

Abstract
We report that expression of the α-COP protein rescues disease phenotype in a severe mouse model of Spinal Muscular Atrophy (SMA).. Lentiviral particles expressing α-COP were injected directly into the testes of genetically pure mouse strain of interest resulting in infection of the spermatagonial stem cells. α-COP was stably expressed in brain, skeletal muscle, and spinal cord without altering SMN protein levels. SMA mice transgenic for α-COP live significantly longer than their non-transgenic littermates, and showed increased body mass and normal muscle morphology at postnatal day 15. We previously reported that binding between SMN and α-COP is required for restoration of neurite outgrowth in cells lacking SMN, and we report similar finding here. Lentiviral-mediated transgenic expression of SMN where the dilysine domain in exon 2b was mutated was not able to rescue the SMA phenotype despite robust expression of the mutant SMN protein in brain, muscle and spinal cord. These results demonstrate that α-COP is a validated modifier of SMA disease phenotype in a mammalian, vertebrate model and is a potential target for development of future SMN-independent therapeutic interventions.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive inherited neurodegenerative disease and is the leading genetic cause of infant mortality in the United States with an incidence of approximately 1 in 10,000 live births and a carrier frequency of approximately 1 in 40 [1]. SMA is caused by low levels of the Survival Motor Neuron protein (SMN) due to loss of or mutations in the SMN1 gene [2]. Disease severity in SMA is dependent on the copy number of the nearly identical SMN2 gene. A single nucleotide change in exon 7 of SMN2 disturbs normal splicing, rendering the gene largely incapable of producing full-length SMN protein [3, 4]. However, each copy of SMN2 generates a minimal amount of full-length SMN protein, and thus SMN2 copy number mediates disease severity, leading to three types of SMA characterized by their time of onset and disease severity [5].
There are reports of discordant families where individuals with identical SMN2 copy number present with differing clinical courses [6, 7]. Analysis of these families identified genetic modifiers of SMA pathology including Plastin3 [8]. Expression of Plastin 3 ameliorated disease phenotype in mild mouse models of SMA as well as in Zebrafish [9, 10], but was unable to rescue disease phenotype in the severe Delta 7 mouse model [11]. Other modifier genes were identified in screens focused on splicing errors that occur in the absence of normal SMN protein levels. Chondrolectin (Chodl), a neuronal protein important in axon guidance, is mis-spliced in SMA mice [12, 13], and its over-expression in zebrafish was able to rescue the motor neuron disease phenotype [14]. These experiments demonstrated that expression of modifier genes could ameliorate SMA disease phenotypes.
Plastin 3 and other modifiers were identified using genetic screens. We chose to look at proteins that interacted with SMN as possible sources of phenotypic modifiers. α-COP, a member of the COPI coatomer complex, was identified by yeast two hybrid screening as a direct binding partner of SMN [15]. In cell culture models, over-expression of human α-COP was able to rescue defects in neurite outgrowth resulting from low SMN levels, and mutants of SMN that were unable to bind α-COP failed to restore neurite length [16]. In a zebrafish model of SMA, normal motor neuron morphology was similarly rescued by expression of human α-COP as with human SMN, while SMN mutants defective for binding to α-COP failed to extend axons properly [17].
