

Summary
Application of single-stranded DNA recombineering for genome editing of species other than enterobacteria is limited by the efficiency of the recombinase and the action of endogenous mismatch repair (MMR) systems. In this work we have set up a genetic system for entering multiple changes in the chromosome of the biotechnologically relevant strain EM42 of Pseudomononas putida. To this end high-level heat-inducible co-transcription of the rec2 recombinase and P. putida's allele mutLE36KPP was designed under the control of the PL/cI857 system. Cycles of short thermal shifts followed by transformation with a suite of mutagenic oligos delivered different types of genomic changes at frequencies up to 10% per single modification. The same approach was instrumental to super-diversify short chromosomal portions for creating libraries of functional genomic segments—e.g., ribosomal-binding sites. These results enabled multiplexing of genome engineering of P. putida, as required for metabolic reprogramming of this important synthetic biology chassis.
Subject Areas: Bioengineering, Metabolic Engineering, Biotechnology, Microbial Biotechnology
Graphical Abstract
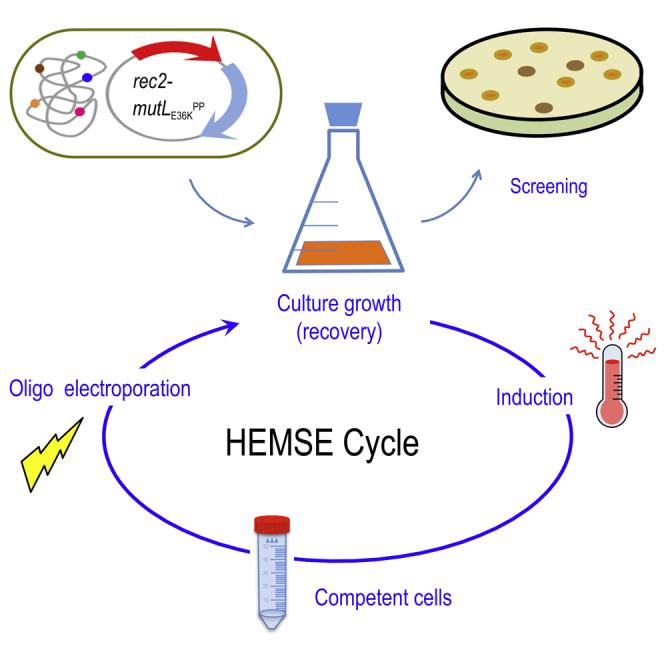
Highlights
-
•
Pseudomonas putida is a useful Synthetic Biology chassis for metabolic engineering
-
•
Co-expression of Rec2 recombinase and mutLE36K allele empowers ssDNA recombineering
-
•
Cyclic DNA replication fork invasion causes up to 10% single-site mutation frequency
-
•
The experimental HEMSE pipeline eases multi-site genome editing of P. putida
Bioengineering; Metabolic Engineering; Biotechnology; Microbial Biotechnology
Introduction
DNA recombineering was first developed in the early 2000s (Datsenko and Wanner, 2000, Yu et al., 2000) as a genetic technology for replacing genomic segments of E. coli with synthetic double-stranded (ds) DNA by means of the DNA exchange mechanism brought about by the Red system of phage lambda. Although the native approach involves three proteins (a β-recombinase, an exonuclease, and the γ protein, which protects free ds ends of DNA from degradation by RecBCD), it turned out that the Red-β protein sufficed to promote invasion of the replication fork by single-stranded (ss) oligonucleotides incorporated as Okazaki fragments (Ellis et al., 2001). If such oligonucleotides were designed to carry mutations, the resulting changes could be inherited at considerable frequencies upon subsequent rounds of DNA segregation. The key value of this approach is that by using cocktails of mutagenic oligonucleotides and either manual or automated cycles of Red expression/oligonucleotide transformation one can enter simultaneous changes in many genomic sites and/or saturate given DNA stretches with specific or random mutations (Wang et al., 2009, Nyerges et al., 2016, Nyerges et al., 2018). This technology gave rise to MAGE (multiplex automated genome engineering) in E. coli, a cycled and multiplexed application of recombineering that exploits the capabilities of the Red system for large-scale reprogramming of cells, i.e., metabolic engineering of lycopene production (Wang et al., 2009) or genome-wide codon replacements (Isaacs et al., 2011). These methods have been improved further by using host strains transiently disabled in mismatch repair (MMR) and by enriching mutants through Cas9/guide RNA-based counterselection of wild-type sequences (Costantino and Court, 2003, Jiang et al., 2013, Nyerges et al., 2014, Nyerges et al., 2016, Ronda et al., 2016, Oesterle et al., 2017). These technologies work well in E. coli, whereas they are difficult to transplant directly to non-enteric bacteria. Yet, their applicability to species such as Pseudomonas putida has a special interest because of the value of environmental microorganisms as useful platforms for metabolic engineering (Nikel et al., 2014, Nikel et al., 2016, Martínez-García and de Lorenzo, 2019). Attempts of functional expression of the lambda Red system in various species of Pseudomonas have been reported, but recombination frequencies were low in the absence of selection (Lesic and Rahme, 2008, Liang and Liu, 2010, Luo et al., 2016, Chen et al., 2018, Yin et al., 2019). Red-like counterparts found in Pseudomonas prophages have been more successful to the same ends. For example, the RecET recombinase/exonuclease pair of P. syringae has been instrumental for executing a suite of manipulations in this species (Swingle et al., 2010a, Bao et al., 2012). Furthermore, bioinformatic mining of Pseudomonas-borne recombinases from known protein families (i.e., Redβ, ERF, GP2.5, SAK, and SAK4; Lopes et al., 2010) followed by experimental validation of the most promising in a standardized recombineering test exposed two new enzymes (Ssr and Rec2: Aparicio et al., 2016, Ricaurte et al., 2018, Aparicio et al., 2020). These recombinases delivered a comparatively high level of activity in the reference strains P. putida KT2440 and its genome-reduced derivative P. putida EM42. Still, numbers were way below those reported for E. coli. Furthermore, the action of the endogenous MMR system of this bacterium impeded single-nucleotide changes (i.e., A to T, mismatch A:A) that were efficiently fixed by the indigenous mutS/mutL device (Aparicio et al., 2016, Aparicio et al., 2019b).
