

Abstract
Incomplete maternal vascular responses to pregnancy contribute to pregnancy complications including intrauterine growth restriction (IUGR) and preeclampsia. We aimed to characterize maternal vascular dysfunction in a murine model of fetal growth restriction as an approach toward identifying targetable pathways for improving pregnancy outcomes. We utilized a murine model of late-gestation hypoxia-induced IUGR that reduced E18.5 fetal weight by 34%. Contrary to our hypothesis, uterine artery blood flow as measured in vivo by Doppler ultrasound was increased in mice housed under hypobaric hypoxia (385 mmHg; 5500 m) vs normoxia (760 mmHg; 0 m). Using wire myography, uterine arteries isolated from hypoxic mice had similar vasodilator responses to the two activators A769662 and acetylcholine as those from normoxic mice, although the contribution of an increase in nitric oxide production to uterine artery vasodilation was reduced in the hypoxic vs normoxic groups. Vasoconstrictor responses to phenylephrine and potassium chloride were unaltered by hypoxia. The levels of activated adenosine monophosphate-activated protein kinase (AMPK) were reduced with hypoxia in both the uterine artery and placenta as measured by western blot and immunohistochemistry. We concluded that the rise in uterine artery blood flow may be compensatory to hypoxia but was not sufficient to prevent fetal growth restriction. Although AMPK signaling was reduced by hypoxia, AMPK was still receptive to pharmacologic activation in the uterine arteries in which it was a potent vasodilator. Thus, AMPK activation may represent a new therapy for pregnancy complications involving reduced uteroplacental perfusion.
Keywords: hypoxia, IUGR, pregnancy, uterine artery
Late-gestation hypoxia in murine pregnancy induces fetal growth restriction while raising uterine artery volumetric blood flow and reducing uteroplacental adenosine monophosphate-activated protein kinase (AMPK) activation.
Introduction
Maternal vascular responses to pregnancy are crucial contributors to both short- and long-term outcomes for both the mother and infant [1, 2]. During normal human pregnancy, uterine artery blood flow increases at least 20-fold [3], thus serving to maintain adequate nutrient and oxygen delivery to the developing fetus [4]. Insufficient oxygen delivery to the uteroplacental circulation and fetal hypoxia adversely affect fetal growth and are common features of the “great obstetrical syndromes” [5], including preeclampsia and intrauterine growth restriction (IUGR). Despite the well-established links between impaired maternal vascular adaptation to pregnancy and poor pregnancy outcomes, the molecular mechanisms governing uteroplacental blood flow are not well understood.
The chronic hypoxia of residence at high altitude (HA, >2500 m) is among the many causes of IUGR and incomplete maternal vascular responses to pregnancy. In humans, HA residence shifts the entire birthweight distribution leftward, increasing the incidence of IUGR threefold [6, 7]. Likewise, hypoxic exposure induces fetal growth restriction in experimental animal models [8, 9]. In human [10, 11] and experimental animal models [8, 9], hypoxia-induced IUGR appears to result, in part, from incomplete maternal vascular adaptation to pregnancy. More than 140 million persons live at HA worldwide [12], of which 4+ million are likely pregnant at any one time, thereby providing a natural laboratory in which to study the physiologic and molecular links between uteroplacental hypoxia and fetal growth restriction in the absence of overt pathology. For this reason, we and others have used HA as a study design with which to address the mechanisms, by which hypoxia influences maternal physiological adaptation to pregnancy and fetal growth [13–16].
Our prior work implicates the adenosine monophosphate-activated protein kinase (AMPK) pathway—a key metabolic regulator and vasodilator—in the regulation of fetal growth under hypoxic conditions. Specifically, Andeans HA residents, who are relatively protected from HA-induced fetal growth restriction [17–19], possess a higher frequency of gene variants predicted to activate AMPK and whose presence was associated with larger uterine artery diameter, implying greater blood flow, and less altitude-associated reduction in infant birth weight [20]. Although such variants were present at a greater frequency in Andeans, they were not unique to the Andean population. Since other factors (e.g., drugs such as metformin and nutritional substances such as resveratrol) can activate AMPK, AMPK activation could be a strategy for increasing uterine artery blood flow and improving fetal growth in any population. The National Institutes of Health and other agencies have called for new drugs for treating pregnancy pathologies given that there has been limited development of drugs that are both safe and effective during pregnancy.
We focus on the potential therapeutic role for AMPK signaling since its activation is an ex vivo vasodilator of the murine uterine artery [21], a drug—metformin—which activates AMPK has been approved for use in pregnancy [22], and AMPK activation may be a contributor to the protection afforded Andean highlanders from hypoxia-associated fetal growth restriction. Here, we aimed to 1) characterize the effects of hypoxia on the vasodilatory effects of AMPK activation and on the levels of uterine artery and placental AMPK activation and 2) determine the role of uterine artery blood flow in hypoxia-induced, late-onset fetal growth restriction. We hypothesized that the hypoxia-induced reduction of fetal weight was due in part to reduced uterine artery blood flow resulting from increased uterine artery contractility and/or decreased vasodilator response, and that these uterine artery changes would be correlated with reduced AMPK activation. Therefore, we exposed pregnant mice to hypoxia or normoxia during the period of rapid late-gestational fetal growth. We undertook studies to characterize the effects of hypoxia exposure on uterine artery vasoreactivity ex vivo by wire myography, uterine artery blood flow in vivo using Doppler ultrasound, and AMPK activation in uterine arteries and placenta by western blot and immunohistochemistry.
Materials and methods
Animals and study conditions
C57Bl/6 mice were obtained from Charles River (Wilmington, MA). Female mice (20–25 g) were mated with males overnight, with a copulatory plug interpreted as evidence of successful breeding at embryonic day (E) 0.5. Animals were housed at the University of Colorado Anschutz Medical Campus’s animal facility altitude (Aurora, 1611 m) until E14.5, when the pregnant mice were placed either in hyperbaric (barometric pressure, PB, ~760 mmHg, 0 m, n = 10) or hypobaric (PB ~385 mmHg, 5500 m, n = 10) chambers to simulate normoxia or hypoxia, respectively. Dam body weights were obtained at E0.5, E14.5, and E18.5, and food intake measured during the period of normoxic or hypoxic exposure. Animals were euthanized by carbon dioxide asphyxiation and cervical dislocation at E18.5 (term = 19.5 days). Right and left uterine arteries were excised, and either used immediately for myography or placed in 4% paraformaldehyde overnight and paraffin embedded within 24 h for histological sectioning. Fetuses and placentas were obtained from all animals, excised, fetal (excluding membranes) and placental weights obtained, and placentas were immediately either placed in 4% paraformaldehyde overnight and paraffin embedded for sectioning within 24 h for histological sectioning, or flash frozen for protein analysis. All animal procedures and protocols were completed in accordance with NIH Guide for the Care and Use of Laboratory Animals [23] and were approved by the Institutional Animal Care and Use Committee at the University of Colorado.