We report here the generation of lentiviral based transgenic mice expressing α-COP, SMN and SMN where the α-COP binding site in exon 2b has been mutated. This is the first report of a lentiviral-based transgenic rescue of disease phenotype in a mouse model of SMA.
Results
Generation and Characterization of lentiviral-mediated α-COP transgenic mice
To begin examining the activity of α-COP in a mouse model of SMA, we first examined its protein level in spinal cord lysates from both Delta7 (Jackson stock 005025) and the so-called “Taiwanese” or “Li” mouse model of SMA (Jackson stock 005058) hereafter referred to as 5058 [18, 19]. Spinal cords were harvested from these SMA strains and healthy siblings at postnatal day 6. Representative blots in Figure 1a and 1b show that α-COP levels are unchanged in SMA spinal cord compared to healthy siblings at this time point.
Figure 1: A-COP levels are unchanged in spinal cord from SMA mouse models.
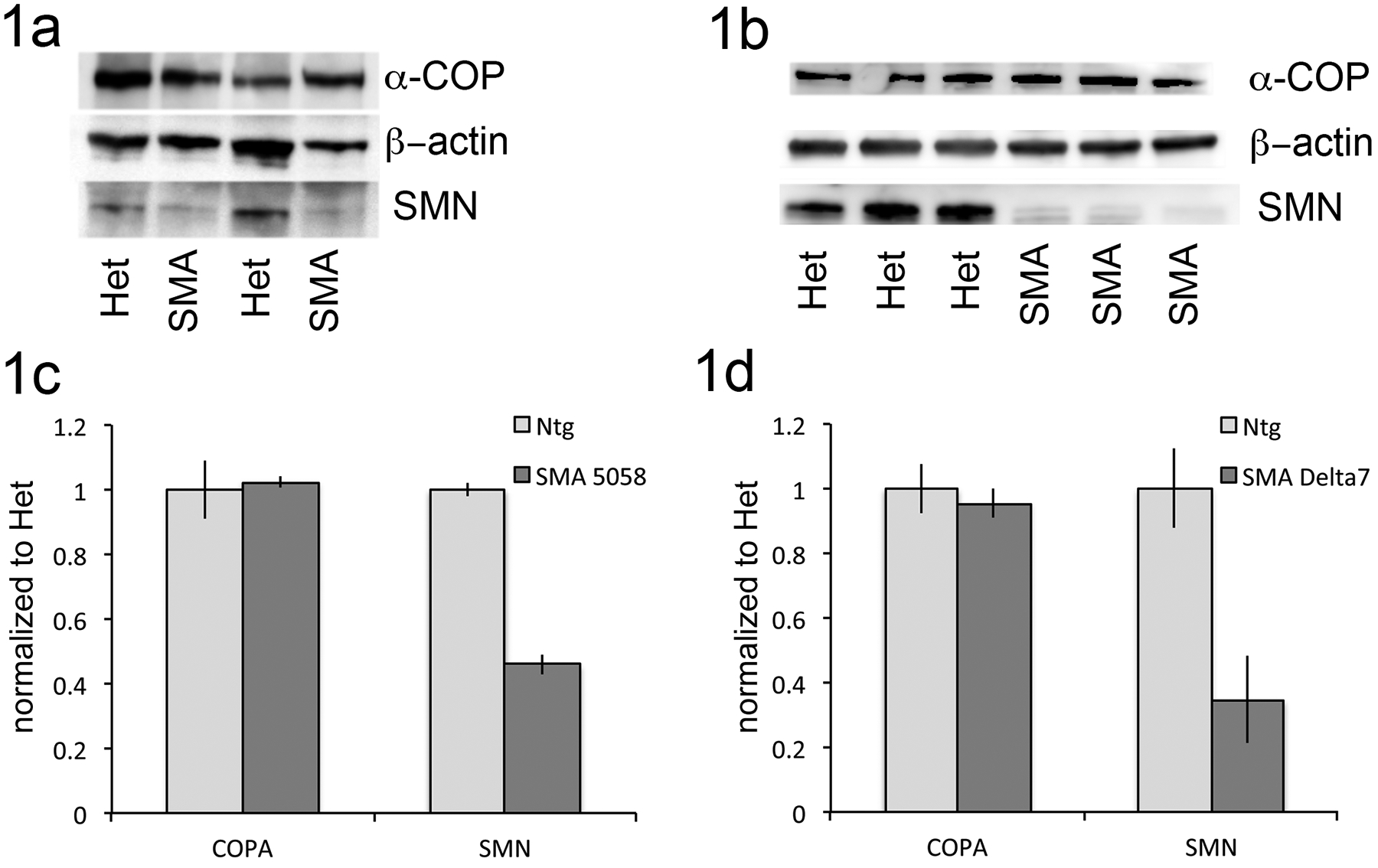
1a) Representative blot of spinal cord lysates from 5058 SMA pups and healthy siblings at postnatal day 6 probed for endogenous α-COP (H3 antibody), total SMN (Mansma2) and β–actin as a loading control. 1b) Representative blot of spinal cord lysates from Delta7 SMA pups and healthy siblings at postnatal day 6 probed for endogenous α-COP (H3 antibody), total SMN (Mansma2) and β–actin as a loading control. 1c,d) Quantification band intensity of SMN and α-COP protein levels from multiple spinal cord lysates (n=6 per genotype) from 5058 and Delta7 mouse models at postnatal day 6. Protein levels were normalized to heterozygous siblings using β–actin as a loading control. Error bars represent SEM.
To determine whether over-expression of α-COP could ameliorate the SMA phenotype in an intact, mammalian model system, we sought to introduce FLAG-tagged human transgene into the Li model of SMA This strain is maintained on a pure FVB/NJ background, given that genetic background has been shown to impact disease severity in models of SMA and other neuromuscular diseases [9, 20, 21]. A lentiviral-mediated transgenic approach was used to infect the spermatagonial stem cells of FVB/NJ male mice [22]. This FLAG-tagged human α-COP was previously used to rescue SMA phenotypes in cultured cells and zebrafish [16, 17] (Figure 2a). Lentivirus was produced in HEK-293TT cells, concentrated, and then injected into the testes of anesthetized FVB/NJ male mice. After recovering for 30 days, the mice were bred to FVB/NJ females. Of the resultant progeny, 4 of 7 F0 pups were positive for the EF1α-FLAG-COPA transgene (Figure 2b).
Figure 2: Generation of α-COP lentiviral-mediated transgenic mice.
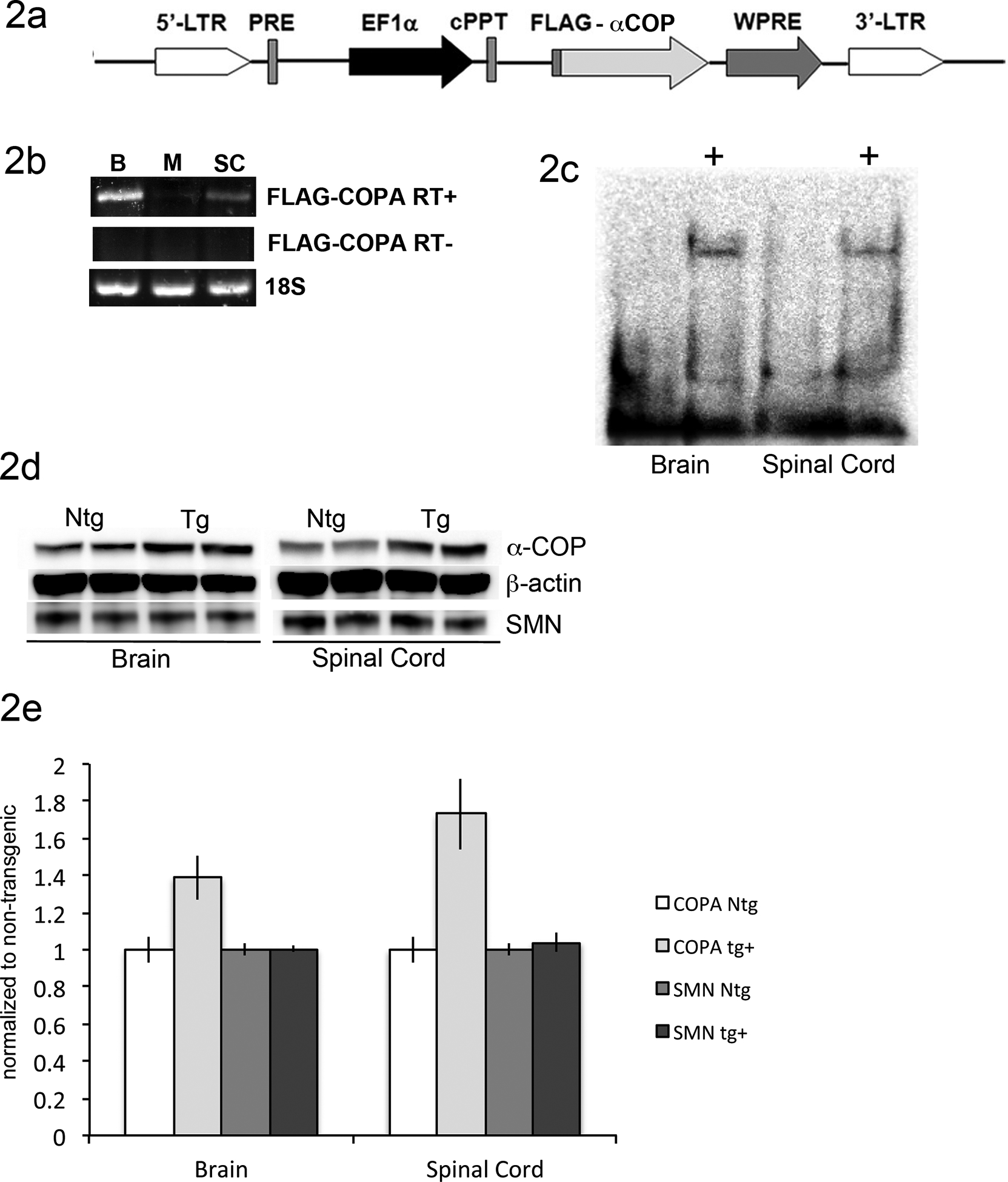
2a) Schematic of EF1a-FLAG-COPA lentiviral vector. Previously characterized FLAG-tagged human α-COP cDNA was sub cloned from pExchange4a into the Afe1 and Not1 sites, replacing the original FLAG-SMN insert. 2b) PCR genotyping results from tail DNA shows 4 of 7 transgene positive F0 progeny in the first litter following injection of the EF1a-FLAG-COPA lentivirus. Primers were anchored in the FLAG tag (forward) and COPA exon 1 (reverse) and the original lentiviral vector was used as the positive control template (+). 2c) RT-PCR shows transgene expression in brain, muscle and spinal cord from transgene positive F1 progeny. RT- samples were generated with no reverse transcriptase enzyme present demonstrating that genomic DNA contamination was not present. Primers against 18S RNA were used as a loading control to show that equal amount of cDNA were present in each reaction. 