

Abstract
Immune checkpoint blockade (ICB) therapy is a treatment strategy for hepatocellular carcinoma (HCC); however, its clinical efficacy is limited to a select subset of patients. Next-generation sequencing has identified the value of tumor mutation burden (TMB) as a predictor for ICB efficacy in multiple types of tumor, including HCC. Specific driver gene mutations may be indicative of a high TMB (TMB-H) and analysis of such mutations may provide novel insights into the underlying mechanisms of TMB-H and potential therapeutic strategies. In the present study, a hybridization-capture method was used to target 1.45 Mb of the genomic sequence (coding sequence, 1 Mb), analyzing the somatic mutation landscape of 81 HCC tumor samples. Mutations in five genes were significantly associated with TMB-H, including mutations in tumor protein 53 (TP53), Catenin®1 (CTNNB1), AT-rich interactive domain-containing protein 1A (ARID1A), myeloid/lymphoid or mixed-lineage leukemia (MLL) and nuclear receptor co-repressor 1 (NCOR1). Further analysis using The Cancer Genome Atlas Liver Hepatocellular Carcinoma database showed that TP53, CTNNB1 and MLL mutations were positively correlated with TMB-H. Meanwhile, mutations in ARID1A, TP53 and MLL were associated with poor overall survival of patients with HCC. Overall, TMB-H and associated driver gene mutations may have potential as predictive biomarkers of ICB therapy efficacy for treatment of patients with HCC.
Keywords: HCC, immune checkpoint blockade, tumor mutation burden, cancer genomics
Introduction
Hepatocellular carcinoma (HCC) is the sixth most prevalent type of cancer and the second leading cause of cancer-associated death worldwide, accounting for ~75% of the primary liver cancer cases due to rapid disease progression and poor prognosis (1). HCC is typically diagnosed at an advanced stage, and as such, treatment options are limited and the 5-year overall survival (OS) rate is only 3–5% (2).
Advances in cancer immunotherapy, particularly the development of immune checkpoint blockades (ICBs), have shown clinical benefit and success in the treatment of several types of cancer, including melanoma, lung cancer and HCC (3–5). However, the clinical efficacy of ICB treatment is limited to a subset of patients, due to high rates of drug resistance (6,7). Efforts are being made to elucidate the underlying mechanisms of drug efficacy and resistance to ICB treatment, which may help provide individualized treatment and improved therapeutic strategies. Programmed cell death ligand 1 (PD-L1) is the most commonly used biomarker in anti-programmed death protein 1 (PD-1) therapy (7,8), but has several limitations, for example immunohistochemistry analysis of PD-L1 expression lacks a standardized format. Biological and technical challenges, such as heterogeneity of PD-L1 expression and variation in the affinities of different anti-PD-L1 antibodies, further complicate PD-L1 expression level testing (8).
Advances in next-generation sequencing (NGS) has identified tumor mutation burden (TMB) as a potential biomarker for predicting the response of patients with multiple tumors to ICB therapy (9,10). The underlying mechanisms of ICB therapy involves the synthesis of large quantities of neoantigens required for activating anti-tumor immune responses (11,12). A recent study evaluated the efficacy of anti-PD-1 antibody treatment combined with anti-angiogenesis therapy for patients with advanced stage HCC, showing that the TMB of individuals who responded favorably was significantly higher compared with patients who showed a less favorable response. In addition, the combined treatment strategy significantly improved the survival of patients with HCC with a higher TMB (13). Therefore, TMB may be a potentially novel predictor of survival for patients with HCC undergoing ICB therapy. High TMB (TMB-H) is frequently observed in patients with mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) (11,14,15). TMB-H has also been identified in patients harboring driver gene mutations associated with different types of cancer with a TMB-H, including breast cancer antigen 2 in melanoma (16) and human epidermal growth factor receptor (HER)-2 and HER3 in urological cancer (17). These findings have provided novel insight into therapeutic strategies, such as combined therapy consisting of ICBs and targeted molecular drugs. In the present study, NGS was performed on a group of HCC tumor samples and matching normal plasma samples were used to identify recurrent mutations associated with TMB-H in patients with HCC.
Materials and methods
Patients and tissue samples
The present study was approved by The Institutional Review Board of Peking University International Hospital (Xi'an, China; approval no. 2016-045). A total of 81 patients with HCC from Peking University International Hospital were enrolled into the study between January 2015 and June 2018 and written informed consent was obtained from all patients. The median age of patients was 57 years (range, 30–85 years) and 30/81 patients (37.04%) were over 60 years old. The majority of patients were male (71 males and 10 females) and clinical characteristics of patients are shown in Table I. Patients had not received ICB therapy at the time of specimen collection. HCC tissue and paired plasma samples were collected to perform NGS (18). HCC formalin fixed paraffin embedded (FFPE) samples were stored at room temperature, and were processed within 72 h of collection. Peripheral blood was collected in EDTA Vacutainer tubes (BD Diagnostics; Becton, Dickinson and Company) at room temperature and processed within 4 h. Peripheral blood lymphocytes (PBLs) and other blood cells were stored at −80°C.
Table I.
