

SUMMARY
DNA-protein crosslinks (DPCs) are a frequent form of DNA lesion and are strongly inhibitive in diverse DNA transactions. Despite recent developments, the biochemical detection of DPCs remains a limiting factor for the in-depth mechanistic understanding of DPC repair. Here, we develop a sensitive and versatile assay, designated ARK, for the quantitative analysis of DPCs in cells. ARK uses sequential chaotropic and detergent-based isolation of DPCs and substantially enhances sample purity, resulting in a 5-fold increase in detection sensitivity and a 10-fold reduction in background reading. We validate the ARK assay with genetic mutants with established deficiencies in DPC repair and demonstrate its robustness by using common DPC-inducing reagents, including formaldehyde, camptothecin, and etoposide. In addition, we show that the Fanconi anemia pathway contributes to the repair of DPCs. Thus, ARK is expected to facilitate various studies aimed at understanding both fundamental biology and translational applications of DNA-protein crosslink repair.
Graphical Abstract
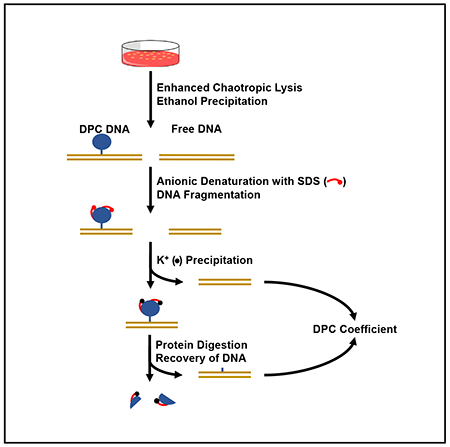
In Brief
Hu et al. develop a protocol to analyze DNA-protein crosslinking (DPC) damage. Designated the ARK assay, this method outperforms widely used assays by allowing the detection of global DPCs with improved sensitivity and expanded readout. Defective DPC repair is detected in Fanconi anemia mutant cells by this protocol.
INTRODUCTION
DNA-protein crosslinks (DPCs) are a form of DNA damage generated when a protein moiety is covalently conjugated to cellular DNA. The exceedingly bulky nature of DPCs poses impassible barriers to essential DNA transactions, including replication, transcription, and recombination. DPCs are cytotoxic and mutagenic and lead to a destabilized genome (Maskey et al., 2014; Tretyakova et al., 2013). Cellular mechanisms of DPC repair are increasingly gaining attention, especially with the characterization of an inheritable, cancer-prone premature aging disease originating from DPC repair deficiencies (Lessel et al., 2014; Maskey et al., 2014, 2017). Exogenous and endogenous DNA-damaging agents generating nuclear DPCs are also abundant (Chválová et al., 2007; Garaycoechea et al., 2012; Langevin et al., 2011; Noguchi et al., 2017; Walport et al., 2012). However, in-depth understanding of the molecular mechanism(s) of DPC repair is lagging behind, not only because of the complexity of the repair process but also due to limited experimental readouts for monitoring cellular DPC repair.
Genotoxic DPCs arise from two distinct mechanisms: enzymatic DPCs and non-enzymatic DPCs. DNA metabolic and modifying enzymes, such as topoisomerases (TOPs), DNA polymerases, and DNA methyltransferases, form transient covalent intermediates with DNA. These catalytic intermediates can become interminable by mutations in the catalytic domain of the enzyme (Centore et al., 2010), by aberrant DNA substrates (Quiñones et al., 2015), or by inhibitors that stabilize covalent enzyme-DNA transient structures (Ide et al., 2018; Stingele and Jentsch, 2015). For example, DPCs form efficiently from entrapped TOP1 and TOP2 cleavage complexes (TOP1cc and TOP2cc, respectively), which are produced by several broadly used cancer chemotherapy drugs such as doxorubicin, camptothecin (CPT), and etoposide (ETO) (Fielden et al., 2018; Pommier et al., 2014; Pommier and Marchand, 2011). These topoisomerase poisons trap TOP1 and/or TOP2 by intercalating into the interface between DNA and the enzymes and consequently prevent re-ligation of the strand breaks, forming stabilized cleavage complexes, which are repaired by tyrosyl-DNA phosphodiesterase 1 and 2 (TDP1 and TDP2) (Pommier et al., 2014).
