

Abstract
Missense mutations in the RNA exosome component exosome component 2 (EXOSC2), also known as ribosomal RNA-processing protein 4 (RRP4), were recently identified in two unrelated families with a novel syndrome known as Short stature, Hearing loss, Retinitis pigmentosa and distinctive Facies (SHRF, #OMIM 617763). Little is known about the mechanism of the SHRF pathogenesis. Here we have studied the effect of mutations in EXOSC2/RRP4 in patient-derived lymphoblasts, clustered regularly interspaced short palindromic repeats (CRISPR)-generated mutant fetal keratinocytes and Drosophila. We determined that human EXOSC2 is an essential gene and that the pathogenic G198D mutation prevents binding to other RNA exosome components, resulting in protein and complex instability and altered expression and/or activities of critical genes, including those in the autophagy pathway. In parallel, we generated multiple CRISPR knockouts of the fly rrp4 gene. Using these flies, as well as rrp4 mutants with Piggy Bac (PBac) transposon insertion in the 3′UTR and RNAi flies, we determined that fly rrp4 was also essential, that fly rrp4 phenotypes could be rescued by wild-type human EXOSC2 but not the pathogenic form and that fly rrp4 is critical for eye development and maintenance, muscle ultrastructure and wing vein development. We found that overexpression of the transcription factor MITF was sufficient to rescue the small eye and adult lethal phenotypes caused by rrp4 inhibition. The autophagy genes ATG1 and ATG17, which are regulated by MITF, had similar effect. Pharmacological stimulation of autophagy with rapamycin also rescued the lethality caused by rrp4 inactivation. Our results implicate defective autophagy in SHRF pathogenesis and suggest therapeutic strategies.
Introduction
The RNA exosome is a highly conserved complex found in yeast, Drosophila and humans. In bacteria, the degradosome complex is involved in similar processes in the processing of ribosome RNAs and degradation of mRNAs and non-coding RNAs (1). In eukaryotes, one of the exosome subunits Rrp4 was first discovered to have a role in 5.8 rRNA maturation in yeast through the exosome complex (2, 3). There are nine core components forming the RNA-binding cap structure and the ring channel for RNAs to pass through. Exosome subunits 1–3 (Csl4, Rrp4, Rrp40) make up the RNA-binding cap structure, and subunits 4–9 (Rrp41, 42, 43, 45, 46 and Mtr3) form the ring channel (4). One catalytically active ribonuclease, Dis3/Rrp44, associated with the ring structure at the base, is mainly localized in the cytoplasm. Another ribonuclease subunit, EXOSC10/Rrp6, is associated with the cap structure and mainly acts in the nucleus. The function of the RNA exosome has been closely linked to RNA metabolism (5). It is known to degrade a range of non-coding RNAs and improperly processed mRNAs, as well as maintain overall mRNA turnover. Disruption of any core exosome components in yeast results in cell cycle arrest and lethality (2).
Mutations in multiple subunits of the RNA exosome complex and their cofactors have been identified in patients with various neurological diseases in recent years. EXOSC3, EXOSC8 and EXOSC9 mutations have been found to cause pontocerebellar hypoplasia (PCH) (OMIM #614678, #616081, #618065) (6–9). Mutations in RBM7, a subunit of the nuclear exosome targeting complex that binds and carries RNAs to the exosome, have been identified in patients with a spinal muscular atrophy-like disorder (10). The pathogenic mechanisms and therapeutic treatments for these diseases remain underexplored. Model organisms promise to facilitate investigation into disease mechanisms. Studies of EXOSC3, 8, 9 and RBM7 loss-of-function models in zebrafish have revealed lethality and brain degeneration phenotypes comparable to PCH patients (6, 7, 11–13), but EXOSC2 has not been studied in any animal models.
Mutations in EXOSC2, one of the cap proteins in the RNA exosome, cause a novel and complex neurological disease called Short stature, Hearing loss, Retinitis pigmentosa and distinctive Facies (SHRF Syndrome, OMIM #617763) (11). SHRF patients exhibit childhood myopia, early onset retinitis pigmentosa, progressive sensorineural hearing loss, short stature, brachydactyly, a recognizable facial gestalt, premature skin aging and mild intellectual disability. EXOSC2 patients also have mild cerebellar atrophy (11). It should be emphasized that the EXOSC2 patient phenotype appears to be distinct from that of PCH patients, the molecular basis of which is currently unclear.
To date, sequencing of SHRF patients from two unrelated families has identified missense mutations in the EXOSC2 gene. Two of the patients have Gly30Val (G30V) homozygous mutations in the N-terminal domain, and one patient carries the above Gly30Val mutation in trans to Gly198Asp (G30V/G198D), with the G198D mutation located in the K Homology (KH) RNA-binding domain, named after the heterogeneous nuclear ribonucleoprotein K (11). The two affected residues are highly conserved evolutionarily. According to published protein structures, the G30V mutation appears to shorten the interatomic distance between this EXOSC2 residue and the neighboring amino acids of EXOSC4 and would require more intermolecular space and thus may conflict with the molecular surface of Asp154 of EXOSC4. The G198D mutation shortens the β-strand from six to four amino acids, thereby affecting the overall β-hairpin structure (11). However, the effects of these two mutations on exosome function and disease pathogenesis remain to be determined.
We hypothesize that EXOSC2-linked pathology is related to loss of RNA exosome function. The mutations in EXOSC2 may affect the binding affinity of the RNA exosome complex to specific mRNAs and result in deregulated RNA metabolism. Certain cell types may be more sensitive to such alterations. We addressed this question using human EXOSC2 patient-derived B-lymphoblast cells, mutant keratinocytes generated by CRISPR and multiple genetic manipulations in the model organism Drosophila. The use of patient B-lymphoblast cells permits an ex vivo analysis of mutant cells while genetic analysis in Drosophila allows comprehensive phenotypic analysis and genetic interaction studies that offer in-depth mechanistic insights and address deficits in our current understanding of SHRF pathogenesis.
Results
The G198D but not G30V mutation affects EXOSC2 protein stability and its interactions with other RNA exosome components
The SHRF patient-associated G30V and G198D mutations are located in the N-terminal and KH domains of EXOSC2, respectively (Fig. 1A). They may exert different pathogenic effects in EXOSC2-linked SHRF. To study the effect on protein expression by each of these mutations, we obtained blood samples of two SHRF subjects and three of their unaffected parents from two independent families (Fig. 1B). We used the EBV virus to transform their B cells into B-lymphoblast cell lines for further study. Western blot analysis revealed that the G30V mutation did not affect overall EXOSC2 protein levels, while cells possessing the G198D allele produced lower EXOSC2 levels, which was noticeable even in the healthy carriers of the G198D mutation (Fig. 1C). In parallel, we examined EXOSC3 and EXOSC10 proteins in patient-derived B-lymphoblast cells. Interestingly, EXOSC3 and EXOSC10 protein levels were much lower in G198D mutation-containing SHRF samples in contrast to the healthy parent samples or G30V mutation-containing SHRF samples (Fig. 1C). This suggests that the overall abundance of exosome in G198D mutation-containing SHRF patients is less than in normal subjects.
Figure 1.
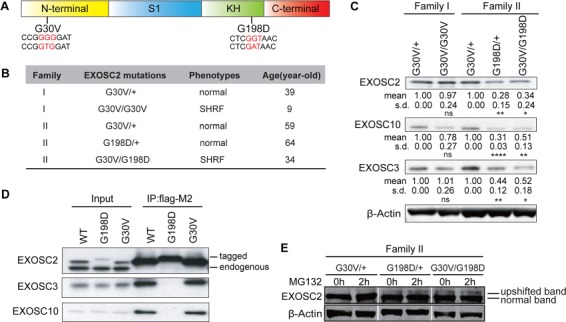
EXOSC2 mutations differentially affect protein stability and protein interactions. (A) EXOSC2 protein schematic domains and mutations. S1, KH domains are RNA-binding domains. G30V mutation (GGG to GTG) and G198D mutation (GGT to GAT) are labeled in red. (B) Family, genotype, phenotype and diagnosed age information of EXOSC2-mutated individuals. (C) Western blot of EXOSC2, EXOSC3 and EXOSC10 protein levels in B-lymphoblast cells from two EXOSC2 mutated families. Quantification of protein levels is provided under each blot. *, ** and **** indicate P < 0.05, P < 0.01 and P < 0.0001, respectively, in Student’s t test (n = 4). n.s, non-significant; s.d, standard deviation. (D) Co-immunoprecipitation between EXOSC2-FLAG and EXOSC3/EXOSC10 in HEK293T cells. (E) Western blot of EXOSC2 level from MG132-treated family II B-lymphoblasts. Note that although a much fainter upper band is visible in the G30V/+ samples, it is likely that this is some non-specific signal recognized by the EXOSC2 antibody rather than some unknown posttranslational modification of EXOSC2 that causes the upshift, since the tagged EXOSC2s do not produce such additional band (D).