Doppler ultrasound
Doppler ultrasounds were performed in the Cardiovascular Pre-Clinical Core at the University of Colorado-Denver Anschutz Medical Campus (1611 m) on E17.5 pregnant mice while breathing room air and under 1–2% isoflurane anesthesia using a 30-MHz pulsed Doppler system (Vevo 2100, Visual Sonics, Toronto, ON). Aortic stroke volume and heart rate were measured to calculate cardiac output (stroke volume in μl/min × heart rate in bpm). The uterine artery was identified at the point where it appears to cross over the external iliac artery (Supplementary Figure S1). Uterine artery peak systolic velocity, end-diastolic velocity, and time-averaged mean velocity were determined by manually tracing and then averaging the values obtained from waveform tracings of good quality from three consecutive cardiac cycles. Uterine artery pulsatility index (PI) was calculated as (systolic – diastolic velocity)/systolic velocity. Uterine artery diameter was approximated using B-mode color Doppler. For each artery, three Doppler images were taken; the diameter was measured in each and the three values were averaged. B-mode color may have exaggerated the actual dimension and flow calculation, and did not allow for differentiation between the lumen and artery wall since the color image fills the entire vessel. Such effects were considered uniform across animals and thus enabled comparisons of uterine artery volumetric blood flow between groups. Uterine artery volumetric flow was calculated as π × uterine artery radius2 × uterine artery mean velocity. One uterine artery was chosen at random for study so volumetric uterine artery blood flow was multiplied by two to approximate the total uterine artery blood flow. The uterine artery index was calculated as total uterine artery blood flow/cardiac output to determine the portion of cardiac output that was flowing through the uterine arteries.
Uterine artery wire myography
Immediately following euthanasia, one or both main uterine arteries were isolated in ice-cold phosphate-buffered saline (PBS), cleaned of connective tissue, and cut into 2 mm segments. Segments were mounted using two stainless steel 40 μm wires for each chamber of a four-channel, small-vessel wire myograph (Danish Myo Technology 610 M, Ann Arbor, MI. The mounted vessels were continuously maintained at 37°C and oxygenated (95% O2/5% CO2; Airgas, Radnor, PA) while submerged in Krebs solution (118.5 mM NaCl, from Sigma-Aldrich, St. Louis, MO; 25 mM NaHCO3, 4.75 mM KCl, 1.2 mM MgSO47H20, 1.2 mM KH2PO4, 2.5 mM CaCl2, 11.1 mM D-Glucose, all from Thermo Fisher Scientific, Waltham, MA). After a 30-min equilibration, the vessels were normalized to an internal diameter of 0.9 of L13.3kPa, and this diameter was maintained for the rest of the protocol. Vessels were then equilibrated for another 30 min before vasoreactivity studies began.
Viability was demonstrated by contractile responses of at least 2 mN to 60 mM potassium chloride (KCl) and 10 μM phenylephrine (PE; Sigma-Aldrich), and intact endothelium was demonstrated in preconstricted vessels by an 80% or greater vasodilator response to 1 μM acetylcholine (ACh; Acros Organics, Pittsburgh, PA). The full contractile response to PE was determined across a 1 nM-100 μM range and repeated after incubation with the AMPK agonist A769662 (30 μM; Tocris Bioscience, Bristol, UK) to determine the effects of AMPK activation on PE contraction. PE response curves were normalized to the maximum KCl response to control for the length of each uterine artery segment being studied. Relaxation directly by AMPK activation was tested by first contracting the vessels with a submaximal (80% of maximal) PE concentration and then determining the response to increasing concentrations of A769662 (1–100 μM). To determine the contribution of nitric oxide (NO) synthase and cyclooxygenase products to the relaxant effects of A769662, the response to A769662 was tested after incubation with 10 μM L-NG-nitroarginin methyl ester (L-NAME) (BioVision Inc., San Francisco, CA) alone or together with 10 μM indomethacin (Sigma-Aldrich), respectively. A769662 response curves were normalized to the initial PE contraction.
Western blotting
At the time of euthanasia, the placenta corresponding to the fetus weighing closest to the average fetal weight of each dam was flash frozen. Protein extracts were prepared from the placental homogenates in Meso Scale lysis buffer (150 mM NaCl, 20 mM Tris pH 7.5, 1 mM ethylenediaminetetraacetic acid, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid), 1% Triton X-100) containing a protease and phosphatase inhibitor cocktail (Halt Inhibitor Cocktail ×100, Thermo Fisher Scientific), and the protein concentration was determined using the Pierce BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL). Proteins (15 μg/lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 4–20% gradient gel and transferred to nitrocellulose membranes. Li-Cor protocols were followed for membrane blocking and incubations (Li-Cor Biosciences, Lincoln, NE). The membranes were incubated with primary antibodies to β-actin, P-AMPK, and AMPK. All primary antibodies were from Cell Signaling Technology (Danvers, MA). After subsequent incubation with Li-Cor fluorescent secondary antibodies (1:20,000), membranes were imaged and analyzed on a Li-Cor Odyssey CLx. Data are presented as fluorescent signal of target protein normalized to β-actin.
Immunohistochemistry and immunofluorescence
Paraffin sections of uterine arteries and placentas were deparaffinized in xylene and rehydrated in decreasing concentrations of ethanol (100–80%) and deionized water. For antigen retrieval, sections were submerged in 10 mM, pH 6.0 citrate buffer and boiled in a pressure cooker for 30 min at 45°C. Sections were blocked in 1% bovine serum albumin (BSA) in ×1 TBS for 30 min. Primary P-AMPK antibody (Cell Signaling Technology) was diluted 1:50 in 1% BSA in ×1 TBS and applied to sections overnight at 4°C. Secondary anti-horseradish peroxidase antibody (Dako, Santa Clara, CA) was applied for 1 h at room temperature. DAB substrate (Dako) was then added and allowed to develop for 5 min. Sections were then counterstained with hematoxylin for 1 min, dehydrated with increasing concentrations of ethanol (70–100%) and xylene, and then covered with cytoseal and cover slips.