Non-enzymatic DPCs are generated by a large variety of endogenous and exogenous agents. Ionizing radiation, ultraviolet light, certain transition metal ions, and reactive compounds, including reactive aldehydes, are capable of covalently conjugating proteins onto DNA through primary amine groups on amino acid and nucleotide residues (Garaycoechea et al., 2012; Ide et al., 2018; Langevin et al., 2011). Notably, formaldehyde (FA), with its widespread environmental presence and constant intracellular presence (Trewick et al., 2002; Walport et al., 2012; Yu et al., 2015), is a potent crosslinking agent and a demonstrated carcinogen (Pontel et al., 2015; Swenberg et al., 1980). Formaldehyde-induced DPCs have been widely used as a model lesion for studying the cellular pathways of DPC repair (Heck et al., 1990; Lai et al., 2016). Bifunctional alkylating drugs, such as cisplatin, are also capable of inducing non-enzymatic DPCs (Ming et al., 2017).
Current approaches for assaying cellular DPCs fall into two categories of direct or indirect measurements. Upon separating protein-crosslinked DNA from free DNA, the direct measurement method quantifies the amount of proteins associated with DNA. This is accomplished by the general detection of proteins via fluorescent labeling. For a particular protein of interest, antibody-based detection is used to assess the amount of DNA-tethered target protein (Kiianitsa and Maizels, 2013; Mórocz et al., 2017; Nakano et al., 2017; Shoulkamy et al., 2012; Stingele et al., 2014).
An indirect assay of DPCs relies on isolating and measuring the amount of protein-bound DNA and calculating its proportion in total DNA as a quantitative reflection of DPCs (Costa et al., 1996; Stingele and Jentsch, 2015; Zhitkovich and Costa, 1992). The K-SDS assay is the conventional platform of indirect DPC measurement. To isolate protein-tethered DNA, SDS is used to dissolve chromatin preparations. Free proteins and DPCs are converted to the precipitated form by the addition of potassium chloride while free DNA remains soluble (Trask et al., 1984). DNA contained in DPCs is released by proteinase digestion to remove the protein moiety. Normalizing the amount of DPC-associated DNA against total sample DNA yields the percentage of genomic DNA bearing DPCs (Costa et al., 1996; de Graaf et al., 2009; Liu et al., 2006; Mórocz et al., 2017; Olin et al., 1996; Stingele et al., 2014, 2016; Vaz et al., 2016; Ye et al., 2013; Zhitkovich and Costa, 1992).
The advantage of the direct DPC assay is that the absolute amount of DNA-linked proteins is determined collectively. However, the process of isolating DNA-tethered proteins from free proteins necessitates repeated CsCl gradient purification and dialysis, which render the procedure lengthy and increases the potential for inconsistency. The K-SDS-based indirect DPC assays are straightforward and better suited to handling a large number of samples. One major unfavorable aspect, however, is the high level of background and, accordingly, moderate signal:noise ratio (Liu et al., 2006; Mórocz et al., 2017; Olin et al., 1996; Ye et al., 2013). In the canonical rapid approach to DNA adduct recovery (RADAR) assay, wherein an antibody is used to detect the DNA-bound protein target, the ethanol precipitation that removes free proteins leads to background issues, and resolution is dependent on the specificity of the antibody (Kiianitsa and Maizels, 2013). DPC isolation using the RADAR procedure can be resolved by SDS-PAGE for the quantification of global DPCs (Vaz et al., 2016).
Attempts to increase the signal:background ratio of the current DPC assays have been described in a number of studies. These include the addition of an RNA digestion step to lower the background DNA reading or the resolution of the DPC samples by gel electrophoresis to reduce free DNA contamination. These additional steps had varying degrees of effectiveness, but success in mitigating the high background level of the K-SDS assay was limited (Mórocz et al., 2017; Stingele et al., 2014; Vaz et al., 2016). In most published K-SDS experiments, the background DPC reading in unperturbed cells is set at 1. Those reported as absolute DPC coefficient can reach as high as 10% and the fold change under physiological doses of DPC-inducing agents is generally <5-fold, restricting the application of this method (Barker et al., 2005; Liu et al., 2006; Olin et al., 1996; Ye et al., 2013).