Clinical characteristics of 81 patients with hepatocellular carcinoma.
| Characteristic | Patients, n (%) |
|---|---|
| Age | |
| <60 years | 48 (59.26) |
| ≥60 years | 30 (37.04) |
| Unknown | 3 (3.70) |
| Sex | |
| Male | 71 (87.65) |
| Female | 10 (12.35) |
DNA extraction from HCC samples
FFPE slides were stored at room temperature. Plasma samples were centrifuged at 1,600 × g at 4°C for 10 min, then transferred to new microcentrifuge tubes and centrifuged at 16,000 × g at 4°C for 10 min to remove the remaining cell debris. Germline genomic DNA was extracted from PBLs using a DNeasy Blood and Tissue kit according to the manufacturer's protocol (Qiagen GmbH). Genomic DNA was extracted from FFPE samples using a Maxwell® RSC DNA FFPE kit according to the manufacturer's protocol (Promega Corporation). DNA concentration was estimated using a Qubit fluorometer and a Qubit double stranded DNA high sensitivity analysis kit according to the manufacturer's protocol (Invitrogen; Thermo Fisher Scientific, Inc.).
NGS of HCC DNA
HCC tumor samples were analyzed using target capture and NGS (18). Genomic DNA libraries were constructed using a KAPA DNA Library kit (Kapa Biosystems; Roche Diagnostics). The capture probe design was based on ~1.45 Mb genomic regions of 1,021 genes frequently mutated in solid tumors (coding sequence, 1 Mb). DNA sequencing was performed using HiSeq 3000 instrument according to the manufacturer's protocol (Illumina, Inc.). Germline DNA extracted from PBLs was used as the non-cancer associated component. Germline mutations of HCC tissues were filtered out using the paired PBLs DNA from the same patient.
Sequencing data analysis
If the proportion of indeterminate bases in a read accounted for >50%, or if the proportion of bases with BaseQ <5 was >50%, then this was considered a low-quality read. Terminal adaptor sequences and low-quality data were removed from the raw data. Clean reads were mapped onto the human genome build GRCh37 using Burrows-Wheeler Aligner (version 0.7.12-r1039) (19). Picard (version 1.98) was used to mark polymerase chain reaction (PCR) duplicates (http://broadinstitute.github.io/picard/). Single nucleotide variants (SNVs) and insertions or deletions (indels) were identified using MutTect2 (version 3.4–46-gbc02625 (20). Somatic copy number variations (CNVs) were detected using CONTRA (version 2.0.8) (21). Structure variations (SVs) were identified using split-read and discordant read-pair using in-house methods (22). Capture baits for SVs were designed according to selected exons and introns of the RET, ALK, ROS1 and neurotrophic tyrosine kinase receptor type 1 (NTRK1) oncogenes based on reported SVs (22).
Calculation of tissue TMB (tTMB)
tTMB was calculated after comprehensive genomic profiling of tissue samples using the 1,021 gene panel on 1 Mb of genomic coding region. The numbers of somatic, coding, SNVs and short indels detected at a frequency of ≥3% were calculated as tTMB, not including synonymous mutations. Patients harboring ≥7 mutations/Mb (the top quartile of tTMB distribution) were classified as the high tTMB (tTMB-H) group and all others were classified as low tTMB (tTMB-L) patients. The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) database was used as the validation set to verify the correlation between TMB and driver genes and the value of predicting the clinical benefit. The data was downloaded from the cBioPortal website (http://www.cbioportal.org/).
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 6.01 (GraphPad Software, Inc.). A χ2 or Fisher's exact test (n<5) were used to compare the mutation status of frequently mutated genes in the patients classed as tTMB-H and tTMB-L. A Mann-Whitney U test was performed to analyze the difference in tTMB between wild-type samples and mutation samples of frequently mutated genes. Kaplan-Meier curves were plotted to assess survival outcomes. Overall survival (OS) and recurrence-free survival (RFS) were accessed using a log-rank test in subgroups classified according to gene mutation. P<0.05 was considered to indicate a statistically significant difference.
Results
Significantly altered genes in HCC
A total of 81 HCC tumor samples were analyzed using a 1,021-gene panel at 1,200 × average sequencing depth. Somatic mutations included SNVs, short indels, CNVs and SVs. Overall, 506 SNVs and 38 CNVs were found in the HCC tumor samples (Fig. 1). The most commonly mutated genes were: Tumor protein 53 (TP53); telomerase reverse transcriptase (TERT); Catenin beta 1 (CTNNB1); RB transcriptional co-repressor 1 (RB1); AT-rich interactive domain-containing protein (ARID)1A; axis inhibition protein 1 (AXIN1); ARID2; cyclin D1 (CCND1); and adenomatous polyposis coli (APC). The TP53 tumor suppressor gene was the most frequently mutated (55.6% of samples) of all the highly mutated genes in the cohort and RB1 gene was mutated in 9.9% of the samples. Inactivation of the TP53-RB pathway was frequently observed in the HCC samples. TERT, a crucial unit of the telomerase complex (23), showed somatic mutations in its promoter region 33.3%. CTNNB1, AXIN1 and APC, which are important components of the wingless-type MMTV integration site family (WNT) signaling pathway (23), showed alteration frequencies of 18.5, 12.4 and 7.4%, respectively. ATP-dependent nucleosome remodelers, ARID1A and ARID2, were mutated in 14.8 and 8.6% of tumors, respectively. CNV mutations were also commonly found in HCC tumors samples and the most frequent CNV was a CCND1 amplification (6.17%). CCND1 encodes cyclin D1, the major downstream target factor of the WNT signaling pathway (23). CCND1 amplification was exclusively mutated in the crucial components of the WNT signaling pathway, including CTNNB1, AXIN1 and APC. Another CNV identified was an amplification of NTRK1 in 3.7% of samples.