2d) Western Blot analysis with antibody against the FLAG tag shows expression of FLAG-α-COP protein in brain and spinal cord of transgenic animals (+) compared to non-transgenic littermates. 2e) Western blot analysis from brain and spinal cord lysates of non-transgenic littermates (Ntg) and FLAG-α-COP transgenic animals (Tg) shows the levels of total α-COP protein as well as SMN, using β–actin as a loading control. 2f) Quantification of α-COP and SMN protein levels from multiple western blots (n=6 per genotype) normalized to non-transgenic with using β–actin as a loading control shows no significant changes in SMN level after α-COP over-expression. Error bars represent SEM.
Two transgene positive F0 progeny were selected and expanded on the 5058 background. The transgene was detected by RT-PCR at high levels in brain and spinal cord and modest levels in skeletal muscle (Figure 2c). Western blot analysis with anti-FLAG antibody was able to detect the FLAG-α-COP protein in brain and spinal cord lysates (Figure 2d). Western blot analysis of brain and spinal cord lysates using an antibody that detects both mouse and human α-COP shows that transgenic animals over-express α-COP at a range of 1.4–1.8-fold over endogenous. Blotting with an antibody against SMN shows that over-expression of α-COP does not increase SMN protein levels. Figure 2e shows a representative blot, Figure 2f shows the quantification of α-COP and SMN protein levels from multiple animals relative to β–actin loading controls.
Expression of α-COP extends survival in an SMA mouse model
After expression of the FLAG-α-COP transgene in brain and spinal cord was confirmed at both the mRNA and protein level, we proceeded to cross the mice onto the Li SMA model [19]. We followed the breeding scheme outlined by the Didonato lab [18] to produce animals that were heterozygous for the SMN targeted deletion, the SMN2 transgene and the FLAG-α-COP transgene ((SMN1tm1Hung/WT, Tg(SMN2)2HungTg/0, FLAG-COPAtg/0)). These mice were crossed to produce “experimental breeding pairs” (SMN1tm1Hung/tm1Hung, Tg(SMN2)2HungTg/Tg × SMN1tm1Hung/WT, Tg(SMN2)2HungTg/0) where one parent was also heterozygous for the FLAG-α-COP transgene. This produced litters that were ~50% SMA, and of those SMA pups, ~50% were positive for the FLAG-α-COP transgene.
SMA pups expressing the FLAG-α-COP transgene were larger beginning at postnatal day 3 (Figure 3a). While SMA pups die around 12 days postnatal, the FLAG-α-COP expressing animals survived into adulthood. Figure 3b shows a Kaplan-Meier analysis (n=50) and expression of FLAG-α-COP statistically extends survival (p<0.01), shifting the median survival from 11 to 18 days. Beginning around postnatal day 21, severe necrosis of the tail and footpads set in (see third panel of Figure 3a) and the animals routinely had to be euthanized to prevent auto-amputation of the hind limbs. Although the FLAG-α-COP rescue animals were smaller than their healthy siblings (Figure 3c weight curve), the animals were alert and demonstrated normal cage activity, strength and mobility. To determine if the muscle structure was preserved in the FLAG-α-COP SMA mice, tibialis anterior (TA) was isolated at postnatal day 15. Although the muscle itself was in proportion to the smaller body size, hematoxylin and eosin staining of paraffin embedded sections shows that the muscle fibers are healthy and of normal diameter (Figure 3d). Taken together, these results demonstrate that similar to results obtained in cell culture and zebrafish model of SMA, expression of α-COP reduces the severity of the SMA phenotype in a mouse model of spinal muscular atrophy.
Figure 3. Expression of FLAG-α-COP extends survival of SMA mouse model.