To localize P-AMPK staining in uterine artery and placental sections, immunofluorescence staining was performed for CD31 or wheat germ agglutinin (WGA) to identify endothelial cells, and smooth muscle actin (α-SMA) to identify vascular smooth muscle cells in uterine arteries. Placenta was categorized by following three major functional areas: decidual zone, junctional zone, and labyrinthine zone with the decidual and labyrinthine zones having dense CD31 staining and the junctional zone intermediate staining. For immunofluorescence, paraffin sections of uterine arteries and placentas were deparaffinized in xylene and rehydrated in decreasing concentrations of ethanol (100–80%) and deionized water. Antigen retrieval was carried out using a sodium citrate solution as described above. Autofluorescence was blocked with 0.8% glycine in PBS at room temperature for 30 min. Non-specific binding was blocked with overnight incubation with 10% normal goat serum (NGS) and 10% BSA in PBS, followed by a 1-h incubation with 10% NGS and 0.1% saponin in PBS. Primary antibodies were diluted in 10% NGS in PBS and incubated at 4°C overnight. Primary antibodies against CD31 and α-SMA were obtained from Cell Signaling Technology (Danvers, MA). Secondary antibodies were applied for 1 h at room temperature, including Alexa Fluor 488 from Invitrogen (Carlsbad, CA), DAPI from Sigma, and WGA-FITC from Invitrogen. Sections are sealed with Hard Mount Aqueous Mount from Scytek Laboratories (Logan, UT) and dried overnight before imaging.
Statistical analysis
Ultrasound measures, dam weight, and fetal weight were compared between normoxic and hypoxic groups using Student unpaired t-test (Graph Pad software). Contractile response curves to PE as measured using myography were expressed as relative to the maximal KCl response in each uterine artery. The best-fit line for each PE curve was compared using two-way analysis of variance (ANOVA) with Sidak multiple comparisons. The EC50, which represents the concentration of drug that gives a half-maximal response, was calculated for each individual curve using GraphPad, averaged for a given animal and then expressed as the mean ± SEM for the normoxic and hypoxic or other specific treatment groups. The maximum PE response and EC50 were compared using Sidak multiple comparisons. Response curves to A769662 were expressed as a percentage of the initial, 80% maximally PE contraction before vasodilation with increasing concentrations of A769662. As mentioned above, the best-fit line for each curve was compared using two-way ANOVA with Sidak multiple comparisons. Curves were further analyzed by comparing maximum % relaxation, EC50, and area under the curve (AUC) using Sidak multiple comparisons. Significance was defined as a two-tailed P < 0.05.
Results
Reduction of fetal weight with late-gestation hypoxia exposure
E18.5 fetal weight was reduced in the hypoxia-exposed litters by an average of 34% (Figure 1A). Maternal food intake, number of pups, and resorptions per litter were similar in the normoxic and hypoxic groups (Table 1), suggesting that the dams tolerated the hyperbaric and hypobaric chambers equally well but that clearly fetal growth was reduced. The lesser maternal weight gain under conditions of hypoxia (Table 1) was likely due to the reduction in fetal growth as judged by the similarity in maternal weight at E18.5 minus the total weight of the excised fetuses and placentas (29.3 ± 0.8 vs 27.8 ± 0.8 g, respectively, P = NS). In contrast to the reduction in fetal weight, placental weight was ~7% higher in the hypoxia-exposed group (Figure 1B), leading to a lower placental efficiency as expressed as the ratio of fetal to placental weight (Figure 1C).
Figure 1.
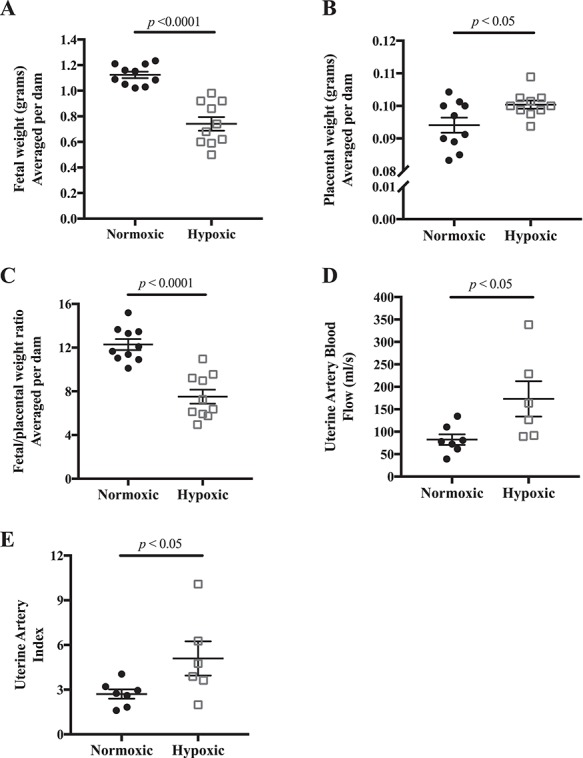
Effects of late-gestation (E14.5–18.5) hypoxia on fetal growth and uterine artery blood flow. A) E18.5 fetal weight was reduced, B) placental weight was increased, and therefore C) the ratio of fetal to placental weight was lower indicating a selective reduction in fetal growth; n = 10 normoxic, 10 hypoxic dams. D) Uterine artery volumetric blood flow was increased with late-gestation hypoxia, and E) uterine index, the ratio of uterine artery blood flow to cardiac output, was higher; n = 7 normoxic, 6 hypoxic dams. Data are presented as mean ± SEM and analyzed by Student unpaired t-test.
Table 1.
Growth parameters and uterine artery hemodynamics
| Normoxic | Hypoxic | P-value | |
|---|---|---|---|
| Growth parameters | |||
| Maternal daily food intake (g) | 6.4 ± 0.3 | 5.8 ± 0.4 | NS |
| Litter size, number of fetuses | 8.7 ± 0.3 | 8.2 ± 0.3 | NS |
| Resorption number | 0.50 ± 0.21 | 0.67 ± 0.30 | NS |
| E0.5 Maternal weight (g) | 23.3 ± 0.6 | 22.6 ± I.I | NS |
| E18.5 Maternal weight gain (g (% increase)) | 15.7 ± 0.9 (69) | 10.9 ± 0.7 (51) | 0.03 |
| Uterine artery hemodynamics | |||
| UtA peak systolic velocity (mm/sec) | 254 ± 37 | 345 ± 40 | NS |
| UtA end-diastolic velocity (mm/sec) | 113 ± 21 | 169 ± 10 | 0.05 |
| UtA mean velocity (mm/s) | 148 ± 21 | 208 ± 23 | 0.02 |
| UtA PI | 0.57 ± 0.03 | 0.49 ± 0.03 | 0.13 |
| UtA diameter (mm) | 0.63 ± 0.02 | 0.70 ± 0.04 | 0.20 |
| Cardiac output (ml/min) | 31 ± 3 | 35 ± 3 | NS |
| Stroke volume (μl) | 64 ± 6 | 67 ± 7 | NS |
| Heart rate (bpm) | 486 ± 6 | 527 ± 7 | 0.001 |
Data presented as mean ± SEM NS = non-significant maternal daily food intake (g) 6.4 ± 0.3 5.8 ± 0.4 NS.
