

Abstract
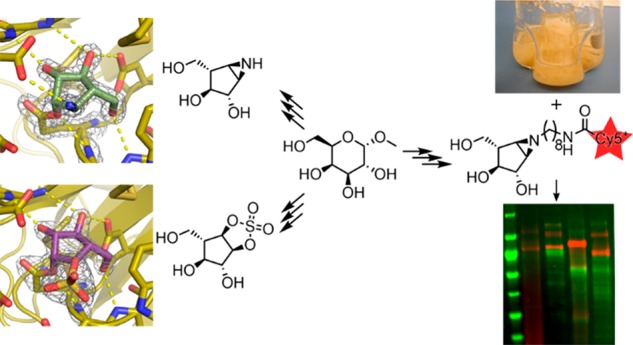
Identifying and characterizing the enzymes responsible for an observed activity within a complex eukaryotic catabolic system remains one of the most significant challenges in the study of biomass-degrading systems. The debranching of both complex hemicellulosic and pectinaceous polysaccharides requires the production of α-l-arabinofuranosidases among a wide variety of coexpressed carbohydrate-active enzymes. To selectively detect and identify α-l-arabinofuranosidases produced by fungi grown on complex biomass, potential covalent inhibitors and probes which mimic α-l-arabinofuranosides were sought. The conformational free energy landscapes of free α-l-arabinofuranose and several rationally designed covalent α-l-arabinofuranosidase inhibitors were analyzed. A synthetic route to these inhibitors was subsequently developed based on a key Wittig–Still rearrangement. Through a combination of kinetic measurements, intact mass spectrometry, and structural experiments, the designed inhibitors were shown to efficiently label the catalytic nucleophiles of retaining GH51 and GH54 α-l-arabinofuranosidases. Activity-based probes elaborated from an inhibitor with an aziridine warhead were applied to the identification and characterization of α-l-arabinofuranosidases within the secretome of A. niger grown on arabinan. This method was extended to the detection and identification of α-l-arabinofuranosidases produced by eight biomass-degrading basidiomycete fungi grown on complex biomass. The broad applicability of the cyclophellitol-derived activity-based probes and inhibitors presented here make them a valuable new tool in the characterization of complex eukaryotic carbohydrate-degrading systems and in the high-throughput discovery of α-l-arabinofuranosidases.
Introduction
Carbohydrate-degrading machinery is a fundamentally important component of the metabolic systems that underpin the global carbon cycle. Our understanding of these systems is dependent on an ability to identify the capacities of the carbohydrate-active enzymes produced by an organism. The growth of genomic libraries has revealed an expansive world of carbohydrate-degrading enzymes, of which only a small fraction have been isolated and probed for catalytic potential.1 Transcriptomic and proteomic experiments comparing the gene expression and protein secretion patterns of organisms grown on different substrates have helped to identify the genetic logic used by these organisms to efficiently degrade recalcitrant biomass.2 However, the underlying chemical rationale for these expression patterns remains obscure without highly detailed experimental work characterizing the role of each enzyme.
Inspired by the work of Withers3−5 and Wright,6 we have been developing cyclophellitol-derived activity-based inhibitors and probes (some aspects of which are reviewed in refs (7−9)) for the rapid detection and identification of specific biomass-degrading glycoside hydrolases within complex systems. The potential of cyclophellitol-derived activity-based probes (ABPs) as tools for the detection and identification of retaining glycoside hydrolases has been well-established.10 Mimicking the half chair conformation of the enzymatic transition state, cyclophellitol and cyclophellitol aziridine derivatives react specifically with the catalytic nucleophile of a retaining glycoside hydrolase, forming a nonhydrolyzable ester linkage through a ring-opening addition.11 This general strategy has been exploited to inhibit and label glycosidases displaying a variety of specificities including α- and β-d-glucosidases,12−14 β-d-glucuronidases,15 and α- and β-d-galactosidases,16,17 among others. Building on this work, we have recently reported the synthesis and validation of a collection of cyclophellitol-derived inhibitors and probes which specifically label retaining β-d-xylanases and β-d-xylosidases.18 These compounds were able to efficiently attach chemical handles for the detection and identification of key secreted xylan-degrading enzymes within an Aspergillus secretome. Expanding this toolbox to target side-chain removal enzymes has remained a challenge, not least for furanoside-active enzymes.
α-l-Arabinofuranoside “side-chains” are commonly found on both hemicellulosic and pectinaceous plant polysaccharides. The efficient removal of α-l-arabinofuranose branches enhances the breakdown of xylan-rich biomass.19 Furthermore, α-l-arabinofuranosidases are an essential part of the polysaccharide utilization loci which ferment arabinan chains in dietary rhamnogalacturonan I and arabinogalactan within the human gut.20 Thus, cyclophellitol-derived ABPs and inhibitors for α-l-arabinofuranosidases could be used to identify the enzymes responsible for the breakdown of a variety of complex polysaccharides. However, it is not currently known whether cyclophellitol derivatives can be effectively extended to target furanosidases.
No route to the synthesis of covalent inhibitors of α-l-arabinofuranosidases has previously been identified. The first synthesis of covalent furanose-configured inhibitors was the preparation of β-d-arabinofuranosyl and α-l-xylofuranosyl aziridines reported by Bols et al. in 2003.21 These were prepared via N–O reduction of cyclopentaisoxazolidines. Due to the inverted stereochemistry of the electrophilic moiety with respect to C4 (carbohydrate numbering), this synthetic strategy cannot be translated to α-l-arabinofuranose analogues, so new synthetic methodologies are needed to expand the scope of synthetically accessible furanoside mimics.
We have designed a collection of putative α-l-arabinofuranosidase inhibitors and ABPs with different electrophilic traps and detection tags. Potential inhibitors were analyzed in silico for their ability to mimic the natural 5-membered ring structure, stereochemistry, and conformational itinerary of retaining α-l-arabinofuranosides. These inhibitors and probes were synthesized following a route inspired by the synthesis of six-membered cyclophellitol derivatives. Inhibition kinetics measured with α-l-arabinofuranosidases from glycoside hydrolase families 51 and 54 (GH51 and GH54), the two major families of retaining α-l-arabinofuranosidases, were measured to validate our predictions. Furthermore, the ability of our α-l-arabinofuranosidase probes to facilitate the selective detection, identification, and characterization of active GH51 and GH54 enzymes within the complex mixture of enzymes secreted by Aspergillus niger was validated. These methods were then extended to the identification of α-l-arabinofuranosidases within the secretomes of basidiomycete fungi grown on complex biomass.
Experimental Section
All chemicals were purchased from Sigma-Aldrich unless otherwise specified.
Design and Synthesis of α-l-Arabinofuranose-Configured Cyclophellitol Derivatives
Detailed protocols for synthesis of compounds 1 to 23 and their NMR characterization can be found in the Supporting Information.
Secretome Production
Aspergillus niger strain N402 was grown as described by Schröder et al.18 with a mixture of 50 mM arabinose, 1% sugar beet arabinan, and 2 mM fructose as the sole carbon source. Samples were collected, 0.2 μm filtered, and snap-frozen after 5 days. Samples were stored at −80 °C until being thawed immediately before use.
The strains Abortiporus biennis BRFM 1215 (A. biennis), Fomes fomentarius BRFM 1323 (F. fomentarius), Hexagonia nitida BRFM 1328 (H. nitida), Leiotrametes menziesii BRFM 1557 (L. menziesii), Polyporus brumalis BRFM 958 (P. brumalis), Trametes ljubarskyi BRFM 957 (T. ljubarskyi) Trametes gibbosa BRFM 952 (T. gibbosa), and Trametes meyenii BRFM 1361 (T. meyenii) were obtained from the CIRM-CF collection (International Centre of Microbial Resources dedicated to Filamentous Fungi, INRA, Marseille, France). All strains were identified by morphological and molecular analysis of ITS (Internal Transcribed Spacer) sequences. The strains were maintained on malt agar slants at 4 °C.
Basidiomycete cultures were grown in 250 mL baffled Erlenmeyer flasks with 100 mL medium containing 2.5 g L–1 of maltose as a starter (except for the maltose control condition; 20 g L–1), 1.842 g L–1 of diammonium tartrate as a nitrogen source, 0.5 g L–1 yeast extract, 0.2 g L–1 KH2PO4, 0.0132 g L–1 CaCl2/2H2O and 0.5 g L–1 MgSO4/7H2O, and as a main carbon source, 15 g L–1 (dry weight) of wheat straw (Triticum aestivum) or Wiley-milled aspen (Populus grandidentata). Cultures were incubated in the dark at 30 °C with shaking at 120 rpm. The cultures were stopped 10 days after inoculation and the culture broths (secretomes) were filtered using 0.2 μm poly(ether sulfone) membrane (Millipore) and then stored at −20 °C until use.
Recombinant Enzyme Production
The coding sequence for Geobacillus stearothermophilus abfA (GsGH51, GenBank: AAD45520) was synthesized with E. coli codon optimization and cloned into pET28a(+) with an N-terminal TEV protease-cleavable 6xhis tag by GenScript. Following transformation of BL21(DE3) Gold, the enzyme was produced in an auto induction medium (1% tryptone, 0.5% yeast extract, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 0.05% glucose, 0.5% glycerol, 0.2% lactose) at 37 °C. The enzyme was purified as described previously22 with an added overnight treatment with his-tagged TEV protease S219 V23 in pH 8 Tris-HCl, 5 mM DTT, 1 mM EDTA at RT followed by inverse histrap purification and desalting into 5 mM Tris-HCl, 1 mM EDTA, pH 8.0.
Aspergillus niger abfA (AnAbfA, GenBank: CAK43424) and Aspergillus kawachii abfB (AkAbfB, GenBank: BAB96816) were produced in P. pastoris X-33. A plasmid encoding AkAbfB in pPICZα with no purification tag was obtained from professors Takuya Koseki and Shinya Fushinobu. AnAbfA was synthesized by IDT as a GBlock and cloned into the vector fragment PCR-amplified from the AkAbfB-pPICZa plasmid using Gibson assembly.24 The AkAbfB (E221Q) mutant was generated using the Q5 site-directed mutagenesis kit (New England Biolabs) with primers designed by the NEBaseChanger tool.
Plasmid DNA for transformation into P. pastoris was linearized with SacI and purified using a PCR cleanup kit (Qiagen) using ultrapure water as the eluent. 100 ng of linearized DNA was electroporated into 80 μL of X-33 electrocompetent cells prepared following the protocol of Wu and Letchworth.25 Nine colonies from each transformation were purified on YPD-Zeocin plates, then grown in 5 mL of BMGY medium. At saturation (OD600 ∼ 20) cells were collected by centrifugation and resuspended in 5 mL of BMMY medium for expression screening at 20 °C. The transformant which gave the highest titer of the target protein with minimal detectable contamination after 3 daily 0.5% MeOH feedings was grown in 500 mL of BMGY in a 2.5 L baffled shaking flask at 30 °C overnight. The culture was then cooled to 20 °C and supplemented with 2.5 mL of 100% MeOH each day for 3 days.