To test whether the mutant EXOSC2 proteins can still interact with other RNA exosome subunits, we performed co-immunoprecipitation for the EXOSC2 protein. In order to study mutant proteins individually without interference from the others, we generated C-terminal FLAG-tagged EXOSC2 G30V and G198D expression plasmids and exogenously expressed them in HEK293T cells. To achieve physiological expression levels, we used an upstream Open Reading Frame (uORF) sequence to limit exogenous EXOSC2 levels to a level similar to the endogenous protein. With this approach, we found that EXOSC2-G30V could interact with the other RNA exosome components EXOSC3 and EXOSC10, but EXOSC2-G198D lost the ability to do so (Fig. 1D). We noted that EXOSC2-G198D displayed a slight upshift on Western blots in contrast to EXOSC2-WT and EXOSC2-G30V (Fig. 1D), presumably based on the negative charge conferred by the G to D mutation. Thus, EXOSC2-G198D but not EXOSC2-G30V fails to correctly interact with RNA exosome components.
Considering that epitope-tagged EXOSC2-G198D expressed in HEK293 cells exhibited an upshift in contrast to EXOSC2-WT and EXOSC2-G30V, we wished to test whether this was also the case in parent B-lymphoblast cells. A very faint upshifted band was visible in G198D/+ and G30V/G198D B-lymphoblast samples, and treatment of cells with the proteasome inhibitor MG132 led to more accumulation of the upshifted band in G198D/+ and G30V/G198D cells but not G30V/+ or G30V/G30V cells (Fig. 1E). This indicates that in G198D/+ and G30V/G198D samples, the normal-sized EXOSC2 bands came from the WT and G30V alleles, and that EXOSC2-G198D might be degraded by the ubiquitin proteasome system (UPS).
EXOSC2-G198D mutant is unstable and cannot rescue the decreased cell proliferation in EXOSC2-deficient keratinocytes
To further assess the stability of EXOSC2-G198D, we performed a protein stability assay of tagged exogenous proteins using cycloheximide to inhibit new protein synthesis, combined with MG132 treatment to inhibit the UPS. We found that EXOSC2-G198D had a much shorter half-life than EXOSC2-WT and -G30V (Fig. 2A and B), supporting that EXOSC2-G198D is inherently less stable than EXOSC2-WT and -G30V. Importantly, EXOSC2-G198D protein level could be partially restored by MG132 treatment (Fig. 2A), further supporting that EXOSC2-G198D stability is at least partially regulated by the UPS.
Figure 2.
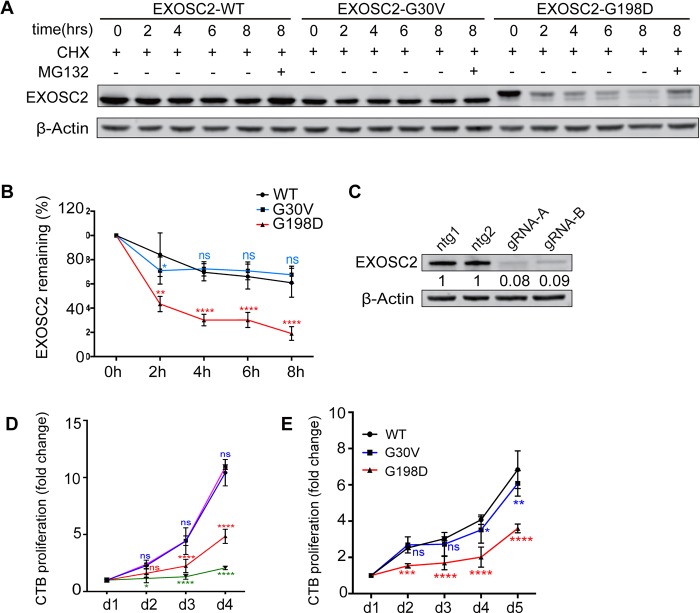
EXOSC2 mutations differentially affect protein stability and cell viability. (A) Western blot analysis of wild-type, G30V and G198D EXOSC2 in HEK293T cells treated with cycloheximide over an 8 h time-course and MG132 added at the 8th hour. (B) Quantification of EXOSC2 protein levels from (A), without MG132. *, ** and **** indicate P < 0.05, P < 0.01 and P < 0.0001, respectively, in two-way ANOVA test (n = 4). n.s, non-significant. (C) Western blot analysis of EXOSC2 knockdown by gRNA-A and gRNA-B using the optimized titer. Data represent at least two biological repeats. (D) CellTiter Blue assay of EXOSC2 KO cells. * and **** indicate P < 0.05 and P < 0.0001, respectively, in two-way ANOVA test (n = 3). n.s, non-significant. (E) Cell proliferation rate of EXOSC2 KO keratinocytes transfected with exogenous WT, G30V or G198D EXOSC2 showing differential rescuing abilities. *, ** *** and **** indicate P < 0.05, P < 0.01, P < 0.001 and P < 0.0001, respectively, in two-way ANOVA test (n = 3). n.s, non-significant.
An interesting and relatively unique phenotype in SHRF is premature skin aging. We were therefore curious whether the EXOSC2-G30V and -G198D mutations might affect cell proliferation in primary human keratinocytes, thus shedding light on the skin aging phenotype. Previously, four RNA exosome subunits were previously shown to exhibit dramatically decreased expression in differentiated keratinocytes, and knockdown of EXOSC9 significantly decreased keratinocyte proliferation (14). We therefore tried to knock out EXOSC2 by CRISPR in a pool of keratinocytes, testing nine different gRNAs targeting EXOSC2 exons 1–4 (Supplementary Material, Fig. S1A). Two gRNAs, gRNA-1 (gRNA-A) and gRNA-9 (gRNA-B), were selected for further targeting experiments (Supplementary Material, Fig. S1B). After titer adjustments, over 90% knockdown efficiency could be achieved 120 h post-lentivirus delivery of gRNA (Fig. 2C). Using the CellTiter-Blue (CTB) assay, we found that both gRNA-targeted cell lines exhibited a dramatically reduced proliferation rate (Fig. 2D). Next, we wished to test whether exogenous EXOSC2-WT, -G30V or -G198D could rescue the decreased cell proliferation caused by loss of endogenous EXOSC2. We transduced primary cultured keratinocytes with gRNA-resistant EXOSC2-WT, -G30V or -G198D and simultaneously knocked down endogenous EXOSC2 using gRNA-B. The CTB assay revealed that EXOSC2-G198D could not rescue the cell proliferation defect, whereas both EXOSC2-WT and -G30V could (Fig. 2E). Western blot analysis of exogenous EXOSC2 showed that EXOSC2-G198D protein levels were much lower than EXOSC2-WT or -G30V (Supplemental Material, Fig. S2A). This implies that EXOSC2-G198V is a non-functional protein and associated with compromised cell proliferation. In contrast, EXOSC2-G30V seems to be a weaker disease allele, and its overexpression may provide sufficient EXOSC2 function at least in this setting.
RNA-Seq analysis of patient lymphoblasts identifies deregulated transcripts in SHRF
Given our hypothesis that RNA metabolism is compromised in the context of the EXOSC2-G30V and -G198D mutations, we expected that certain target RNAs might be deregulated. Among the deregulated RNAs, we hypothesized that certain key RNAs might be responsible for some of the pathogenic effects contributing to SHRF. These may include important transcription factors regulating cell proliferation, development or survival. To identify the deregulated RNAs, we performed bulk RNA-Seq on five EXOSC2 mutated B-lymphoblast samples (Fig. 1B), along with three wild-type B-lymphoblast cell lines from control subjects. We found 46 RNAs that were significantly upregulated and 55 RNAs downregulated in patients in contrast to healthy individuals and parental carriers (Fig. 3A). Gene Ontology (GO) term analysis of upregulated RNAs revealed enrichment for genes involved in tissue development-related pathways (Fig. 3B). One of the most affected pathways was nervous system development, and six neural development-associated RNAs were found highly upregulated in patient samples. Among the six RNAs, three of them, sex determining region Y-box 5 (SOX5), annexin A3 (ANXA3) and endothelin receptor type B (EDNRB), were higher in the G30V/G198D patient sample than in G30V/G30V (Fig. 3C). We also noted that 42 out of 46 of the upregulated mRNAs were expressed at higher levels in the G30V/G198D sample in contrast to the G30V/G30V sample. This suggests that the G198D mutation may have stronger effect on RNA regulation. We next performed qRT-PCR to quantify the RNA levels in B-lymphoblasts and confirmed that SOX5, ANXA3 and EDNRB were more highly expressed in patient samples, supporting that they are candidate targets of EXOSC2 (Fig. 3D). SOX5 is a critical transcription factor in the SOXD family involved in the regulation of cartilage, neuron and melanocyte development, with increased or decreased SOX5 levels both having been shown to lead to developmental defects (15–21). SOX5 mutations have also been linked to late-onset Alzheimer’s disease and amyotrophic lateral sclerosis (22, 23).
Figure 3.