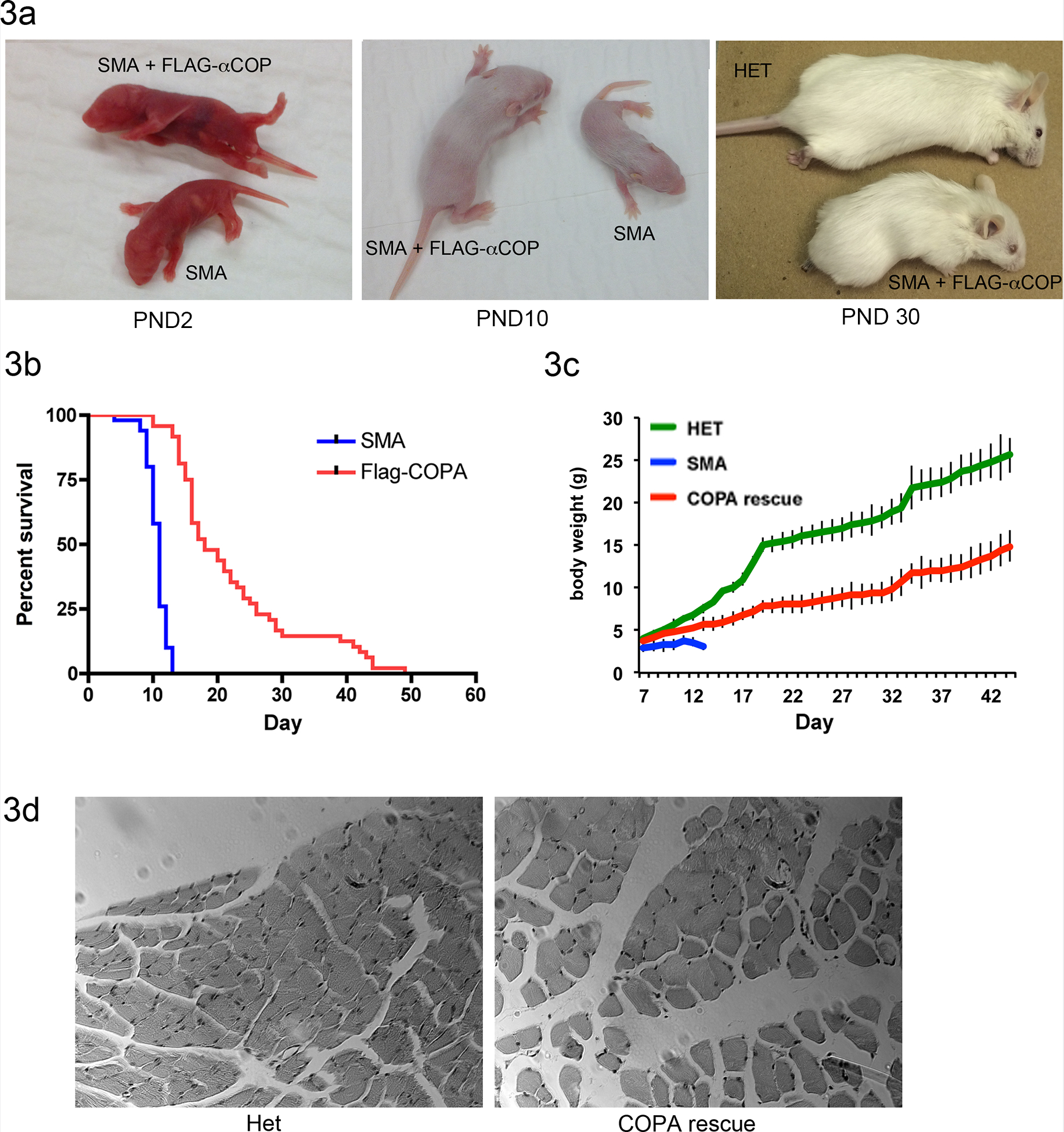
3a) Mice heterozygous for the FLAG-α-COP on the SMA background are larger than their transgene negative SMA siblings at postnatal day 2 and 10. By postnatal day 30 they are significantly smaller than their healthy siblings and have severe tail and footpad necrosis. 3b) Kaplan-Meier survival curve shows that expression of FLAG-α-COP significantly extends survival in SMA mice (p<0.01). 3c) Body weight curves show that although they are significantly smaller than healthy heterozygous animals, FLAG-α-COP expressing SMA animals continue to gain weight until they are euthanized due to severe footpad necrosis. 3d) Hematoxylin and eosin staining of paraffin embedded tibialis anterior harvested at postnatal day 15 shows that FLAG-α-COP SMA have normal muscle fiber size and peripheral nuclei.
Lentiviral-mediated transgenic expression of dilysine mutant SMN
We previously characterized a dilysine motif in exon 2b of the SMN protein that is required for the interaction with α-COP [16]. In a zebrafish model of SMA, expression of these dilysine mutants was unable to restore normal motor neuron morphology [17]. However, not all SMN function is affected by this mutation as SMN with lysines 76, 77, 82 and 83 mutated to alanine (referred to as SMN ΔΔ) was able to restore Golgi morphology to normal in SMA patient fibroblasts despite its inability to bind α-COP [23]. To determine whether the dilysine motif was required the human SMN rescue of SMA disease pathology in the 5058 mouse model, we used site-directed mutagenesis to convert either lysine 76 and 77 (Δ76), lysine 82 and 83 (Δ82) or all four lysines to alanines SMN ΔΔ. The mutations were then sub cloned into the EF1a-driven lentiviral vector (Figure 4a), which had previously been used to rescue defects in iPS-derived SMA motor neurons [24]. Using the lentiviral-mediated transgenic approach described above, high titer lentivirus expressing with wild-type (wt) FLAG-tagged SMN, SMN-Δ76, SMN-Δ82 and SMN-ΔΔ was injected into the testes of FVB/NJ male mice. Following recovery, the males were bred to FVB/NJ females and transgene positive F0 mice were chosen from each line. The SMN-wt injected male only produced 3 F0 progeny, and unfortunately only one of those three was able to transmit the SMN transgene. Total RNA was isolated from brain, muscle and spinal cord of WT-FLAG-SMN expressing animals, and converted to cDNA. PCR primers anchored in the FLAG tag and exon 2a show expression of FLAG-tagged SMN in all three tissues from all lines tested (Figure 4b). However, Western blot analysis of muscle and spinal cord lysates using an antibody against human SMN demonstrated that while there was strong expression of SMN-Δ82 and SMN-ΔΔ FLAG-tagged protein in both tissues, the SMN-wt transgenic could only be detected in muscle (Figure 4c). To confirm this finding, brain, muscle and spinal cord lysates were harvested from additional SMN-wt animals and compared to a non-transgenic sibling, and again transgenic SMN protein could only be detected in muscle (Figure 4d). Despite producing numerous F0 progeny, the two lines of SMN-Δ76 mice chosen failed to produce detectable protein and were not carried forward.
Figure 4: Generation of mice expressing dilysine-mutant SMN.