The culture supernatant was clarified by centrifugation followed by 0.45 μm filtration. A 500 mL portion of medium was concentrated using a KrosFlo tangential flow system fitted with a 30 kDa MWCO mPES filter and then diluted with 9 volumes of 10 mM pH 5 sodium acetate buffer and concentrated again. Protein was then collected onto a 5 mL Q sepharose HP column (GE Healthcare), washed with 3 CV of 50 mM pH 5 sodium acetate buffer, then eluted with a 25 CV gradient from 0 to 0.5 M NaCl in the same buffer. Fractions from the largest UV-active peak were pooled, concentrated to 10–30 mg/mL using a 30 kDa MWCO centrifugal concentrator (Amicon) and purified over Superdex 200 (GE Healthcare) into 50 mM sodium acetate pH 5. Protein-containing fractions were pooled and concentrated to give a colorless 15–25 mg/mL protein solution. Approximately 5 mg of protein was then treated with 1000 U of EndoHf (New England Biolabs) overnight at rt. This was purified using a 5 mL Q sepharose HP column as above. To prepare the sample for crystallization, the eluent from Q sepharose was mixed 1:1 with saturated ammonium sulfate and purified over a 1 mL phenyl sepharose HP column with a 25 CV gradient from 2 M ammonium sulfate to 0 M ammonium sulfate in 50 mM pH 5 sodium acetate buffer. Protein-containing fractions were pooled, desalted into 20 mM sodium acetate pH 5, concentrated to 10–30 mg/mL and frozen at −80 °C.
Enzyme Visualization with ABP 4
ABP 4 was dissolved in DMSO to prepare a 10 mM stock solution which was diluted in ultrapure water. Unless otherwise noted, samples were stained with 10 μM ABP 4 at 37 °C for 30 min at pH 6.5 and proteins were separated at 200 V using either a precast 4–20% (Bio-Rad) or an 8.75% 1 mm miniprotean SDS-PAGE gel. Fluorescence was imaged using a Typhoon 5 laser scanner with the Cy5 laser and filter set. Enzyme molecular weights were estimated using a Pageruler 10–180 kDa prestained protein ladder.
Basidiomycete secretomes were buffered with 0.1 volumes of 1 M NH4OAc pH 5.5. For screening, 17.2 μL of buffered secretome was mixed with 2.8 μL of 60 μM ABP 4 and incubated for 1 h at 30 °C. The sample was then supplemented with 2 μL of 10X glycoprotein denaturing buffer (New England Biolabs), heated to 95 °C for 5 min and split in half. Each half was mixed with 10 μL of 2x PNGaseF Mastermix (2X glycobuffer 2, 2% NP-40 containing either 0 or 7.5 U/μL of PNGaseF) and incubated for 1 h at 37 °C. Samples were then diluted with 6.7 μL of 4X SDS–PAGE loading dye, heated to 95 °C for 5 min and 10 μL was separated through a 4–15% Criterion (Bio-Rad) gel.
For scaled up labeling, 20 μL of 60 μM ABP 4 was added to 100 μL of buffered secretome and incubated at 30 °C for 1 h. 500 μL of acetone was then added and the samples were incubated at −20 °C for 1 h. Precipitate was collected by centrifugation at 10000g for 5 min at 4 °C. The supernatant was discarded, and the sample was left to air-dry to minimize residual acetone. The sample was then resuspended in 20 μL of 1X SDS-PAGE loading dye and heated to 95 °C for 5 min to dissolve. The entire sample was then separated through a 4–20% gel.
In Situ Characterization of Secreted Enzymes
The pH optimum of enzyme labeling was determined by visualization with ABP 4 using the standard protocol (above) with variable buffer solutions including a series of McIlvane buffers prepared at 0.5 M strength (0.28 M citrate, 0.22 M phosphate) from pH 2–7.5 in 0.5 pH unit increments and a series of succinate–phosphate–glycine (SPG) buffers prepared at 0.5 M strength (62.5 mM succinic acid, 219 mM phosphate, 219 mM glycine) from pH 4–10 in 1 pH unit increments. Five μL of each buffer was added to 45 μL of A. niger arabinan secretome immediately prior to ABP addition.
The thermal tolerance of secreted enzymes was assayed at the inhibition optimum (50 mM pH 6.5 phosphate buffer) by incubating the A. niger arabinan secretome at temperatures ranging from RT to 95 °C for 1 h. Secretome samples were then rapidly cooled to 20 °C and enzymes were visualized with ABP 4 using the standard protocol.
Measuring Irreversible Inhibition Kinetics
The kinetics of enzyme inhibition were measured using a continuous assay26,27 at 25 °C in a 384-well plate with 4-methylumbelliferyl α-l-arabinofuranoside (4MU-Araf) as substrate. Kinetic measurements were made in technical quadruplicate. Curve fitting and statistical analysis was performed using OriginPro graphing software. Enzymes were diluted in 50 mM sodium phosphate buffer pH 7.0. Substrate was dissolved in DMSO to give a 100 mM stock which was diluted with ultrapure water. Putative inhibitors were dissolved in and diluted with ultrapure water with the exception of inhibitor 3 which was dissolved in DMSO to give a 50 mM stock, which was diluted with ultrapure water.
Enzyme specific activity was initially assessed by monitoring the hydrolysis of 50 μM 4MU-Araf in pH 7 phosphate for 10 min. Michaelis–Menten parameters for the hydrolysis of 4MU-Araf were estimated by varying the substrate concentration from 4 to 500 μM and fitting a site-saturation kinetic model (v0/[E]t = kcat[S]0/KM + [S]0) to the resulting rate vs substrate concentration data (Supplemental Table 1, Supplemental Figures 1A and 2A). Measurements were made at an excitation wavelength of 390 nm (15 nm bandwidth) to eliminate primary inner filter effects at substrate concentrations as high as 500 μM in our assay format (Supplemental Figure 3). Inhibition kinetics were measured using a substrate concentration of 100 μM and an excitation wavelength of 360 nm, an enzyme concentration of 50 ng/mL, and variable inhibitor concentrations. Each fluorescence vs time curve was fitted with an exponential decay model (F = F∞(1 – e–kappt)). The resulting apparent decay constants were plotted against inhibitor concentration and fitted with a site-saturation kinetic model with correction for competition by the substrate using the measured KM value and the initial substrate concentration (kapp = kinact[I]0/1 + ([S]0/KM) + ([I]0/KI)).
Intact MS Following Enzyme Labeling
GsGH51 or EndoH-treated AkAbfB were diluted to 0.1 mg/mL in their respective SEC elution buffers. Compounds 1, 2, or 6 were added to a final concentration of 50 μM and incubated for 30 min at rt. The treated protein samples were diluted with 4 volumes of 1% formic acid, 10% acetonitrile and 5 μL was injected over an MSPac DS-10 Desalting Cartridge flowing at 30 μL/min using a NanoAcquity HPLC (Waters). Following a 5 min wash with 20% acetonitrile, 0.1% formic acid in water, protein was eluted into a maXis UHR-Tof (Bruker) with a 10 min gradient from 20 to 55% acetonitrile. The column was washed for 2 min with 80% acetonitrile and equilibrated for 3 min with 20% acetonitrile between runs. Following protein signal integration and baseline subtraction, spectra were deconvoluted using the maximum entropy algorithm within Compass to calculate protein mass.
Enzyme Pull-down Using ABP 5
A. niger arabinan secretome was buffered with 50 mM McIlvane buffer pH 6.5 and then treated with 0.1 mM inhibitor 2 or DMSO control for 1 h at 37 °C (inhibitor 6 is also suitable for pretreatment, Supplemental Figure 4). Following this, the secretome was treated with either 20 μM ABP 5 or DMSO control for 30 min at 37 °C. Biotinylated proteins were pulled down, digested, and identified as described by Schröder et al.18 Basidiomycete secretome samples were processed without concentration or lyophilization with three modifications to the protocol: first, protein was precipitated through the addition of 4 volumes of acetone followed by incubation at −20 °C for 1 h; second, following the initial strep mag sepharose bead wash with 0.5% SDS, beads were washed with 2% SDS at 65 °C for 10 min with agitation followed by 2 M urea and then PBS; and last, peptides liberated through on-bead digest were modified with TMT0 following the manufacturer’s instructions prior to LC–MS/MS analysis using an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific). Peptides were identified by mapping onto the predicted proteomes deduced from genome sequence of A. biennis BRFM 1778, F. fomentarius BRFM 1823, L. menziesii BRFM 1781, and T. gibbosa BRFM 1770. For each genome (to be published elsewhere), CAZymes were annotated as in Lombard et al., 2014.1 All genome and proteome data are publicly available on the Mycocosm portal (mycocosm.jgi.doe.gov/mycocosm/home).
Enzyme Crystallization and Diffraction
Crystals of GsGH51 were grown essentially as described by Hövel et al.22 Optimized crystals were grown by mixing 1.2 μL of protein (10 mg/mL in 5 mM Tris-HCl pH 8.0) with 0.6 μL of well solution containing 15% PEG3350, 5% 2-propanol, 0.1 M Tris–HCl pH 7.5, 0.80 M NH4F in a sitting drop at 293 K (Supplemental Figure 5A). To generate inhibitor-bound complexes, crystal-containing droplets were supplemented with 0.1 μL of 2 mM inhibitor in water and incubated overnight prior to cryo-protection in well solution supplemented with 12.5% glycerol and flash freezing in LN2.
Initially, crystals of AkAbfB were grown essentially as described by Miyanaga et al.28 Optimized crystals grew from 0.5 μL of 10 mg/mL AkAbfB in 50 mM pH 5 sodium acetate mixed with 0.5 μL of 100 mM Tris–HCl pH 8.0, 200 mM MgCl2, 400 mM NaCl, 20% PEG6000, 2.5% DMF at 279 K. However, preferential formation of poor-quality needle clusters and poor diffraction of these crystals led us to explore other crystallization conditions. EndoH-deglycosylated AkAbfB or AkAbfB (E221Q) (12 mg/mL in 50 mM sodium acetate pH 5.0) formed slow-growing isolated crystals when mixed 2:1 with 0.2 M lithium sulfate, 0.1 M sodium acetate pH 4.5, 50% PEG400 (Supplemental Figure 5B). Supplementation with 0.2–0.5 M NaCl resulted in more rapid crystal growth. To generate inhibitor-bound complexes, crystals were transferred to mother liquor supplemented with inhibitor 6 or 2 to a final concentration of 0.2 mM, or saturated with PNP-Araf (for AkAbfB (E221Q)). Crystals were soaked for 1 h at RT prior to freezing.
Diffraction data were collected at Diamond Light Source (Harwell, UK) on beamline I04 and automatically processed using the fast_dp29 (GsGH51), autoPROC30 (AkAbfB-2 and AkAbfB-6), or Xia231 (AkAbfB-PNP-Araf) pipelines. Computation was carried out using programs from the CCP4 suite32 unless otherwise stated. All crystal structure figures were generated using Pymol (Schrodinger). Data collection and processing statistics for all structures are given in Supplemental Table 2.