In this work we have set out to overcome the above-mentioned bottlenecks to efficacious recombineering in P. putida. The approach builds on the apparently superior ability of the Rec2 recombinase to promote DNA annealing with exogenous synthetic oligonucleotides during chromosomal replication. By playing with a stringent expression system for rec2, applying multiple cycles of recombinase production/oligonucleotide transformation, and reversibly inhibiting the MMR system during a limited time window we report in the following discussion high-fidelity recombination frequencies that approach those achieved with the archetypal Red-based system (Datsenko and Wanner, 2000). This opens genome editing possibilities in this environmental bacterium that were thus far limited to strains of E. coli, closely related enteric species (Nyerges et al., 2018, Szili et al., 2019), and some lactic acid bacteria (van Pijkeren et al., 2012).
Results
Optimization of Rec2 and MutLE36KPP Delivery for ssDNA Recombineering
The bicistronic gene cassette of pSEVA2514-rec2-mutLE36KPP (Figure 1A) was developed earlier for examining the hierarchy of recognition of different types of single-nucleotide mispairs by the native MMR system of P. putida (Aparicio et al., 2019b). In this construct rec2 and mutLE36KPP were placed under the control of the thermo-inducible PL/cI857 system, in which the product of cI857 represses the PL promoter at 30°C but becomes inactivated at 42°C, triggering the expression of the genes after a short thermal shift. During the course of that work, we noticed that a short, transient thermal induction of the Rec2 recombinase increased very significantly ssDNA recombineering (∼1 order of magnitude) when compared with the same with an expression device responsive to 3-methylbenzoate (i.e., xylS/Pm). Although the reason for this improvement is not entirely clear, it may have resulted from (1) the short-lived, high-level transcription of the otherwise toxic recombinase—when compared with the permanent hyperexpression caused by the chemically inducible system; (2) thermal inactivation of ssDNA nucleases and thus improved survival of the mutagenic oligonucleotides in vivo; or (3) a combination of both. In any case, the average frequency of single-base replacements in just one single-shot recombineering test was in the range of 1 × 10−2 mutants per viable cell. This was high when compared with previous recombineering efforts in this bacterium (Aparicio et al., 2016) but still low for identifying mutations without a selectable phenotype. We, however, speculated that by multi-cycling the procedure with short thermal pulses of recombinase induction and transformation with mutagenic oligos, such frequencies could be added at each cycle, eventually resulting in high nucleotide replacement rates. A second realization (Aparicio et al., 2019b) was that transient co-expression of the dominant allele MutLE36KPP of the MMR system of P. putida along with the rec2 gene in plasmid pSEVA2514-rec2-mutLE36KPP (Figure 1) virtually eliminated recognition of any type of base mispairings in DNA. This allowed entering all classes of nucleotide replacements that would otherwise be conditioned by MMR—without triggering a general mutagenic regime. Yet, note that both activities (Rec2 and MutLE36KPP) were delivered in vivo with a high-copy-number vector with an origin or replication (RSF1010) of unknown thermal sensitivity. This may result in some instability upon thermal cycling of the procedure for boosting recombineering efficiency (see below). To determine the best plasmid frame for rec2-mutLE36KPP transient expression, the cognate DNA segment was recloned in vectors pSEVA2214 (RK2 origin or replication, low copy number) and pSEVA2314 (pBBR1 origin, medium copy number) as shown in Figure 1A. Recombineering tests were then carried out with oligonucleotide NR, which generated a double mutation in gyrA endowing resistance to nalidixic acid (NalR) by means of two MMR-sensitive changes G → A and C → T. In parallel, another MMR-insensitive change A → C was also tested with oligonucleotide SR that mutated rpsL for making cells resistant to streptomycin (SmR), and recombineering assays were run in non-induced and heat-induced cultures. The results of this test indicated that thermal induction of rec2 and mutLE36KPP genes enhances recombineering by 3–4 logs with efficiencies in the range of 1 × 10−2 mutants/viable cell for the three plasmids assayed. Performance comparison points to pSEVA2314-rec2-mutLE36KPP as the preferred construct of reference for the multi-site mutagenesis platform presented in the following discussion. On the basis of this we set out to re-create in P. putida the same conditions that enabled implementation in E. coli of high-efficiency ssDNA recombineering protocols such as MAGE (Wang et al., 2009), DIvERGE (directed evolution with random genomic mutations; Nyerges et al., 2018), and pORTMAGE (portable MAGE; Nyerges et al., 2016)—and thus expand frontline genomic editing methods toward this environmentally and industrially important bacterium.
Figure 1.