Given the advantages and limitations of each DPC assay, improving the signal:background ratio while maintaining the simplicity and consistency of the protocol is highly desirable. In this study, we established a novel procedure of DPC analysis by significantly elevating the stringency of DPC isolation conditions. This new assay, designated ARK (for advanced recovery of K [potassium]-SDS precipitates), increased the readout range of DPC quantification by 5-fold. The simplicity of this assay is similar to that of the current indirect assays and is capable of processing a large number of samples compared to the CsCl gradient-based direct assay. Here, we demonstrate the effectiveness of the ARK assay in detecting exogenous DPCs induced by formaldehyde and topoisomerase inhibition. Using the ARK assay, we detected impaired DPC repair in mutants with disrupted FANCL, suggesting a role for the Fanconi anemia pathway in the repair of DPCs. Thus, this new assay provides a much-improved tool for studying DPC repair mechanisms as well as studies regarding DPC-inducing agents.
RESULTS
Design of the New ARK Assay
An effective approach to improve an indirect DPC assay is to reduce the background while retaining positive signals. We designed the ARK method with several measures to reduce the background reading and improve the recovery of DPCs (Figure 1). We used guanidine thiocyanate (GTC) for cell lysis to apply a stringent condition to eliminate noncovalent DNA-protein association, which constitutes a major source of background. Subsequent to the 50% ethanol cutoff precipitation, recovered DPCs together with free DNA and unbound proteins were dissolved in 1% SDS at denaturing temperature to further disrupt noncovalent DNA-protein associations. Following KCl-mediated precipitation to remove free DNA, the protein-bound DNA was released by proteinase K digestion and quantified against total genomic DNA to obtain the DPC coefficient.
Figure 1. Schematic Illustration of the ARK Assay.
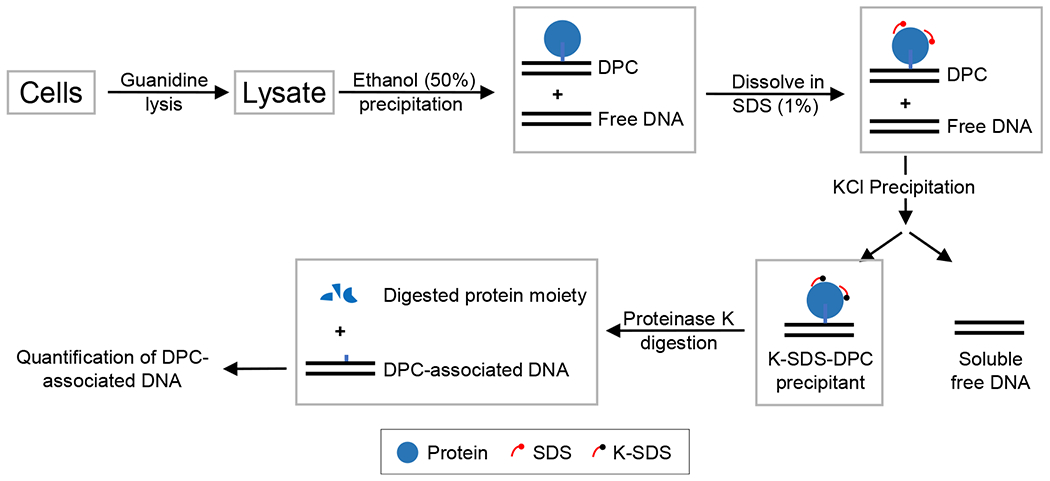
Cells are lysed with 5.6 M GTC to disrupt the noncovalent association between DNA and proteins. Free DNA and DPC-associated DNA are recovered by ethanol precipitation. The DNA-DPC pellet is dissolved in SDS buffer to further eliminate noncovalent DNA-protein interactions and to denature the proteins. The subsequent addition of KCl results in the precipitation of SDS-bound proteins along with DPC-associated DNA, while free DNA remains soluble. The DPCs that are recovered are digested with proteinase K to remove the protein adducts, resulting in the release of DPC-associated DNA for the quantification and calculation of the DPC coefficient.
In addition to the chaotropic- and detergent-based dissociation of noncovalent DNA-protein association, we tested several modifications that further reduce the background reading. Preparing the initial cell lysate with 55°C GTC buffer and shearing DPC samples in the 1% SDS buffer decreased the background reading in 293A and TK6 cells by ~61% ± 5.7% and ~67% ± 5.5%, respectively (Figure 2A). To eliminate the potential interference of RNA in the ARK assay, we treated the DPC samples after proteinase K digestion with the mixed RNases A and T1. As shown in Figure 2B, no detectable differences in DPC background readings were found in the presence or absence of RNase treatment in both HeLa and TK6 cell samples. This result indicates that the interference from RNA contamination is insignificant in the final DNA product of the ARK protocol.