Structure Solution and Refinement
Data for GsGH51 bound to inhibitors 2 and 6 were collected to 1.40 Å. Each structure was solved by molecular replacement using Phaser33 with the known structure (PDBID: 1pz3) as the search model. The resulting solution showed clear density for the bound ligand within the enzyme active site. Ligand coordinates and dictionaries were generated using jLigand34 and built into the model using Coot,35 followed by alternating rounds of manual model building and refinement using Coot and REFMAC5.36
Data for AkAbfB bound to inhibitors 2 and 6 were collected to 1.47 and 1.86 Å, respectively. Each structure was solved by molecular replacement using Phaser with the known structure (PDBID: 1wd3) as the search model. The resulting solution showed clear density for the bound ligand within the enzyme active site. The structures were refined, as above, and the same ligand coordinates and geometries were used.
Data for AkAbfB (E221Q) bound to PNP-Araf were collected to 1.64 Å. The structure was solved by molecular replacement using Phaser with the AkAbfB-2 complex as the search model. The resulting structure showed clear density for two PNP-Araf (ligand ID: KHP) molecules bound to the carbohydrate-binding module. Following several rounds of manual model building and refinement, partial density for an additional PNP-Araf molecule, which was modeled at 60% occupancy, became apparent in the active site.
Conformational Analysis
Conformational free energy landscapes (FELs) were computed for α-l-arabinofuranose and compounds 1, 2, and 6 using Density Functional Theory-based molecular dynamics (MD), according to the Car–Parrinello (CP) method.37 Each molecule was enclosed in an isolated cubic box of 12.5 Å × 12.5 Å × 12.5 Å. A fictitious electron mass of 500 atomic units (a.u.) was used for the CP Lagrangian and a time step of 0.12 fs was used in all CPMD simulations to ensure that the adiabacity of the fictitious kinetic energy of the electrons was smaller than 10–5 a.u./atom. The Kohn–Sham orbitals were expanded in a plane wave basis set with a kinetic energy cutoff of 70 Ry. Ab initio pseudopotentials, generated within the Troullier-Martins scheme, were employed.38 The Perdew, Burke, and Ernzerhoff generalized gradient-corrected approximation39 was selected in view of its good performance40 in previous work on isolated sugars,41 glycosidases, and glycosyltransferases.42 The metadynamics algorithm,43 provided by the Plumed 2 plugin,44 was used to explore the conformational free energy landscape of the systems, taking as collective variables the pseudorotational phase (φ) puckering coordinate,45,46 as well as a dihedral angle accounting for the rotation of the sugar hydroxymethyl group. The energy was projected into the φ coordinate for representation purposes. Initially, the height of these Gaussian terms was set at 0.6 kcal/mol and a new Gaussian-like potential was added every 500 MD steps. Once the whole free energy space was explored, the height of the Gaussian terms was reduced to 0.2 kcal/mol to facilitate convergence of the FEL. The width of the collective variables was set according to their oscillations in the free dynamics which corresponded to 0.035 and 0.1 rad for φ and the hydroxymethyl dihedral angle, respectively. The simulations were stopped when energy differences among wells remain constant, which was further confirmed by a time-independent free energy estimator.47 The exploration of the phase space was extended up to 380, 360, 324, and 474 ps for α-l-arabinofuranose, compound 1, compound 2, and compound 6, respectively. The errors in the principal minima, taken as a standard deviation (SD) from the last 200 ps, are below 0.6 kcal mol–1. Conformational FELs computed using only φ as CV gave very similar results.
The Michaelis complexes of compounds 1, 2, and 6 were modeled using the crystal structures of the adducts obtained for GsGH51 and AkAbfB as a reference. In the case of compounds 1 and 2, the Michaelis complex was reconstructed by removing the covalent bond between the inhibitor and the nucleophile in the protein structure bound to inhibitor 2. The amine group was reverted to an aziridine (compound 2), which was replaced with an oxygen atom to give compound 1.
Molecular dynamics (MD) simulations were set up employing the program LEaP included in the Amber suite48 and the ff14SB protein force field.49 The compounds were parametrized using gaff2.50 The systems were solvated with explicit TIP3P water molecules.51 They were neutralized with 31 and 21 sodium atoms for all neutral compounds in GsGH51 and AkAbfB, respectively. The systems with protonated compound 2 were neutralized with one fewer sodium atom (30 and 20 in GsGH51 and AkAbfB, respectively). MD simulations were performed using Amber16.48 A thermal equilibration to 300 K was done prior to the equilibration of dynamics in the NPT ensemble with a production phase of 51 ns for each system. The SHAKE algorithm, with an integration time step of 2 fs, was used. The binding free energy of the compounds were obtained by using the MMPBSA method52 integrated in the Amber suite.
Results and Discussion
Free Energy Landscape of α-l-Arabinofuranose, And the Conformational Itinerary of Family GH51 and GH54 Retaining α-l-Arabinofuranosidases
To gain insight into the ability of our potential inhibitors to mimic the natural conformational preferences of α-l-arabinofuranosides, we computed the relative energy of all ring conformations of compounds 1, 2, and 6. α-l-Arabinofuranose was also analyzed for comparison. The conformational free energy landscape (FEL) of each molecule was calculated using ab initio metadynamics and the Cremer–Pople puckering coordinates. This approach has recently been successful in predicting the performance of pyranose-like inhibitors.14,18
In contrast to GHs which act on pyranosides (e.g., α/β-glucosidases53 and α/β-mannosidases54), little is known about the catalytic conformational itineraries of α-l-arabinofuranosidases. The computed FEL of α-l-arabinofuranose (Figure 1B) shows that all conformations lie in an energy window of ∼5 kcal/mol. This window is significantly narrower than what is typical for pyranose compounds (∼15 kcal/mol)41,55 and shows that most α-l-arabinofuranose conformations are thermally accessible. The most stable conformation is 1T2. However, this conformation is not catalytically competent since the axial 2-OH group creates steric hindrance with the nucleophile residue located on the “beta” face of the sugar. Conformations between 2E and 4E, being only ∼2 kcal/mol higher in energy, feature an equatorial 2-OH, eliminating this steric hindrance. Thus, the ideal Michaelis complex conformation for an α-l-arabinofuranosidase should be between 2E and 4E (shaded region in Figure 1B).
Figure 1.
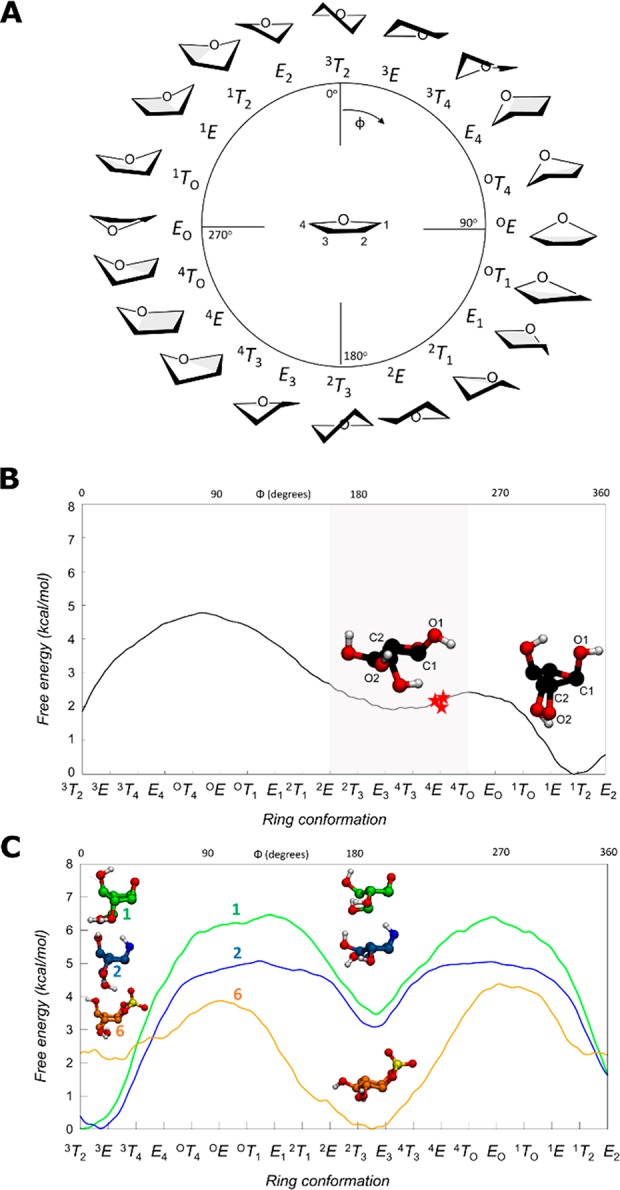
(A) Graphical representation of the conformations of a 5-membered ring according to the Cremer–Pople angle ϕ. (B) Conformational FEL of isolated α-l-arabinofuranose. Conformations observed in Michaelis complexes of α-l-arabinofuranosidases are represented with a red star (PDB 2VRQ and 1QW9 for GH51 and PDB 6SXR, this work, for GH54). The conformational region having an equatorial O2 is shaded. (C) Conformational FEL of α-l-arabinofuranose-configured cyclophellitol (1), aziridine (2), and cyclic sulfate (6).
To determine where on this landscape the observed conformations of enzyme-bound species lie, we surveyed all of the conformations of l-arabinofuranose observed within the active sites of crystallized GH51 and GH54 enzymes. Specific α-l-arabinofuranosidases have been identified within GH families 43, 51, 54, and 62, of which only families 51 and 54 follow the anomeric stereochemistry-retaining Koshland double-displacement mechanism.
The most detailed studies of α-l-arabinofuranosidase mechanisms have been performed using bacterial GH51 enzymes. Paes et al. obtained the structure of an intact branched pentasaccharide substrate bound to the active site of TxAbf, a thermostable GH51 from Thermobacillus xylanilyticus (PDB ID 2VRQ).56 Hövel et al. reported the crystal structure of Geobacillus stearothermophilus AbfA (hereafter referred to as GsGH51) bound to 4-nitrophenyl α-l-arabinofuranoside (PNP-Araf) (PDB ID 1QW9).57 In both of these Michaelis complexes, the α-l-arabinofuranose rings were found in the 4E conformation (Figure 2A). Therefore, similar to observations with GHs acting on pyranose sugars,53 furanosidases distort the −1 sugar to a conformation that is preactivated for catalysis. Thus, the conformational catalytic itinerary for the rate limiting step of the reaction for GH51 family is expected to go through an oxocarbenium ion-like E3 conformer to fulfill the requirement of having C4–O5–C1–C2 planarity.58
Figure 2.

(A) Koshland double-displacement mechanism employed by retaining α-l-arabinofuranosidases, as proposed for GH51 and GH54, showing the conformational reaction itinerary including the (left-to-right) Michaelis complex, transition state 1, covalent substrate-enzyme intermediate, transition state 2, and the hydrolyzed product. (B) Chemical structures of putative α-l-arabinofuranosidase inhibitors 1, 2, 3, and 6 and ABPs 4 and 5.