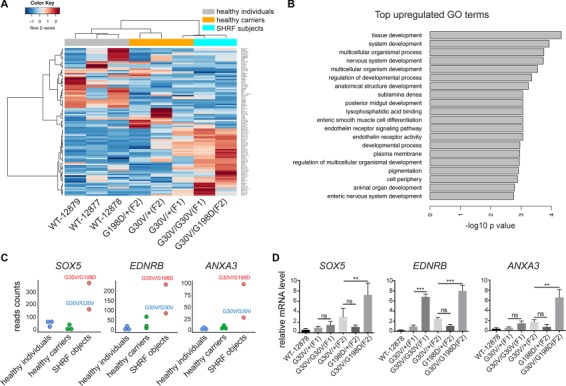
RNA-Seq analysis of B-lymphoblasts identifying several neurodevelopment factors upregulated in SHRF subjects. (A) Heat map of significantly altered transcripts in SHRF subjects only. (B) GO term analysis for significantly upregulated RNAs. (C) SOX5, ANXA3 and EDNRB mRNA levels in three groups—healthy individuals (WT), healthy carriers (GV/+, GD/+) and SHRF subjects (GV/GV, GV/GD). (D) qRT-PCR validation for SOX5, ANXA3 and EDNRB mRNAs in B-lymphoblast cells. **, and *** indicate P < 0.01, and P < 0.001, respectively, in Student’s t test for family I and one-way ANOVA test for family II (n = 3). n.s, non-significant.
To test whether the RNA level changes for SOX5 and other factors were specific to B-lymphoblast cells, we used the EXOSC2 knockout primary cultured keratinocytes to test the RNA levels of SOX5, EDNRB and ANXA3. Again, we chose keratinocytes based on the premature skin aging phenotypes in SHRF patients. qRT-PCR revealed that SOX5, EDNRB and ANXA3 mRNA levels were upregulated in two independent EXOSC2 CRISPR gRNA-infected cells in contrast to two non-targeting gRNA-infected cells (Supplementary Material, Fig. S2B). Thus, these EXOSC2-regulated mRNAs are candidate RNA targets shared in different cell types.
Using Drosophila melanogaster to study rrp4 function in vivo
To complement and build on our human cell culture studies, we wished to make use of the powerful tools available in Drosophila to study EXOSC2 function. A BLASTp search revealed a single ortholog of EXOSC2 in flies known as Rrp4, showing 61.5% identity and 80.9% similarity with human EXOSC2. We obtained an RNAi line for the rrp4 gene from the VDRC stock center and crossed it to the eyeless-GAL4 (ey-Gal4) line expressed in the optic lobes at early developmental stages, as well as the ubiquitously expressed daughterless-GAL4 (da-Gal4) driver. ey-Gal4-driven rrp4 knockdown resulted in small and rough eyes (Fig. 4A), sometimes with small ectopic appendages (data not shown). da-Gal4-driven rrp4 knockdown resulted in lethality at the first instar larval stage (Fig. 4B). To test the hypothesis of fly rrp4 being a counterpart of human EXOSC2, we attempted to rescue the rrp4 RNAi phenotype with a human EXOSC2 (UAS-EXOSC2-WT) transgene that we generated. The small eye phenotype of rrp4 knockdown could be partially rescued by this transgene, but it could not be rescued by a UAS-hEXOSC2 transgene containing the G30V mutation that we generated in parallel (Fig. 4C and D). Interestingly, hEXOSC2-G30V not only could not rescue the fly eye size but also induced a new glossy-eye phenotype (Fig. 4C), which has been seen in some fly mutants with mitochondrial defects (24, 25). Overexpression of hEXOSC2-WT alone with ey-Gal4 was partially toxic, resulting in some flies with small eyes and extra appendages; however, most of them had normal eyes (Supplementary Material, Fig. S3A). Overexpression of hEXOSC2-G30V alone did not show any visible phenotypes (Supplementary Material, Fig. S3A). Moreover, da-Gal4-driven expression of hEXOSC2-WT, but not hEXOSC2-G30V, led to pupal lethality. Together, these data support the hypothesis that human EXOSC2 is a functional homolog of fly Rrp4. Our results also indicate that the G30V mutant is non-functional and may gain toxicity when overexpressed under conditions of rrp4 deficiency and that excessive expression of human EXOSC2 is toxic to flies. Thus, unlike the situation in human keratinocytes where EXOSC2-G30V worked effectively in rescuing the cell proliferation defect in EXOSC2 KO cells, EXOSC2-G30V and -WT exert differential effects in rrp4 RNAi fly eyes. This might be based on a cell-type-specific effect, or different levels of endogenous EXOSC2/Rrp4 knockdown in the two systems. Using a commercial antibody that specifically recognized hEXOSC2 on western blots, we confirmed comparable expression of EXOSC2 protein from the hEXOSC2-WT and –G30V transgenes driven by da-Gal4 (Supplementary Material, Fig. S3B).
Figure 4.
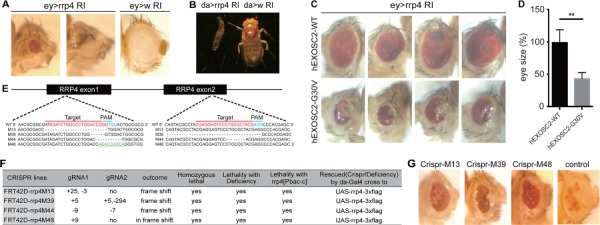
Phenotypic characterization of rrp4 loss-of-function fly mutants. (A) Microphotographs of fly eyes in which the fly rrp4 gene was knocked down by ey-Gal4>UAS-rrp4 RNAi (ey>rrp4 RI); ey-Gal4>UAS-white RNAi (ey>w RI) was used as a control. (B) Whole body images of animals with ubiquitous knockdown of the rrp4 gene in early development using da-Gal4>UAS-rrp4 RI (da>rrp4 RI) or control RNAi (da> w RI). (C) Microphotographs of ey-Gal4-driven rrp4 RNAi flies with co-overexpression of WT and G30V hEXOSC2. Genotypes are ey-Gal4>UAS-rrp4 RNAi; UAS-hEXOSC2 (top) and ey-Gal4>UAS-rrp4 RNAi; UAS-hEXOSC2-G30 V (bottom). (D) Quantification of the eye size of flies shown in (C). ** indicates P < 0.01 in Student’s t test (n = 4). (E) Diagram of deletion alleles from CRISPR-mediated knockout of the fly rrp4 gene. Green labeled sequences are inserted fragment in mutant 48 line. (F) Information on the deleted or inserted nucleotides in the CRISPR lines, and summary of their genetic interaction with various rrp4 alleles. (G) Microphotographs of fly eyes containing eyFLP-generated homozygous clones of the rrp4 M13, M39 or M48 CRISPR knockout alleles. The genotypes are ey-Flp/+; FRT42D, rrp4 M13/FRT42D, w+, cl (rrp4 M13 clone), ey-Flp/+; FRT42D, rrp4 M39/FRT42D, w+, cl (rrp4 M39 clone), or ey-Flp/+; FRT42D, rrp4 M48/FRT42D, w+, cl (rrp4 M48 clone). cl stands for cell lethal.
To generate rrp4 null flies, we used the CRISPR-Cas9 system to disrupt the rrp4 gene. Two gRNAs were designed to target the first and last exons of rrp4 (Fig. 4E). The candidate lines that failed to complement the rrp4 deficiency line were further tested using PCR and sequencing, resulting in the identification of three lines possessing frameshift deletions and one line containing a nine-nucleotide in-frame insertion (Fig. 4E and F). To rule out potential off-target effects, the fly UAS-rrp4 transgene was used to rescue the candidate CRISPR lines. All four candidate lines were homozygous lethal, and the lethality was rescued by da-Gal4-driven expression of the UAS-rrp4-3xFLAG transgenes (Fig. 4F, Supplementary Material, Fig. S3C). Overexpression of fly Rrp4 did not cause visible phenotypes in wild-type background. Next, we made mutant eye clones for three rrp4 CRISPR KO alleles (M13, M39 and M48), with the small white patches being homozygous mutant for the rrp4 CRISPR deletion. They all showed small-eye and glossy-eye phenotypes (Fig. 4G).
In addition, we made use of two independent PBac lines c21 and e463, with transposon insertions in rrp4 3′UTR and referred to as rrp4[c] and rrp4[e], respectively. They were mostly homozygous lethal with few escapers. Crossing them together to generate rrp4c/e flies resulted in variable phenotypes including ectopic wing veins (Supplementary Material, Fig. S3D). We therefore concluded that they are hypomorphic rrp4 alleles. Recombining them onto an FRT42D chromosome and crossing them to an eyFLP line showed that the recombined mutant eye clone had phenotypes similar to the abovementioned rrp4 CRISPR KO lines (Supplementary Material, Fig. S3E).