Figure 1.

Mutation landscape of 81 hepatocellular carcinoma tumor samples. Central matrix shows somatic mutations with colors indicating different types of mutations and genes mutated at high frequency are represented in the right colored bar. The top bar plot shows the number of gene mutations in each sample and the mutation rate of significantly mutated genes is displayed on the left. CNV, copy number variant.
Distribution of tTMB in HCC samples
Calculation of tTMB was performed using a hybridization-capture method and targeted ~1.45 Mb of the genomic sequence (coding sequence, 1 Mb). tTMB was defined as the number of somatic coding SNVs and indels occurring at a frequency of ≥3%/1×106 bases in the 1,021 gene panel. The median tTMB of the HCC cohort was 5 mutations/megabase (muts/Mb). The top quartile (7 muts/Mb) of TMB distribution was used as the cutoff value to define TMB-H (Fig. 2). The maximum of tTMB distribution was 18 muts per Mb. There were 26 patients with HCC and high tumor mutation burden. Subsequently, the influenced factors of tTMB-H were analyzed.
Figure 2.
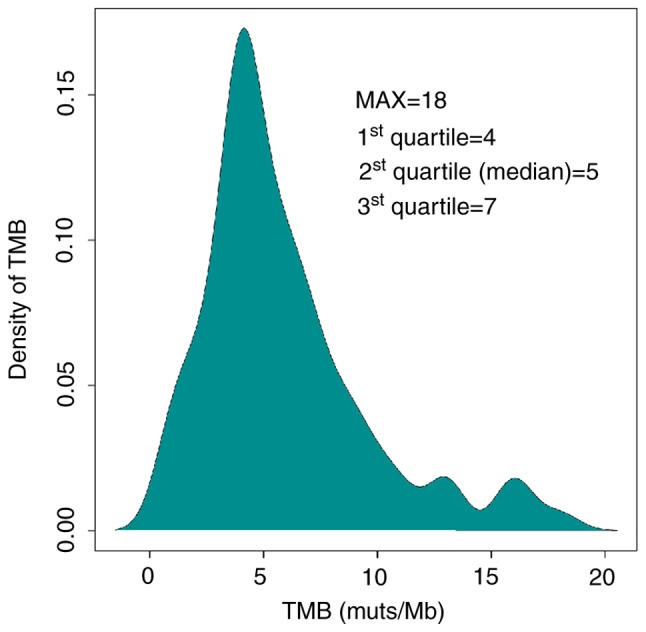
Distribution of tTMB in 81 HCC tumor tissue samples. Density of tTMB represents the percentage of HCC samples at various tTMB levels. tTMB, tissue tumor mutation burden; HCC, hepatocellular carcinoma; muts/Mb, mutations/Mb; MAX, maximum.
Age and TMB distribution in HCC
HCC specimens were stratified according to age, in order to analyze the differences in TMB distribution. From the age of 30–70, there was an increasing trend of tTMB, however this was not statistical significance. A slight decrease in tTMB was observed in patients >70 years (Fig. S1). These results are consistent with a previous study by Podolskiy et al (24) which demonstrated a similar relationship between human aging and the development of tumor mutations.
Sex and TMB distribution in HCC
Stratification by sex showed that the median tTMB values of male and female patients with HCC were 5 muts/Mb and 4 muts/Mb, respectively. The top quartile of tTMB in the male cohort (7 muts/Mb) was higher compared with the female cohort (6.5 muts/Mb). No significant difference in TMB distribution was observed between male and female HCC cohorts (P=0.6917; Fig. S2); however, this may be due to the lower number of HCC specimens from females.
Frequently mutated genes in the TMB-H cohort
To identify recurrently mutated genes in the TMB-H cohort, HCC patients were classified into two groups: tTMB-H (≥7 Muts/Mb) and low tTMB (tTMB-L; <7 Muts/Mb). The results showed that mutations in ARID1A, CTNNB1 and NCOR1 were more frequently detected in HCC samples in the tTMB-H group compared with tTMB-L group (P=0.0013, 0.0152 and 0.0347, respectively; Table II).
Table II.