Beyond the bacterial GH51 enzymes, there is only one retaining α-l-arabinofuranosidases which has been crystallized. The structure of Aspergillus kawachii AbfB (a member of GH54 hereafter referred to as AkAbfB) with arabinose in the active site (PDB ID 1WD4), displays a product complex ring conformation of 4E.59 Unfortunately, no Michaelis complex of this enzyme had been reported to date.
Determination of the Michaelis Complex of AkAbfB
To complete, and thus compare the conformational itineraries of the GH51 and GH54 families, we studied AkAbfB as a model GH54 active site. To observe the Michaelis complex, we soaked crystals of deglycosylated AkAbfB E221Q in a saturated solution of PNP-Araf in mother liquor. The resulting 1.64 Å crystal structure contained 3 PNP-Araf molecules: two full occupancy molecules bound to the carbohydrate binding module and a partial occupancy molecule bound in the active site (Supplemental Figure 6A).
Overall, the Michaelis complex displayed similarity to the product complex published by Miyanaga et al. in 200459 (Supplemental Figure 6B). O2 formed hydrogen bonds with the carbonyl oxygen of Q221 and the backbone amide of D297. O3 formed hydrogen bonds with the backbone amide of G296 and the carboxylate of D219. The ring oxygen formed a hydrogen bond with the backbone amide of N222 and O5 formed hydrogen bonds with the carboxylate of D219 and the backbone amide of N223. The furanose ring was found in a 4E conformation, stacked against a hydrophobic surface formed by W206 and the C176–C177 disulfide linkage. The axial nitrophenyl leaving group pointed out of the active site into a solvent channel. The electrophilic carbon (C1) was positioned 3 Å away from the amide nitrogen, primed for migration away from the nitrophenyl leaving group with support from anti protonation of the glycosidic oxygen by D297, the general acid/base.
Based on this result, and the general observation of one itinerary per family (at least for members active on similar substrates),54 we infer that enzymes within GH54 and GH51 share a common catalytic conformational itinerary (Figure 2A). Following binding in a reactive 4E conformation, the glycone is predicted to pass through an E3 transition state conformation to give a 2E glycosyl-enzyme intermediate. Following exchange of the leaving group with water, the glycone then passes through a second E3 transition state to form a lower energy product-bound complex observed in the low energy E3 – 4T3 region. Therefore, the predicted conformational itinerary for the two half-reactions is 4E → [E3]‡ → 2E (glycosylaton) and 2E → [E3]‡ → E3/4T3 (deglycosylation), as shown in Figure 2.
Conformational Analysis of Potential α-l-Arabinofuranosidase Inhibitors
Having ascertained the FEL for α-l-arabinofuranose and the conformational itinerary of retaining α-l-arabinofuranosidases, we next considered the design and synthesis of covalent inhibitors. As discussed above, both GH51 and GH54 enzymes form Michaelis complexes in the 4E conformation (red stars in Figure 1B). Therefore, a suitable covalent α-l-arabinofuranosidase inhibitor should readily adopt a 4E conformation in which the atom that mimics the anomeric carbon is similarly accessible for nucleophilic attack from the beta face of the sugar ring. Computed FELs for compounds 1, 2, and 6 (Figure 1C) show that conformations around 4E are energetically favored for 6, whereas 1 and 2 instead prefer conformations in which the 2-OH is axial (in the 1T2 – E2 – 3T2 region). Thus, cyclic sulfate 6 was anticipated to be a potentially more potent inhibitor than the epoxide (1) or aziridine (2) for both GH51 and GH54 α-l-arabinofuranosidases.
Synthesis of α-l-Arabinofuranose-Configured Inhibitors and ABPs
To synthesize α-l-arabinofuranosidase inhibitors, we took inspiration from the synthesis of six-membered cyclophellitol derivatives beginning from appropriately functionalized cyclohexene starting materials. α-l-Arabinofuranose-configured cyclopentene was prepared in nine steps from commercial methyl α-d-galactopyranoside in 15% yield. The initial installation of a p-methoxybenzylidene acetal (PMP) at C4 and C6 of methyl α-d-galactopyranoside (carbohydrate numbering) by treatment with anisaldehyde dimethylacetal followed by benzylation at C2 and C3 afforded intermediate 7 in 74% yield over 2 steps (Scheme 1). Selective opening of the PMP-group in compound 7 with Bu2BOTf and BH3·THF, followed by nucleophilic substitution of the primary alcohol with iodine and Vasella fragmentation with activated zinc powder afforded intermediate 10 in 60% yield over three steps. We were able to scale this process up to 56 mmol with moderate yields. Wittig olefination of aldehyde 10 and subsequent ring-closing metathesis (RCM) with second-generation Grubb’s catalyst afforded 12. The PMB group was then selectively removed with DDQ and intermediate 14 was obtained in 80% yield over two steps by subsequent alkylation with freshly synthesized Bu3SnMeI. The key step, a Wittig–Still rearrangement of intermediate 14 with n-BuLi at −78 °C, afforded the desired cyclopentene 15 in 68% yield.
Scheme 1. Synthesis of l-Arabinofuranose-Configured Cyclopentene 15.

Reagents and conditions: (a) (1S)-(+)-10-camphorsulfonic acid, CH3CN, 50 °C, 300 mbar, 2.5 h; (b) BnBr, NaH, TBAI, DMF, 0 °C, rt, 18 h, 74% over two steps; (c) BH3·THF, Bu2BOTf, DMF, 0 °C, 15 min, 90%; (d) I2, TPP, THF, reflux, 3 h, 79%; (e) activated Zn powder, THF, 35 °C, 2 h, 84%; (f) Ph3PCH3Br, n-BuLi, THF, −78 to −20 °C for 1 h, then rt, 18 h, 73%; (g) Grubb’s II cat., DCM, reflux, 18 h, 90%; (h) DDQ, DCM, 0 °C, rt, 2 h, 86%; (i) Bu3SnMeI, KH, dibenzo-18-crown-6, THF, 0 °C, rt, 18 h, 91%; (j) n-BuLi, THF, −78 °C to rt, 18 h, 68%.
The first step toward the designed α-l-epoxide and α-l-aziridine compounds was stereoselective epoxidation of cyclopentene 15 (Scheme 2). We rationalized that treatment of cyclopentene 15 with m-CPBA would lead to predominant β-l-epoxidation where the neighboring primary alcohol would play a directing role by hydrogen bonding with m-CPBA. Indeed, m-CPBA epoxidation at 50 °C overnight resulted in a separable 3.4:1 mixture of β-l- and α-l-epoxides in 62% yield. Cooling the mixture to 4 °C slowed the reaction, and after 4 days, we observed a β-l to α-l ratio of 4.3:1, with a higher reaction yield (91%). To synthesize the α-l-epoxide selectively, cyclopentene 15 was benzylated and subjected to epoxidation with m-CPBA. Although the β-l to α-l ratio was improved to 1:2, it resulted in a chromatographically inseparable mixture. Thus, α-l-arabinofuranose-configured epoxide 1 was obtained by hydrogenation of partially benzylated 17 with Pearson’s catalyst.
Scheme 2. Synthesis of Epoxides.

Reagents and conditions: (a) m-CPBA, DCM, 50 °C, 18 h, 62%, 3.4:1 of 16:17; (b) m-CPBA, DCM, 0 °C, 4 days, 91%, 4.3:1 of 16/17; (c) m-CPBA, DCM, 50 °C, 18 h, 62%, 1:2 of 19/20; (d) H2, Pd(OH)2, MeOH, 18 h, 50%.
Taking advantage of the C2 and C4 stereochemistry of 18, direct aziridination aided by steric hindrance of the vicinal protecting groups was attempted first. No aziridination was observed with 3-amino-2-(trifluoromethyl)quinazolin-4(3H)-one (Q-CF3) as nitrogen donor and phenyliodine(III) diacetate (PIDA) to form the reactive acetylated quinazolinone.60O-(2,4-Dinitrophenyl)hydroxylamine (DPH) and a ruthenium catalyst also gave no aziridination.61 Hypothesizing that the alkene is not accessible enough due to the conformation of cyclopentene and/or steric hindrance of the benzyl groups, we pursued aziridine 2 by benzylation of the primary hydroxyl of epoxide 16 and subsequent SN1 ring opening with sodium azide. This afforded two separable regioisomers in 1:2 (21:22) ratio with 77% yield (Scheme 3). Hydroxyls of 21 and 22 were first tosylated and subsequently treated with triphenylphosphine (TPP) and diisopropylethylamine (DIPEA) at 60 °C to obtain benzylated aziridine 23 in 28% yield over two steps. Aziridine 2 was obtained after deprotection under Birch conditions (sodium and tert-butanol) with an overall yield of 11% from epoxide 16. To synthesize ABPs, aziridine 23 was alkylated with 8-azidooctyl triflate. Following Birch deprotection, amino-octylaziridine 3 was obtained in 54% yield over two steps. Aziridine 3 was then coupled with either Cy5-OSu or biotin-OSu esters in the presence of DIPEA to afford ABPs 4 and 5 following reverse-phase HPLC-MS purification.
Scheme 3. Synthesis of α-l-Aziridines 2–5.

Reagents and conditions: (a) BnBr, NaH, TBAI, DMF, rt, 18 h, 78%; (b) NaN3, LiClO4, DMF, 100 °C, 18 h, 77%; (c) TsCl, DMAP, TEA, DCM, 0 °C, 18 h, 50%; (d) TPP, DIPEA, THF/H2O, reflux, 1.5 h, 56%; (e) Li, NH3, −60 °C, 1 h, 66%; (f) 8-azidooctyl triflate, DIPEA, DCM, 0 °C to rt, 18 h, 57%; (g) Na, NH3, t-BuOH, −60 °C, 1 h, 95%; (h) Cy5-Osu or biotin-OSu, DIPEA, DMF, 18 h, 4: 56% and 5: 19%.
The synthesis of irreversible α-l-arabinofuranose configured cyclic sulfate 6 (Scheme 4) started with the oxidation of 18 with a mixture of NaIO4 and RuCl3·3H2O affording exclusively cis-α-l-diol 25 in 48% yield. Diol 25 was then treated with thionyl chloride and trimethylamine, and the sulfite mixture was then further oxidized with NaIO4 and RuCl3•3H2O to give cyclic sulfate 26. This was deprotected using Pearson’s catalyst to afford final cyclic sulfate 6 in 24% yield from 18.
Scheme 4. Synthesis of Cyclic Sulfate 6.

Reagents and conditions: (a) NaIO4, RuCl3·3H2O, EtOAc/CH3CN/H2O, 0°C, 3 h, 48%; (b) (i) SOCl2, Et3N, DCM, 0 °C, 30 min, (ii) NaIO4, RuCl3·3H2O, EtOAc/CH3CN/H2O, 0 °C, 3 h, 51%; (c) H2, Pd(OH)2, MeOH, 18 h, 24%.