Doppler indices of uterine artery blood flow and cardiac output
Uterine artery blood flow was increased by 110% in the hypoxic compared to normoxic dams (Figure 1D). This was due largely to increased uterine artery end-diastolic velocity, which resulted in a higher uterine artery mean flow velocity (Table 1). The hypoxia group’s larger (by 67 μm) uterine artery diameter also contributed, although it did not reach significance (Table 1). Heart rate was significantly higher in the hypoxic mice but did not result in greater cardiac output (Table 1). The uterine artery index (i.e., the ratio of uterine artery volumetric blood flow to cardiac output) was doubled in hypoxic mice (Figure 1E), indicating that a greater proportion of total blood flow was directed to the uterus with hypoxia exposure.
Effects of hypoxia and ex vivo AMPK activation on uterine artery contractility
We evaluated the effects of hypoxia on isolated uterine arteries by small-vessel wire myography. The contractile response to PE, normalized to the maximum KCl response in each vessel, was similar in uterine arteries from normoxic and hypoxic mice whether assessed by comparing the whole curve (Figure 2A), the maximum PE contractile response, or the EC50 of the PE response curves (Figure 2E). The maximum contractile response to KCl was also similar in the normoxic and hypoxic groups (Figure 2E).
Figure 2.
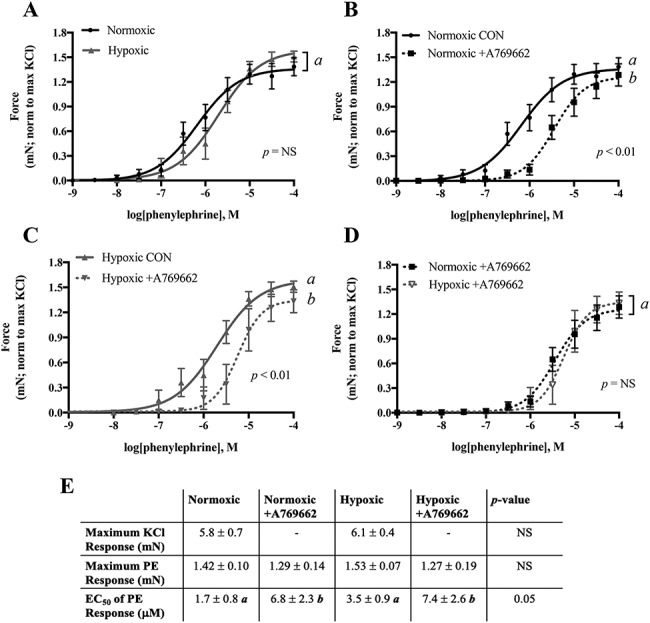
Uterine artery contractility measured by wire myography and the effect of AMPK activation on blunting the contractile response. A) The contractile response to PE was similar in uterine artery from normoxic and hypoxic mice. A769662 blunted the PE response in both B) normoxic and C) hypoxic mice. D) The magnitude of A769662 effects on blunting PE response was similar in both groups. E) Neither the maximum KCl response nor the maximum response to PE was changed by hypoxia or A769662, but sensitivity to PE was reduced by A769662 in vessels from both normoxic and hypoxic groups. The contractile response to PE was normalized to the maximum KCl response in each vessel studied. Data are presented as mean ± SEM and NS = non-significant. Different italicized letters represent significant differences using either the overall result using two-way ANOVA or two-way ANOVA with Sidak multiple comparisons. CON = control untreated. N = Norm CON: 8 mice, 12 vessels; Norm +A769662: 8 mice, 12 vessels; Hx CON: 7 mice, 8 vessels; Hx + A769662: 7 mice, 7 vessels.
AMPK activation by A769662 blunted sensitivity to PE in uterine arteries from both normoxic (Figure 2B) and hypoxic (Figure 2C) mice and did so similarly (Figure 2D). AMPK activation thus increased the EC50 in both groups while maximum PE response remained unaltered (Figure 2E).
Effect of hypoxia on uterine artery vasodilation
In uterine arteries preconstricted to 80% of their maximal PE response, AMPK activation by A769662 resulted in similar dose-dependent vasodilation in the normoxic and hypoxic mice (Figure 3A). In uterine arteries from normoxic mice, inhibition of NO synthase by L-NAME reduced the maximum vasodilatory effects of AMPK activation by 42%, indicating that some but not all AMPK-induced vasodilation was dependent on increased endothelial NO production. The addition of indomethacin to inhibit production of cyclooxygenase products did not further decrease the vasodilatory effects of AMPK activation in the normoxic animals, indicating that there was little, if any, contribution of vasodilator prostaglandins (Figure 3B). In hypoxic mice, inhibition of NO synthase by L-NAME blocked the vasodilatory effects of AMPK activation by 29%, and the addition of the cyclooxygenase inhibitor indomethacin further blunted the effects of AMPK activation by an additional 18%. This indicated that the production of both NO and vasodilator prostaglandins contributed to AMPK activation-induced vasorelaxation in hypoxic mice (Figure 3C). In other words, since the effect of L-NAME on blunting AMPK-induced vasodilation was significantly greater in normoxic compared to hypoxic mice, the vasodilator role of NO synthase was greater in the uterine arteries from the normoxic than hypoxic group. L-NAME and indomethacin blunted the vasodilatory effects of AMPK activation both by reducing the maximum percent relaxation and the overall sensitivity to the effects of AMPK activation as measured by AUC (Figure 3D). The remaining vasodilatory effects of AMPK activation may be due to endothelium-derived hyperpolarizing factor or smooth muscle factors.
Figure 3.