Figure 2. ARK Assay Optimization Yielded Significantly Reduced Background and Increased Sensitivity.
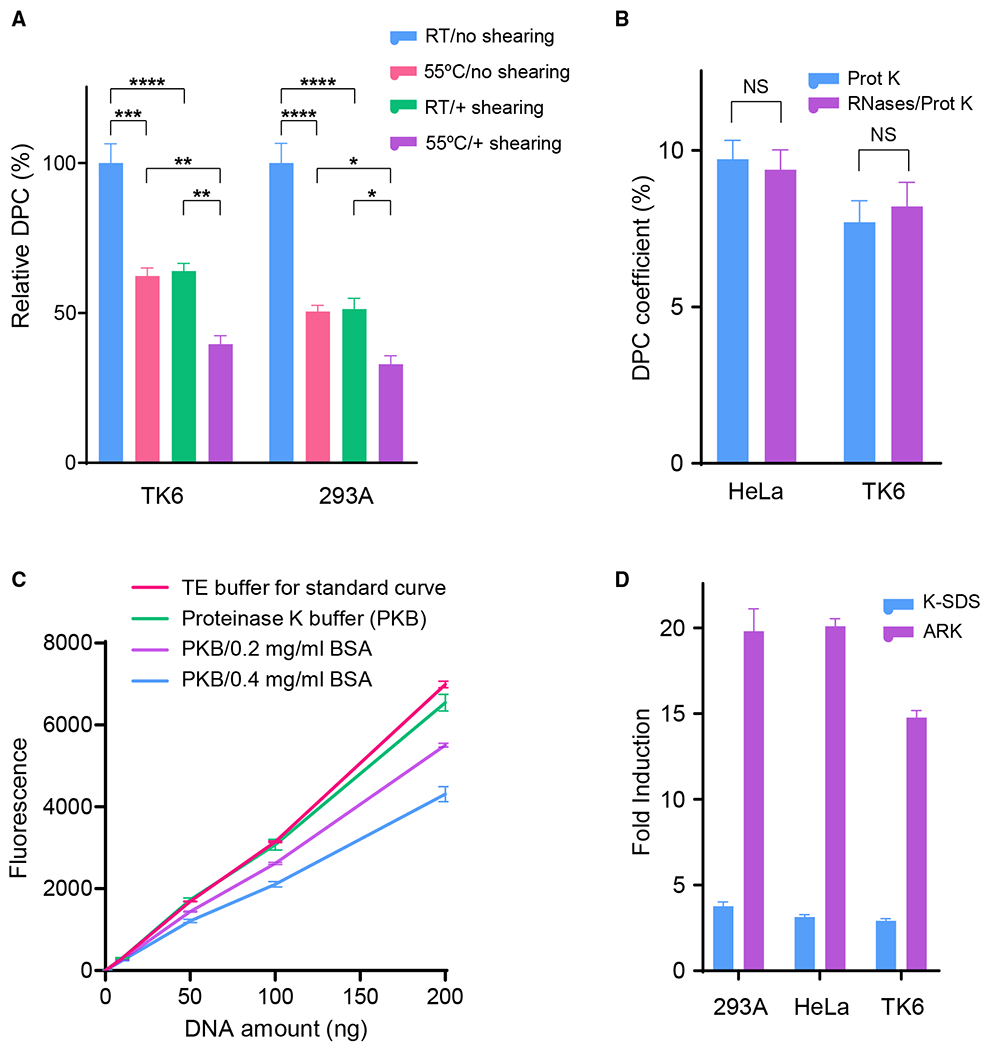
(A) Reduction of DPC coefficient background by pre-warming lysis buffer to 55°C (versus room temperature [RT]) and by syringe shearing after DPC precipitate is dissolved in 1% SDS buffer. The background DPC levels before these optimizations were set to 100% for comparison purposes.
(B) Impact of RNA removal on DPC fold induction reading from HeLa and TK6 cells exposed to 200 μM FA (2 h). DNA samples recovered after proteinase K digestion were treated and mock-treated with RNase A-T1 mix and subsequently measured by PicoGreen quantification.
(C) Effect of BSA in PicoGreen DNA measurement. Recovered DNA samples after proteinase K digestion were subjected to PicoGreen quantification in the presence or absence of the indicated amount of BSA.
