

Summary
Missense mutations may affect proteostasis by destabilizing or over-stabilizing protein complexes and changing the pathway flux. Predicting the effects of stabilizing mutations on protein-protein interactions is notoriously difficult because existing experimental sets are skewed toward mutations reducing protein-protein binding affinity and many computational methods fail to correctly evaluate their effects. To address this issue, we developed a method MutaBind2, which estimates the impacts of single as well as multiple mutations on protein-protein interactions. MutaBind2 employs only seven features, and the most important of them describe interactions of proteins with the solvent, evolutionary conservation of the site, and thermodynamic stability of the complex and each monomer. This approach shows a distinct improvement especially in evaluating the effects of mutations increasing binding affinity. MutaBind2 can be used for finding disease driver mutations, designing stable protein complexes, and discovering new protein-protein interaction inhibitors.
Subject Areas: Protein Folding, Bioinformatics, 3D Reconstruction of Protein
Graphical Abstract
Highlights
-
•
A new method to predict binding affinity changes upon single and multiple mutations
-
•
Improved performance in evaluating the effects of mutations increasing binding affinity
-
•
Generation of the structural model of a mutant complex
Protein Folding; Bioinformatics; 3D Reconstruction of Protein
Introduction
Protein-protein interactions mediate many biological processes, and missense mutations may affect protein interactions and interaction networks leading to dysfunctional proteins and their complexes, pathway dysregulation, and potentially to diseases (Teng et al., 2009, Nishi et al., 2013, Creixell et al., 2015, Tee et al., 2019). Indeed, several recent studies systematically characterized thousands of disease mutations and found that many of them were located on protein-binding interfaces and induced macromolecular interaction perturbations, whereas neutral variants retained most interactions (Nishi et al., 2013, Teng et al., 2009, Creixell et al., 2015, Sahni et al., 2015, Wang et al., 2012, An et al., 2013, Cukuroglu et al., 2014, Tan et al., 2019, Ozdemir et al., 2018). However, not all mutations have severe damaging impacts, and the majority of mutations produce rather subtle effects with unclear clinical significance. Quantification of these effects on specific protein-protein interactions requires assessing the changes in binding affinity induced by mutations. These effects can be quite accurately measured by low-throughput experiments. However, large-scale rapid experimental assays that would allow the assessment of thousands of variants are still limited. The development of reliable computational approaches to predict the effects of missense mutations on protein complexes would enable the prioritization of functionally disrupting mutations and provide a basis for understanding the molecular mechanisms of their impacts.
Several computational approaches have been proposed so far to calculate the changes in binding affinity by mutations (Li et al., 2014, Li et al., 2016b, Dehouck et al., 2013, Petukh et al., 2015, Petukh et al., 2016, Kruger and Gohlke, 2010, Pires et al., 2014, Xiong et al., 2017, Brender and Zhang, 2015, Zhao et al., 2014, Rodrigues et al., 2019, Geng et al., 2019, Jemimah et al., 2019). In the past we developed two methods to address this pressing need. The first method used the modified MM/PBSA (Molecular Mechanics Poisson−Boltzmann Surface Area) approach and structure optimization protocol with an explicit solvent model (Li et al., 2014). Later we came up with another method, MutaBind (Li et al., 2016b). MutaBind was characterized by higher prediction accuracy and speed, making it possible to implement it as a web server, which has been used to quantify the impacts of mutations in a wide range of protein complexes (https://mutabind.org/v1). For instance, it was successfully applied to assess the effects of cancer mutations on binding between CBL ubiquitin ligase and E2 conjugating enzyme where computationally predicted binding affinity changes were compared with the experiments using cancer and non-cancer cell lines (Li et al., 2016a).
The functional effects of mutations decreasing binding affinity are better understood compared with the effects of mutations increasing binding affinity. However, the latter may also have important consequences leading to disruption of proteostasis by over-stabilizing transient protein complexes (Nishi et al., 2013, Stefl et al., 2013, Rutten et al., 2018, Shoichet et al., 1995, Nagatani et al., 2007, Witham et al., 2011, Jubb et al., 2016) and changing the pathway flux. Critically, existing computational methods perform much better for mutations decreasing than for mutations increasing binding affinity. Several studies tried to determine factors contributing to this bias by comparing the methods' performance using experimental data on changes of protein stability (Usmanova et al., 2018, Montanucci et al., 2018, Pucci et al., 2018). These studies concluded that all computational methods produced predictions that were immensely skewed toward higher accuracy for mutations decreasing binding affinity and the amplitude of this bias increased with the number of introduced mutations in a protein (Usmanova et al., 2018). There are several reasons for such predisposition of proteins toward mutations with decreasing effects: proteins and their binding interfaces tend to be optimized in evolution with regard to their stability; the existing experimental sets are enriched with mutations that decrease binding and the methodologies of training procedures employed by many computational methods rely on these experimental sets. Correcting for such bias in performance is demanding as it requires developing new energy functions and enriching the datasets with mutations increasing binding affinity either by using additional experimental data or by calculating the effects of reverse mutations and modeling their mutant structures.