Gene mutation rates in a total of 81 patients with hepatocellular carcinoma, including 26 samples with tTMB-H and 55 samples with tTMB-L.
| Gene | tTMB-H, % | tTMB-L, % | P-value |
|---|---|---|---|
| TP53 | 65.38 | 50.91 | 0.2419 |
| TERT | 42.31 | 29.09 | 0.3135 |
| ARID1A | 34.62 | 5.45 | 0.0013b |
| CTNNB1 | 34.62 | 10.91 | 0.0152a |
| AXIN1 | 11.54 | 12.73 | 1 |
| MLL2 | 11.54 | 9.09 | 0.7071 |
| LRP1B | 11.54 | 7.27 | 0.6749 |
| RB1 | 7.69 | 10.91 | 1 |
| ARID2 | 15.38 | 5.45 | 0.2031 |
| APC | 11.54 | 5.45 | 0.3798 |
| MLL | 15.38 | 3.64 | 0.0803 |
| ATRX | 7.69 | 5.45 | 0.6539 |
| PTEN | 7.69 | 5.45 | 0.6539 |
| PBRM1 | 11.54 | 3.64 | 0.3211 |
| KRAS | 3.85 | 7.27 | 1 |
| ATM | 3.85 | 7.27 | 1 |
| NCOR1 | 15.38 | 1.82 | 0.0347a |
P<0.05
P<0.01. tTMB-L, low tumor tissue mutation burden; tTMB-H, high tumor tissue mutation burden.
Association between gene mutations and TMB distribution in HCC
To further analyze the association between gene mutations and TMB-H status in HCC, the tTMB distribution in HCC samples was compared between the wild-type and mutated genotypes. Frequently mutated genes, which were detected in >5 HCC tumor cases were analyzed for TMB distribution. Median tTMB values and the top quartile values of tTMB were compared in 18 genes. A total of five genes were shown to be significantly different in tTMB distribution between the wild-type and mutated genotypes: TP53; CTNNB1; ARID1A; MLL; and NCOR1 (P=0.0439, 0.0196, 0.0023, 0.0247 and 0.0076, respectively; Fig. 3). tTMB distribution is shown in Fig. S3.
Figure 3.

Association between gene mutation and median tTMB and top quartile tTMB values. Ratio of median tTMB values and the ratio of top quartile values of tTMB in mutated and wild-type samples are indicated by red and blue lines, respectively. A total of 18 genes were mutated in at least 5 samples. tTMB, tissue tumor mutation burden.
TMB distribution based on gene mutation status in The Cancer Genome Atlas Liver Hepatocellular Carcinoma database (TCGA-LIHC)
A retrospective analysis was performed using HCC data obtained from the TCGA-LIHC database. A total of 363 HCC tumor samples were included in the cohort and the data were obtained from cBioPortal (cbioportal.org/). It was found that mutations in TP53, CTNNB1 and MLL were positively correlated with higher TMB (P=0.0009, 0.0016 and 0.0013, respectively; Fig. 4).
Figure 4.

Tumor mutation burden distribution based on gene mutation status in The Cancer Genome Atlas. Blue represents the wild-type genotype and red represents the mutated genotype for (A) TP53, (B) CTNNB1, (C) MLL, (D) ARID1A and (E) NCOR1. TP53, tumor protein 53; CTNNB1, Catenin®1; ARID1A, AT-rich interactive domain-containing protein 1A; MLL, myeloid/lymphoid or mixed-lineage leukemia.
Frequently mutated genes predict overall survival and RFS of HCC in TCGA-LIHC
Kaplan-Meier analysis of OS demonstrated that the ARID1A mutation was significantly correlated with poor survival outcome in the TCGA HCC cohort [median OS (mOS), 27.57 vs. 71.03 months, ARID1A mutations vs. wild-type; mOS ratio, 0.3881, 95% confidence interval (CI), 0.2139–0.7040; log-rank test, P=0.0372; Fig. 5]. Mutations in TP53 predicted a trend towards poor outcome (mOS 46.57 vs. 71.03 months, TP53 mutations vs. wild-type; log-rank test, P=0.0567). No statistically significant differences in survival outcome were observed for CTNNB1, MLL and NCOR1 mutations compared with the wild-type genotypes in the TCGA HCC cohort (Fig. S4). Kaplan-Meier analysis of RFS demonstrated that MLL mutations were significantly correlated with poorer RFS compared with the wild-type genotype (mRFS, 8.93 vs. 29.73 months; mRFS ratio, 0.3004, 95% CI, 0.1470–0.6140; log-rank test, P=0.069, Fig. 5).
Figure 5.

Kaplan-Meier analysis of median OS and RFS based on gene mutation status of (A) ARID1A, (B) TP53 and (C) MLL. OS, overall survival; RFS, recurrence free survival; CI, confidence interval. TP53, tumor protein 53; ARID1A, AT-rich interactive domain-containing protein 1A; MLL, myeloid/lymphoid or mixed-lineage leukemia.
Microsatellite instability status in HCC
Only 1/81 HCC tumor samples (HCC057) in the present study showed MSI-H. This sample also had TMB-H with a tTMB value of 9 muts/Mb, and one TP53 mutation and one ARID1A mutation (data not shown).
Mutational status of genes that may predict efficacy of ICB therapy in HCC
A total of 2/81 HCC patients (2.47%) possessed mutations in POLE (R1324H and I478M; Table SI). The tTMB values of the R1324H and I478 M mutations were 8 muts/Mb and 4 muts/Mb, respectively (Fig. 6). Overall, 2 mutations were observed in PTEN and 1 mutation was observed in DNMT3A. No mutations were detected in MLH1, MLH2, PMS2, MSH6, POLD1, JAK1, JAK2, B2M and STK11. Mutations in POLE and PTEN occurred in the primary clonal mutation and the inactivating mutation in DNMT3A occurred in the sub-clonal mutation.