(D) Parallel comparison of assay readout between the K-SDS and ARK methods. Cells were treated with 400 μM FA for 2 h. DPC levels are represented by fold induction compared to mock-treated cells.
Number of biological repeats: n = 6 for (A), n = 3 for (B)–(D). The error bars depict standard deviations.
In the conventional K-SDS assay, a final concentration of 0.4 mg/mL BSA is included to mitigate the potential interference of free SDS in the DNA samples. However, the presence of BSA is likely to interfere with the fluorometric detection of the PicoGreen assay. To test this, we analyzed the typical range of DPC DNA concentrations with and without the addition of BSA. We observed a significant decrease in the fluorometric readings from samples containing BSA (0.2 and 0.4 mg/mL) (Figure 2C), indicating that the inclusion of BSA in the final DNA samples leads to reduced detection sensitivity. Thus, we eliminated the BSA addition in the K-SDS assay from our ARK assay. Furthermore, DNA purity from the ARK assay, as measured by the 260:280 nm absorbance ratio, is consistently at 1.9, suggesting that the ARK procedure yields DNA with sufficient purity for quantitative analysis.
Integrating the above modifications in the ARK assay design, we tested three model cell lines in direct comparison with the conventional K-SDS assay. Baseline DPC reading from three model cell lines–293A, HeLa, and TK6–were generated in parallel by the ARK assay and by the K-SDS assay, as shown in Figure S1. We found that the ARK protocol has consistently yielded a background DPC coefficient at ~1%, compared to the 8%–14% background readings from the K-SDS assay (Figure S1) (Liu et al., 2006). As a result, the ARK assay provides substantially increased sensitivity and detection range. When testing cells identically exposed to the LC20 dose of formaldehyde (400 μM, 2 h), folds of DPC induction by the ARK assay are 5.3 ± 0.69, 6.4 ± 0.28, and 5.1 ± 0.28 times higher than those obtained with the conventional K-SDS assay for 293A, HeLa, and TK6 cells, respectively (Figure 2D). Moreover, the improved indirect DPC assay can be completed in <7 h.
Detection of Nonenzymatic DPCs Using the ARK Assay
To demonstrate the effectiveness of the ARK assay in the analysis of induced DPCs, we investigated DPC formation in response to treatment with varying doses of FA, a commonly used DPC-inducing agent. As shown in Figure 3A, the ARK assay exhibited near-linear dose dependency in all three cell lines (293A, HeLa, TK6) tested within a physiologically relevant dose range (Olin et al., 1996; Vaz et al., 2016). At a commonly adopted dose of 500 μM FA, the DPC coefficient fold increases in the three cell lines are approximately 23, 23, and 16 in 293A, HeLa, and TK6, respectively. In comparison, the conventional K-SDS assay typically exhibits a 3- to 4.5-fold increase at the same dose (Figure 2D). Recently, a modified K-SDS assay by Mórocz et al. (2017) obtained a DPC induction near 20-fold at 500 μM FA concentration in a serum-free medium. In contrast, the ARK assay was performed at the same dose but in the presence of 10% serum, with much reduced FA potency and allowed the experiment to be carried out at normal culture conditions and closer to a physiological dose range. These results suggest that the ARK assay provides a significantly expanded sensitivity and range of DPC detection, particularly at physiologically relevant doses.
Figure 3. Detection of Nonenzymatic DPCs by the ARK Assay.
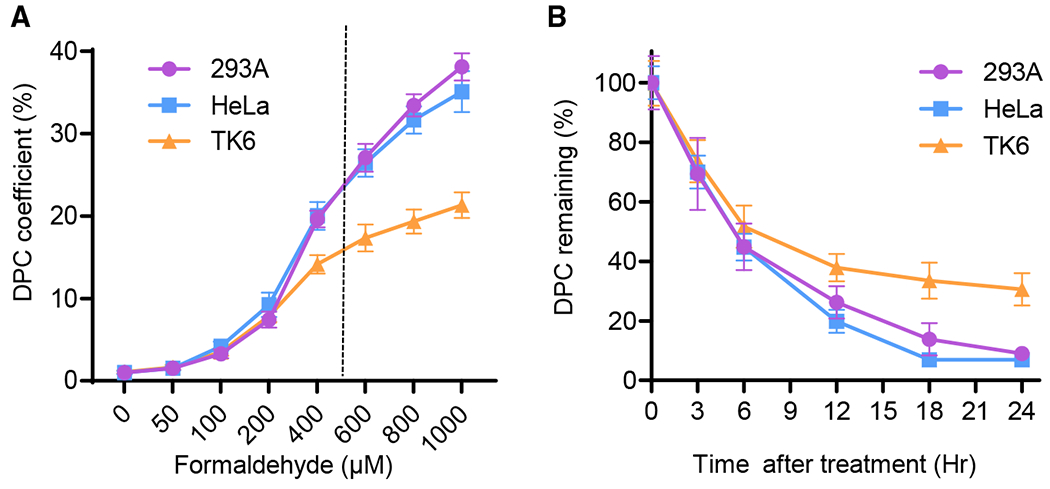
(A) Dose response of 293A, HeLa, and TK6 cells exposed to the indicated concentrations of FA for 2 h. DPC fold inductions were calculated by normalizing DPC-associated DNA to that of mock treatment.