Influence of Plasmid Copy Number on the Editing Efficiency of the Heat-Induced rec2-mutLE36KPP Genes
(A) Genetic map and structure of plasmids used in this study. The figure shows the plasmids tested, all having the same elements with the exception of the origin of replication, represented with “x.” T0 and T1, transcriptional terminators; Km, kanamycin resistance gene; oriT, origin of transfer; cI857-PL, temperature-inducible expression system; rec2, recombinase; mutLE36KPP, dominant-negative allele of mutL; ori x (origin of replication): #2, RK2 (low copy number); #3, pBBR1 (medium copy number); #5, RSF1010 (medium-high copy number). Pictures are not drawn to scale.
(B) Recombineering assays with P. putida EM42: the strain harboring each pSEVA2x14-rec2-mutLE36KPP variant was subjected to recombineering with oligos SR and NR upon heat induction of the cI857-PL expression system and without induction. After overnight recovery, culture dilutions were plated on LB-Sm (SR oligo) and LB-Nal (NR oligo) to estimate the number of allelic changes. Culture dilutions plated on LB allowed viable cell counting. Column values represent mean recombineering frequencies (mutants per 109 viable cells) of two independent experiments with the standard deviation.
Cyclic Pulses of rec2/mutLE36KPP Expression Enable a High Level of Single-Nucleotide Substitutions
The first issue at stake was determining the frequencies of mutations caused by using a cocktail of oligonucleotides targeting five genes representative of diverse genomic locations, different types of nucleotide changes, and associated or not with selectable traits upon multiple ssDNA recombineering cycles. The genes at stake, their position in the chromosomal map, the cognate phenotypes, and the type of replacements brought about by the corresponding mutagenic ssDNAs are summarized in Figure 2. They were all designed to pair sequences in the lagging strand of the replication fork in each of the replichores of the P. putida genome according to Aparicio et al. (2016). Note that the experiments were run with P. putida EM42, not with the archetypal strain KT2440. This is because it is a recA+ derivative of the EM383 genome-streamlined variant that has higher endogenous levels of ATP and NAD(P)H and has thus become a preferred metabolic engineering platform (Martínez-García et al., 2014). Moreover, the modifications entered in P putida EM42 make this strain more tolerant to pulses of high temperature (Aparicio et al., 2019a), as repeatedly applied throughout this work (see later). The cyclic recombineering protocol (see Transparent Methods for details) is summarized in Figure 3, and it basically involves four steps: (1) growing cells, (2) triggering thermal induction of rec2 and mutLE36KPP genes by a short heat shock, (3) preparing competent cells for electroporation with the mutagenic oligonucleotides, and (4) recovering the culture for a new cycle.
Figure 2.

Target Genes and Recombineering Oligonucleotides Used for HEMSE
(A) The five genes selected as targets for recombineering are represented in the chromosomal map of P. putida KT2440 with gene coordinates and strand orientation. oriC and dif regions are shown to define the two replichores in the genomic map. Pictures are not drawn to scale.
(B) The main features of recombineering oligonucleotides used to assay HEMSE are shown: name of oligo, target gene with its locus tag, type of allelic replacement, level of MMR sensitivity of the allelic changes, and the cognate phenotypes produced. See Tables S1 and S3 for additional information.
Figure 3.

Scheme of HEMSE Cycle
The main steps of the procedure are depicted: cultures of P. putida EM42 (pSEVA2314-rec2-mutLE36KPP) grown at OD600 = 1.0 are induced by a heat shock at 42°C/15 min; then competent cells are prepared and transformed with recombineering oligonucleotides. After recovery on fresh media at 30°C/170 rpm, cultures enter into the next round of HEMSE by applying the induction step. Screening of allelic replacements within a given cycle is performed after recovery by plating culture dilutions on the appropriate solid media (see Transparent Methods for details).
The results of applying multiple recombineering cycles to P. putida EM42 (pSEVA2314-rec2-mutLE36KPP) with the oligonucleotides listed in Figure 2B are shown in Figure 4A. Note that the frequencies of mutant appearance increased during the runs from 2.8 × 10−3 (1 cycle) to 9.3 × 10−2 (10 cycles) in the case of gyrA, and from 4.8 × 10−2 to 2.0 × 10−1 for pyrF under the same conditions. In the best-case scenario (i.e., gene rpsL), the frequencies multiplied by 24-fold, reaching a remarkable 21%. After the 10th cycle, these figures are thus close to the rates reported in E. coli with the archetypal Red-β system of phage lambda and also to the theoretical limit of recombineering frequencies (25%) that stems from segregation of one allelic change after two rounds of genome replication (Wang et al., 2009, Nyerges et al., 2016). It is worth mentioning that control strain P. putida EM42 harboring insertless vector pSEVA2314—but transformed with the same mutagenic oligonucleotides—gave rise to recombineering frequencies ∼ 1 × 10−5/1 × 10−6 mutants/viable cells per cycle for single changes (Figure S1). Given that these background levels are higher with thermal induction than with chemical induction (Ricaurte et al., 2018), it is plausible that heat shock intrinsically improves recombineering regardless of the action of heterologous recombinases. As a matter of fact, purely endogenous ssDNA recombineering at significant frequencies has been reported in a variety of Gram-negative bacteria, including E. coli and Pseudomonas syringae (Swingle et al., 2010b), a fact that plays in our favor for establishing the methodology in P. putida.
Figure 4.