Next, we performed transmission electron microscopy (TEM) on rare rrp4[c] adult escaper flies in order to examine eye and muscle ultrastructure. We found a generally normal-appearing retina, while the lamina was severely disorganized with cartridges lacking appropriate photoreceptor structure and outlines (Fig. 5A). Examination of the muscle revealed disrupted the Z line and significantly increased mitochondrial mass and sarcomeric shortening (Fig. 5B and C), consistent with findings in patients with mutations in another Exosome subunit, EXOSC3 (26). To confirm our finding of rrp4-associated mitochondria defects in another genetic background, we examined the effect of rrp4 RNAi in the muscle driven by Mhc-GAL4 in the presence of a mitochondria-targeted GFP (mito-GFP) reporter and observed mitochondrial aggregation under confocal microscopy (Fig. 5D). Interestingly, we noted that the rrp4[c] mutant muscle was filled with a dysmorphic material compatible with glycogen lake formation and lysosomal rupture (Fig. 5E), as seen in lysosomal disorders like Pompe disease (29). Together, these data suggested a potential autophagy defect in rrp4-deficient flies.
Figure 5.
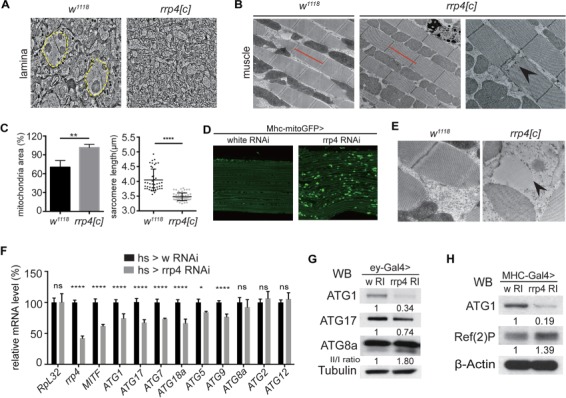
Autophagy is significantly affected in rrp4 loss-of-function flies. (A) Transmission electron microscopy (TEM) micrographs of the lamina eye tissues of control (w1118) and homozygous rrp4[c] escaper flies. Dotted yellow lines mark the photoreceptor cells. (B) Transmission electron microscopy (TEM) micrographs of the muscle of control (w1118) and homozygous rrp4[c] escaper flies. Red lines indicate the length of wild-type muscle sarcomere. Black arrowhead marks disrupted Z line. (C) Quantification of the mitochondrial area and sarcomere length in (B). ** and **** indicate P < 0.01 and P < 0.0001, respectively, in Student’s t test. The mitochondria and sarcomere data are obtained from three different flies, and more than 20 sarcomeres were measured. (D) Immunostaining of GFP-labeled mitochondria in Mhc-Gal4>rrp4 RNAi fly muscle. (E) TEM pictures of glycogen lake formation and lysosomal rupture (black arrow) in homozygous rrp4[c] escaper fly muscle. w1118 fly muscle is used as the control. (F) qRT-PCR of rrp4, MITF and ATG mRNAs in hs-Gal4>UAS-rrp4 RNAi third instar larvae. hs-Gal4>UAS-white RNAi (w RI) was used as control. * and **** indicate P < 0.05 and P < 0.0001, respectively, in one-way ANOVA test (n = 4). n.s, non-significant. (G) Western blots of ATG1, ATG17 and ATG8 proteins in ey-Gal4>rrp4 RNAi fly eye tissue. Expression levels were normalized with hs-Gal4>UAS-white RNAi (w RI) control. (H) Western blots of ATG1 and p62 in Mhc-Gal4>UAS-rrp4 RNAi fly muscle. Expression level was normalized with Mhc-Gal4>UAS-white RNAi (w RI) control.
Autophagy is significantly affected in rrp4 loss-of-function flies
SOX102F is the well-characterized fly ortholog of human SOX5 (27), the gene that was found overexpressed in SHRF patient cells in our RNA-Seq studies. The other two genes that were found upregulated in patient cells, ENDRB and ANXA3, do not have highly conserved homologs in flies. We therefore focused on analyzing SOX102F. When overexpressed in a wild-type background using either ey-Gal4 or da-Gal4, SOX102F caused embryonic lethal phenotypes, consistent with previous studies (35). On the other hand, knocking down SOX102F in rrp4 RNAi background did not significantly change the rrp4 RNAi-induced eye size reduction (data not shown) or lethality phenotypes (Supplementary Material, Fig. S4A). These results, together with the fact that loss- or gain-of-SOX102F function both affect Drosophila development (23, 35), led us to choose downstream effectors of SOX5/SOX102F to perform genetic interactions with Rrp4. A previous study in human melanoma cells found that SOX5 binds to the Microphthalmia-associated Transcription Factor (MITF) promoter and downregulates its expression, and this is important for melanoma development (28). Given that loss of MITF is associated with small eyes (microphthalmia), as seen with rrp4 knockdown, and that it is an important transcription factor for the autophagy pathway, we hypothesized that MITF and autophagy genes might be altered in rrp4 mutant flies. Moreover, if MITF is a key downstream effector of Rrp4, its overexpression might suppress rrp4 mutant phenotypes. We tested the first part of our hypothesis using heat shock (hs)-Gal4 to induce ubiquitous knockdown of rrp4 at the late third instar larval stage and found that this resulted in significant downregulation of MITF mRNA levels, when the rrp4 mRNA level was inhibited by 60% (Fig. 5F). Meanwhile, ATG1, ATG17, ATG7, ATG18a, ATG5 and ATG9 mRNAs all showed decreased expression in hs-Gal4 driven rrp4 knockdown flies, whereas ATG8a, ATG2 and ATG12 mRNA levels were not affected (Fig. 5E). ATG1 and ATG17 are core factors involved in initiating autophagy, and their protein levels were significantly downregulated in rrp4 RNAi flies (Fig. 5G and H). ATG1 mRNA level was also decreased in ey-Gal4-driven rrp4 knockdown eye tissue (Supplementary Material, Fig. S4B).
Autophagy activation can rescue rrp4 RNAi phenotypes in vivo
Considering that ey-Gal4-driven rrp4 RNAi results in semi-lethality, we took advantage of this sensitized background to test whether the mTORC1-inhibiting, autophagy-inducing drug rapamycin delivered to flies through feeding could improve viability. Confirming our hypothesis, we found that the viability of ey>rrp4 RNAi flies increased significantly in the presence of rapamycin (Fig. 6A). ATG1 protein level was restored by rapamycin (Fig. 6A), which was presumably based on inhibition of mTORC1-mediated phosphorylation and destabilization of ATG1, or some other unknown mechanism, by rapamycin. Next, we tested whether genetic manipulation of the autophagy pathway could also rescue rrp4 RNAi phenotypes. We found that MITF overexpression effectively rescued the eclosion rate and small eye phenotypes caused by rrp4 knockdown (Fig. 6B–D). UAS-yellow, which was used as a control for UAS-MITF, was unable to rescue the eye phenotype (Fig. 6B–D), demonstrating that the rescue by MITF is specific and not caused by titration of Gal4 protein by another UAS transgene. Overexpression of MITF itself had no obvious effect on eye morphology (Supplementary Material, Fig. S4C). These results support that the SOX5/MITF axis may play an important role in the pathogenesis associated with EXOSC2 loss of function.
Figure 6.
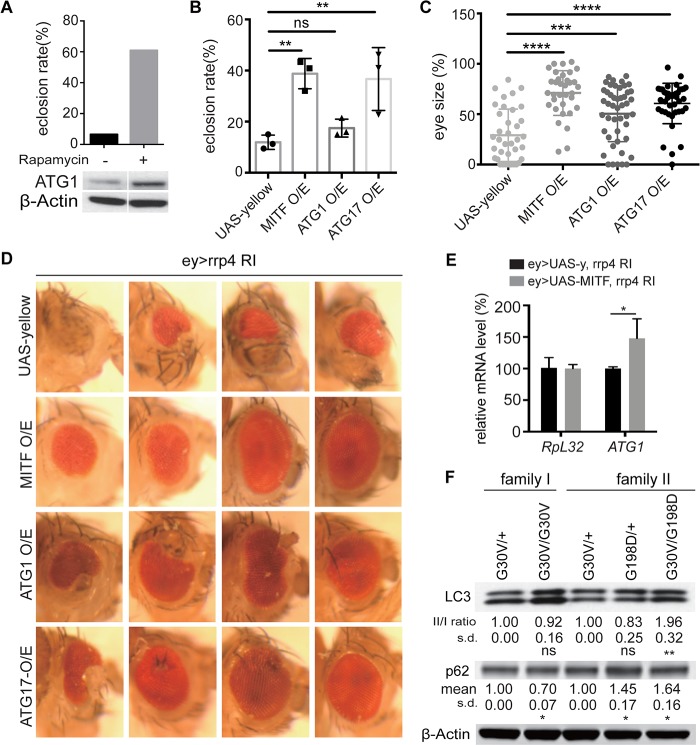
Rescue of rrp4 loss-of-function phenotypes by activation of autophagy. (A) Rescue of the eclosion rate of ey-Gal4>rrp4 RNAi flies by rapamycin treatment. Theoretical Mendelian rate is 50%. Western blot shows induction of ATG1 expression in rapamycin-treated ey-Gal4>UAS-rrp4 RNAi fly eye tissue. Data represent two biological repeats. (B) Rescue of the eclosion rate of ey-Gal4>UAS-rrp4 RNAi flies by UAS-MITF, UAS-ATG1 or UAS-ATG17 co-overexpression; UAS-yellow serves as control. The Mendelian rate is 50%. * and ** indicate P < 0.05 and P < 0.01, respectively, in one-way ANOVA test (n = 3). n.s, non-significant. Each group has 50–200 flies. (C) Quantification of ey-Gal4>UAS-rrp4 RNAi fly eye size after UAS-MITF, UAS-ATG1 or UAS-ATG17 co-overexpression. UAS-yellow serves as control. ** and **** indicate P < 0.01 and P < 0.0001, respectively, in one-way ANOVA test (n = 3). (D) Microphotographs of representative fly eyes of the indicated genotypes described previously. (E) qRT-PCR analysis of ATG1 mRNA level in ey-Gal4>UASrrp4 RNAi fly eyes rescued by UAS-MITF co-expression. * indicates P < 0.05 in Student’s t test (n = 3). (F) Western blot analysis of LC3 and p62 in B-lymphoblast cells from two EXOSC2-mutated families. Quantification of normalized LC3 II/I and p62 levels is shown under each panel. * and ** indicate P < 0.05 and P < 0.01, respectively, in Student’s t test (n = 4). n.s, non-significant; s.d, standard deviation.
