M., Mager-Heckel A.M. and Remy P. (1999) Xl erg: expression pattern and overexpression during development plead for a role in endothelial cell differentiation. Dev. Dyn., 216, 420–433. [DOI] [PubMed] [Google Scholar]
- 49. Liu F. and Patient R. (2008) Genome-wide analysis of the zebrafish ETS family identifies three genes required for hemangioblast differentiation or angiogenesis. Circ. Res., 103, 1147–1154. [DOI] [PubMed] [Google Scholar]
- 50. Han R., Pacifici M., Iwamoto M. and Trojanowska M. (2015) Endothelial Erg expression is required for embryogenesis and vascular integrity. Organogenesis, 11, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Torsney E., Mandal K., Halliday A., Jahangiri M. and Xu Q. (2007) Characterisation of progenitor cells in human atherosclerotic vessels. Atherosclerosis, 191, 259–264. [DOI] [PubMed] [Google Scholar]
- 52. Ryer E.J., Garvin R.P., Schworer C.M., Bernard-Eckroth K.R., Tromp G., Franklin D.P., Elmore J.R. and Kuivaniemi H. (2015) Proinflammatory role of stem cells in abdominal aortic aneurysms. J. Vasc. Surg., 62, 1303, e1304–1311. [DOI] [PubMed] [Google Scholar]
- 53. Zhang L., Issa Bhaloo S., Chen T., Zhou B. and Xu Q. (2018) Role of resident stem cells in vessel formation and arteriosclerosis. Circ. Res., 122, 1608–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salata K., Syed M., Hussain M.A., Eikelboom R., de Mestral C., Verma S. and Al-Omran M. (2018) Renin-angiotensin system blockade does not attenuate abdominal aortic aneurysm growth, rupture rate, or perioperative mortality after elective repair. J. Vasc. Surg., 67, 629-636e622. [DOI] [PubMed] [Google Scholar]
- 55. Kokje V.B., Hamming J.F. and Lindeman J.H. (2015) Editor's choice - pharmaceutical management of small abdominal aortic aneurysms: a systematic review of the clinical evidence. Eur. J. Vasc. Endovasc. Surg., 50, 702–713. [DOI] [PubMed] [Google Scholar]
- 56. Bergqvist D. (2011) Pharmacological interventions to attenuate the expansion of abdominal aortic aneurysm (AAA) - a systematic review. Eur. J. Vasc. Endovasc. Surg., 41, 663–667. [DOI] [PubMed] [Google Scholar]
- 57. Guessous I., Periard D., Lorenzetti D., Cornuz J. and Ghali W.A. (2008) The efficacy of pharmacotherapy for decreasing the expansion rate of abdominal aortic aneurysms: a systematic review and meta-analysis. PLoS One, 3, e1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salata K., Syed M., Hussain M.A., de Mestral C., Greco E., Mamdani M., Tu J.V., Forbes T.L., Bhatt D.L., Verma S. et al. (2018) Statins reduce abdominal aortic aneurysm growth, rupture, and Perioperative mortality: a systematic review and meta-analysis. J. Am. Heart Assoc., 7, e008657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang Q., Yang H., Lin Q., Hu M., Meng Y. and Qin X. (2018) Effect of Statin therapy on survival after abdominal aortic aneurysm repair: a systematic review and meta-analysis. World J. Surg., 42, 3443–3450. [DOI] [PubMed] [Google Scholar]
- 60.van de Werken H.J., de Vree P.J., Splinter E., Holwerda S.J., Klous P., de Wit E. and de Laat W. (2012) 4C technology: protocols and data analysis. Methods Enzymol., 513, 89–112. [DOI] [PubMed] [Google Scholar]
- 61. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.R Core Team. (2013) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 63. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M. and Haussler D. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. The Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 65. Aronesty E. (2011) Ea-utils: Command-Line Tools for Processing Biological Sequencing Data. ExpressionAnalysis, Durham, NC, Vol. 2011. [Google Scholar]
- 66. Andrews S. FastQC: A Quality Control tool for High Throughput Sequence Data.
- 67. Li H. and Durbin R. (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Williams R.L.,.J., Starmer J., Mugford J.W., Calabrese J.M., Mieczkowski P., Yee D. and Magnuson T. (2014) fourSig: a method for determining chromosomal interactions in 4C-Seq data. Nucleic Acids Res., 42, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rosenbloom K.R., Sloan C.A., Malladi V.S., Dreszer T.R., Learned K., Kirkup V.M., Wong M.C., Maddren M., Fang R., Heitner S.G. et al. (2013) ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res., 41, D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang Y., Song F., Zhang B., Zhang L., Xu J., Kuang D., Li D., Choudhary M.N.K., Li Y., Hu M. et al. (2018) The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol., 19, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cheneby J., Gheorghe M., Artufel M., Mathelier A. and Ballester B. (2018) ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res., 46, D267–D275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Consortium, G (2013) The genotype-tissue expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.