

Abstract
In a consanguineous Pakistani family with two affected individuals, a homozygous variant Gly399Val in the eighth transmembrane domain of the taurine transporter SLC6A6 was identified resulting in a hypomorph transporting capacity of ~15% compared with normal. Three-dimensional modeling of this variant has indicated that it likely causes displacement of the Tyr138 (TM3) side chain, important for transport of taurine. The affected individuals presented with rapidly progressive childhood retinal degeneration, cardiomyopathy and almost undetectable plasma taurine levels. Oral taurine supplementation of 100 mg/kg/day resulted in maintenance of normal blood taurine levels. Following approval by the ethics committee, a long-term supplementation treatment was introduced. Remarkably, after 24-months, the cardiomyopathy was corrected in both affected siblings, and in the 6-years-old, the retinal degeneration was arrested, and the vision was clinically improved. Similar therapeutic approaches could be employed in Mendelian phenotypes caused by the dysfunction of the hundreds of other molecular transporters.
Introduction
Transporters constitute a large number of cell membrane proteins involved in the uptake of small molecules into cells (1). The current human gene catalog contains 423 protein-coding genes classified as solute carrier (SLC) (1) (https://www.genenames.org/data/genegroup/#!/group/752). Members of this large gene family are involved in a number of human disorders; OMIM contains 268 entries with dominant, recessive, and X-linked Mendelian phenotypes caused by pathogenic variants in these genes (24sep19 search https://www.omim.org/).
Genome or exome sequencing provided the opportunity to identify novel causative links between pathogenic variants of genes and Mendelian phenotypes. This is particularly important for the discovery of novel autosomal recessive genes since the majority of those are still unknown. Consanguinity, which is practiced in a considerable fraction of world’s populations, provides a means to identify novel recessive genes because of the large genomic regions of homozygosity in the offspring of closely related parents (2). The identification of pathogenic variants in novel genes causing recessive disorders provide a better understanding of the molecular pathophysiology of the resulting phenotype, and the opportunity for improved diagnostic services to the affected families and populations; occasionally, new therapeutic options could be offered based on the underlying molecular mechanism.
As part of the Swiss–Pakistani consanguinity project, to identify novel genes for visual impairment or intellectual disability (3, 4), we present a novel gene for childhood progressive retinal degeneration and cardiomyopathy in one family. The taurine transport defect identified due to a hypomorph homozygous pathogenic variant in the SLC6A6 gene, has provided the opportunity for treatment with long-term taurine supplementation. After 24 months of treatment the cardiomyopathy was corrected in both affected siblings, and in the 6-years-old, the retinal degeneration was arrested and the vision was clinically improved.
Results
Genetic analysis
We used exome sequencing and genotyping of more than 200 Pakistani consanguineous families with multiple affected individuals to identify candidate genes and high impact variants responsible for recessive visual impairment. In one of these consanguineous families, F315, from the Kohat region of Pakistan with two affected individuals, we have identified a homozygous deleterious variant Gly399Val (NM_003043.5:c.1196G > T) in the taurine transporter SLC6A6 (MIM:186854) that segregated with the phenotype of progressive retinal degeneration (Fig. 1A and B). Gly399 of SLC6A6 is well-conserved in all vertebrates (Fig. 1C), and 3D molecular modeling (Fig. 1D) predicted that the Val399 substitution causes a displacement of Tyr138 side chain, important for the recognition and transport of the ligand. Blood taurine levels in the two affected individuals IV:1 and IV:3 were almost undetected (6–7 μmol/l).
Figure 1.
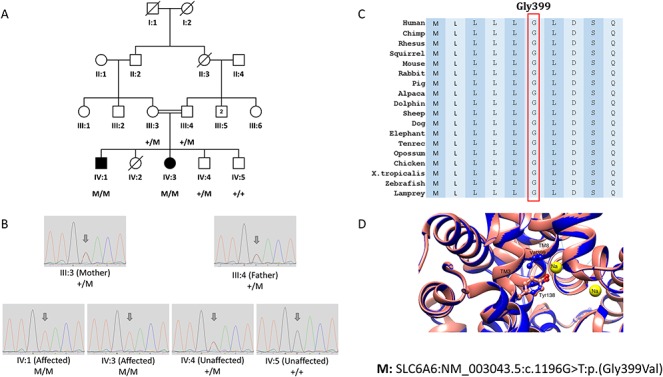
The segregation, conservation and 3D modeling of the SLC6A6 pathogenic variant Gly399Val in family F315. (A) Pedigree of consanguineous family F315 showing the segregation of the homozygous pathogenic variant NM_003043.5:c.1196G > T:p.(Gly399Val) in the SLC6A6 gene. (B) Chromatograms of Sanger sequencing showing the segregation of the SLC6A6 variant c.1196G > T:p.(Gly399Val) in all family members tested. (C) Amino acid alignment in various species showing that Gly399 is well conserved. (D) Molecular modeling of the SLC6A6 variant Gly399Val indicating that Val399 is predicted to cause the displacement of the Tyr138 (TM3) side chain, which is important for recognition and transport of the ligand.