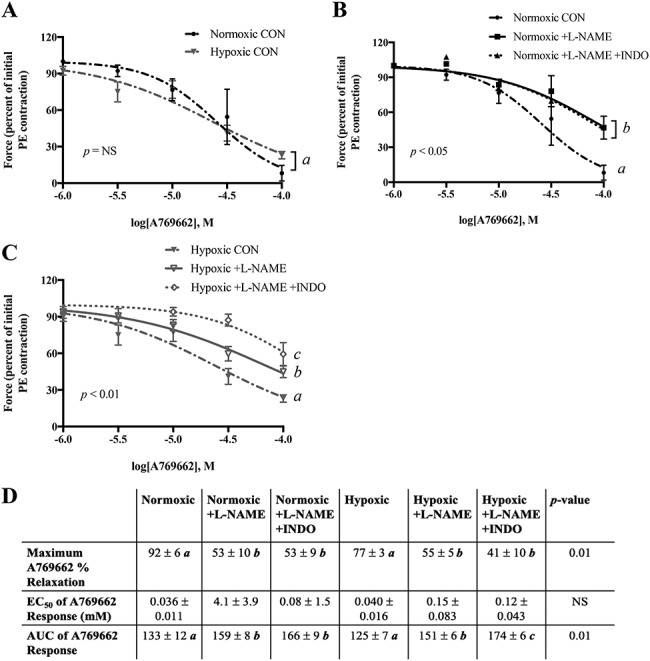
Vasodilatory effect of AMPK activation in preconstricted uterine artery. A) Preconstricted uterine arteries from normoxic and hypoxic mice similarly vasodilated in response to the AMPK activator A769662. B) Incubation with L-NAME reduced the vasodilatory effect of A769662 in uterine arteries from normoxic mice and C) did so to a lesser degree in hypoxic mice. Addition of indomethacin did not further reduce the effect of A769662 in normoxic mice but did so in hypoxic mice. D) Comparison of curves by maximum relaxation, EC50, and AUC. Data are presented as mean ± SEM and normalized to initial PE contraction. Response curves were analyzed by two-way ANOVA. In part D, the overall ANOVAs are shown, and different italicized letters represent results of Sidak multiple comparisons. NS = non-significant. CON = control untreated. N = Norm CON: 7 mice, 9 vessels; Norm +L-NAME: 7 mice, 8 vessels; Norm +L-NAME +INDO: 3 mice, 3 vessels; Hx CON: 7 mice, 8 vessels; Hx + L-NAME: 7 mice, 7 vessels; Hx + L-NAME +INDO: 6 mice, 6 vessels.
The vasodilatory responses to ACh were similar in uterine arteries from both normoxic and hypoxic mice (Supplementary Figure S2). In normoxic mice, L-NAME significantly blunted ACh vasodilation, as analyzed by the maximum % relaxation and the AUC of the ACh response curves. However, L-NAME did not significantly blunt ACh vasodilation in hypoxic animals, indicating that hypoxic exposure decreased the contribution of increased NO production to ACh-stimulated uterine artery vasodilation. Further mechanisms such as cyclooxygenase products or endothelial-derived hyperpolarizing factor could be augmented by hypoxia to maintain a similar ACh vasodilation as in the normoxic group.
Less AMPK activation in uteroplacental tissues with hypoxia
Immunostaining for P-AMPK in uterine arteries from normoxic mice indicated AMPK activation in the endothelial cells, while P-AMPK immunostaining was not present in the uterine artery from hypoxic mice (Figure 4A). Qualitative assessment of the uterine artery WGA and α-SMA staining suggested that the normoxic (n = 3) compared to hypoxic (n = 4) mice had layers of fibrocollagenous tissue within the tunica intima, as there was a distinctive layer between the endothelium and smooth muscle layer.
Figure 4.
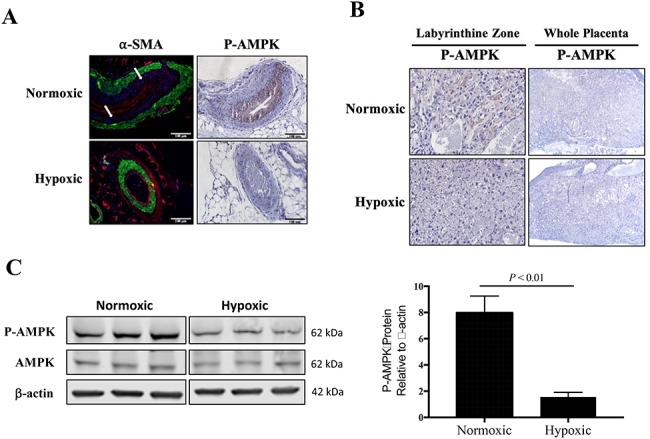
Characterization of placental and uterine artery AMPK activation. A) By immunohistochemistry, P-AMPK was pronounced in the uterine arterial smooth muscle cells from normoxic mice (n = 3) but not visible in the uterine artery from hypoxic mice (n = 4). In normoxic mice, there was a distinctive layer between the vascular smooth muscle cells (α-SMA, green staining) and endothelium (stained in red with WGA), pointed to by white arrows. B) Immunohistochemistry staining for placental P-AMPK revealed high AMPK activation in the labyrinthine zone of placentas from normoxic mice but was largely absent from hypoxic placentas. C) By western blot, AMPK activation was lower in the total placental homogenate from hypoxic compared to normoxic mice, as compared by Student unpaired t-test (n = 6 each).
We also evaluated changes in placental AMPK signaling. P-AMPK was localized to the junctional zone of the placenta from normoxic mice and not detectable in placenta from hypoxic mice, as visualized using immunohistochemistry (Figure 4B). P-AMPK protein expression was higher in the placenta from normoxic compared to hypoxic dams, while total AMPK protein expression levels were unchanged, as measured by western blot of total homogenized placenta (Figure 4C).
Discussion
This is, to the best of our knowledge, the first report of volumetric uterine artery blood flow in a murine, hypoxia-induced model of late-gestation fetal growth restriction. We found that late-gestation hypoxia reduced fetal weight and increased uterine artery blood flow. Neither of these changes is associated with alterations in uterine artery contractile responses to PE nor dilator responses to AMPK activation or ACh but were paralleled by lesser AMPK activation in uterine arteries and placental tissues. Our observations suggest that while the rise in uterine artery blood flow under hypoxic conditions may serve to partially compensate for reduced ambient oxygen availability, it is insufficient to prevent fetal growth restriction.