By testing whether different autophagy-related factors might act as potential genetic modifiers of rrp4, we found that overexpression of ATG1 and another of the early autophagy-inducing factor FIP200/ATG17, which forms a complex with ATG1, could partially rescue the small eye phenotype as well as semi-lethality of rrp4 RNAi flies (Fig. 6B–D). Overexpression of FIP200/ATG17 alone did not produce any visible eye phenotypes, whereas overexpression of ATG1 occasionally resulted in flies with slightly irregular eyes (Supplementary Material, Fig. S4C). This might explain why the rescue effect of ATG1 was not as strong as MITF or ATG17, since its overexpression caused certain toxicity. Additionally, ATG1 mRNA level was restored in the MITF-rescued fly eyes (Fig. 6E), consistent with MITF acting through the ATG pathway to rescue rrp4 loss-of-function effect.
Autophagy defects in EXOSC2-deficient human cells
Seeking to confirm that there was an autophagy defect in EXOSC2 mutant human cells, we performed biochemical analysis of autophagy-related markers in EXOSC2 CRISPR KO keratinocytes and EXOSC2 siRNA-treated HEK293 cells. We observed increased ratio of lipidated LC3-II to unmodified LC3-I in these cells (Supplementary Material, Fig. S4D and E). In patient-derived B-lymphoblasts, we also found increased LC3-II/LC3-I ratio in G30V/G198D patient samples of family II (Fig. 6F). We also found that the autophagy receptor p62 level was increased (Fig. 6F). Similar effects were observed in rrp4 RNAi flies (Fig. 5G and H). The increased LC3-II/LC3-I ratio and increased p62 level are consistent with a slowdown of autophagic flux. Considering that the patient samples only possess missense mutations and the G30V mutant protein is quite stable, they might not show as strong autophagy defects as in EXOSC2/rrp4 KO or RNAi conditions. Together, these data support our hypothesis that there is an autophagy defect in EXOSC2/Rrp4-deficient mammalian cells and fly model.
Discussion
The EXOSC2-associated SHRF disease has many pleiotropic symptoms including hearing loss, retinitis pigmentosa, cognitive impairment, skeletal, facial and digital phenotypes and premature skin aging. This constellation of physical findings is intriguing and may provide clues to disease pathogenesis. One possibility is that such a collection of clinical features may be caused by dysregulation of a pleiotropic and critical developmental pathway. Supporting such a scenario, the RNA exosome components EXOSC8 and EXOSC9 were recently shown to affect erythroid proliferation and maturation through increased transcription by the transcription factor c-Kit (30).
In our study, we found SOX5, EDNRB and ANXA3 mRNAs upregulated in SHRF parent samples. These are all very critical neurodevelopmental factors in mammals. For example, SOX5 is one of the SOX transcription factors and is important for embryonic development, cell fate determination and chondrogenesis. SOX5 mutations have been found in patients with ALS and developmental delay, intellectual disability and dysmorphic features (31, 32). In prior fly studies, too much or too little SOX102F expression was both shown to cause neurodevelopmental defects. Loss of SOX102F in motor neurons results in a markedly reduced number of boutons and loss of bouton structures at the neuromuscular junction and also leads to significantly reduced locomotion function (23). Overexpression of SOX102F in Drosophila also results in small eyes and folded wing phenotypes (33) similar to what we saw with rrp4 knockdown. This suggests that fine-tuned SOX102F dosage is important for development and may explain why we did not see clear-cut genetic interactions between SOX102F and rrp4.
MITF is one of the transcription factors in the Microphthalmia family (including MITF, TFEB, TFE3 and TFEC), and it is a potential downstream target of SOX5. A previous study showed that SOX5 binds to the MITF promoter region and that knocking down SOX5 upregulates MITF (28). MITF mutations have been identified in a disease called Waardenburg syndrome 2A, which interestingly can also be caused by mutations in EDNRB (34), another of our top hit deregulated RNAs. Although there is no clear fly homolog of EDNRB, the pathogenic role of EDNRB and other upregulated human genes found in the microarray study in SHRF cannot be excluded. Loss of function of MITF has been shown to cause retinal degeneration in mice (35). In flies, expression of dominant-negative MITF in the larval disc interferes with eye development (36). We found decreased levels of MITF mRNA in rrp4 RNAi flies. Overexpression of MITF in the fly eye in wild-type background did not show any visible phenotypes. This makes it possible to perform genetic interactions with rrp4. We observed that MITF overexpression effectively rescued the phenotypes caused by rrp4 loss of function, allowing us to conclude that MITF is a genetic modifier for Rrp4. Further studies are required to test whether a SOX5-MITF axis exists and mediates Rrp4 function in Drosophila.
The Microphthalmia family of transcription factors (MITF, TFEB, TFE3 and TFEC) is well known to promote the transcription of lysosomal genes, autophagy genes and other targets such as genes involved in oxidative metabolism and the oxidative stress response. MiT/TFE transcription factors can also regulate the mTORC1 and Wnt/β-catenin pathways. In Drosophila, MITF is the only homolog of the four microphthalmia family members, and fly MITF can regulate the V-ATPase and the lysosomal-autophagic pathway (37). Consistently, we observed induction by MITF of the key autophagy regulator ATG1 (Fig. 6E). Autophagy has been linked to various neurodegenerative diseases (13, 38). One of the most interesting findings we made in the course of this study was that autophagy was affected in both patient cells and fly tissue in rrp4 mutants, with a dramatic glycogen lake phenotype and an aberrant mitochondrial morphology phenotype visible in hypomorphic fly mutants. The aberrant mitochondrial morphology is likely based on accumulation of defective organelles that fail to be removed by the autophagy machinery. Mitochondrial dysfunction is commonly linked with neuromuscular diseases, presumably based on higher demand for energy and thus mitochondrial function in neuromuscular tissues. Future studies using patient-derived muscle or neural tissues will test whether mitochondrial defects are common in SHRF.
In conclusion, we have identified the MITF/ATG1/17 pathway as significantly dysregulated in human patient cells, keratinocyte cell culture and in vivo fly models of EXOSC2/Rrp4 dysfunction and found that the mutant phenotypes could be suppressed by increasing autophagy activity through genetic and pharmaceutical means. The fact that overexpression of MITF, a key regulator of the lysosomal pathway or two autophagy-initiating factors ATG1 and ATG17/FIP200, as well as treatment with the autophagy-inducing drug rapamycin all proved beneficial to rrp4 knockdown flies, suggests that Rrp4 dysfunction may compromise different steps of the autophagy pathway, without blocking them, such that activation of different steps of the pathway is beneficial. Determining precisely how EXOSC2 mutations affect this pathway, either through MITF and/or ATG1/17 mRNA binding and degradation or some non-canonical mechanisms, remains an important future direction. Our findings suggest induction of autophagy as a potential therapeutic intervention for patients with SHRF or related disorders. This is an exciting area, as there is increasing interest in understanding the value of caloric restriction, intermittent fasting, resveratrol, polyphenols and other autophagy-inducing regimes in the treatment of a wide range of diseases, and the benefits may be broad (39–41).
Materials and Methods
Clinical information and ethics statement
Informed consent for the patient lymphoblasts and research work was obtained from the patients’ families.
Constructs for mammalian cell studies
EXOSC2-WT cDNA fragment was cloned into the plex-uORF-flag-HA-6His vector, and G30V, G198D mutations were generated by Q5 Site-Directed Mutagenesis Kit (NEB). Cas9 and gRNA plasmids information is described previously (42). Ten gRNAs designed to target exons 1–4 of EXOSC2 are as follows:
gRNA1: CCAAGATGGCGATGGAGATG; gRNA2: GATGGAGATGAGGCTTCCAG;
gRNA3: AAGCCTCTTAGCGAGAGACT; gRNA4: CTAGATGTTTCTTAGTGTCG;
gRNA5: CGACACTAAGAAACATCTAG; gRNA6: AGAAACATCTAGTGGTGCCG;
gRNA7: GAAACATCTAGTGGTGCCGG; gRNA8: TCCGTAGTGATTGTATCCCC;
gRNA9: TCAACAGAAGAGGTGGAAGG;
gRNA9 (gRNA B) proof sequence is TCAGCAAAAGCGATGGAAAG, the A in bold font destroys the PAM sequence and the underlined nucleotides block gRNA/DNA annealing.