Functional analysis
Transient and stable transfection of the Gly399Val variant in HEK-293 cells resulted in a hypomorph with transport capacity of ~15% compared with normal as determined by single point (Supplementary Material, Fig. S2) and saturation (Supplementary Material, Fig. S3 and Table S6) radioactive taurine uptake analyses. Fibroblasts from affected and carrier individuals of the family showed similar results, where single point uptake revealed significant taurine uptake deficits in the affected individuals compared with carriers (Supplementary Material, Fig. S4) consistent with kinetic data showing a significant reduction in VMAX (Fig. 2A and Supplementary Material, Table S7). Plasma membrane expression analysis in HEK-293 cells and fibroblasts strongly support that the transport deficit is functional rather than a result of decreased surface protein (Supplementary Material, Fig. S5 and Fig. 2B and C). In HEK-293 cells, taurine transport KM values were 3.4-fold lower in SLC6A6 Gly399Val compared with the normal transporter (Supplementary Material, Table S5) and whereas this could contribute to decreased transport capacity in HEK cells, KM values were unchanged between affected and carrier fibroblasts (Supplementary Material, Table S7) indicating the reduced taurine uptake in the affected patients originates from reduced transporter cycling and not substrate recognition.
Figure 2.
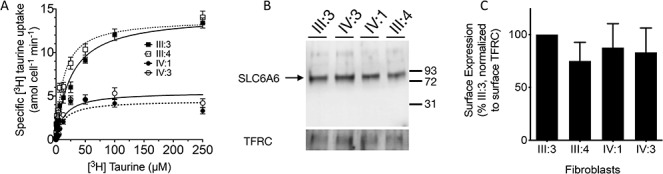
Functional characterization of SLC6A6 activity from patient-derived fibroblasts. (A) Specific taurine saturation uptake analysis in patient-derived fibroblasts. Fibroblasts of affected individuals IV:1 (filled circle), IV:3(open circle) and parents III:3 (closed square) and III:4 (open square) were incubated with [3H] taurine concentrations from 0.05 to 250 μM for 10 min. Non-specific counts were determined by inhibition of SLC6A6 with β-alanine (25 mM) and subtracted from total counts. (B) Western Blot detection of SLC6A6 in the plasma membrane from patient-derived fibroblasts. Surface protein was purified through biotinylation with a cell impermeant crosslinker and detected using anti-SLC6A6 antibody. (C) Percent surface expression was calculated from band densitometry for each sample and normalized to TFRC and plotted. *P < 0.05.
Clinical evaluation
Clinical examinations in the University Hospitals of Geneva revealed a cone-rod retinopathy and cardiomyopathy. The older 15-years-old male IV:1 had light perception vision due to advanced macular atrophy with severe peripheral alterations including pseudo-osteoblast formation and peripheral atrophy. There was no retinal response in the electroretinogram (ERG) and extensive loss of photoreceptors was noted in the optical coherence tomography (OCT). The younger 6-years-old female IV:3 had vision of counting fingers due to foveal-spearing macular atrophy. There were less marked peripheral retinal changes (salt-and-pepper fundus pigmentation), no response in global ERG, and minimal electrical focal macular response in the multifocal ERG. The OCT showed atrophy of the photoreceptors with persistence of residual photoreceptors in the central area (Supplementary Material, Fig. S1). Echocardiography showed mild hypokinetic cardiomyopathy in both affected individuals with systolic dysfunction (shortening fraction 24–27%) and systolic dilatation of the left ventricle (Fig. 3B); the effort test was however within normal limits. In both patients, brain MRI and hepatic ultrasound was normal (Supplementary Material, Table S1). Plasma amino acids showed very low levels of taurine in the affecteds and intermediate levels in the carrier parents (Fig. 3A).