To address this issue, we developed a new method, MutaBind2, with significantly improved performance (https://mutabind.org/). MutaBind2 uses a new minimization protocol and scoring function composed of seven terms. In addition to single mutations, it can predict the effects of multiple mutations on protein binding affinity. MutaBind2 can be applied to a large number of tasks, including, but not limited to, finding disease driver mutations and understanding their molecular mechanisms, assessing the effects of sequence variants on protein fitness, structural modeling of mutant complexes, and designing protein interaction inhibitors (Goncearenco et al., 2017).
Results and Discussion
By developing a new version of MutaBind2 we tried to achieve the following goals: (1) to improve the overall performance, especially for mutations increasing binding affinity; (2) to avoid overfitting; and (3) to allow for multiple mutations predictions. To do this, first we designed a scoring function with seven terms instead of 10 used in the previous MutaBind version (detailed in Supplemental Information, Transparent Methods). Second, we trained our models on a much larger dataset from SKEMPI2, which encompassed 1.7 times more mutations and 3.3 times more complexes compared with SKEMPI used for training of MutaBind. Third, to enrich the existing dataset with mutations increasing binding affinity and to build a more balanced training dataset, we produced structural models of complexes with reverse mutations and estimated the corresponding values of each term of the scoring function. Finally, we added a new functionality of predicting the effects of multiple mutations (up to 10 mutations) on binding affinity to account for possible cooperative and epistatic effects.
Evaluating the Performance of MutaBind2 Using Cross-Validation
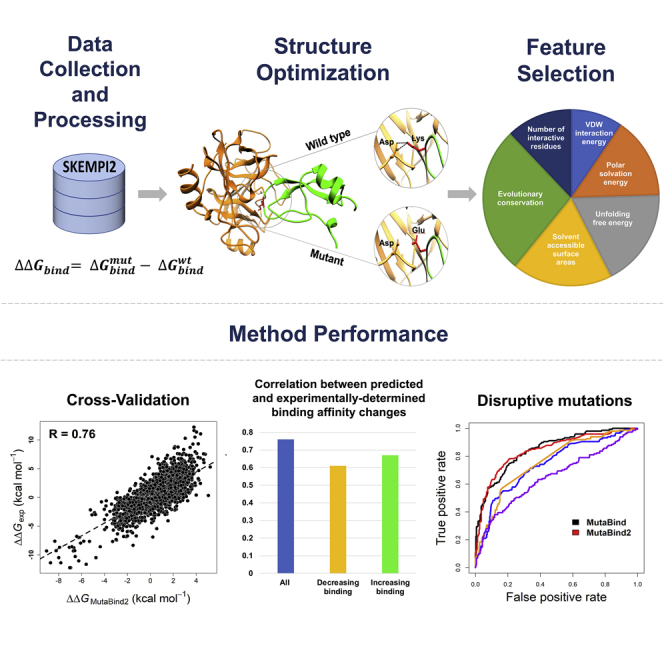
Avoiding overfitting is one of our major concerns while developing a computational method that should make predictions with high accuracy for diverse sets of single or multiple mutations. Overfitting of model parameters may occur while minimizing the mean square deviations of predicted from experimental values in the training set, which could indicate the loss of generalization in the model (Dehouck et al., 2013). To overcome this issue, a cross-validation can be applied, which allows to estimate the future performance of the method on previously unseen data. Five types of cross-validation were performed in our study (explained in more detail in the Methods section). Figure 1 shows the Pearson correlation coefficients between experimental and calculated of the first three types of cross-validation procedures. The correlation coefficients of each cross-validation round exceed 0.80 for “CV1” and “CV2” and about 0.70 for “CV3” cross-validation for both single and multiple mutations.
Figure 1.