Editing Efficiencies HEMSE
(A) Editing efficiencies of single changes were assayed applying 10 cycles of HEMSE to P. putida EM42 (pSEVA2314-rec2-mutLE36KPP) using an equimolar mixture of oligos SR, NR, RR, PR, and CR. After recovery steps of cycles no 1 (C-I), no 5 (C-V), and no 10 (C-X), appropriate dilutions of the cultures were plated on LB to estimate viable cells and also on LB solid media supplemented with Sm, Nal, Rif, 5FOA-Ura, or benzoate to enumerate allelic replacements. Colonies growing on Sm, Nal, Rif, or 5FOA-Ura were counted as allelic changes, whereas brown, catechol-accumulating colonies growing in LB-benzoate were counted as catA-I- clones (see phenotype in Figure S2). Recombineering frequencies of single replacements at each cycle were normalized to 109 viable cells, and the media of two independent replicas were plotted with standard deviations. Absolute recombineering frequencies (mutants per viable cell) are also depicted over the bars.
(B) From the same experiments, editing efficiencies of multiple changes were analyzed. Dilutions of C-I, C-V, and C-X were plated on LB-SmNalRif-5FOA-Ura and LB-SmNalRif-5FOA-Ura-benzoate solid media, allowing the estimation of, respectively, quadruple (SmR NalR RifRpyrF) and quintuple (SmR NalR RifRpyrF−catA-I-) editions. Results are represented as in (A).
(C) A representative plate of quintuple screening at C-X is shown. The zoom-up shows colonies with the characteristic dark brown phenotype of catA-I- clones.
The most remarkable outcome of the operations shown in Figure 4A was that such high figures enabled manual screening of inconspicuous mutations, thus avoiding the need of adding a genetic counterselection device (e.g., CRISPR/Cas9) for identifying rare changes. As these results accredited the value of multi-cycling thermoinduction of the bicistronic rec2-mutLE36KPP operon of pSEVA2314-rec2-mutLE36KPP for raising ssDNA recombineering efficiency, the next obvious question was whether the high figures could afford simultaneous multi-site genomic editing with mixtures of mutagenic oligos, in a fashion reminiscent of the MAGE process available for E. coli.
Multi-site Editing of Non-Adjacent Genomic Locations
Given average individual mutation rates of 10% after 10 thermal recombineering cycles and assuming they are separately maintained when cells face a cocktail of mutagenic oligonucleotides one can predict frequencies of 1% double changes all the way to 0.001% mutants (1 × 10−5) of genomes with all the five changes in the absence of any phenotypic advantage. To test this prediction, we subjected a culture of P. putida EM42 (pSEVA2314-rec2-mutLE36KPP) to 10 cycles of thermo-induced recombineering (see Transparent Methods) with re-transformation in each cycle with an equimolar mixture of oligos SR, NR, RR, PR, and CR (Figure 2B; Table S1) so that all possible changes could be entered in the same cells. Emergence of multiple (i.e., quadruple and quintuple) mutations in the population was then monitored at cycles I, V, and X and their frequencies recorded. Figure 4B shows the results of such a procedure. The data exposed a good match between the theoretical expectation of multiple changes and the actual figures, although the evolution of the mutation rates was not linear. At cycle #1, single changes showed recombineering frequencies barely below 1 × 10−2 mutants per viable cell. If we take that as a reference, theoretical frequencies of acquisition of four and five changes would be 1 × 10−8 and 1E−10 respectively, whereas the actual numbers were way higher (6 × 10−6 and 2 × 10−7). By cycle #5, single changes reached average recombineering above 5 × 10−2. The gross theoretical prediction for simultaneous appearance of four and five changes would be as low as 6 × 10−6 and 3 × 10−7. Yet, again, the actual experiments yield 1.3 × 10−4 and 2.3 × 10−6 mutants per viable cell for quadruple and quintuple mutants. By cycle #10, however, the scenario was different. Single changes appeared at frequencies ∼ 1 × 10−1, close to the theoretical maximum of recombineering efficiency (2.5 × 10−1). In this case, predicted frequencies for four and five changes were 1 × 10−4 and 1 × 10−5, which were very similar to the actual numbers delivered by the experiment, i.e., 2 × 10−4 and 6 × 10−6.
The aforementioned results suggested that during the first five recombineering cycles a strong co-selection phenomenon occurs. Appearance of multiple mutations is 2–3 logs higher than expected, suggesting that cells undergoing ssDNA incorporation in specific loci are more prone to incorporate changes in other genomic locations. This phenomenon, which has been observed before (Carr et al., 2012, Gallagher et al., 2014), could be due to differences in the ability of single cells in a population to take up exogenous ssDNA upon electroporation. Regardless of the specific mechanisms, the results of Figure 4B show that multi-cycle recombineering boosts mutagenic frequencies through single to quintuple changes. Yet, whereas 10 cycles appear to reach saturation at single sites, it is plausible that additional runs could enrich further the population in multi-edited bacterial cells. Taken together, the experiments of Figure 4 document the power of the hereby described method for simultaneously targeting five genomic sites of P. putida for desired mutations. On this basis we propose to call the entire workflow high-efficiency multi-site genomic editing (or HEMSE). The method is conceptually comparable to MAGE developed for E. coli (Wang et al., 2009), but it lacks (thus far) the automation aspect.
As a growing culture of P. putida in lysogeny broth (LB) typically ranges from 108 to 109 cells/mL from early exponential to early stationary phase, we speculated that the maximum number of genes that could be edited in a HEMSE experiment of this sort with mixed oligos in the absence of any selective advantage or phenotypic screening could be ∼ 8–9. This is clearly not enough for massive changes of the sort necessary, e.g., for recoding a whole genome (Isaacs et al., 2011) or reassigning/erasing specific triplets (Ostrov et al., 2016). Fortunately, in most typical metabolic engineering endeavors, the issue is not so much entering many defined mutations in given chromosomal sites but fostering the system to explore a solution space by letting it come up with many combinations—the most successful of which can be enriched and subject to further mutation rounds. This effect can be exacerbated if the mutagenic oligos boost the diversification of, e.g., regulatory sequences, so their combination generates fluctuations in the stoichiometry of a multi-gene pathway (Hueso-Gil et al., 2020)—or they create variants of the same protein with different activities by diversifying specific segments. The technical issue shared by all these scenarios is the focusing of the diversification in a defined sequence window of the genomic DNA. In this context the question is whether the above-described HEMSE is instrumental to this end also—as the recombineering-based method to the same end called DIvERGE is in E. coli and related enterobacteria (Nyerges et al., 2018).