Figure 3.
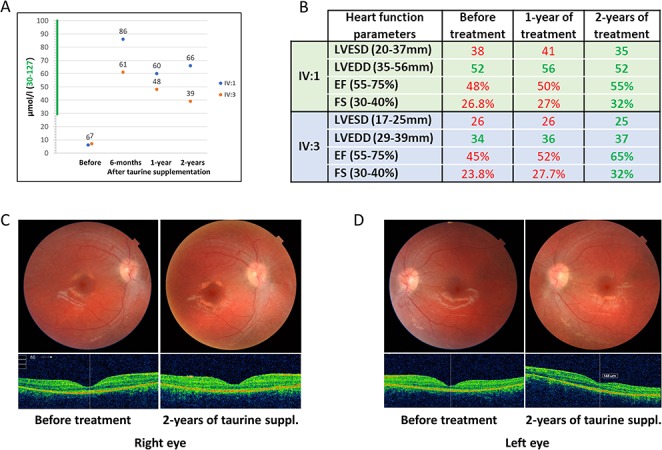
Results of taurine supplementation therapy for 24-months. (A) Taurine levels in the blood of both affected individuals (IV:1 and IV:3) before and after 6-months, 1-year and 2-years of taurine supplementation. (B) Results of the echocardiography show that the cardiomyopathy of both affected individuals (IV:1 and IV:3) has been corrected after 2-years of taurine supplementation. LVESD: left ventricular end-systolic diameter, LVEDD: left ventricular end diastolic diameter, EF: ejection fraction, FS: fractional shortening. Green and red numbers represent normal and abnormal values respectively. (C, D) Fundus photographs and macular OCT of the right eye (C) and left eye (D) of the patient IV:3 at baseline and after 24 months of taurine supplementation; anatomical stability with preservation of foveal photoreceptors can be noted.
Taurine supplementation treatment
The oral taurine loading test and subsequent supplementation at 100 mg/kg/day resulted in maintenance of normal blood taurine levels in both affected individuals (more than 40 μmol/l in each patient) (Supplementary Material, Fig. S6, Fig. 3A). We hypothesized that taurine administration may be beneficial in this family. Following approval by the ethics committee of the University Hospitals of Geneva (protocol #CER 11–036), a long-term oral supplementation treatment of 100 mg/kg/day taurine divided in three doses was introduced upon return of the family to Pakistan. Remarkably, after 24 months of treatment, the cardiomyopathy was corrected in both affected siblings. The heart function objectified by echocardiography measurements were within normal limits; indicatively, the fractional shortening of the left ventricle in both affected children was 32% (normal range of 30–40%) (Fig. 3B). In the female IV:3 (now 8-years-old), we have noted an improved visual performance with the visual acuity of 20/100 in the right eye and 20/160 in the left eye, while the ophthalmological exams showed stability of the anatomy of the central retina, suggesting an arrest in the further degeneration of the retina (Fig. 1C and D). Note that the elder brother (IV:1) had a complete visual loss at the age of 8-years. No side effects from the taurine supplementation were noted, as previously reported (5).
Discussion
Taurine is the most abundant amino acid in the retina, important in photoreceptor survival and protection from oxidative stress and light damage (6, 7). Mice with targeted disruption of the Taut/Slc6a6 gene develop degenerative retinal disease (8) similar to that observed in the family F315. Furthermore, chemically-induced taurine deficiency in mice following a treatment with a taurine transporter inhibitor guanidoethane sulfonate, resulted in photoreceptor degeneration and retinal ganglion cell loss (7, 9, 10). In addition, taurine deficiency in cats and dogs (11) causes cardiomyopathy (12). Dogs diagnosed with taurine deficiency and dilated cardiomyopathy had significant improvement in their echocardiographic parameters and normalization of taurine concentrations following diet change and taurine supplementation (11).
In this study, we present a family with taurine deficiency due to a homozygous amino acid substitution in third transmembrane domain (TM3) of the taurine transporter SLC6A6. The identification of the functional defect of the taurine transporter SLC6A6 in this consanguineous family which altered taurine homeostasis provided an opportunity for treatment. Two years of oral taurine supplementation resulted in complete reversal of the systolic cardiomyopathy in both affected children, and non-progression of the retinopathy in the younger sibling. We propose the continuation of the taurine supplementation with the objective to stabilize the retinal damage. Additional families with this novel SLC6A6 retinopathy and cardiomyopathy are necessary to establish the therapeutic value of oral taurine supplementation. This study emphasizes the contribution of each novel Mendelian gene in the understanding of disease etiology, and provides the opportunity to investigate nutritional or pharmaceutical therapy for severe Mendelian disorders due to the large family of 423 transporter-encoding genes (1). In addition, the identification of novel genes for autosomal recessive disorders provides the opportunity for carrier detection in the extended pedigree and family planning.