Pearson Correlation Coefficients between Experimental and Calculated for Three Types of Cross-Validation Tests on the S4191 (Single Mutations) and M1707 (Multiple Mutations) Sets
See also Table S1.
The Pearson correlation coefficient between experimental and computed values using the “leave-one-complex-out” (“CV4”) procedure reaches 0.76 for single mutations and 0.74 for multiple mutations (Table 1 and Figure 2). In addition, we performed a validation by leaving one binding site out of the training set (“CV5” cross-validation). According to this validation, the model was parameterized and tested using completely different non-overlapping sets of binding sites. Nevertheless, the correlation coefficient remained statistically significant, being equal to 0.69 for single mutations and 0.71 for multiple mutations (Table 1 and Figure S4D). From the evaluation of the performance of MutaBind2 using cross-validation, we can conclude that the MutaBind2 for single mutations significantly outperforms the previous version of MutaBind, which had R = 0.68 and R = 0.57 for “CV4” and “CV5,” respectively (Li et al., 2016b) (see Table 1 for RMSE values).
Table 1.
Comparison of Methods' Performance for Single and Multiple Mutations
| Training/Test Set | Model | All Mutations |
Decreasing |
Increasing |
||||
|---|---|---|---|---|---|---|---|---|
| R | RMSE | Slope | R | RMSE | R | RMSE | ||
| Single mutations | ||||||||
| Skempi + Reverse/S1748 | MutaBind2ˆ | 0.63 | 1.25 | 0.83 | 0.45 | 1.17 | 0.77 | 1.52 |
| Skempi/S1748 | MutaBind | 0.38∗ | 1.51 | 0.72 | 0.44 | 1.11 | – | 2.43 |
| BeAtMuSiC | 0.30∗ | 1.58 | 0.55 | 0.43 | 1.14 | −0.25∗ | 2.57 | |
| Test: S1748 | FoldX | 0.42∗ | 1.57 | 0.52 | 0.41 | 1.37 | 0.26∗ | 2.12 |
| Test: S4191 | MutaBind2 CV4 | 0.76 | 1.34 | 1.11 | 0.61 | 1.31 | 0.67 | 1.39 |
| MutaBind2 CV5 | 0.69 | 1.50 | 1.18 | 0.54 | 1.41 | 0.47 | 1.65 | |
| Multiple mutations | ||||||||
| Test: M1707 | MutaBind2 CV4 | 0.74 | 2.13 | 1.09 | 0.51 | 2.04 | 0.60 | 2.26 |
| MutaBind2 CV5 | 0.71 | 2.24 | 1.00 | 0.47 | 2.18 | 0.56 | 2.33 | |
| Test: M1337 | FoldX | 0.49 | 2.43 | 0.52 | 0.37 | 2.49 | 0.24 | 2.21 |
MutaBind2ˆ: MutaBind2 was retrained on “Skempi + Reverse” set.
∗Significant difference between MutaBind2 and other methods with p value < 0.01 calculated on a test set S1748 (implemented in R package cocor).
R, Pearson correlation coefficient between experimental and predicted ΔΔG values; RMSE (kcal mol−1), root-mean-square error, the standard deviation of the residuals (prediction errors); Slope, the slope of the regression line between experimental and predicted ΔΔG values. All presented values of correlation coefficients are statistically significantly different from zero (p value << 0.01). The details about datasets are shown in Table S1.
Figure 2.

Experimental and Predicted Values of Changes in Binding Affinity for All Mutations in the S4191 (Single Mutations) and M1707 (Multiple Mutations) Sets Using “Leave-One-Complex-Out” (CV4) Cross-Validation
See also Table S1.
To better understand MutaBind2's limitations and strengths, we analyzed 5% and 1% outliers and evaluated the performance after removing outliers using leave-one-complex-out validation (CV4) on S4191. Studentized residuals are used in detecting outliers. Figure S11A shows that the performance is improved significantly after removing 5% outliers. Moreover, we found that the outliers are more likely to appear in complexes with higher protein-protein binding affinity and at the mutated sites with higher number of hydrogen bonds (Figure S11B). Consistent with the observation of Pires et al. (Pires et al., 2016), the outliers usually correspond to those mutations with extreme experimental values (highly decreasing/increasing binding affinity), and MutaBind2 could correctly classify them as either increasing or decreasing binding affinity (Figures S11C and S11D).