Diversification of the SD Motif Context Creates New Functional RBSs in P. putida
To have a tractable proxy of generation in vivo of large libraries of functional DNA sequences in the P. putida genome, the experimental setup shown in Figure 5 was developed. In it, a Tn7 mini-transposon vector was inserted with the gfp gene downstream of the constitutive promoter PEM7 but lacking a recognizable Shine-Dalgarno (SD) sequence for translation initiation. The hybrid transposon was subsequently inserted in the cognate attTn7 site of the P. putida EM42 chromosome (see Transparent Methods) from which it was expectedly unable to produce any detectable fluorescence. The resulting strain (P. putida TA245, Table S2) was transformed with pSEVA2314-rec2- mutLE36KPP and used in recombineering experiments with oligonucleotides designed for creating ribosomal binding site (RBSs) variants. The business parts of such oligonucleotides are shown in Figure 5B. As controls we used oligos named RBS-C6 and RBS-C9. These ssDNA enter, respectively, a short and an extended SD sequence, 8 bp upstream of the start codon of the gfp gene, using as a reference the P. putida 16S ribosomal gene and containing the core for optimal translation of 5′-GAGG-3′ (Shine and Dalgarno, 1975, Kozak, 1983, Farasat et al., 2014). For RBS diversification we used oligonucleotides RBS-Deg6 and RBS-Deg9 (Figure 5B), which include soft randomized sequences with discrete changes R (A or G) that cover six degenerated positions with a potential to generate 64 (= 26) combinations. This was expected to create a large population of RBS of different efficacies, which could be quantified through fluorescent emission of individual cells.
Figure 5.

Diversification of the gfp Shine-Dalgarno Motif
(A) A mini-Tn7 transposon bearing the gfp gene devoid of its original SD sequence and under the control of the constitutive PEM7 promoter was constructed. The elements depicted are: Tn7-L and Tn7-R, left and right Tn7 sites; T0 and T1, transcriptional terminators; GmR, gentamicin resistance gene; PEM7, constitutive promoter; GFP, green fluorescent protein gene. A blue arrow represents the target region of recombineering oligonucleotides aimed to reconstruct the gfp ribosome-binding site (shown as a red square).
(B) Partial sequence of the four recombineering oligonucleotides (Table S1) designed to introduce SD motifs upstream of the gfp gene. RBS-C6 and RBS-C9 insert, respectively, the semi-canonical AGGAGG and the canonical TAAGGAGGT SD motifs eight nucleotides upstream the ATG start codon of the gfp (underlined); RBS-Deg6 and RBS-Deg9 insert, the randomized sequences RRRRRR and TARRRRRRT, where R stands for A (adenine) or G (guanine). Each degenerated oligonucleotide comprises a pool of 64 variants (26) with all possible combinations A/G.
(C) The mini-Tn7 device was inserted in the attTn7 site of P. putida EM42. Upon transformation with pSEVA2314-rec2-mutLE36KPP, the resulting strain P. putida TA245 (pSEVA2314-rec2-mutLE36KPP) was subjected to one HEMSE cycle with the recombineering oligos in independent experiments. After plating in LB-GmKm-charcoal, GFP-positive clones were identified. A plate from the screening of RBS-Deg9 is also shown, with a magnification of a fluorescent colony.
For the experiments described in the following discussion, P. putida TA245 (pSEVA2314-rec2-mutLE36KPP) was separately subject to one recombineering cycle with each of four oligos of Figure 5B, after which cells were diluted and plated in charcoal-LB agar for easing visual detection of colonies emitting low fluorescence on a black background. Positive controls RBS-C6 and RBS-C9 allowed the estimation of editing frequencies as GFP+ cells/total number of cells, which resulted in 5.9 × 10−4 and 9.9 × 10−4, respectively. Those values were relatively low when compared with the recombineering efficiencies reported earlier for single changes (∼1 × 10−2). This could be possibly due to the shorter homology arms of the oligos (30 nucleotides [nts]) and the extended sequence inserted between them (Aparicio et al., 2020). Yet, these figures provided a reference for subsequent quantification of the effect of soft-randomized oligos RBS-Deg6 and RBS-Deg9. After treatment with these, cultures were diluted and plated for inspection of ∼9,000 colonies resulting of each recombineering experiment. Visual screening of the colonies revealed the appearance of 67 and 53 fluorescent clones coming, respectively, from experiments with RBS-Deg6 and RBS-Deg9. These 120 clones were picked up for further analysis. PCR and sequencing of the region upstream the gfp gene allowed identification of 14 variants of RBS-Deg6 and 17 variants of RBS-Deg9, the GFP levels of which were measured by flow cytometry. The results plotted in Figure 6 show that the different variants delivered emissions ranging from very low to high fluorescent levels across a 20-fold change span. It is worth highlighting that the best RBS of the series (Strain Code #33, Figure 6) has a perfect match with the complementary sequence of the last 9 nts of the 16S ribosomal RNA of P. putida (PP_16SA). Other clones (e.g., #32; RBS = 5′-AAGGAG-3′) also displayed high fluorescence levels. Interestingly, a few productive variants contained the same 6-nt sequence in the degenerated region regardless of the type of randomized oligo (e.g., #32 = #30, #26 = #16, #14 = #11, and #23 = #10). Although most high-signal variants belong to the longer RBS-C9-borne clones, a comparison of signals does not support the hypothesis that longer complementarity to the 16S ribosomal sequence correlates with more efficient translation. Other factors have been proposed to affect translation efficacy of RBS variants, such as the stability and secondary structure of RNA and transcriptional efficiency (Chen et al., 1994, Salis et al., 2009). Regardless of the possible biological significance of the results, the data of Figure 6 certifies the efficacy of the HEMSE platform to generate diversity in specific genomic segments—a welcome feature that can doubtless be multiplexed to other chromosomal locations as required.