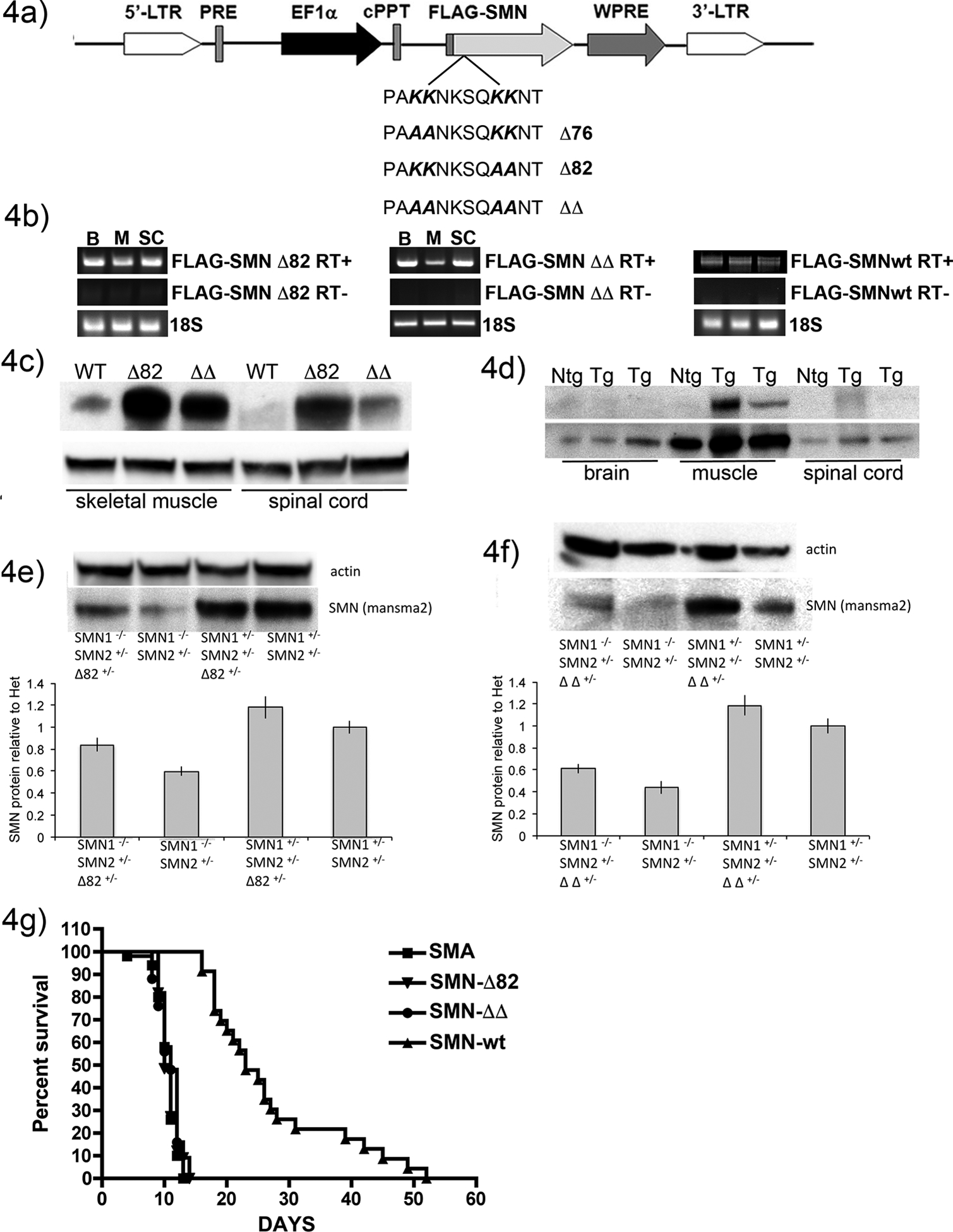
4a) Schematic of EF1aFLAG-SMN lentiviral vector. 4b) RT-PCR shows transgene expression in brain, muscle and spinal cord from transgene positive F1 progeny in each line. RT- samples were generated with no reverse transcriptase enzyme present demonstrating that no genomic DNA contamination is present. Primers against 18S RNA were used as a loading control to show that equal amount of cDNA were present in each reaction. 4c) Western blot analysis with an antibody specific to human SMN shows FLAG-SMN protein is present in skeletal muscle from all three lines, but only SMN-Δ82 and SMN-ΔΔ protein was detected in spinal cord. 4d) Western blot analysis of brain, muscle and spinal cord lysates from SMN-wt transgenic mice and a non-transgenic sibling confirms that FLAG-SMN protein is only detected in skeletal muscle lysates. 4e) Representative Western blot showing total SMN protein levels in a litter of mice with and without the SMN-Δ82 transgene. SMA pups with the SMN-Δ82 transgene displayed increased levels of SMN although not as high as their healthy siblings. Quantification from multiple experiments (6 animals per genotype) demonstrates a significant increase in SMN protein levels in SMA pups expressing the FLAG-SMN-Δ82 compared to the levels in SMA pups without the transgene (p<0.01 by Student’s t-test). Levels were normalized to non-transgenic healthy littermates using β–actin as a loading control. Error bars represent SEM. 4f) Representative Western blot shows total SMN protein levels in a litter of mice with and without the SMN-ΔΔ transgene. SMA pups with the SMN-ΔΔ transgene show increased levels of SMN, although not as high as their healthy siblings. Quantification from multiple experiments (6 animals per genotype) shows a significant increase in SMN protein levels in SMA pups expressing the FLAG-SMN-ΔΔ compared to the levels in SMA pups without the transgene. (p<0.01 by Student’s t-test) Levels are normalized to non-transgenic healthy littermates using β–actin as a loading control. Error bars represent SEM. 4g) Kaplan-Meier analysis shows that neither SMN-Δ82 nor SMN-ΔΔ transgenes were able to extend survival in the 5058 mouse model. Skeletal muscle expression of FLAG-SMN wt was sufficient to extend survival from 11 to 14 days.
Dilysine mutant SMN does not rescue disease phenotype in a mouse model of SMA
Given the strong expression of SMN-Δ82 and SMN-ΔΔ FLAG-tagged protein in disease relevant tissues, we bred the three transgenic lines onto the 5058 background using the breeding scheme described above. Figures 4e and 4f show Western blot analysis of total SMN protein in SMA pups and their heterozygous siblings showing that both the SMN-Δ82 and SMN-ΔΔ lines were able to significantly increase levels of SMN compared to non-transgenic siblings. Kaplan-Meier survival analysis showed that despite robust transgene expression, neither dilysine-mutant line was able to increase survival in the SMA background (Figure 4g). In contrast, despite the fact that detectable FLAG-tagged SMN wt protein was limited to skeletal muscle, the SMN-wt line statistically increased survival.