Validation of MutaBind2 on Independent Test Sets
To check if enriching the training set with mutations increasing binding affinity improved the performance, we constructed an independent “unseen” test set consisting of complexes and mutations that were present in SKEMPI2 but were absent from SKEMPI (S1748 set). The original version of MutaBind, which was trained on binding affinity changes dataset from SKEMPI (referred to as the “Skempi” set), yielded a Pearson correlation coefficient of R = 0.44 between predicted and experimentally determined values for mutations decreasing binding affinity on an independent S1748 set, whereas it did not yield statistically significantly predictions for mutations increasing binding affinity (Table 1 and Figure S4A). However, after applying a model trained on the SKEMPI set enriched with mutations increasing binding affinity (“Skempi + Reverse” set) using MutaBind2 features and protocol, the performance on an independent S1748 set improved considerably (the correlation coefficient increased from 0.38 to 0.63, and root-mean-square error decreased from 1.51 to 1.25 kcal mol−1). Moreover, predictions for mutations increasing binding affinity were significantly improved without compromising the accuracy for predicting mutations decreasing binding affinity (Table 1).
For single mutations, we also compared MutaBind2 with four other methods, BeAtMuSiC (Dehouck et al., 2013), FoldX (Guerois et al., 2002), iSEE (Geng et al., 2019), and mCSM-PPI2 (Rodrigues et al., 2019). BeAtMuSiC is a machine learning method, which uses a combination of different statistical potentials to predict values and is parameterized on mutations from SKEMPI. FoldX uses an empirical energy function, which is parametrized on experimental changes of unfolding free energy. iSEE is parameterized on the SKEMPI set and uses several dozens of interface, structure, evolution, and energy-based features. iSEE is not available as a server or a standalone version, so it could not be applied to the S1748 set. mCSM-PPI2 uses several dozens of features such as graph-based signatures, evolutionary conservation, and interaction energy between two partners calculated from FoldX and also incorporates features derived from reverse mutations. It has been trained on 8,338 mutations from the SKEMPI2 dataset, which includes almost all mutations from the MutaBind2 training dataset S4191.
For comparison with iSEE, we used the S487 dataset obtained from the iSEE article (Geng et al., 2019) where the MutaBind2 model was retrained after removing S487 from the S4191 training set. As can be seen in Table 2, the MutaBind2 model parameterized on this training set shows the best performance on S487 compared with other methods (more comparisons can be found in Table S5). We did not have an independent set for comparing the predictions between MutaBind2 and mCSM-PPI2, therefore we used the same training protocol and retrained MutaBind2 on the dataset of S8338 (a training dataset of mCSM-PPI2), even though our feature selection was not based on this dataset. We obtained comparable correlation coefficients with mCSM-PPI2 using the CV4 and CV5 cross-validations (Table 2), which were slightly lower than results reported for the original MutaBind2 model on the S4191 (Table 1). Additional comparisons with mCSM-PPI2 are shown in Table S6, which points to a slightly better performance for MutaBind2 in terms of the slope of the regression line indicating that predicted and experimental values are on the same scale.
Table 2.
Comparison of Methods' Performance on Different Datasets
| Test Set | Method | R | RMSE |
|---|---|---|---|
| S487 | MutaBind2 | 0.41 | 1.25 |
| MutaBind | 0.29∗∗ | 1.63 | |
| BeAtMuSiC | 0.35 | 1.28 | |
| FoldX | 0.34∗ | 1.53 | |
| iSEE | 0.25∗∗ | 1.32 | |
| S8338 | MutaBind2 CV4 | 0.74 | 1.37 |
| MutaBind2 CV5 | 0.66 | 1.53 | |
| mCSM-PPI2 CV4 | 0.75 | 1.30 | |
| mCSM-PPI2 CV5 | 0.67 | 1.39 |
∗ and ∗∗ indicate statistically significant difference between MutaBind2 and other methods in terms of R with p value < 0.05 and p value < 0.01, respectively, calculated on test set S487 (implemented in R package cocor).
R, Pearson correlation coefficient; RMSE, root-mean-square error.
R and RMSE values were taken from the mCSM-PPI2 article (Rodrigues et al., 2019). For testing on S487 set, MutaBind2 was retrained after removing S487 from the training dataset. For testing on S8338 set, MutaBind2 was retrained on S8338. See also Table S6.