Figure 6.
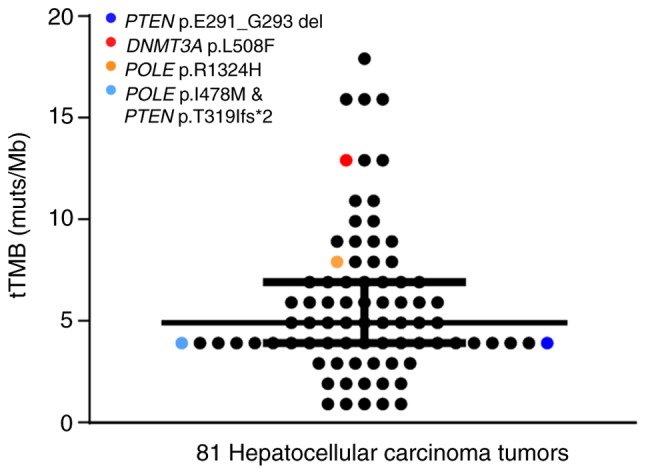
Mutations in genes that predict efficacy of ICB therapy in hepatocellular carcinoma. Each dot represents a patient and immunotherapy-associated variants were detected in four colored patients. Two variants, POLE I478M and PTEN T319, were detected simultaneously in the light blue patient.
Discussion
HCC is an aggressive disease and previous genetic characterization of this disease has revealed significant heterogeneity among tumors; even within a single tumor lesion, the allelic frequency can be as low as 13% (25). Previous studies have identified several signaling pathways, which are disrupted in HCC, including the WNT, hypoxia-inducible factor 1, mechanistic target of rapamycin, and several metabolic related pathways (26,27). Actionable target genes found in other tumors, especially lung adenocarcinoma, such as those encoding protein kinases and other crucial enzymes are not significantly mutated in HCC (26–29). It has been reported that no single protein kinase is mutated at a frequency >5% in HCC (28,29). The majority of targeted therapy (tyrosine kinase inhibitors or monoclonal antibodies) for HCC has demonstrated minimal or no clinical efficacy (30).
HCC typically manifests in patients with previous liver damage, commonly caused by chronic hepatitis infection (31). Chronic hepatitis and subsequent inflammation are reported to induce immune evasion via prolonged activation of the interferon γ signaling pathway (32,33). The immune system serves an important role in the development of HCC. Research has demonstrated that ICB therapy may be an effective treatment strategy, which results in effective and long-lasting responses in patients with HCC. For example, in phase I/II of the CheckMate 040 trial evaluating the safety and efficacy of nivolumab as a monotherapy in patients with advanced HCC, the response rate was 20% and the disease control rate was 64% (34). Although the CheckMate 040 trial was non-randomized, it led to the approval of nivolumab by the Food and Drug Administration (FDA) for the treatment of HCC. Despite advances in ICB therapy, the clinical efficacy of this treatment is limited due to drug resistance (6,7). Even for melanoma, one of the tumors most sensitive to ICB, ~60% of patients display primary resistance and ~50% of individuals who respond favorably are likely to develop acquired resistance after 3 years of treatment (6,7). To provide individualized treatment, improve current treatment modalities and to investigate potentially novel treatment strategies, researchers have been trying to identify factors that predict drug efficacy. Previous studies have reported tumor cell mutations associated with drug efficacy observed in patients undergoing ICB-based immunotherapy and that these mutations may have value as biomarkers (35,36). Mutations in MLH1, MLH2, PMS2 and MSH6 are associated with dMMR and result in MSI (37). The FDA approved MSI status as a biomarker for immunotherapy of pan-cancerous species (38). Mutations in POLE and POLD1 are associated with extremely high TMB (39), and inactivation mutations in JAK1, JAK2, B2M and PTEN are associated with immunotherapy resistance (35,40). Meanwhile, mutations in DNMT3A are associated with hyper-progression in immunotherapy (36). In the present study, the somatic mutation landscape of HCC tumors was characterized, with a particular focus on mutations associated with TMB status, to improve our understanding of the role of the tumor cell intrinsic factors on the efficacy of immunotherapy for HCC.
The genetic alterations identified in the present study are consistent with previously published studies (23,41,42). The results of the present study showed that most genes were mutated in <20% of the samples analyzed, supporting the genetic heterogeneity of HCC. Regarding tumor intrinsic genetic aberrations which may be associated with the clinical efficacy of ICB therapy, 2 (2.47%) mutations in POLE, 2 mutations (2.47%) in PTEN, 1 mutation (1.23%) in DNMT3A were detected as well as only 1 sample with MSI-H. No evidence of dMMR was observed in any of the samples. It is possible that none of the factors investigated in the present study, which have been studied in other tumor types (43,44), are suitable targets for anti-HCC therapy. The underlying mechanisms of immune evasion in HCC may be different compared with other types of cancer. Overall, potential predictors of clinical efficacy of ICB therapy in patients with HCC needs to be further explored.