Taken together, these results demonstrate that expression of α-COP can reduce disease pathology in cell culture, vertebrate and mammalian models of SMA. The failure of dilysine-mutant SMN to rescue survival despite robust protein expression in brain, muscle and spinal cord supports our previous findings in cell culture models that the interaction between SMN and α-COP is required to support proper motor neuron function.
Discussion
α-COP is a member of the heptameric COPI coatomer complex involved primarily in Golgi-ER retrograde transport, endosomal trafficking and macro autophagy [25]. We show here that expression of α-COP is sufficient to increase survival in a mouse model of SMA. One potential mechanism of action for this rescue would be that interactions between SMN and α-COP would stabilize the minimal levels of SMN protein present in this model of SMA, however no increases in SMN protein levels were detected following expression of α-COP. We believe that expression of α-COP is redistributing the low levels of SMN protein available and directing its ability to support motor neuron function. We recently showed that in motor neuron-like NSC-34 cells, SMN protein is absent from the growth cone following depletion of α-COP, indicating that α-COP expression is required for normal distribution of SMN, and echoing finding from a previous publication which reported that a portion of the SMN protein was sequestered in the trans-Golgi network following knockdown of α-COP [23, 26].
A second possibility is that COPI coatomer function is impaired under conditions of low SMN, and by over-expressing α-COP, we are restoring some previously un-appreciated loss of COPI coatomer function. Western blot analysis of spinal cord lysates from two different SMA mouse models showed that levels of α-COP were unchanged under conditions of low SMN. However, we have not yet investigated other members of the COPI complex. COPI coatomer integrity has been implicated in other motor neuron diseases including Amyotrophic Lateral Sclerosis (ALS). In mice expressing mutant SOD1, which causes a dominantly inherited form of ALS, levels of beta-COP protein were reduced [27]. This paper mentions a disturbance in Stathmin 1/2 levels resulting in unstable microtubules, and a similar finding has been reported in cells with depleted SMN [28]. Cells expressing mutant SOD1 also have a similar dispersal of the Golgi apparatus to what we have reported in SMN depleted NSC-34 cells, so there may be some shared pathology between SOD1 ALS and SMA. Further experiments will need to examine the levels of all members of the COPI complex, but it may be that by over-expressing α-COP, we are stabilizing the COPI coatomer and promoting increased trafficking between the Golgi apparatus and the endoplasmic reticulum.
It was originally reported that in the absence of SMN, Tmem41b/Stasimon is spliced incorrectly, and that over-expression of Stasimon was sufficient to rescue synaptic defects in Drosophila models of SMA. Stasimon is expressed at high levels in mouse neural tissues, and studies recently showed that it is expressed at point of contact between the endoplasmic reticulum (ER) and mitochondria. Over-expression of GFP-tagged Stasimon in HEK293T cells followed by immunoprecipitation identified α-COP as a Stasimon interacting protein [29]. This finding opens up the possibility that by expressing α-COP, we are impacting Stasimon function in a way that is beneficial to SMA mice, perhaps by enhancing its function at the interface between the ER and mitochondria. We have not yet validated the interaction between α-COP and Stasimon in our own preparations, but it is worth noting that under the conditions used by Alstyne et. al, SMN did not co-immunoprecipitate with Stasimon and α-COP, indicating that they may have isolated a complex that is SMN-independent. We have not assessed levels of Stasimon protein in the 5058 model of SMA, but future experiments will examine the levels of this protein in the presence and absence of the α-COP transgene. Experiments in our NSC-34 cell model of SMN depletion could determine whether Stasimon protein levels or intracellular location are altered under conditions of low SMN and whether the expression of α-COP alters either of these.
The ability to quickly and reproducibly express a gene as large as α-COP using a lentiviral vector allows us a wide array of future experiments in other motor neuron diseases. The SOD1 model of familial ALS is notoriously sensitive to genetic background, and the ability to over-express α-COP directly into the F0 progeny provides a relatively simple experiment to determine if a similar rescue can be produced in this mouse model of motor neuron disease, given the similarities that have been reported between SMA and ALS [30, 31].
Materials and Methods:
Plasmids.
Flag-α-COP was amplified from pExchange4a using the forward primer to add an AfeI site: 5’ – ATCGAGCGCTACCATGGATTACAAG – 3’. The reverse primer included the NotI site and the stop codon: 5’ – GCATGCGGCCGCCCTTAGCGAAAC – 3’. The EF1a-FLAG-SMN lentiviral vector (a generous gift of Xue-Jun Li at Univ. of Connecticut) was cut with PmeI and Not1 to remove the FLAG-SMN. The AfeI-FLAG-COPA-NotI insert was ligated into the purified PmeI/NotI vector backbone and transformed into Stbl3 e. coli. For site-directed mutagenesis of the dilysine domain in exon 2b, FLAG-SMN was excised from the EF1a lentiviral vector using BamHI and NotI and cloned into the pcDNA3 with the same enzymes. Sequential site-directed mutagenesis was performed to add convert the dilysines at amino acid 76 and 77 or 82 and 83 to alanines using the following primer pairs: 76/77 KK➔AA forward 5’ CACCTAAAAGAAAACCTGCTCAGCAGAATAAAAGCCAAAAGAAG 3’, reverse 5’ CTTCTTTTGGCTTTTATTCTGCTGAGCAGGTTTTCTTTTAGGTG 3’ and 82/83 KK➔AA forward 5’ CTAAGAAGAATAAAAGCCAAGCCGCCAATACTGCAGCTTCCTTAC 3’, reverse 5’GTAAGGAAGCTGCAGTATTGGCGGCTTGGCTTTTATTCTTCTTAG 3’. Following sequence verification of all mutations, the FLAG-SMN was ligated back into the parental EF1a lentiviral vector.
Lentivirus production and infections.