(B) DPC repair time course in 293A, HeLa, and TK6 cells exposed to 400 μM FA treatment for 2 h. DPC coefficient of each cell line after treatment is set as 100%.
n = 4 for (A) and n = 6 for (B). The error bars depict standard deviations.
To determine whether the ARK assay is capable of monitoring the kinetics of DPC repair, a time course study at 400 μM was performed to assess the removal of DPCs in 293A, HeLa, and TK6 cells (Figure 3B). In 293A and HeLa cells, steady declines of DPCs were observed, and the level of residual DPCs reached the background level after 24 h of recovery time. In TK6 cells, ~30% of formaldehyde-induced DPCs remained at 24 h. This is consistent with published data that show that complete removal of DPCs from lymphocytes can take up to 3 days (Quievryn and Zhitkovich, 2000). The recovery curves generated by the ARK assay also matched the biphasic repair pattern, as observed with the CsCI-gradient-based DPC purification method (Shoulkamy et al., 2012). Approximately 60% DPCs in 293A and HeLa cells and 50% DPCs in TK6 cells were removed after 6 h, followed by a slower phase of DPC repair.
DPC half-life, derived from the ARK method, is 5 h for 293A and HeLa cells. This result is very consistent with the 4.8-h half-life as determined by the direct method using sensitive fluorescein isothiocyanate (FITC) labeling to measure DNA-conjugated proteins (Shoulkamy et al., 2012). To exclude the potential confounding factor of cell death during the time course, cell viability was determined at the corresponding time points for DPC detection. The highest variation of cell viability among all of the time points was <18% (Figure S2), suggesting that cell death is not a significant factor in the DPC coefficient reading from the ARK assay. These results suggest that the ARK assay is of higher sensitivity and broader readout range than the present methods and that it is able to measure DPC repair comparable to the more complex direct assay.
Detection of Enzymatic DPCs Using the ARK Assay
TOP inhibitors are a major class of enzymatic DPC-inducing agents, which account for nearly half of the present chemotherapeutic regimens. Among them, CPT and ETO are model agents for enzymatic DPCs by generating TOP1cc and TOP2cc. To determine whether the ARK assay is capable of measuring TOP1cc and TOP2cc, which are formed by 2 defined proteins, 293A, HeLa, and TK6 cells were exposed to serial doses of CPT for 1 h and analyzed by the ARK assay to obtain the DPC coefficient. As shown in Figure 4A, typical dose-response patterns in the tested cell lines were observed. The dose-response approached a plateau at 20 μM for CPT, presumably at the point at which the TOP targets became saturated. At 10 μM CPT, a dose used in most cellular experiments, the increase in DPC coefficients from the 3 tested cell lines ranged from 12.3- to 15.3-fold, whereas the RADAR assay with similar treatment yielded a fold increase of <10 (Kiianitsa and Maizels, 2013).
Figure 4. Detection of Enzymatic DPCs Induced by Topoisomerase Inhibitors with the ARK Assay.