The association between TMB and patient response to ICB therapy was originally indicated by melanoma studies and ICB-sensitive melanomas were found to comprise tumors with the highest mutation burden (11,14). The association between TMB and ICB efficacy was subsequently confirmed in colorectal cancer (CRC). Only hyper-mutated CRCs with dMMR or MSI-H tend to respond to ICB therapy (15). A recently published study evaluated the efficacy of SHR-1210 anti-PD-1 antibody combined with the anti-angiogenesis agent, apatinib, for patients with advanced stages of tumors including adenocarcinoma of the stomach and gastroesophageal function, and HCC (13). Among the 18 patients with HCC enrolled in this study, the TMB of individuals who responded favorably was significantly higher compared with those who did not respond favorably, and this treatment significantly improved the survival outcomes of patients with HCC who possessed a large number of mutations (13). Therefore, TMB is a potentially novel predictor of the efficacy of ICB therapy for patients with HCC. This may be due to an increased number of tumor-associated antigens or neoantigens expressed by cancer cells with high TMB, which are critical for the activation of anti-tumor immune responses. Increased numbers of neoantigens are known to increase recruitment of various types of T cells, particularly CD8+ T lymphocytes. Tumor neoantigens that are generated by mutations, especially frameshift-mutation-derived peptides have the highest immunogenicity (45,46).
The DNA damage response (DDR) system is essential for preserving genomic integrity by repairing damaged DNA (47). Dysfunction in this system may induce MSI-H or hyper-mutational phenotypes and DDR deficiency is considered to be a primary cause of TMB-H and MSI-H (47,48). There are 8 pathways included in the DDR system, of which mismatch repair (MMR) has been extensively studied and is a commonly used predictor in a clinical setting (48,49). The present study did not detect dMMR in any of the 81 samples that were analyzed, including the sample that harbored the highest TMB (18 muts/Mb); however, five genes, including TP53, CTNNB1, ARID1A, MLL and NCOR1, were found to be significantly associated with TMB-H. In addition, only 1/81 HCC samples showed MSI-H and TMB-H (9 muts/Mb). This sample also harbored a TP53 mutation and an ARID1A mutation.
TP53 is one of the most frequently mutated genes in HCC (23,42). It is a transcription factor controlling the expression of genes involved in cell cycle arrest, apoptosis and senescence in response to hypoxic stress, DNA damage and oncogenic activation. TP53 is also a tumor suppressor gene, functioning in the preservation of genomic integrity during hypoxia, which is a common phenomenon in HCC (50,51). ARID1A is also frequently mutated in HCC, as demonstrated in the present study. ARID1A binds to other subunits such as BRG1/BRM, forming a switch/sucrose non-fermentable chromatin remodeling complex. This complex uses energy from ATP to mobilize nucleosomes and to regulate DNA accessibility to various cellular machinery, including DNA replication and DNA-damage repair machinery (52). In a proteomic screen, Shen et al (53) found that ARID1A interacts with MMR protein MSH2, recruiting MSH2 to chromatin during DNA replication and promoting MMR. Conversely, ARID1A inactivation compromised MMR and increased mutagenesis. ARID1A deficiency was associated with an MSI genomic signature, a predominant C>T mutation pattern and increased mutation load across several types of cancer. Tumors formed using an ARID1A-deficient ovarian cancer cell line in syngeneic mice displayed increased mutation load (53). MLL belongs to the family of histone H3 lysine 4 methyltransferases and is a chromatin regulatory enzyme (25). NCOR1 also serves an important role in regulating a variety of nuclear factors and in chromatin remodeling (54). CTNNB1 encodes β-catenin, a subunit of the cadherin protein complex which functions as a signaling molecule in the WNT signaling pathway and regulates cellular proliferation (55). It is possible that factors which influence genetic stability, facilitate DNA error generation or regulate cell proliferation may all contribute to TMB-H. Further studies are required to elucidate the underlying mechanisms contributing to TMB-H development. The result of the present study showing no association between ARID1A and NCOR1 with TMB in TCGA cohort may be due to biased sampling from regional differences.
PD-L1 is the most commonly used clinical biomarker for ICBs, but it has several limitations (8). The ability of NGS to reveal the TMB status of patients provides another potential predictor of ICB-therapy efficacy, as shown in clinical trials investigating other tumor types (11,13–15). Therefore, TMB-H may also serve as biomarker complementary to PD-L1. However, several key questions need to be answered: How many genes (the whole genome, targeted panel, or only expressed mutations) should be included to define TMB status? What is the optimal threshold for TMB-H? Is there a consensus between the different diagnostic assays? Whether crucial driver gene mutations associated with high mutation load could serve as potential predictive biomarkers in patients with HCC treated with ICB therapy? Further studies are required to establish uniform diagnostic standards.
The present study has several limitations. First, there was no cohort treated with ICB therapy. Second, it was only demonstrated that gene mutations associated with TMB-H are present in patients with HCC, but the underlying mechanisms of this association remains to be investigated in vitro and in vivo. In addition, the majority of patients enrolled in the present study were male, accounting for 87.65% of the entire cohort. This was higher compared with the sex distribution of patients shown in a different study on HCC (56).