Lentiviral vectors were transfected into HEK 293-TT cells along with the packaging vectors Pax2 and MD2.G at a ratio of 20:15:6. Supernatant was harvested 60 hours post-transfection then concentrated by centrifugation at 35,000g over a 20% sucrose cushion for 2 hours at 4°C. The viral particles were resuspended in 250μl sterile PBS tinted with 0.04% trypan blue.
8-week-old male FVB/NJ mice were obtained from Jackson laboratories (stock # 001800). Mice were given pre-surgical analgesic treatment with subcutaneous injection of buprenorphine and carprofen. Mice were anesthetized by inhaling isofluorane gas, and the lower abdomen was shaved and sterilized. A 1.5cm lateral incision was made just anterior to the penis, and the testes were removed from the scrotal sac using sterile forceps. Lentiviral particles were introduced to the seminiferous tubules by injection into the intertubular space using a 31g needle until the tubule network was visually confirmed filled by the blue dye (usually 100μl/animal). The testis was returned to the scrotal sac and the incision sutured closed and secured with dermabond. The animals were allowed to recover on a heated pad until they resumed normal activity, and pain was managed with carprofen for 48 hours following surgery. After 30 days recovery to clear any un-infected sperm, the animals were introduced to breeding partners FVB/NJ. F0 progeny were genotyped from tail DNA using the following primers: COPA forward 5’-GATAAGATCATGTTAACCAAATTCG-3’, COPA reverse 5’-GTCATTGAGAGTGCACATCC-3’, SMN forward, 5’-CGATAAGATGGCGATGAGC -3’, SMN reverse, 5’-AATGCAACCGTCTTCTGACC -3’. Transgene positive progeny were bred to 005058 animals to generate triple heterozygotes, which were then intercrossed to generate experimental crosses, which yield 50% SMA pups and 50% healthy siblings.
Animal husbandry and histology.
The animal study protocols were approved by the Institutional Animal care and Use Committee of Indiana University and the Methodist Research Institute and conform to the Guide for the Care and Use of Laboratory Animals. For genotyping, DNA was isolated from tail snip (0.5cm) at weaning (postnatal day 21) using the DNeasy Blood and Tissue kit (Qiagen). SMN1tm1 and SMN2 genotyping was performed as directed by Jackson laboratory using their suggested primers. For muscle histology, animals were euthanized by CO2 inhalation at postnatal day 15. The tibialis anterior was dissected from both hind limbs of three FLAG-COPA SMA mice and three healthy siblings, pinned and fixed in 4% PFA. Fixed tissue was blocked in paraffin and sectioned followed by staining with hematoxylin and eosin.
Endpoint justification:
The mouse model of SMA (005058) we routinely use is very severe, and these animals die around 12 days of respiratory failure as the motor neuron innervating the diaphragm degrade. To study the impact of α-COP overexpression on this SMA mouse model, we use time until death as an endpoint, thus there are animals in USDA category “E”. Mice that reach PND16 are a clear rescue of the SMA phenotype. At this stage, we begin to employ the following humane endpoint for humane removal from the study. If the animal reaches a score a 2/5 on body composition score as suggested on page 2 of http://researchcompliance.iu.edu/iacuc/iacuc_docs/iupui/Guidelines_Tumor_Induction.pdf, if it was unable to right itself within 30 seconds, or had advanced necrosis that is impinging upon the footpads, the animal was humanely euthanized and removed from the study. Animals were monitored daily for body mass and overall health. For animals that survived beyond PND 16, animals were monitored twice daily, and any animal that reached qualifications for humane endpoint discussed above was immediately euthanized by CO2 inhalation followed by cervical dislocation. For Kaplan-Meier analysis, a total of 98 animals used death as an endpoint. 146 animals survived to be removed from the study upon reaching the declared humane endpoint. This study design and all endpoints as discussed was approved by the IUSM IACUC, protocol 11348. The total duration of the survival study was 53 days. All study personnel were trained by the IUSM Laboratory Animal Research Committee (LARC) in proper animal husbandry and methods of euthenasia.
Western blotting.
Tissues were minced in lysis buffer (150mM NaCl, 50mM Tris-HCl, 1% NP-40, 0.1% SDS) followed by passage through and 18g needle and a 25g needle to completely dissociate tissue. Lysates were spun at 16,000rpm to remove debris and protein quantification was performed on the resulting supernatant. 25ug protein per lane was separated by SDS-PAGE and transferred to PVDF membrane. Membranes were blocked in 5% milk (in PBST) and probed over night with the following antibodies: FLAG (Sigma, F3165), α-COP (Santacruz, sc-398099), beta-actin (Sigma, A2228), human SMN (Cell Signaling, 12976S), total SMN (Mansma2, Developmental Studies Hybridoma Bank, Iowa).
mRNA isolation and RT-PCR.