Recently the impacts of 2,009 missense mutations across 2,185 human protein-protein interactions (4,797 mutation-interaction pairs) were measured by yeast two hybrid experiments (Fragoza et al., 2019), and 903 mutations were identified as interaction-disruptive mutations. A mutation was defined as disruptive if it damaged one or more protein-protein interactions and was defined as non-disruptive otherwise. Among 4,797 mutation-interaction pairs, 451 mutations, including 147 interaction disruptive mutations, could be mapped on corresponding protein-protein complexes with the known 3D structures. Then we calculated binding affinity changes for these mutations using different methods. Figures 3 and S5 show excellent performance of MutaBind and MutaBind2 in distinguishing interaction-disruptive from other mutations.
Figure 3.

Receiver Operating Characteristic Curves for Predicting Mutations Disrupting Protein-Protein Interactions Using Different Methods
As one mutation/interaction could be mapped to several Protein DataBank structures, the maximum predicted value of each method was used for each interaction-disruptive mutation and the minimum predicted values were used for those mutations that do not disrupt interactions. More details are shown in Figure S5.
Prediction of Mutations Highly Decreasing and Increasing Binding Affinity
The previous version, MutaBind, could predict single mutations highly decreasing binding affinity relatively well but failed to annotate mutations highly increasing affinity. Table 3 and Figure S6 demonstrate the high performance of MutaBind2 in predicting mutations highly decreasing and highly increasing binding affinity (see Methods for details). MutaBind2 further improves the performance for both interfacial and non-interfacial mutations compared with the previous version and outperforms other methods on the S1748 set (Figure S7). We subdivided complexes with multiple mutations into different categories based on the number of mutations and the number of mutated chains involved (see Table S9 for more details). We found that MutaBind2 performed well for almost all categories; the worst performance was observed for three or more mutations introduced on the same chain (R = 0.61 in CV4 validation), and the best performance was achieved for double mutations on multiple chains (R = 0.85 in CV4 validation).
Table 3.
Comparative Performance of MutaBind2 and Three Methods for Predicting Mutations Highly Decreasing and Increasing Binding Affinity on the Independent Test Set of S1748
| MutaBind2 | MutaBind | BeAtMuSiC | FoldX | |
|---|---|---|---|---|
| Highly decreasing | ||||
| Sensitivity | 0.75 | 0.86 | 0.73 | 0.58 |
| Specificity | 0.89 | 0.82 | 0.87 | 0.94 |
| MCC | 0.63 | 0.63 | 0.58 | 0.57 |
| AUC | 0.82 | 0.82 | 0.79 | 0.79 |
| Highly increasing | ||||
| Sensitivity | 0.55 | 0 | 0 | 0.44 |
| Specificity | 0.99 | 1.00 | 1.00 | 0.99 |
| MCC | 0.64 | −0.01 | 0 | 0.51 |
| AUC | 0.86 | 0.65∗ | 0.56∗ | 0.74∗ |
MutaBind2 was retrained on the dataset “Skempi + Reverse.”
MCC, Matthews correlation coefficient.
∗p value < 0.01 calculated by Delong test (DeLong et al., 1988) comparing AUC (area under the ROC curve) produced by a given method and AUC produced by MutaBind2; points to significant differences in performance. See also Figure S6.
Next we would like to elucidate the main differences between predictors like BeAtMuSiC, iSEE, and mCSM-PPI2 and methods like FoldX, MutaBind, and MutaBind2. The first group of methods uses powerful machine learning approaches with several dozens of features to calculate the changes in binding affinity and does not provide contribution of each feature for each mutation. On the other hand, methods like FoldX, MutaBind, and MutaBind2 use very few interpretable energy terms and perform structure optimization and energy calculations. This allows the construction of actual molecular models of mutant structures and to evaluate changes in binding affinity for these mutants, potentially accounting for structural changes that cannot be captured by machine learning methods. The molecular models of mutants have been used extensively by researchers to understand the molecular mechanisms of disease mutations, to design drugs, to identify drug targets, to predict driver mutations, and to decipher the mechanisms of drug-resistant mutations (Figure S8). Importantly, MutaBind and MutaBind2, unlike other methods mentioned earlier, estimate interactions of a protein with the solvent, which is one of the most important terms together with the site evolutionary conservation and thermodynamic stability of a protein complex and each binding partner (Table S2). In addition, FoldX and MutaBind2 provide predictions and structural models for multiple mutations introduced at the same time in a protein complex.