Inhibition of Recombinant α-l-Arabinofuranosidases
With 1, 2, and 6 in hand, we first assessed the potency of these putative inhibitors against their intended targets. To test the effectiveness of each inhibitor, inhibition kinetics were measured with a collection of recombinantly produced retaining α-l-arabinofuranosidases including G. stearothermophilus GH51 (GsGH51, a bacterial enzyme from GH51), A. niger AbfA (AnAbfA, a fungal enzyme from GH51), and A. kawachii AbfB (AkAbfB, a fungal enzyme from GH54). Initial overnight incubations of each enzyme with compounds 2 and 6 resulted in the complete loss of activity, while no loss of activity was observed with compound 1. Intact MS of GsGH51 and AkAbfB treated with each compound confirmed complete 1:1 labeling with compounds 2 and 6, and no labeling with compound 1 (Supplemental Figures 7 and 8).
As predicted by our conformational analysis, compound 6 is a potent inhibitor of retaining α-l-arabinofuranosidases. Inhibitor 6 reacted rapidly with the catalytic nucleophile of both AkAbfB and AnAbfA with a kinact well above 1 min–1 (estimated from the limited speed of our assay). However, the lack of any apparent nonlinearity in the kapp vs [I] curve for either AkAbfB or AnAbfA suggested poor initial binding (Supplemental Figures 1 and 2). In spite of this, inhibitor 6 has a performance constant of 170 M–1 s–1 with AnAbfA and 250 M–1 s–1 with AkAbfB (Table 1), comparable to the inhibition of TmGH1 with cyclophellitol reported by Gloster et al.11 (290 M–1 s–1).
Table 1. Kinetic Parameters for Covalent Inhibition of AnAbfA and AkAbfB by α-l-Arabinofuranosidase Compounds 1, 2, 3, and 6a.
| compd | KI (μM) | kinact (min–1) | kinact/KI (s–1 M–1) |
|---|---|---|---|
| AnAbfA (GH51) | |||
| 1 | nd | nd | <0.1 |
| 2 | 140 ± 20 | 0.33 ± 0.02 | 39 |
| 3 | 210 ± 30 | 0.09 ± 0.01 | 7.1 |
| 6 | nd | nd | 160 ± 20 |
| AkAbfB (GH54) | |||
| 1 | nd | nd | <0.1 |
| 2 | 320 ± 50 | 0.54 ± 0.07 | 28 |
| 3 | 320 ± 40 | 0.12 ± 0.01 | 6.2 |
| 6 | nd | nd | 240 ± 30 |
For reactions with compound 6, it was not possible to obtain distinct kinact and KI parameters; only the combined kinact/KI parameter determined from the slope of the kapp vs [I] curve is shown for these cases. nd: not determined.
Contrary to our prediction, compound 2 also proved to be a potent inhibitor of both AkAbfB and AnAbfA, having performance constants only 8-fold and 4-fold lower than inhibitor 6 with AkAbfB and AnAbfA, respectively (Table 1). In contrast to inhibitor 6, inhibition kinetics with inhibitor 2 provided evidence of stronger initial binding in both enzyme active sites, having KI values of 0.1–0.3 mM. The addition of an alkyl chain to generate inhibitor 3 did not hinder initial binding with either AnAbfA or AkAbfB and caused only a 4-fold reduction in kinact, demonstrating that alkylation of the aziridine is a well-tolerated method for generating α-l-arabinofuranosidase ABPs.
The lack of measurable inhibition kinetics for compound 1 allowed us to establish a maximum value for the putative inhibitor’s performance constant (kinact/KI) of approximately 0.1 M–1 s–1 based on the length and sensitivity of the assay and the maximum inhibitor concentration tested (Supplemental Figures 1 and 2). Similarly, no reversible inhibition was observed at concentrations as high as 0.25 mM. Together, these results confirmed that, as predicted from our conformational analysis, compound 1 was not an inhibitor of retaining α-l-arabinofuranosidases at concentrations up to 0.25 mM.
Structural Analysis of Inhibitors 2 and 6 Bound to α-l-Arabinofuranosidases
To determine whether inhibitors 2 and 6 both interact with α-l-arabinofuranosidases as effective α-l-arabinofuranose mimics, we sought to understand how the inhibitors bind to the enzyme active site. Soaking GsGH51 with inhibitors 2 and 6 overnight at room temperature resulted in the formation of full occupancy covalent complex between E294, the known catalytic nucleophile, and each inhibitor (Figure 3A,B). These structures are similar to the complexes reported by Hövel et al.57 They reported a covalent substrate-enzyme intermediate trapped in a 2E conformation (PDB ID 1PZ2); however, poor electron density at the anomeric center of the Hövel structure limits the confidence with which this ligand conformation can be interpreted. Nevertheless, it is clear that the structure of the active site varies between the Hövel complexes and the complexes with inhibitors 2 and 6 that we obtained (Supplemental Figure 9).
Figure 3.

Crystal structures of complexes between inhibitors 2 (B, D, green) and 6 (A, C, purple), and GsGH51 (A, B, blue) and AkAbfB (C, D, yellow). 2F0 – Fc electron density is shown for both the ligand and the catalytic nucleophile as a gray mesh contoured at 2σ. The polypeptide is shown in cartoon form with active site residues shown as sticks. Apparent hydrogen bonding interactions are shown as dotted yellow lines.
Complexes with both inhibitors 2 and 6 are characterized by the positioning of the E294 side chain away from R69, toward the unliganded (PDBID: 1PZ3) position of Y246, which, in place of a hydrogen bond to the ring oxygen of α-l-arabinofuranose, forms a hydrogen bond with O5 of the inhibitor (carbohydrate numbering). While the resulting displacement of Y246 has only a minimal impact on the protein structure when bound to inhibitor 6, binding to inhibitor 2 results in the dramatic displacement of Y246, and consequently W298 and N302, creating sufficient space for the side chains of I356 and L318 to pack into a different position and for a glycerol molecule to bind. The presence of the bulky charged sulfate group following reaction with inhibitor 6 appears to repel E175, the general acid/base residue, displacing H244 and, through steric interactions, the S215-R218 loop. To investigate whether E175 is interfering with the binding of inhibitor 6, we simulated the Michaelis complex of inhibitor 6 with the E175A and E175G mutants, and calculated the binding energy of 6. Binding energy was less favorable with either mutant (+4 kcal/mol for E175A and +8 kcal/mol for E175G). We interpret this as indicating that the displacement of E175 likely occurs following the reaction of the cyclic sulfate with the covalent nucleophile. We speculate that the observed active site rearrangement occurs following the addition reaction and is not relevant to initial inhibitor binding. Overall, while inhibitors 2 and 6 both bind in a manner mimicking the cognate substrate of GsGH51, their labeling of the catalytic nucleophile appears to induce significant rearrangement of the active site structure.
Interestingly, following reaction with the catalytic nucleophile, the conformation of inhibitor 2, but not inhibitor 6, appears to represent the glycone conformation expected of the glycosyl enzyme intermediate. Reacted inhibitor 2 was found in the 2E conformation, forming hydrogen bonds from O2 to N174, from O3 to N74 and E29, and from O5 to Q351 and Y246. Inhibitor 6 formed the same complement of ligand-protein interactions but sat in the active site in an unusual E1 conformation. We attribute this to a combination of electrostatic repulsion and steric bulk pushing the sulfate group out of the active site, promoting an extended conformation for the bonds connecting Oε1 of E294 to the sulfate group.
To generate covalent GH54 complexes, we soaked crystals of AkAbfB with 0.2 mM of inhibitor 2 or 6 for 1 h. Both inhibitors bound to E221 in almost identical positions and conformations (Figure 3C,D), forming hydrogen bonds from O2 to G296 and the sulfur of M195, from O3 to N297 and D219, and from O5 to D219 and N223. The interaction between the ring oxygen and N222 found in the product complex (PDBID: 1WD4) cannot be formed, but the axial amine presents an additional hydrogen bond with D297, the general acid/base. In contrast to the complex with GsGH51, the interactions between both inhibitors 2 and 6 and the active site of AkAbfB cause no significant change in the protein structure. The active site appears to be sufficiently open to accommodate the sulfate of inhibitor 6 without any steric clashes. Thus, we believe that these complexes are good representations of the glycosyl-enzyme intermediate structure. The ring in each covalent complex is found in the 2E conformation. This consensus conformation represents a 1.2 Å migration of C1 from its position in the AkAbfB Michaelis complex toward E221 coupled with a ∼15° axial rotation of the ring around C3 (Supplemental Figure 10).
Probing the A. niger Arabinan Secretomes with ABP 4
Building on the success of inhibitors 2 and 3 as covalent inhibitors of both GH51 and GH54 α-l-arabinofuranosidases, we set out to detect and identify α-l-arabinofuranosidases within complex fungal secretomes. As a validation of this approach, we chose to work with the well-studied secretome of A. niger grown on arabinan.
Multiple α-l-arabinofuranosidases have been purified from the A. niger secretome and characterized.62−64 These include AnAbfA, the fungal GH51 that we produced recombinantly, and AnAbfB, a GH54 enzyme 98% identical to AkAbfB. Thus, we hypothesized that our ABPs could be used to identify the α-l-arabinofuranosidases that are produced by A. niger in response to a specific carbon source.
The treatment of the A. niger arabinan secretome with inhibitor 2 resulted in the complete loss of activity against 4MU-Araf, suggesting that all of the α-l-arabinofuranosidase activity in our secretome sample could be attributed to retaining glycosidases.
Visualization of α-l-arabinofuranosidases using ABP 4 revealed two distinct bands, one running at ∼105 kDa and the other running at ∼65 kDa (Figure 4A). Deglycosylation with PNGaseF under denaturing conditions resulted in a shift of the 105 kDa band down to ∼70 kDa and a shift of the ∼65 kDa band down to ∼60 kDa. Based on similar results obtained with recombinant AnAbfA and AkAbfB, we hypothesized that the top band was one of the A. niger GH51 enzymes and the bottom band was AnAbfB, the only A. niger GH54 enzyme.
Figure 4.

Activity-based protein profiling of fungal secretomes with ABPs 4 and 5. (A) Fluorescence imaging of the secretome isolated from A. niger grown on arabinan, stained with ABP 4, and treated with (PNG+) or without (PNG-) PNGaseF under denaturing conditions prior to separation on an 8.75% SDS-PAGE gel. L indicates the ladder lanes. (B) Label-free quantification of the top eight proteins pulled down from the A. niger arabinan secretome. For each protein (identified by NRRL3 number and common name), integrated peptide intensity is plotted for nonconflicting peptides from the pull-down with ABP 5 (PD, black), from the total secretome (TS, blue), and from the pull-down with ABP 5 following pretreatment with inhibitor 2 (PT, red). Error bars represent the standard deviations of three measurements. (C) Cy5 fluorescence (red) and Coomassie staining (green) of basidiomycete secretomes following staining with ABP 4 and acetone precipitation. L indicates the ladder lane. The BRFM number for the strain from which the secretome was isolated is given above each lane. (D) Plot of total spectral counts in the pull-down sample vs the ratio of spectral counts in the pull-down sample to spectral counts in the total secretome for all of the proteins for which at least 2 peptides were observed with an FDR of 1% in the pull-downs from L. menziesii (BRFM 1557), F. fomentarius (BRFM 1323), T. gibbosa (BRFM 952), and A. biennis (BRFM 1215). Points corresponding to GH51 enzymes are shown in orange, points corresponding to other putative retaining GH enzyme with peptide molecular weights >90 kDa are shown in red. The labels shown include the species abbreviation and the Mycocosm amino acid sequence number.