Primers for mutagenesis:
G30V-mutagenesis-F: AGTGGTGCCGtGGGATACAATC;
G30V-mutagenesis-R: AGATGTTTCTTAGTGTCGCG;
G198D-mutagenesis-F: TGAGGCACCACATGGCAA;
G198D-mutagenesis-R: TGAGGCACCACATGGCAA;
Lentivirus generation, transduction and siRNA transfection
Lentiviruses were produced by transfecting HEK293T cells with 5 μg plex lentiviral expression construct, 5 μg pCMVΔ8.91 and 1.6 μg pUC MDG using Lipofectamine 2000 (Invitrogen) in a 10 cm plate. Viruses were harvested 48 h after transfection and filtered through 0.45 μm filters to remove cell debris and concentrated using a Lenti-X concentrator (Clontech). All cell lines were transduced overnight in 2 μg/ml polybrene and selected with 1 μg/ml puromycin or blasticidin after at least 24 h. The cas9-infected keratinocytes were selected with 1 μg/ml blasticidin for 3 days. gRNA virus transduction was performed afterwards, and cells were selected with 1 μg/ml puromycin for another 4–5 days. The non-transduced cells were used as controls, which all died after the selection.
EXOSC2 Stealth siRNAs were purchased from Invitrogen (HSS118715, HSS118716). We mixed them 1:1 and used 30 pm total siRNA with Lipofectamine RNAiMAX Reagent by following the standard protocol and harvested the cells 90 h after transfection.
B-Lymphoblast cell line generation and keratinocyte isolation
EXOSC2-mutated B cells were immortalized with Epstein–Barr virus (EBV), resulting in establishment of lymphoblast cell lines (LCLs) in vitro. B-LCLs were cultured in RPMI-1640 supplemented with 0.2 mm L-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin and 10% FBS. The defrosted cells were immediately recovered in 20% FBS RPMI-1640, with gradual decrease to 10% FBS RPMI-1640.
Primary human keratinocytes were isolated from fresh biopsy samples approved by the Stanford University institutional review board. Keratinocytes were isolated from fresh surgically discarded skin and cultured in Keratinocyte-SFM (Life Technologies 17005-142) and Medium 154 (Life Technologies M-154-500).
CellTiter Blue assay
Approximately 2000 cells were seeded into each well of a 24-well plate. From the second day, the medium was replaced by CellTiter Blue reagent premixed with full media (1:5 ratio). The cells were incubated for 2 h in the dark in a 37oC incubator and fluorescence measured at 560 nm/590 nm for the next 5 days.
Protein stability assay
Approximately 800 000 HEK293T cells were seeded into a 6-well plate overnight. The second day, cells were treated with 100 μg/ml cycloheximide (Sigma) for 0, 2, 4, 6 and 8 h. One well of cells were also treated with cycloheximide and 10 μm MG132 (Sigma) for 8 h. Cycloheximide solution was made in DMSO. Four biological replicates were performed. The protein expression level was measured by western blot using flag-M2 and beta-actin antibodies.
RNA sequencing and GO term analysis
RNA-Seq libraries were prepared by ribosome reduction method and sequenced on Illumina HiSeq 4000 (75 million reads/sample) at Yale Center for Genomic Analysis. Adaptors were trimmed by BBDuk. The RNA-Seq data was aligned by STAR, using hg19 and dm6 as reference genomes, respectively. Gene read counts were generated using RSEM. The Bioconductor R package edgeR was used to normalize and analyze the RNA read count matrix. Negative binomial generalized linear models (GLMs) with Wald tests were used for identifying genes with significant changes in expression between disease and healthy groups. The Benjamini–Hochberg method was used to adjust for multiple comparisons and assess false discovery rate (FDR). Gene ontology (GO) analysis was used to identify the GO terms significantly associated with the most affected genes.
Western blots
The following antibodies were used: EXOSC2 (ab156698, Abcam), EXOSC10 (ab95028, Abcam), EXOSC3 (ab235390, Abcam), β-actin (MAB1501, EMD Millipore), dATG1 and dATG17/FIP200 (gifts from Dr Jun Hee Lee), dATG8 (ABC974; Millipore), LC3 (2775, Cell Signaling), Ref2p/p62 (ab178440; Abcam), FLAG (M2, Sigma, F3165) and GFP (ab13970, Abcam). Protein levels were quantified using the LiCOR Odyssey system or ImageJ. Western blots were performed according to standard protocols, and most Western blots were repeated at least three times.
Co-immunoprecipitation
The FLAG M2 affinity gel (Sigma) was used for pull-down of Flag-tagged proteins. The beads were washed twice in 1× TBS buffer at room temperature. The cells were washed in PBS once and then lysed in lysis buffer (50 mm Tris HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1× Protease Inhibitor Cocktail) for 20 min at 4oC. The lysed cells were spun for 10 min at highest speed at 4oC to get rid of cell debris, and the cell lysates were then incubated with FLAG beads for 2 hours at 4oC. Beads were washed thrice with wash buffer (50 mm Tris–HCl, pH 7.5, 3 mm EDTA, 0.5% NP-40, 10% glycerol, 500 mm NaCl, 1× Protease Inhibitor Cocktail, 1 mm DTT), 10 min each time at room temperature. The beads were boiled in 1× SDS loading buffer (Invitrogen 4× SDS loading buffer with reducing reagent) for 10 min.
Light and electron microscopy
Light microscopy images were recorded digitally using a Nikon FDX-35 camera fitted on a Nikon Eclipse E1000M microscope and Spot Advanced software (Diagnostic Instruments). Immunofluorescent sections of Drosophila were imaged using a Nikon Eclipse Ti inverted laser scanning confocal microscope. Specimens for TEM were fixed in phosphate-buffered 2.5% glutaraldehyde and 2% paraformaldehyde, post-fixed in 1% osmium tetroxide and embedded in epoxy resin. Sections were stained with lead citrate and uranyl acetate and examined using a Hitachi EM-300 or H-7650 electron microscope (Hitachi, Tokyo, Japan).
Immunostaining and confocal imaging
All muscles are dissected in PBS, and fixed in PBST (1× PBS, 0.3% Triton) with 4% paraformaldehyde for 30 min at room temperature, followed by twice PBST washing, 10 min each. The dissected muscles were blocked in blocking buffer (PBST, 5% normal goat serum) for 1 h at room temperature. The muscles were then stained with primary GFP antibody diluted 1:5000 in blocking buffer overnight at 4oC. The second day, muscles were washed three times, 20 min each in PBST. The muscles were stained with fluorescence-labeled secondary antibody (1:200 dilution) in the dark overnight at 4oC, then washed three times in the dark at room temperature with PBST. Muscles were placed in Antifade (Invitrogen) and mounted on slides with circles drawn with nail polish to give physical support to prevent tissue crushing. Confocal imaging was performed with the Leica SP8 microscope.
Quantitative RT-PCR
All reverse transcription reactions were performed using the iScript cDNA Synthesis Kit (Bio-Rad). All qPCR reactions were performed using SYBR Green (Invitrogen) in LightCycler® 480 Instrument II (Roche Life Science). The qPCR primers are listed in the following.
Human-SOX5-F/R: GTGCCATAGGAGCTGTGCATG/GTTGGTCCTTCATTTGCCGAGC.
Human-EDNRB-F/R: CTGGCCATTTGGAGCTGAGA/CAGAACCACAGAGACCACCC.
Human-ANXA3-F/R: CCGCGCTTTGGATTAGTGTG/TGCTCAAAGTGGCCAGAGAG.
Human-RpL32-F/R: AGGCATTGACAACAGGGTTC/GTTGCACATCAGCAGCACTT.
Fly-RRP4-F/R: AGAACTACGACGGAGATCGG/GGAGCCCTCCTCAAAGATGTT.
Fly-MITF-F/R: AGGGGCTTATGGAATCGGAAT/GGCTCCACTTGACGAATCAATTA.
Fly-ATG1-F/R: GGATGAGCTACTGAAGAACACG/CAAACGCTTCTCAACGGC.
Fly-ATG2-F/R: CACATATATACCCGCCCACAG/CCACGTTCGCCTTCTTTG.
Fly-ATG5-F/R: ATGGAGCTGTGTGGTTCG/ATGTCCTCGGGGAACTTG.
Fly-ATG7-F/R: AAGAGGGTCATGCTTTCCTG/CTAATCCTCGTCGCTATCGG.
Fly-ATG9-F/R: CTGGAGAGGAGCACATACTGTC/ACCACAAAGAGGAACTGCATGA.