Consistent with other studies of hypoxic rodent pregnancy [21, 24], we saw a 34% reduction in E18.5 fetal weight with late-gestation hypoxia equivalent to 10.5% inspired O2. Profound hypoxia is necessary to induce IUGR in small animal models as shown by inspired oxygen levels of 12 or 16% (equivalent to 4500 or 2100 m respectively) failing to reduce fetal weight in pregnant guinea pigs or mice [25, 26]. Conversely, in humans, marked reductions in birth weight are seen at such altitudes [6, 27, 28] with grading effects being seen with increasing altitude [19]. Similar to previous reports, we found a slight increase in placental weight [25, 26] and a reduction in placental efficiency, calculated as the ratio of fetal to placental weight, with hypoxia [24]. Human studies of gestational hypoxia, on the other hand, report inconsistent changes in placental weight, with increases [29], decreases [30, 31], or no change [32, 33] reported.
Based on prior human work demonstrating that HA residence decreases the pregnancy-associated rise in uterine artery blood flow and correlates with reduced fetal weight [10] and uterine artery diameter [11, 34], we expected our murine model of hypoxia-induced fetal growth restriction to show lower uterine artery blood flows. In contrast, volumetric uterine artery blood flow was greater in dams exposed to hypoxia compared to their normoxic counterparts despite a 34% lower E18.5 fetal weight. Greater uterine artery blood flow among hypoxic dams was not due to increased cardiac outputs but was associated with higher end-diastolic velocity and a lower PI, resulting in almost twice as great a proportion of the cardiac output flowing through the uterine artery. This is the opposite of what has been seen in normotensive high- vs low-altitude pregnant women [34] or in early-onset, high-altitude preeclamptic women in whom there was less uterine artery relative to total blood flow [35] than seen in normotensive low-altitude or normotensive high-altitude pregnant women respectively. Other differences among species and the timing and duration of hypoxia also likely affect vascular responses to pregnancy given that the decreased PI we observed was consistent with a rat model of early-onset hypoxia-induced IUGR [36] but unlike a report using guinea pigs in which mid gestation-onset hypoxia-induced fetal growth restriction and was accompanied by an increased PI [24].
In terms of the effects of hypoxia on uterine artery vasoreactivity, like other reports we found that neither the contractile response to PE nor the vasodilator response to ACh was affected by hypoxia [9, 21, 37]. However, while hypoxic exposure exaggerated the vasodilator response to AMPK in mice exposed to 10% oxygen in a previous study [21], we found no such effect. Consistent with previous experimental animal [9, 36, 38] or human [39] observations, we found that AMPK- or ACh-induced vasodilation was less dependent on increased NO production, indicating that the greater uterine artery blood flow seen in our hypoxic mice was not likely due to upregulation of NO signaling.
To determine whether differences in uterine artery structure may have contributed to the hypoxia-associated increase in uterine artery blood flow and larger uterine artery diameter observed, we qualitatively assessed the number of cell layers in the uterine arteries from the hypoxic and normoxic mice. We noted that there were fewer cell layers in the intimal layer surrounding the vessel lumen in hypoxic mice, suggesting less fibrocollagenous cells and possibly a more distensible phenotype with hypoxia. Strengthening this possibility, previous studies by our group in guinea pigs found that chronically-hypoxic animals had more than twice as much uterine artery distensibility or half the stiffness when compared to normoxic pregnant animals [40]. We speculate that the increased uterine artery blood flow we observed at HA may be due, in part, to increased uterine artery distensibility and provide a rationale for future study on the effects of hypoxia on uterine artery compliance. Downstream vascular remodeling is also another and perhaps more likely a possibility given that neither uterine artery diameter nor the PI were significantly altered in the hypoxic compared to the normoxic group, which would have been expected had distensibility been greater.
Our study benefited from certain strengths but also had limitations. First, utilizing a murine model allowed for tight control of the severity and duration of hypoxia exposure without other pregnancy complications. While this late-gestation hypoxia model does not fully mimic the human HA condition since the hypoxic exposure was initiated late in pregnancy, our protocol has the advantage of enabling having pregnancies for study since, in our experience, hypoxia throughout gestation in the murine model frequently results in resorption of the entire litter. Additionally, the timing of our late-gestation model more closely resembles hypoxic disorders of pregnancy insofar as IUGR and preeclampsia are thought to involve uteroplacental hypoxia beginning mid- or late-gestation [41] and therefore may be more clinically relevant. Second, we were able to determine uterine artery diameter using B-mode color Doppler, therefore allowing for calculation of volumetric uterine artery flow, for which we believe that these were the first such measurements in hypoxia-exposed mice. Prior to initiating our study, we carefully considered the possibility that color Doppler ultrasound might overestimate vessel diameter as the color appears to “bleed” over the non-color image. However a prior study compared B-mode color Doppler measurements of murine uterine artery diameter to those obtained from vessel casts, and concluded that these two methods yielded similar values [42], suggesting that our values were not overestimates. Moreover, even if there were an overestimation of vessel diameter, this would be similar in both normoxic and hypoxic groups and thus would not prevent their comparison. Disadvantages of our study, however, were that given the small size of murine vessels, we were unable to study vessels downstream of the main uterine artery that are the primary sites of uterine vascular resistance. Future studies in humans or larger animal models will be beneficial to determine the contribution of downstream vessels to hypoxia-associated alterations in uterine artery blood flow. Finally, a technical limitation of both the ultrasound and myography measurements was that these measurements were done under moderate altitude (Aurora, CO, 1611 m) and hyperoxia (95% O2/5% CO2 bubbling in the chambers), respectively, rather than keeping the mice or their uterine arteries under study conditions; however, this is standard protocol [21, 24, 42].
One factor that may be contributing to the fetal growth restriction that we observed with hypoxia is the inhibition of the mechanistic target of rapamycin (mTOR), thought to be involved in regulating fetal growth in response to maternal nutrient and oxygen availability [43]. Although AMPK can inhibit mTOR [44], we did not find elevated AMPK activation but rather, less AMPK activation in placenta tissues in the presence of reduced fetal growth, suggesting that the lesser AMPK activation in the placenta was not protective. However, since mTOR can be downregulated by hypoxia independently of P-AMPK [45, 46], evaluation of mTOR signaling is justified to determine whether upregulation of mTOR may be contributing to the hypoxia-associated fetal growth restriction observed such as has been reported in human pregnancy [47]. Another factor that may be contributing to hypoxia-induced fetal growth restriction is higher placental vascular resistance; although uterine artery resistance was reduced with hypoxia, it is possible that placental resistance might be greater, so that the higher uterine artery blood flow with hypoxia was not actually reaching the fetuses. Consistent with this possibility, we measured lower P-AMPK in placentas of hypoxic mice compared to normoxic mice, which could indicate a loss of P-AMPK-induced vasodilatory effects in the placenta.