Online Web Server
MutaBind2 is available at https://mutabind.org/v2. The main requirement of the webserver is the availability of the 3D structure of a protein-protein complex, which can be provided by the Protein DataBank accession or by a file with the coordinates uploaded by the researcher. In either case, the structure file should contain at least two protein chains. In the next step two interaction partners should be defined. It is possible to assign one chain or multiple chains to either “Partner 1” or “Partner 2,” and only assigned chains will be considered during the calculation. If the interface size between assigned partners is smaller than 100 Å2, an error message is displayed. The interface size is calculated as a difference between the solvent-accessible surface areas of assigned chains in a complex and unbound partner. The final step is to select mutations. We provide three options to allow users to do large-scale mutational scanning (Figure S9).
-
•
An option “Upload file” allows to submit a list of mutations specified in the uploaded file
-
•
The “alanine scanning” option allows to perform alanine scanning for all contact residues between interaction partners. Contact residues here are defined as those with inter-atomic distances less than 6 Å between any heavy atom of interaction partners. MutaBind2 provides the contact residues list for download
-
•
Contact residues are shown in orange in the residue list of “Specify One or More Mutations,” which allows to view contact residues in the 3D structure
For each mutation on a protein-protein complex, the MutaBind2 server provides the following results:
-
-
ΔΔG (kcal mol−1): predicted change in binding affinity induced by mutations (positive and negative signs correspond to mutations decreasing and increasing binding affinity, respectively)
-
-
The location on interface (yes/no): indicating whether the residue is located on a protein-protein interface in the case when a residue's solvent accessibility in the complex is lower than in the corresponding unbound partners
-
-
Coordinates of the minimized mutant structure
-
-
Deleterious (yes/no), a mutation is classified as deleterious if ≥ 1.5 or ≤ −1.5 kcal mol−1
-
-
The contribution of each term of the target function for every mutation
-
-
Homologous binding sites: the Inferred Biomolecular Interactions Server (Shoemaker et al., 2012) is used to identify the binding sites in protein-protein complexes homologous to the query
Limitation of the Study
-
1.
Requirement of the 3D structure of a protein-protein complex. Six features out of seven in our model are calculated using 3D structure of a protein-protein complex, which limits the application to those mutations that could not be mapped to the structural complex.
-
2.
Multiple mutations instances with more than 10 mutations. As the number of multiple mutations with more than 10 mutations is small in our training dataset and prediction accuracy for these multiple mutations is low, the upper limit of 10 mutations was used in the study. Therefore, our model cannot be applied to the multiple mutation instances with more than 10 mutations.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant No. 31701136), Natural Science Foundation of Jiangsu Province, China (Grant No. BK20170335), and the Priority Academic Program Development of Jiangsu Higher Education Institutions. A.G. and R.V.A. were supported by the Intramural Research Program of the National Library of Medicine at the US National Institutes of Health. A.R.P. was in part supported by the Intramural Research Program of the National Library of Medicine at the US National Institutes of Health and by the Department of Pathology and Molecular Medicine, Queen's University, Canada. A.R.P. is the recipient of a Senior Canada Research Chair in Computational Biology and Biophysics and a Senior Investigator award from the Ontario Institute of Cancer Research, Canada. We would like to thank Dr. Thomas Madej for proofreading of the manuscript.
Author Contributions
Conceptualization, A.R.P. and M.L.; Methodology, N.Z., Y.C., M.L., and A.R.P.; Software, N.Z., Y.C., and A.G.; Validation, N.Z., Y.C., A.G., and M.L.; Formal Analysis, N.Z. and Y.C.; Investigation, N.Z., Y.C., M.L., and A.R.P.; Data Curation, N.Z. and Y.C.; Writing – Original Draft, M.L. and A.R.P.; Writing – Review & Editing, M.L. and A.R.P.; Visualization, H.L., F.Z., R.V.A., and A.G.; Supervision, A.R.P. and M.L.; Project Administration, M.L.; Funding Acquisition, M.L.
Declaration of Interests
The authors declare no competing interests.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100939.
Contributor Information
Anna R. Panchenko, Email: panch@ncbi.nlm.nih.gov.
Minghui Li, Email: minghui.li@suda.edu.cn.
Data and Code Availability
MutaBind2 is available at https://mutabind.org/v2, and the training and test datasets are available for download from the server.