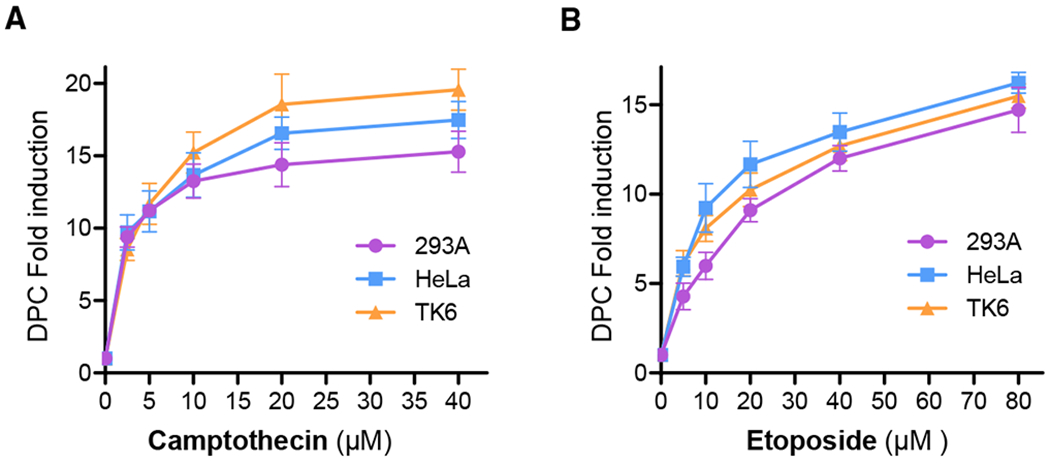
(A) 293A, HeLa, and TK6 cells were exposed to various doses of CPT for 1 h and analyzed by the ARK assay to generate DPC fold induction by normalizing the DPC coefficient of each sample against that of the mock-treated control.
(B) HeLa, 293A, and TK6 cells were exposed to various doses of etoposide for 1 h and analyzed by the ARK assay to generate the DPC fold induction by normalizing the DPC coefficient of each sample against that of the mock-treated control.
Each data point was generated from no less than 5 biological repeats with triplication. The error bars depict standard deviations.
Similarly, TOP2cc DPCs induced by ETO in the three tested cell lines exhibited typical dose-response curves, as measured by the ARK assay (Figure 4B). The increase in DPC coefficients at the commonly used treatment concentration (20 μM) was 9.1- to 11.7-fold, while the dose range extended to 40 μM. These results suggest that the ARK assay is capable of detecting enzymatic DPCs with a comparable sensitivity and broad dose range of readout without relying on protein target-specific antibodies, allowing its application to a much broader range of enzymatic DPCs.
ARK Assay Validation Using DPC Repair-Deficient Genetic Models
To validate the capability of the ARK assay in established genetic models, we analyzed DPC repair activities in TK6 and its isogenic SPRTN knockout mutant (Figures S3A–S3C). SPRTN possesses DNA-dependent proteolytic activity and is important for DPC repair in higher eukaryotes (Duxin et al., 2014; Larsen et al., 2019; Stingele et al., 2014, 2016; Vaz et al., 2016). The DPC repair function of SPRTN appears to be non-redundant to the ubiquitination/proteasome degradation-mediated DPC processing (Larsen et al., 2019; Stingele et al., 2016). Thus, the SPRTN knockout mutant most likely exhibits partially, not fully, impaired DPC repair.
We exposed the parental TK6 cells and 2 independent knockout mutants, SPRTN KO1 and KO2, to 400 μM FA for 2 h and measured the DPC removal at various time points. We found that both SPRTN KO mutants showed a significantly (p <0.001 or 0.0001) higher level of DPC retention at all 3 time points compared to the wild-type TK6 cells (Figure 5A). After 24 h of FA treatment, DPCs in wild-type TK6 cells decreased to 32.0% ± 5.0% of the initial level. The SPRTN mutants combined, however, retained an average of 70.3% ± 7.4% of the DPCs, which is 2.2-fold higher than the wild-type parental cells. This result shows that the ARK assay effectively detects the partial impairment of DPC repair from SPRTN deficiency.
Figure 5. Analysis of DPC Repair Deficiency in SPRTN and TDP1/TDP2 Knockout Mutants with the ARK Assay.