Figure 6.

Characterization of the Diversified Library of SD Sequences
The screening of the HEMSE experiments performed in P. putida TA245 (pSEVA2314-rec2-mutLE36KPP) with RBS-Deg6 and RBS-Deg9 yielded 31 variants in the ribosome-binding site of the gfp gene. The GFP expression of this library was analyzed by flow cytometry including two negative controls: P. putida EM42 (Strain #1), in which there is no gfp gene, and the ancestral strain P. putida TA245 (pSEVA2314-rec2-mutLE36KPP), harboring the mini-Tn7 and the gfp gene but lacking the original SD sequence (Strain #2). The plot shows the mean fluorescent emission of individual clones from two biological replicas with standard deviations. A Strain Code was assigned to each variant analyzed, and the putative SD sequence identified 8 nts upstream of the gfp start codon is shown. Variants showing identical sequence in the randomized region are highlighted with the same color. A plate of LB-charcoal agar with streaks of the two controls and three representative clones exhibiting low, medium, and high signal rates (#7, #25, and #33, respectively) is also depicted under UV light.
Discussion
In this work we have merged and adapted to P. putida and in a single platform three of the most efficacious genetic tools available to metabolic engineers for generating diversity in vivo focused on a predetermined number of chromosomal DNA segments: ssDNA recombineering (Wang et al., 2009), portable MAGE (Nyerges et al., 2016), and DIvERGE (Nyerges et al., 2018). Although conceptually identical to such methods already applied to E. coli, their recreation in a non-enterobacterial species involved (1) the search and testing of functional equivalents of the parts involved but recruited from Pseudomonas genomes and (2) adaptation and optimization to the distinct physiology of the species and strain at stake. Although we have not made a side-by-side comparison of the frequencies resulting from standard MAGE in E. coli and the ones presented in this work, numbers in the range of 10% replacements after 10 recombineering cycles could be sufficient to implement the same powerful method in P. putida. We are reluctant, however, to use the same acronym, because the automation feature is not in sight and the multiplexing is still problematic with the current efficiencies.
One can envision various ways through which HEMSE could be further improved. End-terminal degradation of the mutagenic oligos in vivo does not seem to be an issue: performance of 5′-phosphorothioated ssDNA (which cannot be degraded by exonucleases, Wang et al., 2009) is indistinguishable from non-phosphorothioated equivalents (Aparicio et al., 2020). However, the nature and origin of the recombinase that catalyzes invasion of the DNA replication fork by the synthetic oligo makes a considerable difference (Chang et al., 2019). It is possible that such recombinases act in concert with additional endogenous proteins that could be characteristic of each species (Caldwell et al., 2019, Yin et al., 2019). It seems thus desirable that future alternatives to the Rec2 activity encoded in pSEVA2314-rec2-mutLE36KPP (Figure 1) are mined in species-specific Pseudomonas genomes and phages—by themselves or in combination with other complementary genes. It should be straightforward to then replace the rec2 of pSEVA2314-rec2-mutLE36KPP by the improved counterparts, should they appear (e.g., Wannier et al., 2020), while maintaining the rest of the hereby described HEMSE protocol.
Limitations of the Study
As stated, one key bottleneck for implementing ssDNA recombineering in non-enteric bacteria is the efficacy of the core recombinase adopted in the experimental workflow. The work presented in this article is based on the one named Rec2, which emerged as the best in a limited bioinformatic and wet survey of ssDNA-binding proteins able to promote ssDNA invasion of the chromosomal replication fork (Ricaurte et al., 2018). However, it is most likely that other recombinases, whether naturally occurring or purposely engineered, may work better and could easily replace the Rec2 variant in our genome-editing pipeline whenever available. A second limitation is the somewhat poor ability of P. putida to capture exogenously added DNA, let alone ssDNA, as required for the hereby described method. Any progress in making this species—or any other—a better recipient of synthetic DNA will instantly translate in improved recombineering frequencies. Finally, the whole experimental workflow for multiple-site ssDNA recombineering is thus far restricted to manual implementation of the corresponding cycles. Despite the potential for automation claimed in the earliest description of MAGE (Wang et al., 2009), the cell separation step necessary for transforming bacteria with the mutagenic oligonucleotides still remains as a technical bottleneck that needs to be satisfactorily solved.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors are indebted to Sebastian S. Cocioba for posting in Twitter the useful recipe of LB-charcoal medium (https://twitter.com/ATinyGreenCell/status/1037332606432555009) and to Csaba Pal (Institute of Biochemistry, Biological Research Center, Szeged) for his continued support. This work was funded by the MADONNA (H2020-FET-OPEN-RIA-2017-1-766975), BioRoboost (H2020-NMBP-BIO-CSA-2018), SYNBIO4FLAV (H2020-NMBP/0500), and MIX-UP (H2020-Grant 870294) contracts of the European Union and the S2017/BMD-3691 InGEMICS-CM Project of the Comunidad de Madrid (European Structural and Investment Funds).