Supplemental Information
References
- An O., Gursoy A., Gurgey A., Keskin O. Structural and functional analysis of perforin mutations in association with clinical data of familial hemophagocytic lymphohistiocytosis type 2 (FHL2) patients. Protein Sci. 2013;22:823–839. doi: 10.1002/pro.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brender J.R., Zhang Y. Predicting the effect of mutations on protein-protein binding interactions through structure-based interface profiles. PLoS Comput. Biol. 2015;11:e1004494. doi: 10.1371/journal.pcbi.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creixell P., Schoof E.M., Simpson C.D., Longden J., Miller C.J., Lou H.J., Perryman L., Cox T.R., Zivanovic N., Palmeri A. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell. 2015;163:202–217. doi: 10.1016/j.cell.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukuroglu E., Engin H.B., Gursoy A., Keskin O. Hot spots in protein-protein interfaces: towards drug discovery. Prog. Biophys. Mol. Biol. 2014;116:165–173. doi: 10.1016/j.pbiomolbio.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Dehouck Y., Kwasigroch J.M., Rooman M., Gilis D. BeAtMuSiC: prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013;41:W333–W339. doi: 10.1093/nar/gkt450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong E.R., DeLong D.M., Clarke-Pearson D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- Fragoza R., Das J., Wierbowski S.D., Liang J., Tran T.N., Liang S., Beltran J.F., Rivera-Erick C.A., Ye K., Wang T.-Y. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun. 2019;10:4141. doi: 10.1038/s41467-019-11959-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C., Vangone A., Folkers G.E., Xue L.C., Bonvin A.M.J.J. iSEE: interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations. Proteins. 2019;87:110–119. doi: 10.1002/prot.25630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncearenco A., Li M., Simonetti F.L., Shoemaker B.A., Panchenko A.R. Exploring protein-protein interactions as drug targets for anti-cancer therapy with in silico workflows. Methods Mol. Biol. 2017;1647:221–236. doi: 10.1007/978-1-4939-7201-2_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerois R., Nielsen J.E., Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Jemimah S., Sekijima M., Gromiha M.M. ProAffiMuSeq: sequence-based method to predict the binding free energy change of protein-protein complexes upon mutation using functional classification. Bioinformatics. 2019;35:462–469. doi: 10.1093/bioinformatics/btz829. [DOI] [PubMed] [Google Scholar]
- Jubb H.C., Pandurangan A.P., Turner M.A., Ochoa-Montaño B., Blundell T.L., Ascher D.B. Mutations at protein-protein interfaces: small changes over big surfaces have large impacts on human health. Prog. Biophys. Mol. Biol. 2016;128:3–13. doi: 10.1016/j.pbiomolbio.2016.10.002. [DOI] [PubMed] [Google Scholar]
- Kruger D.M., Gohlke H. DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 2010;38:W480–W486. doi: 10.1093/nar/gkq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Kales S.C., Ma K., Shoemaker B.A., Crespo-Barreto J., Cangelosi A.L., Lipkowitz S., Panchenko A.R. Balancing protein stability and activity in cancer: a new approach for identifying driver mutations affecting CBL ubiquitin ligase activation. Cancer Res. 2016;76:561–571. doi: 10.1158/0008-5472.CAN-14-3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Petukh M., Alexov E., Panchenko A.R. Predicting the impact of missense mutations on protein-protein binding affinity. J. Chem. Theor. Comput. 2014;10:1770–1780. doi: 10.1021/ct401022c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Simonetti F.L., Goncearenco A., Panchenko A.R. MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 2016;44:W494–W501. doi: 10.1093/nar/gkw374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanucci L., Savojardo C., Martelli P.L., Casadio R., Fariselli P. On the biases in predictions of protein stability changes upon variations: the INPS test case. Bioinformatics. 2018;35:2525–2527. doi: 10.1093/bioinformatics/bty979. [DOI] [PubMed] [Google Scholar]
- Nagatani R.A., Gonzalez A., Shoichet B.K., Brinen L.S., Babbitt P.C. Stability for function trade-offs in the enolase superfamily “catalytic module”. Biochemistry. 2007;46:6688–6695. doi: 10.1021/bi700507d. [DOI] [PubMed] [Google Scholar]
- Nishi H., Tyagi M., Teng S., Shoemaker B.A., Hashimoto K., Alexov E., Wuchty S., Panchenko A.R. Cancer missense mutations alter binding properties of proteins and their interaction networks. PLoS One. 2013;8:e66273. doi: 10.1371/journal.pone.0066273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir E.S., Gursoy A., Keskin O. Analysis of single amino acid variations in singlet hot spots of protein-protein interfaces. Bioinformatics. 2018;34:i795–i801. doi: 10.1093/bioinformatics/bty569. [DOI] [PubMed] [Google Scholar]