Overall, our studies demonstrated that despite an increase in uterine artery blood flow, fetal weight was reduced by late-gestation hypoxia in mice. There was no change in uterine artery vasoreactivity but, based on an apparent reduction in the number of layers of fibrocollagenous tissue within the uterine artery tunica intima, we speculate that there was greater downstream vascular remodeling and possibly greater uterine artery compliance with hypoxia. As in a previous study, AMPK activation ex vivo opposed vasoconstriction [21]. Although we found here that hypoxia reduced AMPK activation in the uterine artery and placental tissues, the uterine arteries remained responsive to ex vivo AMPK activation, indicating that these pathways were still functional and could be targeted pharmacologically. Ongoing studies will evaluate whether in vivo AMPK modulation affects fetal growth in our model of hypoxic murine pregnancy and, if so, whether such effects are accompanied by changes in uterine artery blood flow or other vascular parameters.
Supplementary data
Supplementary data are available at BIOLRE online.
Supplementary Material
Acknowledgments
The authors would like to thank Sara Wennersten with the University of Colorado Cardiovascular Pre-Clinical Imaging Core, veterinarians and animal care technicians in the University of Colorado Vivarium, and the University of Colorado Cardiovascular and Pulmonary Research Laboratory for their support in this project.
References
- 1. Roberts JM, Gammill H. Pre-eclampsia and cardiovascular disease in later life. The Lancet 2005; 366. doi: 10.1016/S0140-6736(05)67349-7. [DOI] [PubMed] [Google Scholar]
- 2. Peixoto AB, Rolo LC, Nardozza LMM, Araujo JE. Epigenetics and preeclampsia: programming of future outcomes. Methods Mol Biol 1710; 2018:73–83. [DOI] [PubMed] [Google Scholar]
- 3. Browne VA, Julian CG, Toledo-Jalden L, Cioffi-Ragan D, Vargas E, Moore LG. Uterine artery blood flow, fetal hypoxia and fetal growth. Philos Trans R Soc Lond B Biol Sci 2015; 370:20140068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caton D, Crenshaw C, Wilcox CJ, Barron DH. O2 delivery to the pregnant uterus: its relationship to O2 consumption. Am J Physiol 1979; 237:R52–R57. [DOI] [PubMed] [Google Scholar]
- 5. Romero R. Prenatal medicine: The child is the father of the man. J Matern Fetal Neonatal Med 2009; 22:636–639. [DOI] [PubMed] [Google Scholar]
- 6. Lichty JA, Ting RY, Bruns PD, Dyar E. Studies of babies born at high altitude. I. Relation of altitude to birth weight. A.M.A. J Dis Children 1957; 93:666–669. [DOI] [PubMed] [Google Scholar]
- 7. Jensen GM, Moore LG. The effect of high altitude and other risk factors on birthweight: independent or interactive effects? Am J Public Health 1997; 87:1003–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rockwell LC, Keyes LE, Moore LG. Chronic hypoxia diminishes pregnancy-associated DNA synthesis in guinea pig uteroplacental arteries. Placenta 2000; 21:313–319. [DOI] [PubMed] [Google Scholar]
- 9. Mateev SN, Sillau AH, Mouser R, McCullough RE, White MM, Young DA, Moore LG. Chronic hypoxia opposes pregnancy-induced increase in uterine artery vasodilator response to flow. Am J Physiol Heart Circ Physiol 2003; 284:H820–H829. [DOI] [PubMed] [Google Scholar]
- 10. Zamudio S, Palmer SK, Droma T, Stamm E, Coffin C, Moore LG. Effect of altitude on uterine artery blood flow during normal pregnancy. J Appl Physiol 1995; 79:7–14. [DOI] [PubMed] [Google Scholar]
- 11. Zamudio S, Palmer SK, Stamm E, Coffin C, Moore LG. Uterine blood flow at high altitude In: Sutton JR. (ed.), Hypoxia and the Brain. Burlington, VT: Queen City Printers Inc.; 1995. [Google Scholar]
- 12. Moore LG, Charles SM, Julian CG. Humans at high altitude: hypoxia and fetal growth. Respir Physiol Neurobiol 2011; 178:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rockwell LC, Dempsey EC, Moore LG. Chronic hypoxia diminishes the proliferative response of Guinea pig uterine artery vascular smooth muscle cells in vitro. High Alt Med Biol 2006; 7:237–244. [DOI] [PubMed] [Google Scholar]
- 14. Itani N, Salinas CE, Villena M, Skeffington KL, Beck C, Villamor E, Blanco CE, Giussani DA. The highs and lows of programmed cardiovascular disease by developmental hypoxia: studies in the chicken embryo. J Physiol 2018; 596:2991–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reyes LM, Shah A, Quon A, Morton JS, Davidge ST. The role of the tumor necrosis factor (TNF)-related weak inducer of apoptosis (TWEAK) in offspring exposed to prenatal hypoxia. J Dev Orig Health Dis 2018; 9:661–669. [DOI] [PubMed] [Google Scholar]
- 16. Thompson LP, Chen L, Polster BM, Pinkas G, Song H. Prenatal hypoxia impairs cardiac mitochondrial and ventricular function in guinea pig offspring in a sex-related manner. Am J Physiol Regul Integr Comp Physiol 2018; 315:R1232–R1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Julian CG, Vargas E, Armaza JF, Wilson MJ, Niermeyer S, Moore LG. High-altitude ancestry protects against hypoxia-associated reductions in fetal growth. Arch Dis Child Fetal Neonatal Ed 2007; 92:F372–F377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Julian CG, Hageman JL, Wilson MJ, Vargas E, Moore LG. Lowland origin women raised at high altitude are not protected against lower uteroplacental O2 delivery during pregnancy or reduced birth weight. Am J Hum Biol 2011; 23:509–516. [DOI] [PubMed] [Google Scholar]
- 19. Soria R, Julian CG, Vargas E, Moore LG, Giussani DA. Graduated effects of high-altitude hypoxia and highland ancestry on birth size. Pediatr Res 2013; 74:633–638. [DOI] [PubMed] [Google Scholar]