Author Contributions
T.A., E.M.-G., A.N., and V.d.L. designed the study. T.A. performed the experiments. T.A., E.M.-G., and V.d.L. wrote the manuscript.
Declaration of Interests
Authors declare no conflict of interest.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100946.
Contributor Information
Esteban Martínez-García, Email: emartinez@cnb.csic.es.
Víctor de Lorenzo, Email: vdlorenzo@cnb.csic.es.
Supplemental Information
References
- Aparicio T., Jensen S.I., Nielsen A.T., de Lorenzo V., Martinez-Garcia E. The Ssr protein (T1E_1405) from Pseudomonas putida DOT-T1E enables oligonucleotide-based recombineering in platform strain P. putida EM42. Biotechnol. J. 2016;11:1309–1319. doi: 10.1002/biot.201600317. [DOI] [PubMed] [Google Scholar]
- Aparicio T., de Lorenzo V., Martinez-Garcia E. Improved thermotolerance of genome-reduced Pseudomonas putida EM42 enables effective functioning of the PL/cI857 system. Biotechnol. J. 2019;14:e1800483. doi: 10.1002/biot.201800483. [DOI] [PubMed] [Google Scholar]
- Aparicio T., de Lorenzo V., Martínez-García E. A broad host range plasmid-based roadmap for ssDNA-based recombineering in gram-negative bacteria. In: de la Cruz F., editor. Horizontal Gene Transfer: Methods and Protocols. Springer US; 2020. pp. 383–398. [DOI] [PubMed] [Google Scholar]
- Aparicio T., Nyerges A., Nagy I., Pal C., Martinez-Garcia E., de Lorenzo V. Mismatch repair hierarchy of Pseudomonas putida revealed by mutagenic ssDNA recombineering of the pyrF gene. Environ. Microbiol. 2019;22:45–58. doi: 10.1111/1462-2920.14814. [DOI] [PubMed] [Google Scholar]
- Bao Z., Cartinhour S., Swingle B. Substrate and target sequence length influence RecTE(Psy) recombineering efficiency in Pseudomonas syringae. PLoS One. 2012;7:e50617. doi: 10.1371/journal.pone.0050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell B.J., Zakharova E., Filsinger G.T., Wannier T.M., Hempfling J.P., Chun-Der L., Pei D., Church G.M., Bell C.E. Crystal structure of the Redbeta C-terminal domain in complex with lambda Exonuclease reveals an unexpected homology with lambda Orf and an interaction with Escherichia coli single stranded DNA binding protein. Nucleic Acids Res. 2019;47:1950–1963. doi: 10.1093/nar/gky1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr P.A., Wang H.H., Sterling B., Isaacs F.J., Lajoie M.J., Xu G., Church G.M., Jacobson J.M. Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection. Nucleic Acids Res. 2012;40:e132. doi: 10.1093/nar/gks455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y., Wang Q., Su T., Qi Q. The efficiency for recombineering is dependent on the source of the phage recombinase function unit. bioRxiv. 2019:745448. [Google Scholar]
- Chen H., Bjerknes M., Kumar R., Jay E. Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res. 1994;22:4953–4957. doi: 10.1093/nar/22.23.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Zhang Y., Zhang Y., Pi Y., Gu T., Song L., Wang Y., Ji Q. CRISPR/Cas9-based genome editing in Pseudomonas aeruginosa and cytidine deaminase-mediated base editing in Pseudomonas species. iScience. 2018;6:222–231. doi: 10.1016/j.isci.2018.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino N., Court D.L. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc. Natl. Acad. Sci. U S A. 2003;100:15748–15753. doi: 10.1073/pnas.2434959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis H.M., Yu D., DiTizio T., Court D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U S A. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farasat I., Kushwaha M., Collens J., Easterbrook M., Guido M., Salis H.M. Efficient search, mapping, and optimization of multi-protein genetic systems in diverse bacteria. Mol. Syst. Biol. 2014;10:731. doi: 10.15252/msb.20134955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher R.R., Li Z., Lewis A.O., Isaacs F.J. Rapid editing and evolution of bacterial genomes using libraries of synthetic DNA. Nat. Protoc. 2014;9:2301–2316. doi: 10.1038/nprot.2014.082. [DOI] [PubMed] [Google Scholar]
- Hueso-Gil A., Nyerges Á., Pál C., Calles B., de Lorenzo V. Multiple-site diversification of regulatory sequences enables inter-species operability of genetic devices. ACS Synth. Biol. 2020;9:104–114. doi: 10.1021/acssynbio.9b00375. [DOI] [PubMed] [Google Scholar]
- Isaacs F.J., Carr P.A., Wang H.H., Lajoie M.J., Sterling B., Kraal L., Tolonen A.C., Gianoulis T.A., Goodman D.B., Reppas N.B. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 1983;47:1–45. doi: 10.1128/mr.47.1.1-45.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesic B., Rahme L.G. Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol. Biol. 2008;9:20. doi: 10.1186/1471-2199-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R., Liu J. Scarless and sequential gene modification in Pseudomonas using PCR product flanked by short homology regions. BMC Microbiol. 2010;10:209. doi: 10.1186/1471-2180-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes A., Amarir-Bouhram J., Faure G., Petit M.-A., Guerois R. Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51 and Gp2. 5 remote homologs. Nucleic Acids Res. 2010;38:3952–3962. doi: 10.1093/nar/gkq096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X., Yang Y., Ling W., Zhuang H., Li Q., Shang G. Pseudomonas putida KT2440 markerless gene deletion using a combination of lambda Red recombineering and Cre/loxP site-specific recombination. FEMS Microbiol. Lett. 2016;363:fnw014. doi: 10.1093/femsle/fnw014. [DOI] [PubMed] [Google Scholar]