Mice were euthanized by CO2 inhalation followed by cervical dislocation. Brain, skeletal muscle (quadriceps) and spinal cord were dissected and either stored at −80°C or immediately processed for total RNA using the Purelink RNA micro kit (Invitrogen, 12183018A) with on-column DNAase treatment. cDNA was generated using the SuperScript III system (Invitrogen, 18080051) with or without SSIII (RT+ or RT-). FLAG-COPA and FLAG-SMN transcript was amplified using the genotyping primers, which are anchored in the FLAG-tag to prevent amplification of the endogenous transcripts, and primers against 18S RNA were used as loading controls (forward, 5-GTAACCCGTTGAACCCCATT-3’, reverse 5’-CCATCCAATCGGTAGTAGCG-3’)
Acknowledgments:
Many thanks to the Developmental Studies Hybridoma Bank at University of Iowa for the Mansma antibodies to SMN protein. Funding for this work was provided by the CureSMA foundation (Custer) and NINDS (Androphy, 1R01NS082284)
References
- 1.Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20(1):27–32. doi: 10.1038/ejhg.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65. [DOI] [PubMed] [Google Scholar]
- 3.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–83. [DOI] [PubMed] [Google Scholar]
- 5.Taylor JE, Thomas NH, Lewis CM, Abbs SJ, Rodrigues NR, Davies KE, et al. Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur J Hum Genet. 1998;6(5):467–74. doi: 10.1038/sj.ejhg.5200210. [DOI] [PubMed] [Google Scholar]
- 6.Helmken C, Hofmann Y, Schoenen F, Oprea G, Raschke H, Rudnik-Schoneborn S, et al. Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet. 2003;114(1):11–21. doi: 10.1007/s00439-003-1025-2. [DOI] [PubMed] [Google Scholar]
- 7.Capon F, Levato C, Merlini L, Angelini C, Mostacciuolo ML, Politano L, et al. Discordant clinical outcome in type III spinal muscular atrophy sibships showing the same deletion pattern. Neuromuscul Disord. 1996;6(4):261–4. [DOI] [PubMed] [Google Scholar]
- 8.Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524–7. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ackermann B, Krober S, Torres-Benito L, Borgmann A, Peters M, Hosseini Barkooie SM, et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet. 2013;22(7):1328–47. doi: 10.1093/hmg/dds540. [DOI] [PubMed] [Google Scholar]
- 10.Kaifer KA, Villalon E, Osman EY, Glascock JJ, Arnold LL, Cornelison DD, et al. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight. 2017;2(5):e89970. doi: 10.1172/jci.insight.89970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGovern VL, Massoni-Laporte A, Wang X, Le TT, Le HT, Beattie CE, et al. Plastin 3 Expression Does Not Modify Spinal Muscular Atrophy Severity in the 7 SMA Mouse. PLoS One. 2015;10(7):e0132364. doi: 10.1371/journal.pone.0132364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumer D, Lee S, Nicholson G, Davies JL, Parkinson NJ, Murray LM, et al. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009;5(12):e1000773. doi: 10.1371/journal.pgen.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong Z, Ohnmacht J, Reimer MM, Bach I, Becker T, Becker CG. Chondrolectin mediates growth cone interactions of motor axons with an intermediate target. J Neurosci. 2012;32(13):4426–39. doi: 10.1523/JNEUROSCI.5179-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sleigh JN, Barreiro-Iglesias A, Oliver PL, Biba A, Becker T, Davies KE, et al. Chondrolectin affects cell survival and neuronal outgrowth in in vitro and in vivo models of spinal muscular atrophy. Hum Mol Genet. 2014;23(4):855–69. doi: 10.1093/hmg/ddt477. [DOI] [PubMed] [Google Scholar]
- 15.Peter CJ, Evans M, Thayanithy V, Taniguchi-Ishigaki N, Bach I, Kolpak A, et al. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum Mol Genet. 2011;20(9):1701–11. doi: 10.1093/hmg/ddr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Custer SK, Todd AG, Singh NN, Androphy EJ. Dilysine motifs in exon 2b of SMN protein mediate binding to the COPI vesicle protein α-COP and neurite outgrowth in a cell culture model of spinal muscular atrophy. Hum Mol Genet. 2013;22(20):4043–52. doi: 10.1093/hmg/ddt254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Custer SK, Gilson T, Hao le T, Beattie CE, Androphy EJ. α-COP binding to the survival motor neuron protein SMN is required for neuronal process outgrowth. Hum Mol Genet. 2015;24(25):7295–307. doi: 10.1093/hmg/ddv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gogliotti RG, Hammond SM, Lutz C, Didonato CJ. Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 2010;391(1):517–22. doi: 10.1016/j.bbrc.2009.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24(1):66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 20.Eshraghi M, McFall E, Gibeault S, Kothary R. Effect of genetic background on the phenotype of the Smn2B/- mouse model of spinal muscular atrophy. Hum Mol Genet. 2016;25(20):4494–506. doi: 10.1093/hmg/ddw278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfohl SR, Halicek MT, Mitchell CS. Characterization of the Contribution of Genetic Background and Gender to Disease Progression in the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis: A Meta-Analysis. J Neuromuscul Dis. 2015;2(2):137–50. doi: 10.3233/JND-140068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sehgal L, Thorat R, Khapare N, Mukhopadhaya A, Sawant M, Dalal SN. Lentiviral mediated transgenesis by in vivo manipulation of spermatogonial stem cells. PLoS One. 2011;6(7):e21975. doi: 10.1371/journal.pone.0021975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Custer SK, Foster JN, Astroski JW, Androphy EJ. Abnormal Golgi morphology and decreased COPI function in cells with low levels of SMN. Brain Res. 2018. doi: 10.1016/j.brainres.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang ZB, Zhang X, Li XJ. Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res. 2013;23(3):378–93. doi: 10.1038/cr.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beck R, Rawet M, Wieland FT, Cassel D. The COPI system: molecular mechanisms and function. FEBS Lett. 2009;583(17):2701–9. doi: 10.1016/j.febslet.2009.07.032. [DOI] [PubMed] [Google Scholar]
- 26.Ting CH, Wen HL, Liu HC, Hsieh-Li HM, Li H, Lin-Chao S. The spinal muscular atrophy disease protein SMN is linked to the Golgi network. PLoS One. 2012;7(12):e51826. doi: 10.1371/journal.pone.0051826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellouze S, Baillat G, Buttigieg D, de la Grange P, Rabouille C, Haase G. Stathmin 1/2-triggered microtubule loss mediates Golgi fragmentation in mutant SOD1 motor neurons. Mol Neurodegener. 2016;11(1):43. doi: 10.1186/s13024-016-0111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen HL, Lin YT, Ting CH, Lin-Chao S, Li H, Hsieh-Li HM. Stathmin, a microtubule-destabilizing protein, is dysregulated in spinal muscular atrophy. Hum Mol Genet. 2010;19(9):1766–78. doi: 10.1093/hmg/ddq058. [DOI] [PubMed] [Google Scholar]
- 29.Van Alstyne M, Lotti F, Dal Mas A, Area-Gomez E, Pellizzoni L. Stasimon/Tmem41b localizes to mitochondria-associated ER membranes and is essential for mouse embryonic development. Biochem Biophys Res Commun. 2018;506(3):463–70. doi: 10.1016/j.bbrc.2018.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soltic D, Bowerman M, Stock J, Shorrock HK, Gillingwater TH, Fuller HR. Multi-Study Proteomic and Bioinformatic Identification of Molecular Overlap between Amyotrophic Lateral Sclerosis (ALS) and Spinal Muscular Atrophy (SMA). Brain Sci. 2018;8(12). doi: 10.3390/brainsci8120212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chi B, O’Connell JD, Iocolano AD, Coady JA, Yu Y, Gangopadhyay J, et al. The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex. Nucleic Acids Res. 2018;46(22):11939–51. doi: 10.1093/nar/gky1093. [DOI] [PMC free article] [PubMed] [Google Scholar]