Materials and Methods
Family ascertainment
Family (F315) was ascertained and sampled by the Institute of Basic Medical Sciences, Khyber Medical University, Peshawar, Pakistan and was studied at the Department of Genetic Medicine and Development, University of Geneva, Switzerland. The study was approved by the ethical committee of the Khyber Medical University, Peshawar, Pakistan and by the Bioethics Committee of the University Hospitals of Geneva (Protocol number: CER 11-036). Informed consent was signed by the guardians of this family. Blood samples were obtained from all individuals of the family including affecteds, unaffected siblings and both parents. Genomic DNA was extracted from blood samples.
Genetic analysis
Exome sequencing of the proband (IV:3) was performed as described previously (13). All the family members including affecteds (IV:1 and IV:3), unaffected siblings (IV:4 and IV:5) and both parents (III:3 and III:4) were genotyped to identify the runs of homozygosity (ROH). Screening of all the ROHs segregating with the disease phenotype, identifying variants from the exome sequencing present in the segregating ROHs, and filtering of the selected variants were performed by using CATCH (14). Filtering and prioritization of likely pathogenic variants was performed as described previously (4, 13). All candidate variants were validated by Sanger sequencing (Fig. 1B).
[3H Taurine] Uptake Assay and Surface Biotinylation
Human embryonic kidney (HEK-293) cell lines were transfected with standard TransIT-LT1 protocol to stably express either wildtype taurine transporter SLC6A6 (formerly called TauT) or a SLC6A6 containing the SNP Glu399Val. HEK-293 cells transiently transfected or stably expressing SLC6A6 or the Glu399Val mutation (100 000 cells/well) or primary fibroblasts (50 000 cells/well) were plated in a 24 well culture plate (culturplate-24, Perkin-Elmer, Inc.) and grown for 24 h. Single point uptake of 30 nM or 5 μM [3H] taurine (Perkin Elmer) with vehicle or indicated concentrations of cold taurine was carried out for 10 min at 37°C. About 5 μM was obtained by mixing [3H] taurine with non-labeled taurine. Uptake was terminated with three washes of ice-cold KR buffer of pH 7.4. Radioactivity remaining in cells was measured by liquid scintillation. Non-specific uptake was determined for HEK-293 cells by subtracting radioactivity obtained with parental, non-transfected cells. Non-specific uptake for primary fibroblasts was determined by uptake in the presence of 25 mM non-labeled β-alanine. For saturation analysis, [3H] taurine was diluted into non-labeled taurine to obtain the necessary concentrations. Statistical significance was determined using unpaired T-test and one-way ANOVA (post-Tukey test) with significance set at P < 0.05.
The protocol for surface biotinylation is described in the supplementary data. SLC6A6 expression was normalized to TFRC for each sample.
Taurine loading test and supplementation
A taurine loading test was performed in the Pediatrics clinical research unit of the University hospitals of Geneva after approval by the ethics committee. We have administered an oral bolus dose of 100 mg/kg of Taurine to the two affected individuals and their heterozygous parents on day 2, as this dose was recommended by the literature as non-toxic (5). Taurine was provided in tablets, commercialized by Burgerstein Pharmaceuticals. Repeated measurements of taurine in blood and urine were subsequently performed. On days 3 and 4, the patients received a 100 mg/kg/day, administered in three doses (33 mg/kg/q 8 h).
Supplementary Material
Acknowledgements
We thank the grants from the ProVisu foundation and European Research Council (ERC) 219968 to S.E.A., and National Institutes of Health USA (NIH) DA027845 to L.K.H. We thank the family members for their contribution and Mr M. Akbar for assistance. We thank F. Sloan Bena, J. Fluss, H. Cao Van, S. Hanquinet, F. Marechal and M. Rodriguez, for clinical care and laboratory assistance.
Competing interests
The authors declare no competing interests.