- Petukh M., Dai L., Alexov E. SAAMBE: webserver to predict the charge of binding free energy caused by amino acids mutations. Int. J. Mol. Sci. 2016;17:547. doi: 10.3390/ijms17040547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukh M., Li M., Alexov E. Predicting binding free energy change caused by point mutations with knowledge-modified MM/PBSA method. PLoS Comput. Biol. 2015;11:e1004276. doi: 10.1371/journal.pcbi.1004276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D.E., Ascher D.B., Blundell T.L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30:335–342. doi: 10.1093/bioinformatics/btt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D.E.V., Blundell T.L., Ascher D.B. mCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci. Rep. 2016;6:29575. doi: 10.1038/srep29575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucci F., Bernaerts K.V., Kwasigroch J.M., Rooman M. Quantification of biases in predictions of protein stability changes upon mutations. Bioinformatics. 2018;34:3659–3665. doi: 10.1093/bioinformatics/bty348. [DOI] [PubMed] [Google Scholar]
- Rodrigues C.H.M., Myung Y., Pires D.E.V., Ascher D.B. mCSM-PPI2: predicting the effects of mutations on protein–protein interactions. Nucleic Acids Res. 2019;47:W338–W344. doi: 10.1093/nar/gkz383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutten L., Lai Y.-T., Blokland S., Truan D., Bisschop I.J.M., Strokappe N.M., Koornneef A., van Manen D., Chuang G.-Y., Farney S.K. A universal approach to optimize the folding and stability of prefusion-closed HIV-1 envelope trimers. Cell Rep. 2018;23:584–595. doi: 10.1016/j.celrep.2018.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni N., Yi S., Taipale M., Fuxman Bass J.I., Coulombe-Huntington J., Yang F., Peng J., Weile J., Karras G.I., Wang Y. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015;161:647–660. doi: 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker B.A., Zhang D., Tyagi M., Thangudu R.R., Fong J.H., Marchler-Bauer A., Bryant S.H., Madej T., Panchenko A.R. IBIS (Inferred Biomolecular Interaction Server) reports, predicts and integrates multiple types of conserved interactions for proteins. Nucleic Acids Res. 2012;40:D834–D840. doi: 10.1093/nar/gkr997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet B.K., Baase W.A., Kuroki R., Matthews B.W. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. U S A. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefl S., Nishi H., Petukh M., Panchenko A.R., Alexov E. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013;425:3919–3936. doi: 10.1016/j.jmb.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z.W., Tee W.V., Guarnera E., Booth L., Berezovsky I.N. AlloMAPS: allosteric mutation analysis and polymorphism of signaling database. Nucleic Acids Res. 2019;47:D265–D270. doi: 10.1093/nar/gky1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee W.V., Guarnera E., Berezovsky I.N. On the allosteric effect of nsSNPs and the emerging importance of allosteric polymorphism. J. Mol. Biol. 2019;431:3933–3942. doi: 10.1016/j.jmb.2019.07.012. [DOI] [PubMed] [Google Scholar]
- Teng S., Madej T., Panchenko A., Alexov E. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys. J. 2009;96:2178–2188. doi: 10.1016/j.bpj.2008.12.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usmanova D.R., Bogatyreva N.S., Arino Bernad J., Eremina A.A., Gorshkova A.A., Kanevskiy G.M., Lonishin L.R., Meister A.V., Yakupova A.G., Kondrashov F.A. Self-consistency test reveals systematic bias in programs for prediction change of stability upon mutation. Bioinformatics. 2018;34:3653–3658. doi: 10.1093/bioinformatics/bty340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Wei X., Thijssen B., Das J., Lipkin S.M., Yu H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat. Biotechnol. 2012;30:159–164. doi: 10.1038/nbt.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witham S., Takano K., Schwartz C., Alexov E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins. 2011;79:2444–2454. doi: 10.1002/prot.23065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong P., Zhang C., Zheng W., Zhang Y. BindProfX: assessing mutation-induced binding affinity change by protein interface profiles with pseudo-counts. J. Mol. Biol. 2017;429:426–434. doi: 10.1016/j.jmb.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N., Han J.G., Shyu C.R., Korkin D. Determining effects of non-synonymous SNPs on protein-protein interactions using supervised and semi-supervised learning. PLoS Comput. Biol. 2014;10:e1003592. doi: 10.1371/journal.pcbi.1003592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MutaBind2 is available at https://mutabind.org/v2, and the training and test datasets are available for download from the server.