Investigations of the effects of pH on labeling efficiency revealed that AnAbfB reacted with our probe efficiently over a pH range (2–9), which extended further into the acidic range than the GH51 enzyme (5–8) (Supplemental Figure 11). Notably, both enzymes were labeled optimally at pH 6.5–7, which is significantly above pH 4, at which the enzymes are optimally active. While similar discrepancies between optimal hydrolytic and inhibition pH have been reported previously,15,18 the difference of 2.5–3 pH units that we observed is unusually large, suggesting that the optimal protonation states of active site residues for inhibition by compound 2 and glycoside hydrolysis are different.
This is supported by the trends observed in the binding energies calculated for the modeled Michaelis complexes of inhibitor 1, 2, and 6 in the GH51 and GH54 active sites (Supplemental Figures 12 and 13). The binding energy of inhibitor 2 was calculated in 3 different situations in each active site: deprotonated inhibitor 2 with protonated acid/base residue, protonated inhibitor 2 with protonated acid/base residue, and protonated inhibitor 2 with deprotonated acid/base residue. Protonated compound 2 (with the optimal acid/base residue protonation) binds better than all the other compounds in both enzymes (Supplemental Figure 14); thus, not requiring the donation of a proton from the general acid/base residue for the reaction to take place. Also, the importance of the protonation state of the acid/base residue seems to be different in both enzymes. GH54 with an extended acidic range of labeling efficiency seems to have similar binding energies with either the protonated or deprotonated general acid/base; whereas in GH51, 2 binds much better when it is deprotonated, explaining the more restricted pH range of labeling efficiency.
To investigate the thermal stability of arabinofuranosidases within the A. niger arabinan secretome, we preincubated the secretome at various temperatures for 1 h prior to visualization with ABP 4. This revealed enzyme recovery from surprisingly high temperature treatments (Supplemental Figure 15A,C). Both enzymes were stable up to 60 °C. Increasing the temperature to 65 °C resulting in a complete loss of GH54 staining and raising it to 67 °C resulted in a complete loss of both GH51 and GH54 labeling, suggesting complete denaturation. However, increasing the preincubation temperature beyond 67 °C resulted in a partial recovery of GH54 staining. Preincubation at 86.5 °C resulted in a ∼50% recovery of fluorescence intensity relative to the RT control (estimated by band integration using ImageQuant software (GE)). To determine the role of disulfide bonding in the stability AbfA and AbfB and the apparent refolding of AbfB, we repeated the experiment with 5 mM DTT (Supplemental Figure 15B,C). The addition of DTT had minimal impact on the apparent stability of AbfA, yet significantly reduced the apparent stability of AbfB, causing a near complete loss of staining at 56 °C. This suggests that the four disulfide bonds found in the structure of AkAbfB (conserved in AnAbfB) are critical for enzyme stability, but that disulfide bonds are not important for AbfA stability. Enzyme recovery from elevated temperatures was reduced, but still occurred in spite of the reduction of disulfide bonds.
To determine whether the recovery of AbfB staining was genuinely related to the recovery of active enzyme, we measured hydrolytic activity of the DTT treated secretome samples toward 4MU-Araf (Supplemental Figure 15D). This confirmed that the loss of AbfB staining at 56 °C correlated with an ∼80% reduction in activity and that the subsequent loss of AbfA staining at 67 °C corresponded with a complete loss of activity. At higher temperatures we observed a small recovery of activity which correlated with the recovery of AbfB staining. Thus, visualization with ABP 4 facilitates the identification of thermally resilient enzymes within the context of their native fungal secretome.
Identification of A. niger α-l-Arabinofuranosidases by Pull-down with ABP 5
Based on molecular weight and glycosylation state, we hypothesized that the enzymes stained by ABP 4 were a GH51 and a GH54. However, it was not clear which of the GH51 enzymes produced by A. niger was expressed. A previous report has identified AbfA, AbfB, and AbfC as the major α-l-arabinofuranosidases produced by A. niger in response to growth on arabinan-rich sugar beet pectin.62 The genome of A. niger encodes two other GH51 genes: abfD, which is not expressed during growth on arabinan, and the more recently identified abfE, for which expression has not been investigated in response to arabinan.
On-bead digestion of proteins pulled down following treatment of the secretome with ABP 5 yielded peptides from AbfB (GH54, GenBank: CAK42333), AbfA (GH51, GenBank: CAK43424), and AbfE (GH51, GenBank: ACE00420) as well as a small collection of other proteins not known to be α-l-arabinofuranosidases (Figure 4B). We did not observe AbfC or AbfD in our analysis of the pull-down total secretome, indicating that these were not produced in our culture. Preincubation of the secretome with inhibitor 2 followed by treatment with ABP 5 and pull-down significantly reduced signal for peptides from AbfA, AbfB, and AbfE without causing a significant reduction in signal for any other detected proteins. Although we cannot exclude that ABP 5 has specific targets beyond arabinofuranosidases that are incapacitated by inhibitor 2, these results reveal the utility of ABP 5 in activity-based protein profiling to identify and annotate retaining arabinofuranosidases from secretomes derived from microorganisms grown on arabinofuranose-containing biopolymers.
Screening Basidiomycetes for α-l-Arabinofuranosidase Production
Following the success of the detection and identification of A. niger α-l-arabinofuranosidases, we applied ABPs 4 and 5 to the detection and identification of α-l-arabinofuranosidases secreted by basidiomycetes grown on complex biomass. We selected a sampling of eight basidiomycetes, all known to be proficient biomass-degrading fungi (Supplemental Table 3). The genomes of these fungi encode no apparent GH54 enzymes and either one (A. biennis and T. gibbosa) or two apparent GH51 enzymes. To identify the GH51(s) produced during growth on complex biomass, these fungi were cultured on maltose, aspen pulp, or wheat straw for 10 days prior to secretome collection.
α-l-Arabinofuranosidases were visualized by treatment of secretome samples with ABP 4 at pH 5.5, 30 °C for 1 h followed by denaturation, deglycosylation, and separation on SDS–PAGE. Glycoproteins migrating at 70–80 kDa were observed in secretomes collected from T. gibbosa (the top biomass digestion-enhancing strain identified in a sampling of French biomass-degrading fungi65), F. fomentarius (a white-rot fungus which grows on hardwood trees66), and L. menziesii and A. biennis (both known to be effective in biomass pretreatment67,68) when grown on aspen pulp (Supplemental Figure 16A). T. gibbosa and L. menziesii secretomes gave the same band following growth on wheat straw while the secretomes of A. biennis and F. fomentarius did not (Supplemental Figure 16B). T. gibbosa, F. fomentarius, and A. biennis did not produce any apparent α-l-arabinofuranosidase when grown on maltose. However, surprisingly, L. menziesii did. Coomassie staining showed very little total protein present in any of the secretome samples (Supplemental Figure 16C,D), demonstrating the remarkable sensitivity of ABP 4.
Based on these results, T. gibbosa, L. menziesii, A. biennis, and F. fomentarius were selected for follow-up studies. Staining 100 μL of secretome followed by acetone precipitation gave much higher band intensity compared to the effective loading of ∼2.8 μL in the screening experiment. This revealed a collection of 1–3 bands running between 55 and 130 kDa (Figure 4C and Supplemental Figure 17). Coomassie staining of the same gel revealed a broad range of bands, few of which appeared to comigrate with the bands detected by visualization with ABP 4 (Figure 4C).
Bands stained with ABP 4 in these samples were identified by pull-down using the same protocol as for the A. niger secretome, but with an added 10 min wash of the beads with 2% SDS at 65 °C to more strictly eliminate any proteins nonspecifically bound to the beads. Comparing proteomic analyses of the secretome and pull-down samples, the vast majority of proteins found within the secretome were rendered undetectable by our washing protocol. In spite of this, the proteins confidently observed (at least two unique peptides identified with an FDR of 1%) were still predominantly not enzymes phylogenetically related to known α-l-arabinofuranosidases. Considering the abundance of GH7 enzymes apparent in the total secretome and in the pull-down, and the apparent staining of the same enzymes by ABP 4 (∼50–60 kDa bands in Figure 4C), we believe that many of these hits represent nonspecific labeling of abundant species within the secretome (e.g., GH7 enzymes Lmen|932922, Ffom|431808, and Tgib|1002594).
Thus, we combined the metrics of total spectral counts from the pull-down (SC(PD), a rough measure of abundance) and the ratio of spectral counts from the pull-down to spectral counts from the digestions of the total secretome (SC(PD)/SC(TS), a rough measure of selectivity) to give the plot shown in Figure 4D. Three GH51 enzymes (JGI ProtIDs Lmen|915930, Tgib|1320025, and Ffom|1458192) appear as distinct targets of ABP 5 with elevated SC(PD) and SC(PD)/SC(TS). These GH51 enzymes had masses correlated with the masses of the most intense bands observed by visualization with ABP 4 (those between 70 and 100 kDa). A single GH51 enzyme in the same mass range from A. biennis (Abien|540325) was detected in the pull-down sample. However, the signal was weak, with only 4 spectral counts detected.
Based on the limited number of hits in the pull-down samples with predicted molecular weights above 90 kDa, we believe that the higher molecular weight bands (100–120 kDa) observed in the SDS–PAGE of secretomes from T. gibbosa, A. biennis, and F. fomentarius are GH3, GH31, or GH35 enzymes. Most of these enzymes were observed with poor SC(PD) and SC(PD)/SC(TS) values (red dots in Figure 4D), however a single GH3 enzyme (Tgib|1466933) appeared to be a target of ABP 5. While this may represent substrate flexibility within these GH families, it remains to be determined whether these enzymes display significant α-l-arabinofuranosidase activity.
Conclusions
The discovery of the mechanism-based covalent α-l-arabinofuranosidase inhibitors 2 and 6 expand the library of tools available for the characterization of enzymes expressed during plant biomass degradation. The unexpectedly high efficiency of inhibitors 2 and 3 provided a platform on which ABPs for α-l-arabinofuranosidases could be synthesized. The potential of ABPs 4 and 5 in the discovery, identification, and characterization of α-l-arabinofuranosidases from fungal secretomes grown on both arabinose-rich biomass and complex woody biomass has been demonstrated. We envision that the ability to efficiently screen samples of interest for levels of multiple active α-l-arabinofuranosidases will facilitate and accelerate a variety of applications including enzyme discovery, bioprocess monitoring, and the investigation of plant-pathogen interactions.
Acknowledgments
We thank Professors Takuya Koseki and Shinya Fushinobu for providing the expression plasmid for AkAbfB. We thank Diamond Light Source for beam time (proposal 18598) and the staff of beamline I04 for assistance with crystal testing and data collection. Proteomics data were collected at the York Centre of Excellence in Mass Spectrometry, which was created thanks to a major capital investment through Science City York, supported by Yorkshire Forward with funds from the Northern Way Initiative, and subsequent support from EPSRC (EP/K039660/1; EP/M028127/1). Wiley-milled aspen (Populus grandidentata) was kindly provided by Dan Cullen (Forest Product Laboratory, USDA, Madison, WI). We are grateful to Igor V Grigoriev, Joint Genome Institute, for providing access to genome sequence data for Trametes gibbosa BRFM 1770, Abortiporus biennis BRFM 1778, Hexagonia nitida BRFM 1802, Trametes ljubarskyi BRFM 1659, Leiotrametes menziesii BRFM 1781, Fomes fomentarius BRFM 1823 and Trametes meyenii BRFM 1810.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b11351.