Fly-ATG8a-F/R: TCATTCCACCAACATCGGC/TCATTCCGATGCATCCC.
Fly-ATG12-F/R: GAACCTGGACCGTAGATCCC/TGATTATCTGATCCGGGGC.
Fly-ATG17-F/R: GAAGCTGCACAACATCCCG/GCTGAGTAGCACGACACTTGGFly-ATG18a-F/R: GGAGTGCGCAGGATGTG/ATAGTGGTCCTCCAGCCG.
Fly-RpL32-F/R: CTTCATCCGCCACCAGTC/CGACGCACTCTGTTGTCG.
The carefully collated FlyPrimerBank (https://www.flyrnai.org/flyprimerbank) was used for ordering primers for RT-PCR (43).
Fly strains
Unless indicated, stocks were obtained from the Bloomington Drosophila Stock Center (BDSC) and are listed in FlyBase (http://flybase.bio.indiana.edu). Flies were raised on standard cornmeal food at the indicated temperatures. The following stocks from the Bloomington Stock Center and Harvard Stock Center were used in this study:
PBac lines: rrp4-PBac-c21/CyO (rrp4[c]), rrp4-PBac-e463/CyO (rrp4[e]).
The rrp4 deficiency line: Df(2R)BSC661/CyO.
UAS lines: UAS-HA-MITF (96E), UAS-yellow (UAS-y+), UAS-mito-GFP.
Tool lines: FRT42D, nos-Cas9, Sp/CyO; Ly/TM6B.
Gal4 lines: eyeless-Gal4/CyO (abbreviated ey-Gal4/CyO), daughterless-Gal4 (da-Gal4), Myosin Heavy Chain-Gal4 (MHC-Gal4), hs-Gal4.
rrp4-KK/102375 RNAi line was obtained from the Vienna Drosophila RNAi Center.
Fly drug experiments
Drugs were dissolved in Formula 4–24 instant medium (Carolina Biological Supply Company) at the designated concentrations. Flies were crossed in normal food vials for at least 1 day to guarantee they had mated before moving them to drug-treated food. Vials were kept at a 25-centigrade incubator, and the progeny were scored for the first 4–5 days after eclosion. The drug rapamycin (2 μm, Sigma) was used.
Generation of UAS transgenic flies and rrp4 CRISPR-KO flies
A FLAG-tagged and untagged construct for fly rrp4 was cloned into the UAS-attB construct using Gibson Assembly kit (NEB) and a full-length rrp4 cDNA (Drosophila Genomics Resource Center, stock 2892) and sent to BestGene for injection in the VK27 attP insertion site. A pUAS-attB-hEXOSC2 construct with or without the G30V mutation was similarly generated using a full-length human EXOSC2 cDNA (GE Dharmacon, MHS6278-202825962) and also injected in the VK27 site.
Two gRNA insertions were designed in the pCDF4 plasmid. These two gRNAs target the beginning and end of exons 1 and 2 of fly rrp4.
gRNA1 sequence: AGATCTGGCCCTGGACCGGGTGG;
gRNA2 sequence: CGAGGAGTCCCTGCGCTACGAGG.
The plasmid was used for fly embryo injection, and the vermilion marker was used for clone selection. Transgenic flies were generated at BestGene, Inc. FRT42D, nos-Cas9 and gRNA flies were combined in a single fly to generate the EXOSC2 deletions in the female germline. Next, the double-balanced line Sp/CyO; Ly/TM6B was used to remove the gRNA and Cas9 chromosomes, and 70 male flies were used to select for targeting events by further single male double-balancing crosses. Candidate deletion events were selected based on homozygous lethality and failure of complementation when crossed to deficiency chromosome that removes rrp4. The molecular lesions induced in the CRISPR alleles were determined by PCR and DNA sequencing.
Clonal analysis
To perform mosaic analysis, confirmed FRT42D rrp4-crispr/Cyo flies were crossed to ey-FLP; FRT42D w+ cl/Cyo, Kr-GFP flies to generate the final ey-FLP/+; and FRT42D rrp4-crispr/FRT42D w+ cl genotyped flies. To perform clonal analysis with rrp4-PBac alleles, we used an isogenized homozygous line with the following genotype: w; FRT42D, which has white eyes. We crossed this line to w; rrp4[PBac]/CyO to generate female virgins with the genotype w/w; FRT42D/rrp4[PBac], by selecting against the Cyo balancer chromosome. As the rrp4[PBac] insertion has an orange eye color, the w/w; FRT42D/rrp4[PBac] flies have orange eyes. We next crossed these flies to w; Sp/CyO males. As recombination occurs in females, the progeny will have recombination events between FRT42D and rrp4[PBac] insertion. We then identified approximately 40 male orange-eyed CyO progenies and crossed them again individually to virgin balancer flies (w; Sp/CyO) and generated stocks from them (selecting against the Sp phenotype). These 40 stocks were all orange eyes, meaning that they all had the rrp4[PBac] transgene. In order to determine if they had the FRT42D site as well, we crossed virgins of the genotype w eyFLP; FRT42D w+ cl/CyO to these flies (w; FRT42D? rrp4[PBac]/CyO). Only those crosses that had progeny with clones, i.e. red and orange patches in their eyes, red from the w+ and orange from the rrp4[PBac], indicated that their parent stock had both the FRT42D and the PBac of interest. The orange patches are homozygous mutant for the rrp4[PBac] insertion. We kept these stocks for subsequent analysis. The numbers of male orange-eyed CyO progeny used were based on the estimated recombination distance between the FRT42D site and the rrp4 locus. FRT42D is located at a recombination map location of 2–55 and rrp4 at 2–103 (FlyBase), indicating that they are approximately 48 cM apart, indicating that they have an almost 50% recombination rate. We used 40 because we knew that 20 of them would be recombinants, but we also wanted to choose the healthiest stock for all future experiments.
Statistical analysis
Statistical analysis was performed using Excel (Microsoft) and Prism (GraphPad). Unpaired two-tailed Student t tests, one-way ANOVA and two-way ANOVA were used. Standard deviation is shown as s.d. Gene alignment was performed using NCBI’s Blast program.
Funding
National Institutes of Health (MH111234 and NS084412 to B.L.)
Supplementary Material
Acknowledgements
We gratefully acknowledge the patients and their family members who participated in this study. We would like to thank Dr Ching-Yi Chen (UAB), Jun Hee Lee (Korea Advanced Institute of Science and Technology), Guang-Chao Chen (National Taiwan University), Gabor Juhasz (Eötvös Loránd University), Thomas Lloyd (Johns Hopkins), Jorg Hohfeld (University of Bonn), Wei Wei (Stanford), Robert Tanguay (Université Laval), Francesca Pignoni (SUNY), Martine Simonelig, Leonard Rabinow (Harvard), Bill Saxton (UC-Santa Cruz), Paul Lasko (University of Montreal) and Hugo Bellen (BCM) for the reagents. We would like to thank our interns for their joyful assistance, particularly Julianna Glafkides and Bonnie Yu. We would like to thank the Lu Lab members, Can Cenik, Cain Yam (BestGene), Molly Jia (GenScript), Su Guo (UCSF), Xinnan Wang (Stanford), Gerald Tiu (Stanford), Kotaro Fujii (Stanford), Shinya Yamamoto (BCM), Chandresh Gajera (Stanford) and Pankaj Kapahi (Buck Institute) for the helpful discussions. We would like to thank the Pathology Department of Stanford School of Medicine, and in particular the Neuropathology lab, especially Dr Hannes Vogel, Martin Estrada and Amarjeet Grewala, for their assistance.
Conflict of Interest statement. The authors declare no conflict of interest.