References
- 1. Hediger M.A., Clemencon B., Burrier R.E. and Bruford E.A. (2013) The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol. Aspects Med., 34, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamamy H., Antonarakis S.E., Cavalli-Sforza L.L., Temtamy S., Romeo G., Kate L.P., Bennett R.L., Shaw A., Megarbane A., van Duijn C. et al. (2011) Consanguineous marriages, pearls and perils: Geneva international consanguinity workshop report. Genet. Med., 13, 841–847. [DOI] [PubMed] [Google Scholar]
- 3. Ansar M., Chung H.L., Taylor R.L., Nazir A., Imtiaz S., Sarwar M.T., Manousopoulou A., Makrythanasis P., Saeed S., Falconnet E. et al. (2018) Bi-allelic loss-of-function variants in DNMBP cause infantile cataracts. Am. J. Hum. Genet., 103, 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ansar M., Ullah F., Paracha S.A., Adams D.J., Lai A., Pais L., Iwaszkiewicz J., Millan F., Sarwar M.T., Agha Z. et al. (2019) Bi-allelic variants in DYNC1I2 cause Syndromic microcephaly with intellectual disability, cerebral malformations, and dysmorphic facial features. Am. J. Hum. Genet., 104, 1073–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ghandforoush-Sattari M., Mashayekhi S., Krishna C.V., Thompson J.P. and Routledge P.A. (2010) Pharmacokinetics of oral taurine in healthy volunteers. J. Amino Acids, 2010, 346237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Froger N., Moutsimilli L., Cadetti L., Jammoul F., Wang Q.P., Fan Y., Gaucher D., Rosolen S.G., Neveux N., Cynober L. et al. (2014) Taurine: the comeback of a neutraceutical in the prevention of retinal degenerations. Prog. Retin. Eye Res., 41, 44–63. [DOI] [PubMed] [Google Scholar]
- 7. Froger N., Jammoul F., Gaucher D., Cadetti L., Lorach H., Degardin J., Pain D., Dubus E., Forster V., Ivkovic I. et al. (2013) Taurine is a crucial factor to preserve retinal ganglion cell survival. Adv. Exp. Med. Biol., 775, 69–83. [DOI] [PubMed] [Google Scholar]
- 8. Warskulat U., Heller-Stilb B., Oermann E., Zilles K., Haas H., Lang F. and Haussinger D. (2007) Phenotype of the taurine transporter knockout mouse. Methods Enzymol., 428, 439–458. [DOI] [PubMed] [Google Scholar]
- 9. Gaucher D., Arnault E., Husson Z., Froger N., Dubus E., Gondouin P., Dherbecourt D., Degardin J., Simonutti M., Fouquet S. et al. (2012) Taurine deficiency damages retinal neurones: cone photoreceptors and retinal ganglion cells. Amino Acids, 43, 1979–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hadj-Said W., Froger N., Ivkovic I., Jimenez-Lopez M., Dubus E., Degardin-Chicaud J., Simonutti M., Quenol C., Neveux N., Villegas-Perez M.P. et al. (2016) Quantitative and topographical analysis of the losses of cone photoreceptors and retinal ganglion cells under Taurine depletion. Invest. Ophthalmol. Vis. Sci., 57, 4692–4703. [DOI] [PubMed] [Google Scholar]
- 11. Kaplan J.L., Stern J.A., Fascetti A.J., Larsen J.A., Skolnik H., Peddle G.D., Kienle R.D., Waxman A., Cocchiaro M., Gunther-Harrington C.T. et al. (2018) Taurine deficiency and dilated cardiomyopathy in golden retrievers fed commercial diets. PLoS One, 13, e0209112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pion P.D., Kittleson M.D., Rogers Q.R. and Morris J.G. (1987) Myocardial failure in cats associated with low plasma taurine: a reversible cardiomyopathy. Science, 237, 764–768. [DOI] [PubMed] [Google Scholar]
- 13. Makrythanasis P., Nelis M., Santoni F.A., Guipponi M., Vannier A., Bena F., Gimelli S., Stathaki E., Temtamy S., Megarbane A. et al. (2014) Diagnostic exome sequencing to elucidate the genetic basis of likely recessive disorders in consanguineous families. Hum. Mutat., 35, 1203–1210. [DOI] [PubMed] [Google Scholar]
- 14. Santoni F.A., Makrythanasis P. and Antonarakis S.E. (2015) Catching putative causative variants in consanguineous families. BMC Bioinform., 16, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.