Supplemental figures and table; synthetic methods and characterization for compounds 1–23 (PDF)
Author Contributions
∇ N.G.S.M., M.A., and A.N.-H. contributed equally to this work.
We thank the Natural Sciences and Engineering Research Council of Canada (Post-Doctoral Fellowship to N.G.S.M.), the Royal Society (Ken Murray Research Professorship to G.J.D.), the Biotechnology and Biological Sciences Research Council (BBSRC) (grant BB/R001162/1 to G.J.D.), The Spanish Ministry of Science and Innovation (MICINN) (CTQ2017-85496-P and MDM-2017-076) to C.R., The Netherlands Organization for Scientific Research (NWO TOP grant 2018-714.018.002 to H.S.O.), the European Research Council (ERC-2011-AdG-290836 “Chembiosphing” to H.S.O.), the Generalitat de Catalunya (FI-AGAUR PhD scholarship to A.N.-H. and grant SGR2017-1189 to C.R.), the computer resources at MareNostrum IV and MinoTauro and the technical support provided by the Barcelona Supercomputing Center (BCV-2016-2-0002 and BCV-2016-3-0005), and the computational support from the University of York High Performance Computing service, Viking and the Research Computing team (chem-menz-2019). Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors declare no competing financial interest.
The authors declare no competing financial interest.
Notes
Crystallographic structural models and diffraction data can be accessed at www.rcsb.org under the PDB IDs 6SXV, 6SXU, 6SXT, 6SXS, and 6SXR. Proteomic LC–MS/MS data sets have been deposited with the MassIVE database (massive.ucsd.edu, UCSD) with the identifiers MSV000084886 (A. niger arabinan secretome) and MSV000084877 (Basidiomycete secretomes) and can be accessed via FTP.
Supplementary Material
References
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. The Carbohydrate-Active Enzymes Database (CAZy) in 2013. Nucleic Acids Res. 2014, 42 (D1), D490–D495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi S.; Navarro D.; Grisel S.; Chevret D.; Berrin J. G.; Rosso M. N. The Integrative Omics of White-Rot Fungus Pycnoporus Coccineus Reveals Co-Regulated CAZymes for Orchestrated Lignocellulose Breakdown. PLoS One 2017, 12 (4), e0175528 10.1371/journal.pone.0175528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. J.; Hekmat O.; Withers S. G. Synthesis and Testing of Mechanism-Based Protein-Profiling Probes for Retaining Endoglycosidases. ChemBioChem 2006, 7 (1), 116–124. 10.1002/cbic.200500279. [DOI] [PubMed] [Google Scholar]
- Hekmat O.; Kim Y. W.; Williams S. J.; He S.; Withers S. G. Active-Site Peptide “Fingerprinting” of Glycosidases in Complex Mixtures by Mass Spectrometry: Discovery of a Novel Retaining β-1,4-Glycanase in Cellulomonas Fimi. J. Biol. Chem. 2005, 280 (42), 35126–35135. 10.1074/jbc.M508434200. [DOI] [PubMed] [Google Scholar]
- Hekmat O.; He S.; Warren R. A. J.; Withers S. G. A Mechanism-Based ICAT Strategy for Comparing Relative Expression and Activity Levels of Glycosidases in Biological Systems. J. Proteome Res. 2008, 7 (8), 3282–3292. 10.1021/pr7008302. [DOI] [PubMed] [Google Scholar]
- Chauvigné-Hines L. M.; Anderson L. N.; Weaver H. M.; Brown J. N.; Koech P. K.; Nicora C. D.; Hofstad B. A.; Smith R. D.; Wilkins M. J.; Callister S. J.; Wright A. T. Suite of Activity-Based Probes for Cellulose-Degrading Enzymes. J. Am. Chem. Soc. 2012, 134 (50), 20521–20532. 10.1021/ja309790w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Fredrickson J. K.; Sadler N. C.; Nandhikonda P.; Smith R. D.; Wright A. T. Advancing Understanding of Microbial Bioenergy Conversion Processes by Activity-Based Protein Profiling. Biotechnol. Biofuels 2015, 8 (1), 156. 10.1186/s13068-015-0343-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Armstrong Z.; Schröder S. P.; de Boer C.; Artola M.; Aerts J. M.; Overkleeft H. S.; Davies G. J. An Overview of Activity-Based Probes for Glycosidases. Curr. Opin. Chem. Biol. 2019, 53, 25–36. 10.1016/j.cbpa.2019.05.030. [DOI] [PubMed] [Google Scholar]
- Rosnow J. J.; Anderson L. N.; Nair R. N.; Baker E. S.; Wright A. T. Profiling Microbial Lignocellulose Degradation and Utilization by Emergent Omics Technologies. Crit. Rev. Biotechnol. 2017, 37, 626–640. 10.1080/07388551.2016.1209158. [DOI] [PubMed] [Google Scholar]
- Kuo C. L.; van Meel E.; Kytidou K.; Kallemeijn W. W.; Witte M.; Overkleeft H. S.; Artola M. E.; Aerts J. M.. Activity-Based Probes for Glycosidases: Profiling and Other Applications; Academic Press, 2018; Vol. 598, pp 217–235. [DOI] [PubMed] [Google Scholar]
- Gloster T. M.; Madsen R.; Davies G. J. Structural Basis for Cyclophellitol Inhibition of a β-Glucosidase. Org. Biomol. Chem. 2007, 5 (3), 444–446. 10.1039/B616590G. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Kuo C. L.; Wu L.; Franke C.; Kallemeijn W. W.; Florea B. I.; Van Meel E.; Van Der Marel G. A.; Codée J. D. C.; Boot R. G.; Davies G. J.; Overkleeft H. S.; Aerts J. M. F. G. Detection of Active Mammalian GH31 α-Glucosidases in Health and Disease Using in-Class, Broad-Spectrum Activity-Based Probes. ACS Cent. Sci. 2016, 2 (5), 351–358. 10.1021/acscentsci.6b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte M. D.; Kallemeijn W. W.; Aten J.; Li K. Y.; Strijland A.; Donker-Koopman W. E.; Van Den Nieuwendijk A. M. C. H.; Bleijlevens B.; Kramer G.; Florea B. I.; Hooibrink B.; Hollak C. E. M.; Ottenhoff R.; Boot R. G.; Van Der Marel G. A.; Overkleeft H. S.; Aerts J. M. F. G. Ultrasensitive in Situ Visualization of Active Glucocerebrosidase Molecules. Nat. Chem. Biol. 2010, 6 (12), 907–913. 10.1038/nchembio.466. [DOI] [PubMed] [Google Scholar]
- Artola M.; Wu L.; Ferraz M. J.; Kuo C. L.; Raich L.; Breen I. Z.; Offen W. A.; Codée J. D. C.; Van Der Marel G. A.; Rovira C.; Aerts J. M. F. G.; Davies G. J.; Overkleeft H. S. 1,6-Cyclophellitol Cyclosulfates: A New Class of Irreversible Glycosidase Inhibitor. ACS Cent. Sci. 2017, 3 (7), 784–793. 10.1021/acscentsci.7b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Jiang J.; Jin Y.; Kallemeijn W. W.; Kuo C. L.; Artola M.; Dai W.; Van Elk C.; Van Eijk M.; Van Der Marel G. A.; Codée J. D. C.; Florea B. I.; Aerts J. M. F. G.; Overkleeft H. S.; Davies G. J. Activity-Based Probes for Functional Interrogation of Retaining β-Glucuronidases. Nat. Chem. Biol. 2017, 13 (8), 867–873. 10.1038/nchembio.2395. [DOI] [PubMed] [Google Scholar]
- Willems L. I.; Beenakker T. J. M.; Murray B.; Scheij S.; Kallemeijn W. W.; Boot R. G.; Verhoek M.; Donker-Koopman W. E.; Ferraz M. J.; Van Rijssel E. R.; Florea B. I.; Codée J. D. C.; Van Der Marel G. A.; Aerts J. M. F. G.; Overkleeft H. S. Potent and Selective Activity-Based Probes for GH27 Human Retaining α-Galactosidases. J. Am. Chem. Soc. 2014, 136 (33), 11622–11625. 10.1021/ja507040n. [DOI] [PubMed] [Google Scholar]
- Marques A. R. A.; Willems L. I.; Herrera Moro D.; Florea B. I.; Scheij S.; Ottenhoff R.; van Roomen C. P. A. A.; Verhoek M.; Nelson J. K.; Kallemeijn W. W.; Biela-Banas A.; Martin O. R.; Cachón-González M. B.; Kim N. N.; Cox T. M.; Boot R. G.; Overkleeft H. S.; Aerts J. M. F. G. A Specific Activity-Based Probe to Monitor Family GH59 Galactosylceramidase, the Enzyme Deficient in Krabbe Disease. ChemBioChem 2017, 18 (4), 402–412. 10.1002/cbic.201600561. [DOI] [PubMed] [Google Scholar]
- Schröder S. P.; De Boer C.; McGregor N. G. S.; Rowland R. J.; Moroz O.; Blagova E.; Reijngoud J.; Arentshorst M.; Osborn D.; Morant M. D.; Abbate E.; Stringer M. A.; Krogh K. B. R. M.; Raich L.; Rovira C.; Berrin J. G.; Van Wezel G. P.; Ram A. F. J.; Florea B. I.; Van Der Marel G. A.; Codée J. D. C.; Wilson K. S.; Wu L.; Davies G. J.; Overkleeft H. S. Dynamic and Functional Profiling of Xylan-Degrading Enzymes in Aspergillus Secretomes Using Activity-Based Probes. ACS Cent. Sci. 2019, 5 (6), 1067–1078. 10.1021/acscentsci.9b00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezalel L.; Shoham Y.; Rosenberg E. Characterization and Delignification Activity of a Thermostable α-l-Arabinofuranosidase from Bacillus Stearothermophilus. Appl. Microbiol. Biotechnol. 1993, 40 (1), 57–62. 10.1007/BF00170429. [DOI] [Google Scholar]
- Cartmell A.; Muñoz-Muñoz J.; Briggs J. A.; Ndeh D. A.; Lowe E. C.; Baslé A.; Terrapon N.; Stott K.; Heunis T.; Gray J.; Yu L.; Dupree P.; Fernandes P. Z.; Shah S.; Williams S. J.; Labourel A.; Trost M.; Henrissat B.; Gilbert H. J. A Surface Endogalactanase in Bacteroides Thetaiotaomicron Confers Keystone Status for Arabinogalactan Degradation. Nat. Microbiol. 2018, 3 (11), 1314–1326. 10.1038/s41564-018-0258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez O. L.; Fernández-Bolaños J. G.; Lillelund V. H.; Bols M. Aziridines as a Structural Motif to Conformational Restriction of Azasugars. Org. Biomol. Chem. 2003, 1 (3), 478–482. 10.1039/b210038j. [DOI] [PubMed] [Google Scholar]
- Hövel K.; Shallom D.; Niefind K.; Baasov T.; Shoham G.; Shoham Y.; Schomburg D. Crystallization and Preliminary X-Ray Analysis of a Family 51 Glycoside Hydrolase, the α-L-Arabinofuranosidase from Geobacillus Stearothermophilus T-6. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2003, 59 (5), 913–915. 10.1107/S0907444903004037. [DOI] [PubMed] [Google Scholar]