The present study provides novel insight into gene signatures, which may predict the clinical efficacy of ICB therapy in patients with HCC. The five genes identified showed recurrent mutations which were significantly correlated with high mutation load: TP53; CTNNB1; ARID1A; MLL; and NCOR1. These findings may provide a novel understanding of the underlying mechanisms of HCC to aid the development of therapeutic strategies, such as combined therapy with ICBs and molecule-targeted drugs. Limitations of the present study include the use of a small study cohort and the retrospective nature of the analysis. Further investigation is required to evaluate the association of TMB-H and crucial driver gene with high mutation load and the potential of these genes as predictive biomarkers of ICB therapy treatment in patients with HCC.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Michael J. Overman (Department of Gastrointestinal Medical Oncology, The University of Texas MD Anderson Cancer Center) for his advice regarding the interpretation of the manuscript.
Glossary
Abbreviations
- HCC
hepatocellular carcinoma
- ICBs
immune checkpoint blockades
- PD-1
programmed death protein 1
- PD-L1
programmed cell death ligand 1
- TMB
tumor mutation burden
- TMB-H
high TMB
- dMMR
mismatch repair deficiency
- MSI-H
high microsatellite instability
- SNVs
single nucleotide variants
- indels
insertions or deletions
- CNVs
copy number variants
- SVs
structure variations
- TP53
tumor protein 53
- CTNNB1
Catenin®1
- RB1
RB transcriptional co-repressor 1
- AXIN1
axis inhibition protein 1
- ARID
AT-rich interactive domain-containing protein
- CCND1
cyclin D1
- APC
adenomatous polyposis coli
- WNT
wingless-type MMTV integration site family
- TERT
telomerase reverse transcriptase
- NTRK1
neurotrophic tyrosine kinase receptor type 1
- MLL
myeloid/lymphoid or mixed-lineage leukemia
- NCOR1
nuclear receptor co-repressor 1
- tTMB
tissue TMB
- TCGA-LIHC
Cancer Genome Atlas Liver Hepatocellular Carcinoma
- OS
overall survival
- mOS
median OS
- RFS
recurrence-free survival
- mRFS
median RFS
- FDA
Food and Drug Administration
- CRC
colorectal cancer
- DDR
DNA damage response
Funding
The present study was funded by The National Nature Science Foundation of China (grant no. 81372632).
Availability of data and materials
The data that support the findings of the present study are available from Geneplus-Beijing Institute, but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission from Geneplus-Beijing Institute.
Authors' contributions
LJ conceptualized the study. LL, ZW, XR designed the study. Software was designed by WX. LL, WX and ZW collected the data. LL, ZW, XD, YY, YG, YC and JW analyzed the data. LL, XR, XD, XW, WX and CM obtained the resources necessary for the study. LL, WX, LJ and XR wrote, reviewed and edited the manuscript. All authors approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by The institutional Review Board of Peking University International Hospital and written informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Yu SJ. A concise review of updated guidelines regarding the management of hepatocellular carcinoma around the world: 2010–2016. Clin Mol Hepatol. 2016;22:7–17. doi: 10.3350/cmh.2016.22.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sprinzl MF, Galle PR. Current progress in immunotherapy of hepatocellular carcinoma. J Hepatol. 2017;66:482–484. doi: 10.1016/j.jhep.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol. 2018;29:71–83. doi: 10.1093/annonc/mdx686. [DOI] [PubMed] [Google Scholar]
- 6.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Q, Wu X. Primary and acquired resistance to PD-1/PD-L1 blockade in cancer treatment. Int Immunopharmacol. 2017;46:210–219. doi: 10.1016/j.intimp.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Zhu J, Armstrong AJ, Friedlander TW, Kim W, Pal SK, George DJ, Zhang T. Biomarkers of immunotherapy in urothelial and renal cell carcinoma: PD-L1, tumor mutational burden, and beyond. J Immunother Cancer. 2018;6:4. doi: 10.1186/s40425-018-0314-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–779. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 10.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–2501. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 12.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J, Zhang Y, Jia R, Yue C, Chang L, Liu R, Zhang G, Zhao C, Zhang Y, Chen C, et al. Anti-PD-1 antibody SHR-1210 combined with apatinib for advanced hepatocellular carcinoma, gastric or esophagogastric junction cancer: An open-label, dose escalation and expansion study. Clin Cancer Res. 2019;25:515–523. doi: 10.1158/1078-0432.CCR-18-2484. [DOI] [PubMed] [Google Scholar]
- 14.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pal SK, Agarwal N, Choueiri TK, Stephens PJ, Ross JS, Miller VA, Ali SM, Chung J, Grivas P. Comparison of tumor mutational burden (TMB) in relevant molecular subsets of metastatic urothelial cancer (MUC) Ann Oncol. 2017;28(Suppl-5):v295–v329. doi: 10.1093/annonc/mdx371.004. [DOI] [Google Scholar]