- 20. Bigham AW, Julian CG, Wilson MJ, Vargas E, Browne VA, Shriver MD, Moore LG. Maternal PRKAA1 and EDNRA genotypes are associated with birth weight, and PRKAA1 with uterine artery diameter and metabolic homeostasis at high altitude. Physiol Genomics 2014; 46:687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skeffington KL, Higgins JS, Mahmoud AD, Evans AM, Sferruzzi-Perri AN, Fowden AL, Yung HW, Burton GJ, Giussani DA, Moore LG. Hypoxia, AMPK activation and uterine artery vasoreactivity. J Physiol 2016; 594:1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hyer S, Balani J, Shehata H. Metformin in pregnancy: mechanisms and clinical applications. Int J Mol Sci 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals Guide for the Care and Use of Laboratory Animals. 8th edition Washington (DC): National Academies Press (US)2011. Available from: https://www.ncbi.nlm.nih.gov/books/NBK54050/. doi: 10.17226/12910. [DOI] [PubMed] [Google Scholar]
- 24. Turan S, Aberdeen GW, Thompson LP. Chronic hypoxia alters maternal uterine and fetal hemodynamics in the full-term pregnant Guinea pig. Am J Physiol Regul Integr Comp Physiol 2017; 313:R330–R339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thompson LP, Pence L, Pinkas G, Song H, Telugu BP. Placental hypoxia during early pregnancy causes maternal hypertension and placental insufficiency in the hypoxic Guinea pig model. Biol Reprod 2016; 95:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matheson H, Veerbeek JHW, Charnock-Jones DS, Burton GJ, Yung HW. Morphological and molecular changes in the murine placenta exposed to normobaric hypoxia throughout pregnancy. J Physiol 2016; 594:1371–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCullough RE, Reeves JT, Liljegren RL. Fetal growth retardation and increased infant mortality at high altitude. Obstet Gynecol Surv 1977; 32:596–598. [DOI] [PubMed] [Google Scholar]
- 28. Zamudio S, Moore LG. Altitude and fetal growth: current knowledge and future directions. Ultrasound Obstet Gynecol 2000; 16:6–8. [DOI] [PubMed] [Google Scholar]
- 29. Frisancho AR. Developmental responses to high altitude hypoxia. Am J Phys Anthropol 1970; 32:401–408. [DOI] [PubMed] [Google Scholar]
- 30. Khalid MEM, Ali ME. Ali KZM, Full-term birth weight and placental morphology at high and low altitude. Int J Gynecol Obstetr 1997; 57:259–265. [DOI] [PubMed] [Google Scholar]
- 31. Chabes A, Pereda J, Hyams L, Barrientos N, Perez J, Campos L, Monroe A, Mayorga A. Comparative morphometry of the human placenta at high altitude and at sea level. I. The shape of the placenta. Obstet Gynecol 1968; 31:178–185. [DOI] [PubMed] [Google Scholar]
- 32. Kruger H, Arias-Stella J. The placenta and the newborn infant at high altitudes. Am J Obstet Gynecol 1970; 106:586–591. [DOI] [PubMed] [Google Scholar]
- 33. Reshetnikova OS, Burton GJ, Milovanov AP. Effects of hypobaric hypoxia on the fetoplacental unit: the morphometric diffusing capacity of the villous membrane at high altitude. Am J Obstet Gynecol 1994; 171:1560–1565. [DOI] [PubMed] [Google Scholar]
- 34. Julian CG, Galan HL, Wilson MJ, Desilva W, Cioffi-Ragan D, Schwartz J, Moore LG. Lower uterine artery blood flow and higher endothelin relative to nitric oxide metabolite levels are associated with reductions in birth weight at high altitude. Am J Physiol Regul Integr Comp Physiol 2008; 295:R906–R915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Browne VA, Toledo-Jalden L, Davila RD, Lopez LP, Yamashiro H, Cioffi-Ragan D, Julian CG, Wilson MJ, Bigham AW, Shriver MD, Honigman B, Vargas E et al. . High-end arteriolar resistance limits uterine artery blood flow and restricts fetal growth in preeclampsia and gestational hypertension at high altitude. Am J Physiol Regul Integr Comp Physiol 2011; 300:R1221–R1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aljunaidy MM, Morton JS, Cooke CM, Davidge ST. Maternal vascular responses to hypoxia in a rat model of intrauterine growth restriction. Am J Physiol Regul Integr Comp Physiol 2016; 311:R1068–R1075. [DOI] [PubMed] [Google Scholar]
- 37. Hu XQ, Yang S, Pearce WJ, Longo LD, Zhang L. Effect of chronic hypoxia on alpha-1 adrenoceptor-mediated inositol 1,4,5-trisphosphate signaling in ovine uterine artery. J Pharmacol Exp Ther 1999; 288:977–983. [PubMed] [Google Scholar]
- 38. White MM, McCullough RE, Dyckes R, Robertson AD, Moore LG. Chronic hypoxia, pregnancy, and endothelium-mediated relaxation in guinea pig uterine and thoracic arteries. Am J Physiol Heart Circ Physiol 2000; 278:H2069–H2075. [DOI] [PubMed] [Google Scholar]
- 39. Lorca RA, Lane SL, Bales ES, Nsier H, Yi H, Donnelly MA, Euser AG, Julian CG, Moore LG. High altitude reduces NO-dependent myometrial artery vasodilator response during pregnancy. Hypertension 2019; 73:1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mateev SN, Mouser R, Young DA, Mecham RP, Moore LG. Chronic hypoxia augments uterine artery distensibility and alters the circumferential wall stress-strain relationship during pregnancy. J Appl Physiol 2006; 100:1842–1850. [DOI] [PubMed] [Google Scholar]
- 41. Krampl E, Lees C, Bland JM, Dorado JE, Moscoso G, Campbell S. Fetal biometry at 4300 m compared to sea level in Peru. Ultrasound Obstet Gynecol 2000; 16:9–18. [DOI] [PubMed] [Google Scholar]
- 42. Kulandavelu S, Whiteley KJ, Qu D, Mu J, Bainbridge SA, Adamson SL. Endothelial nitric oxide synthase deficiency reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant mice. Hypertension 2012; 60:231–238. [DOI] [PubMed] [Google Scholar]
- 43. Gupta MB, Jansson T. Novel roles of mechanistic target of rapamycin signaling in regulating fetal growth. Biol Reprod 2018; 0:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 2008; 22:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004; 18:2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yung HW, Cox M, Tissot van Patot M, Burton GJ. Evidence of endoplasmic reticulum stress and protein synthesis inhibition in the placenta of non-native women at high altitude. FASEB J 2012; 26:1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.