- Tropea J. E.; Cherry S.; Waugh D. S. Expression and Purification of Soluble His6-Tagged TEV Protease. Methods Mol. Biol. 2009, 498, 297–307. 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R. Y.; Venter J. C.; Hutchison C. A.; Smith H. O. Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 2009, 6 (5), 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Wu S.; Letchworth G. J. High Efficiency Transformation by Electroporation of Pichia Pastoris Pretreated with Lithium Acetate and Dithiothreitol. BioTechniques 2004, 36 (1), 152–154. 10.2144/04361DD02. [DOI] [PubMed] [Google Scholar]
- Liu W.; Tsou C. lu. Determination of Rate Constant for the Irreversible Inhibition of Acetylcholine Esterase by Continuously Monitoring the Substrate Reaction in the Presence of the Inhibitor. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1986, 870 (2), 185–190. 10.1016/0167-4838(86)90220-7. [DOI] [PubMed] [Google Scholar]
- Gray P. J.; Duggleby R. G. Analysis of Kinetic Data for Irreversible Enzyme Inhibition. Biochem. J. 1989, 257 (2), 419–424. 10.1042/bj2570419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanaga A.; Koseki T.; Matsuzawa H.; Wakagi T.; Shoun H.; Fushinobu S. Expression, Purification, Crystallization and Preliminary X-Ray Analysis of α-L-Arabinofuranosidase B from Aspergillus Kawachii. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60 (7), 1286–1288. 10.1107/S0907444904010108. [DOI] [PubMed] [Google Scholar]
- Winter G.; McAuley K. E. Automated Data Collection for Macromolecular Crystallography. Methods 2011, 55 (1), 81–93. 10.1016/j.ymeth.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. Data Processing and Analysis with the AutoPROC Toolbox. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 293–302. 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; Lobley C. M. C.; Prince S. M. Decision Making in Xia2. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69 (7), 1260–1273. 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40 (4), 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev A. A.; Young P.; Isupov M. N.; Moroz O. V.; Vagin A. A.; Murshudov G. N. JLigand: A Graphical Tool for the CCP4 Template-Restraint Library. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68 (4), 431–440. 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Car R.; Parrinello M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55 (22), 2471–2474. 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- Troullier N.; Martins J. L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B: Condens. Matter Mater. Phys. 1991, 43 (3), 1993–2006. 10.1103/PhysRevB.43.1993. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Marianski M.; Supady A.; Ingram T.; Schneider M.; Baldauf C. Assessing the Accuracy of Across-the-Scale Methods for Predicting Carbohydrate Conformational Energies for the Examples of Glucose and α-Maltose. J. Chem. Theory Comput. 2016, 12 (12), 6157–6168. 10.1021/acs.jctc.6b00876. [DOI] [PubMed] [Google Scholar]
- Biarnés X.; Ardèvol A.; Planas A.; Rovira C.; Laio A.; Parrinello M. The Conformational Free Energy Landscape of β-D-Glucopyranose. Implications for Substrate Preactivation in β-Glucoside Hydrolases. J. Am. Chem. Soc. 2007, 129 (35), 10686–10693. 10.1021/ja068411o. [DOI] [PubMed] [Google Scholar]
- Ardèvol A.; Rovira C. Reaction Mechanisms in Carbohydrate-Active Enzymes: Glycoside Hydrolases and Glycosyltransferases. Insights from Ab Initio Quantum Mechanics/Molecular Mechanics Dynamic Simulations. J. Am. Chem. Soc. 2015, 137 (24), 7528–7547. 10.1021/jacs.5b01156. [DOI] [PubMed] [Google Scholar]
- Laio A.; Parrinello M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (20), 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185 (2), 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Huang M.; Giese T. J.; Lee T. S.; York D. M. Improvement of DNA and RNA Sugar Pucker Profiles from Semiempirical Quantum Methods. J. Chem. Theory Comput. 2014, 10 (4), 1538–1545. 10.1021/ct401013s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer D.; Pople J. A. A General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97 (6), 1354–1358. 10.1021/ja00839a011. [DOI] [Google Scholar]
- Tiwary P.; Parrinello M. A Time-Independent Free Energy Estimator for Metadynamics. J. Phys. Chem. B 2015, 119 (3), 736–742. 10.1021/jp504920s. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Betz R. M.; Cerutti D. S.; Cheatham T. E.; Darden T.; Duke R. E.; et al. AMBER16, 2016.
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25 (9), 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Miller B. R.; McGee T. D.; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8 (9), 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Davies G. J.; Planas A.; Rovira C. Conformational Analyses of the Reaction Coordinate of Glycosidases. Acc. Chem. Res. 2012, 45 (2), 308–316. 10.1021/ar2001765. [DOI] [PubMed] [Google Scholar]
- Speciale G.; Thompson A. J.; Davies G. J.; Williams S. J. Dissecting Conformational Contributions to Glycosidase Catalysis and Inhibition. Curr. Opin. Struct. Biol. 2014, 28 (1), 1–13. 10.1016/j.sbi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardèvol A.; Biarnés X.; Planas A.; Rovira C. The Conformational Free-Energy Landscape of β-d-Mannopyranose: Evidence for a 1S5 → B2,5 → OS2 Catalytic Itinerary in β-Mannosidases. J. Am. Chem. Soc. 2010, 132 (45), 16058–16065. 10.1021/ja105520h. [DOI] [PubMed] [Google Scholar]
- Paës G.; Skov L. K.; O’Donohue M. J.; Rémond C.; Kastrup J. S.; Gajhede M.; Mirza O. The Structure of the Complex between a Branched Pentasaccharide and Thermobacillus Xylanilyticus GH-51 Arabinofuranosidase Reveals Xylan-Binding Determinants and Induced Fit. Biochemistry 2008, 47 (28), 7441–7451. 10.1021/bi800424e. [DOI] [PubMed] [Google Scholar]
- Hövel K.; Shallom D.; Niefind K.; Belakhov V.; Shoham G.; Baasov T.; Shoham Y.; Schomburg D. Crystal Structure and Snapshots along the Reaction Pathway of a Family 51 α-L-Arabinofuranosidase. EMBO J. 2003, 22 (19), 4922–4932. 10.1093/emboj/cdg494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rijssel E. R.; Van Delft P.; Lodder G.; Overkleeft H. S.; Van Der Marel G. A.; Filippov D. V.; Codée J. D. C. Furanosyl Oxocarbenium Ion Stability and Stereoselectivity. Angew. Chem., Int. Ed. 2014, 53 (39), 10381–10385. 10.1002/anie.201405477. [DOI] [PubMed] [Google Scholar]
- Miyanaga A.; Koseki T.; Matsuzawa H.; Wakagi T.; Shoun H.; Fushinobu S. Crystal Structure of a Family 54 α-L-Arabinofuranosidase Reveals a Novel Carbohydrate-Binding Module That Can Bind Arabinose. J. Biol. Chem. 2004, 279 (43), 44907–44914. 10.1074/jbc.M405390200. [DOI] [PubMed] [Google Scholar]
- Artola M.; Wouters S.; Schröder S. P.; de Boer C.; Chen Y.; Petracca R.; van den Nieuwendijk A. M. C. H.; Aerts J. M. F. G.; van der Marel G. A.; Codée J. D. C.; Overkleeft H. S. Direct Stereoselective Aziridination of Cyclohexenols with 3-Amino-2-(Trifluoromethyl)Quinazolin-4(3H)-One in the Synthesis of Cyclitol Aziridine Glycosidase Inhibitors. Eur. J. Org. Chem. 2019, 2019 (6), 1397–1404. 10.1002/ejoc.201801703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jat J. L.; Paudyal M. P.; Gao H.; Xu Q. L.; Yousufuddin M.; Devarajan D.; Ess D. H.; K̈urti L.; Falck J. R. Direct Stereospecific Synthesis of Unprotected N-H and N-Me Aziridines from Olefins. Science 2014, 343 (6166), 61–65. 10.1126/science.1245727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens-Uzunova E. S.; Schaap P. J. Assessment of the Pectin Degrading Enzyme Network of Aspergillus Niger by Functional Genomics. Fungal Genet. Biol. 2009, 46 (1), S170–S179. 10.1016/j.fgb.2008.07.021. [DOI] [PubMed] [Google Scholar]
- Flipphi M. J. A.; Visser J.; van der Veen P.; de Graaff L. H. Cloning of the Aspergillus Niger Gene Encoding α-l-Arabinofuranosidase A. Appl. Microbiol. Biotechnol. 1993, 39 (3), 335–340. 10.1007/BF00192088. [DOI] [PubMed] [Google Scholar]
- Flipphi M. J. A.; van Heuvel M.; van der Veen P.; Visser J.; de Graaff L. H. Cloning and Characterization of the AbfB Gene Coding for the Major α-l-Arabinofuranosidase (ABF B) of Aspergillus Niger. Curr. Genet. 1993, 24 (6), 525–532. 10.1007/BF00351717. [DOI] [PubMed] [Google Scholar]
- Berrin J. G.; Navarro D.; Couturier M.; Olivé C.; Grisel S.; Haon M.; Taussac S.; Lechat C.; Courtecuisse R.; Favel A.; Coutinho P. M.; Lesage-Meessen L. Exploring the Natural Fungal Biodiversity of Tropical and Temperate Forests toward Improvement of Biomass Conversion. Appl. Environ. Microbiol. 2012, 78 (18), 6483–6490. 10.1128/AEM.01651-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze F. Wood Rotting Fungi: Fomes Fomentarius (L.: Fr.) Fr. Hoof or Tinder Fungus. Top. Catal. 1994, 8 (1), 32–34. 10.1016/S0269-915X(09)80679-4. [DOI] [Google Scholar]
- Zhou S.; Raouche S.; Grisel S.; Navarro D.; Sigoillot J. C.; Herpoël-Gimbert I. Solid-State Fermentation in Multi-Well Plates to Assess Pretreatment Efficiency of Rot Fungi on Lignocellulose Biomass. Microb. Biotechnol. 2015, 8 (6), 940–949. 10.1111/1751-7915.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandropoulou M.; Antonopoulou G.; Fragkou E.; Ntaikou I.; Lyberatos G. Fungal Pretreatment of Willow Sawdust and Its Combination with Alkaline Treatment for Enhancing Biogas Production. J. Environ. Manage. 2017, 203, 704–713. 10.1016/j.jenvman.2016.04.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.