- 18.Lv X, Zhao M, Yi Y, Zhang L, Guan Y, Liu T, Yang L, Chen R, Ma J, Yi X. Detection of rare mutations in CtDNA using next generation sequencing. J Vis Exp. 2017 doi: 10.3791/56342. doi: 10.3791/56342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–221. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL. CONTRA: Copy number analysis for targeted resequencing. Bioinformatics. 2012;28:1307–1313. doi: 10.1093/bioinformatics/bts146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nong J, Gong Y, Guan Y, Yi X, Yi Y, Chang L, Yang L, Lv J, Guo Z, Jia H, et al. Circulating tumor DNA analysis depicts subclonal architecture and genomic evolution of small cell lung cancer. Nat Commun. 2018;9:3114. doi: 10.1038/s41467-018-05327-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- 24.Podolskiy DI, Lobanov AV, Kryukov GV, Gladyshev VN. Analysis of cancer genomes reveals basic features of human aging and its role in cancer development. Nat Commun. 2016;7:12157. doi: 10.1038/ncomms12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao Y, Ruan J, Yeh SH, Lu X, Wang Y, Zhai W, Cai J, Ling S, Gong Q, Chong Z, et al. Rapid growth of a hepatocellular carcinoma and the driving mutations revealed by cell-population genetic analysis of whole-genome data. Proc Natl Acad Sci USA. 2011;108:12042–12047. doi: 10.1073/pnas.1108715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breuhahn K, Gores G, Schirmacher P. Strategies for hepatocellular carcinoma therapy and diagnostics: Lessons learned from high throughput and profiling approaches. Hepatology. 2011;53:2112–2121. doi: 10.1002/hep.24313. [DOI] [PubMed] [Google Scholar]
- 27.Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM. Cancer gene discovery in hepatocellular carcinoma. J Hepatol. 2010;52:921–929. doi: 10.1016/j.jhep.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–648. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 30.da Motta Girardi D, Correa TS, Crosara Teixeira M, Dos Santos Fernandes G. Hepatocellular carcinoma: Review of targeted and immune therapies. J Gastrointest Cancer. 2018;49:227–236. doi: 10.1007/s12029-018-0121-4. [DOI] [PubMed] [Google Scholar]
- 31.Gomaa AI, Khan SA, Toledano MB, Waked I, Taylor-Robinson SD. Hepatocellular carcinoma: Epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14:4300–4308. doi: 10.3748/wjg.14.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elsegood CL, Tirnitz-Parker JE, Olynyk JK, Yeoh GC. Immune checkpoint inhibition: Prospects for prevention and therapy of hepatocellular carcinoma. Clin Transl Immunology. 2017;6:e161. doi: 10.1038/cti.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wieder T, Eigentler T, Brenner E, Röcken M. Immune checkpoint blockade therapy. J Allergy Clin Immunol. 2018;142:1403–1414. doi: 10.1016/j.jaci.2018.02.042. [DOI] [PubMed] [Google Scholar]
- 34.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling TH Rd, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242–4250. doi: 10.1158/1078-0432.CCR-17-1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site-when a biomarker defines the indication. N Engl J Med. 2017;377:1409–1412. doi: 10.1056/NEJMp1709968. [DOI] [PubMed] [Google Scholar]
- 39.Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, Davidson S, Edwards M, Elvin JA, Hodel KP, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171:1042–1056.e10. doi: 10.1016/j.cell.2017.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN promotes resistance to t cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693–1702. doi: 10.1002/hep.26540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JS. The mutational landscape of hepatocellular carcinoma. Clin Mol Hepatol. 2015;21:220–229. doi: 10.3350/cmh.2015.21.3.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braun DA, Burke KP, Van Allen EM. Genomic approaches to understanding response and resistance to immunotherapy. Clin Cancer Res. 2016;22:5642–5650. doi: 10.1158/1078-0432.CCR-16-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teng F, Meng X, Kong L, Yu J. Progress and challenges of predictive biomarkers of anti PD-1/PD-L1 immunotherapy: A systematic review. Cancer Lett. 2018;414:166–173. doi: 10.1016/j.canlet.2017.11.014. [DOI] [PubMed] [Google Scholar]
- 45.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15:857–865. doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JK, Choi YL, Kwon M, Park PJ. Mechanisms and consequences of cancer genome instability: Lessons from genome sequencing studies. Annu Rev Pathol. 2016;11:283–312. doi: 10.1146/annurev-pathol-012615-044446. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, Zhao J, Wang G, Zhang F, Zhang Z, Zhang F, Zhang Y, Dong H, Zhao X, Duan J, et al. Co-mutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Cancer Res. 2018;78:6486–6496. doi: 10.1158/0008-5472.CAN-18-1814. [DOI] [PubMed] [Google Scholar]
- 49.Scarbrough PM, Weber RP, Iversen ES, Brhane Y, Amos CI, Kraft P, Hung RJ, Sellers TA, Witte JS, Pharoah P, et al. A cross-cancer genetic association analysis of the DNA repair and DNA damage signaling pathways for lung, ovary, prostate, breast, and colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2016;25:193–200. doi: 10.1158/1055-9965.EPI-15-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muller PA, Vousden KH. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumor suppression. Nat Rev Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson BG, Roberts CW. SWI/SNF nucleosome remodelers and cancer. Nat Rev Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 53.Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, Nagel ZD, Zou J, Wang C, Kapoor P, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24:556–562. doi: 10.1038/s41591-018-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Battaglia S, Maguire O, Campbell MJ. Transcription factor co-repressors in cancer biology: Roles and targeting. Int J Cancer. 2010;126:2511–2519. doi: 10.1002/ijc.25181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of the present study are available from Geneplus-Beijing Institute, but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission from Geneplus-Beijing Institute.