- Martínez-García E., de Lorenzo V. Pseudomonas putida in the quest of programmable chemistry. Curr. Opin. Biotechnol. 2019;59:111–121. doi: 10.1016/j.copbio.2019.03.012. [DOI] [PubMed] [Google Scholar]
- Martínez-García E., Nikel P.I., Aparicio T., de Lorenzo V. Pseudomonas 2.0: genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb. Cell Fact. 2014;13:159. doi: 10.1186/s12934-014-0159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P.I., Martinez-Garcia E., De Lorenzo V. Biotechnological domestication of pseudomonads using synthetic biology. Nat. Rev. Microbiol. 2014;12:368–379. doi: 10.1038/nrmicro3253. [DOI] [PubMed] [Google Scholar]
- Nikel P.I., Chavarria M., Danchin A., de Lorenzo V. From dirt to industrial applications: Pseudomonas putida as a synthetic biology chassis for hosting harsh biochemical reactions. Curr. Opin. Chem. Biol. 2016;34:20–29. doi: 10.1016/j.cbpa.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Nyerges A., Csorgo B., Nagy I., Latinovics D., Szamecz B., Posfai G., Pal C. Conditional DNA repair mutants enable highly precise genome engineering. Nucleic Acids Res. 2014;42:e62. doi: 10.1093/nar/gku105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyerges A., Csorgo B., Nagy I., Balint B., Bihari P., Lazar V., Apjok G., Umenhoffer K., Bogos B., Pósfai G. A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. Proc. Natl. Acad. Sci. U S A. 2016;113:2502–2507. doi: 10.1073/pnas.1520040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyerges A., Csorgo B., Draskovits G., Kintses B., Szili P., Ferenc G., Révész T., Ari E., Nagy I., Bálint B. Directed evolution of multiple genomic loci allows the prediction of antibiotic resistance. Proc. Natl. Acad. Sci. U S A. 2018;115:E5726–E5735. doi: 10.1073/pnas.1801646115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterle S., Wuethrich I., Panke S. Toward genome-based metabolic engineering in bacteria. Adv. Appl. Microbiol. 2017;101:49–82. doi: 10.1016/bs.aambs.2017.07.001. [DOI] [PubMed] [Google Scholar]
- Ostrov N., Landon M., Guell M., Kuznetsov G., Teramoto J., Cervantes N., Zhou M., Singh K., Napolitano M.G., Moosburner M. Design, synthesis, and testing toward a 57-codon genome. Science. 2016;353:819–822. doi: 10.1126/science.aaf3639. [DOI] [PubMed] [Google Scholar]
- Ricaurte D.E., Martinez-Garcia E., Nyerges A., Pal C., de Lorenzo V., Aparicio T. A standardized workflow for surveying recombinases expands bacterial genome-editing capabilities. Microb. Biotechnol. 2018;11:176–188. doi: 10.1111/1751-7915.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronda C., Pedersen L.E., Sommer M.O., Nielsen A.T. CRMAGE: CRISPR optimized mage recombineering. Sci. Rep. 2016;6:19452. doi: 10.1038/srep19452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis H.M., Mirsky E.A., Voigt C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shine J., Dalgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254:34–38. doi: 10.1038/254034a0. [DOI] [PubMed] [Google Scholar]
- Swingle B., Bao Z., Markel E., Chambers A., Cartinhour S. Recombineering using RecTE from Pseudomonas syringae. Appl. Environ. Microbiol. 2010;76:4960–4968. doi: 10.1128/AEM.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingle B., Markel E., Costantino N., Bubunenko M.G., Cartinhour S., Court D.L. Oligonucleotide recombination in Gram-negative bacteria. Mol. Microbiol. 2010;75:138–148. doi: 10.1111/j.1365-2958.2009.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szili P., Draskovits G., Révész T., Bogar F., Balogh D., Martinek T., Daruka L., Spohn R., Vásárhelyi B.M., Czikkely M. Rapid evolution of reduced susceptibility against a balanced dual-targeting antibiotic through stepping-stone mutations. Antimicrob. Agents Chemother. 2019;63:e00207–e00219. doi: 10.1128/AAC.00207-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pijkeren J.-P., Neoh K.M., Sirias D., Findley A.S., Britton R.A. Exploring optimization parameters to increase ssDNA recombineering in Lactococcus lactis and Lactobacillus reuteri. Bioengineered. 2012;3:209–217. doi: 10.4161/bioe.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.H., Isaacs F.J., Carr P.A., Sun Z.Z., Xu G., Forest C.R., Church G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannier T.M., Nyerges A., Kuchwara H.M., Czikkely M., Balogh D., Filsinger G.T., Borders N.C., Gregg C.J., Lajoie M.J., Rios X. Improved bacterial recombineering by parallelized protein discovery. bioRxiv. 2020 doi: 10.1073/pnas.2001588117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J., Zheng W., Gao Y., Jiang C., Shi H., Diao X., Li S., Chen H., Wang H., Li R. Single-stranded DNA-binding protein and exogenous RecBCD inhibitors enhance phage-derived homologous recombination in Pseudomonas. iScience. 2019;14:1–14. doi: 10.1016/j.isci.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D., Ellis H.M., Lee E.C., Jenkins N.A., Copeland N.G., Court D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U S A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.