References
- 1. Cho K.H. (2017) The structure and function of the gram-positive bacterial RNA degradosome. Front. Microbiol., 8, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mitchell P., Petfalski E., Shevchenko A., Mann M. and Tollervey D. (1997) The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′-->5′ exoribonucleases. Cell, 91, 457–466. [DOI] [PubMed] [Google Scholar]
- 3. Mitchell P., Petfalski E. and Tollervey D. (1996) The 3′ end of yeast 5.8S rRNA is generated by an exonuclease processing mechanism. Genes Dev., 10, 502–513. [DOI] [PubMed] [Google Scholar]
- 4. Schneider C. and Tollervey D. (2013) Threading the barrel of the RNA exosome. Trends Biochem. Sci., 38, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogami K., Chen Y. and Manley J.L. (2018) RNA surveillance by the nuclear RNA exosome: mechanisms and significance. Noncoding RNA, 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boczonadi V., Muller J.S., Pyle A., Munkley J., Dor T., Quartararo J., Ferrero I., Karcagi V., Giunta M., Polvikoski T. et al. (2014) EXOSC8 mutations alter mRNA metabolism and cause hypomyelination with spinal muscular atrophy and cerebellar hypoplasia. Nat. Commun., 5, 4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burns D.T., Donkervoort S., Muller J.S., Knierim E., Bharucha-Goebel D., Faqeih E.A., Bell S.K., AlFaifi A.Y., Monies D., Millan F. et al. (2018) Variants in EXOSC9 disrupt the RNA exosome and result in cerebellar atrophy with spinal motor neuronopathy. Am. J. Hum. Genet., 102, 858–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eggens V.R., Barth P.G., Niermeijer J.M., Berg J.N., Darin N., Dixit A., Fluss J., Foulds N., Fowler D., Hortobagyi T. et al. (2014) EXOSC3 mutations in pontocerebellar hypoplasia type 1: novel mutations and genotype-phenotype correlations. Orphanet. J. Rare Dis., 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eggens V.R.C., Barth P.G. and Baas F. (1993-2019) EXOSC3-related pontocerebellar hypoplasia In Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K. and Amemiya A. (eds), GeneReviews [Internet]. University of Washington, Seattle, WA. [PubMed] [Google Scholar]
- 10. Giunta M., Edvardson S., Xu Y., Schuelke M., Gomez-Duran A., Boczonadi V., Elpeleg O., Muller J.S. and Horvath R. (2016) Altered RNA metabolism due to a homozygous RBM7 mutation in a patient with spinal motor neuropathy. Hum. Mol. Genet, 25, 2985–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di N., Neuhann T., Kahlert A.K., Klink B., Hackmann K., Neuhann I., Novotna B., Schallner J., Krause C., Glass I.A. et al. (2016) Mutations in EXOSC2 are associated with a novel syndrome characterised by retinitis pigmentosa, progressive hearing loss, premature ageing, short stature, mild intellectual disability and distinctive gestalt. J. Med. Genet, 53, 419–425. [DOI] [PubMed] [Google Scholar]
- 12. Januszyk K. and Lima C.D. (2014) The eukaryotic RNA exosome. Curr. Opin. Stru. Biol, 24, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jaiswal M., Sandoval H., Zhang K., Bayat V. and Bellen H.J. (2012) Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu. Rev. Genet, 46, 371–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mistry D.S., Chen Y. and Sen G.L. (2012) Progenitor function in self-renewing human epidermis is maintained by the exosome. Cell Stem Cell, 11, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nishimura R., Hata K., Takahata Y., Murakami T., Nakamura E. and Yagi H. (2017) Regulation of cartilage development and diseases by transcription factors. J Bone Metab., 24, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu C.F., Samsa W.E., Zhou G. and Lefebvre V. (2017) Transcriptional control of chondrocyte specification and differentiation. Sem. Cell Dev. Biol., 62, 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ji E.H. and Kim J. (2016) SoxD transcription factors: multifaceted players of neural development. Int. J Stem. Cells, 9, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Akiyama H. and Lefebvre V. (2011) Unraveling the transcriptional regulatory machinery in chondrogenesis. J Bone Miner. Metab., 29, 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harris M.L., Baxter L.L., Loftus S.K. and Pavan W.J. (2010) Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res., 23, 496–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lefebvre V. (2010) The SoxD transcription factors--Sox5, Sox6, and Sox13--are key cell fate modulators. Int. J. Biochem. Cell Biol., 42, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leone D.P., Srinivasan K., Chen B., Alcamo E. and McConnell S.K. (2008) The determination of projection neuron identity in the developing cerebral cortex. Curr. Opin. Neurobiol., 18, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones A.R., Troakes C., King A., Sahni V., De S., Bossers K., Papouli E., Mirza M., Al-Sarraj S., Shaw C.E. et al. (2015) Stratified gene expression analysis identifies major amyotrophic lateral sclerosis genes. Neurobiol. Aging, 36(2006), e2001–e2009. [DOI] [PubMed] [Google Scholar]
- 23. Li A., Hooli B., Mullin K., Tate R.E., Bubnys A., Kirchner R., Chapman B., Hofmann O., Hide W. and Tanzi R.E. (2017) Silencing of the Drosophila ortholog of SOX5 leads to abnormal neuronal development and behavioral impairment. Hum. Mol. Genet., 26, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yarosh W., Monserrate J., Tong J.J., Tse S., Le P.K., Nguyen K., Brachmann C.B., Wallace D.C. and Huang T. (2008) The molecular mechanisms of OPA1-mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet., 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Olahova M., Yoon W.H., Thompson K., Jangam S., Fernandez L., Davidson J.M., Kyle J.E., Grove M.E., Fisk D.G., Kohler J.N. et al. (2018) Biallelic mutations in ATP5F1D, which encodes a subunit of ATP synthase, cause a metabolic disorder. Am. J. Hum. Genet., 102, 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pinto M.M., Monges S., Malfatti E., Lubieniecki F., Lornage X., Alias L., Labasse C., Madelaine A., Fardeau M., Laporte J. et al. (2019) Sarcomeric disorganization and nemaline bodies in muscle biopsies of patients with EXOSC3-related type 1 pontocerebellar hypoplasia. Muscle Nerve, 59, 137–141. [DOI] [PubMed] [Google Scholar]
- 27. Fukuda T., Ewan L., Bauer M., Mattaliano R.J., Zaal K., Ralston E., Plotz P.H. and Raben N. (2006) Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Annal. Neurol., 59, 700–708. [DOI] [PubMed] [Google Scholar]
- 28. Li A., Ahsen O.O., Liu J.J., Du C., McKee M.L., Yang Y., Wasco W., Newton-Cheh C.H., O'Donnell C.J., Fujimoto J.G. et al. (2013) Silencing of the Drosophila ortholog of SOX5 in heart leads to cardiac dysfunction as detected by optical coherence tomography. Hum. Mol. Genet., 22, 3798–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kordass T., Weber C.E., Oswald M., Ast V., Bernhardt M., Novak D., Utikal J., Eichmuller S.B. and Konig R. (2016) SOX5 is involved in balanced MITF regulation in human melanoma cells. BMC Med. Genom., 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McIver S.C., Katsumura K.R., Davids E., Liu P., Kang Y.A., Yang D. and Bresnick E.H. (2016) Exosome complex orchestrates developmental signaling to balance proliferation and differentiation during erythropoiesis. eLife, 5, e17877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Daoud H., Valdmanis P.N., Gros-Louis F., Belzil V., Spiegelman D., Henrion E., Diallo O., Desjarlais A., Gauthier J., Camu W. et al. (2011) Resequencing of 29 candidate genes in patients with familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol., 68, 587–593. [DOI] [PubMed] [Google Scholar]
- 32. Lamb A.N., Rosenfeld J.A., Neill N.J., Talkowski M.E., Blumenthal I., Girirajan S., Keelean-Fuller D., Fan Z., Pouncey J., Stevens C. et al. (2012) Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum. Mut., 33, 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singari S., Javeed N., Tardi N.J., Marada S., Carlson J.C., Kirk S., Thorn J.M. and Edwards K.A. (2014) Inducible protein traps with dominant phenotypes for functional analysis of the Drosophila genome. Genetics, 196, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Attie T., Till M., Pelet A., Amiel J., Edery P., Boutrand L., Munnich A. and Lyonnet S. (1995) Mutation of the endothelin-receptor B gene in Waardenburg-Hirschsprung disease. Hum. Mol. Genet., 4, 2407–2409. [DOI] [PubMed] [Google Scholar]
- 35. Chen Y., Yang J., Geng H., Li L., Li J., Cheng B., Ma X., Li H. and Hou L. (2019) Photoreceptor degeneration in microphthalmia (Mitf) mice: partial rescue by pigment epithelium-derived factor. Dis. Model Mech., 12, dmm035642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hallsson J.H., Haflidadottir B.S., Stivers C., Odenwald W., Arnheiter H., Pignoni F. and Steingrimsson E. (2004) The basic helix-loop-helix leucine zipper transcription factor Mitf is conserved in Drosophila and functions in eye development. Genetics, 167, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bouche V., Espinosa A.P., Leone L., Sardiello M., Ballabio A. and Botas J. (2016) Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy, 12, 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deal S.L. and Yamamoto S. (2018) Unraveling novel mechanisms of neurodegeneration through a large-scale forward genetic screen in drosophila. Front. Genet., 9, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Byun S., Lee E. and Lee K.W. (2017) Therapeutic implications of autophagy inducers in immunological disorders, infection, and cancer. Int. J Mol. Sci., 18,. pii, E1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kou X. and Chen N. (2017) Resveratrol as a natural autophagy regulator for prevention and treatment of Alzheimer’s disease. Nutrients, 9, pii: E927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bagherniya M., Butler A.E., Barreto G.E. and Sahebkar A. (2018) The effect of fasting or calorie restriction on autophagy induction: a review of the literature. Ageing Res. Rev., 47, 183–197. [DOI] [PubMed] [Google Scholar]
- 42. Rubin A.J., Parker K.R., Satpathy A.T., Qi Y., Wu B., Ong A.J., Mumbach M.R., Ji A.L., Kim D.S., Cho S.W. et al. (2019) Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell, 176, e317, 361–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu Y., Sopko R., Foos M., Kelley C., Flockhart I., Ammeux N., Wang X., Perkins L., Perrimon N. and Mohr S.E. (2013) FlyPrimerBank: an online database for Drosophila melanogaster gene expression analysis and knockdown evaluation of RNAi reagents. G3, 3, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.