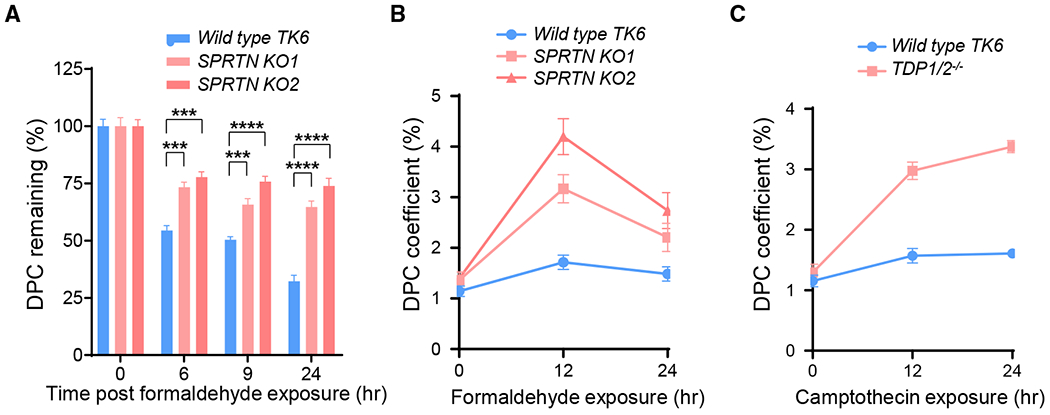
(A) Removal of DPCs in wild-type TK6 cells and 2 SPRTN knockout derivatives (KO1 and KO2). Cells were exposed to 400 μM FA for 2 h. DPC coefficients were determined for each cell line at the indicated time points and normalized against the 0 time point to arrive at the percentages of DPCs remaining.
(B) DPC accumulation in wild-type TK6 cells and 2 SPRTN knockout derivatives continuously exposed to low-dose FA (50 μM) for 12 and 24 h.
(C) DPC accumulation in wild-type TK6 cells and a TDP1/2−/− double-knockout derivative continuously exposed to CPT (75 nM) for 12 and 24 h.
Each data point in the plots and in the bar graphs was derived from no less than 5 biological repeats with duplicates or triplicates. One-way ANOVA analyses for the three time points 6, 9, and 24 h generated p < 0.0001 in (A) for the F-test and interested pairwise Tukey test results are indicated. Number of biological repeats with triplication = 3. The error bars depict standard deviations.
In addition to the acute FA treatment, we analyzed DPC accumulation under continuous exposure of FA at a dose near mammalian plasma concentration (50 μM) (Ridpath et al., 2007). Following 12- and 24-h exposures to 50 μM FA, wild-type and SPRTN mutants were collected, and the amount of DPCs in each sample was analyzed by the ARK assay (Figure 5B). In wild-type TK6 cells, the constant exposure to 50 μM FA resulted in a slight increase in DPC accumulation, suggesting that under physiological FA concentrations, wild-type TK6 cells possess a sufficient repair capacity to prevent the accumulation of DPCs. In the SPRTN mutants, the amount of accumulated DPCs in KO1 and KO2 showed a 3.2 ± 0.5-fold (p = 0.005) and a 4.9 ± 0.8-fold (p = 0.0001) increase at 12 h and a 2.5 ± 0.6-fold (p = 0.05) and a 3.9 ± 1.0-fold (p = 0.04) increase at 24 h, respectively, compared to the mock-treated samples. Lower levels of DPC accumulation at the 24-h time points are likely attributable to the diminishing potency of FA in the culture medium containing 10% serum. This result suggests that when exposed to plasma FA concentrations, loss of the SPRTN gene results in significantly increased DPC accumulation and that the ARK assay is capable of capturing such accumulation.
To validate the ARK assay in genetic models deficient in enzymatic DPC repair, we examined a TDP1/TDP2 double knockout mutant (TDP1/2−/−) generated in the TK6 background (Figures S3D and S3E). TDP1 and TDP2 display redundant functions in TOP1cc and TOP2cc repair, respectively, by cleaving the enzyme-DNA phosphotyrosyl linkage (Zeng et al., 2012). As a result, the loss of both genes impairs DPC removal and leads to the accumulation of TOP1cc and TOP2cc (Hoa et al., 2016). We exposed the parental TK6 cells and the TDP1/2−/− knockout mutant to 75 nM CPT and analyzed DPC coefficients after 12 and 24 h using the ARK assay. We found that DPC accumulation in wild-type TK6 cells at 12 and 24 h increased by 36.3% ± 5.2% and 39.7% ± 4.34%, respectively, compared to mock-treated samples. In the TDP1/2−/− knockout cells, the DPC coefficients increased to 129.9% ± 11.1% and 160.7% ± 9.8%, respectively at 12 and 24 h (Figure 5C). Thus, the ARK assay has detected a 3.6 ± 0.3-fold (p < 0.001) and a 4.0 ± 0.2-fold (p < 0.001) increase in DPCs as a result of TDP1 and TDP2 deletion. To verify that cell death resulting from DPC induction is not a significant confounding factor in the above results and that the doses of FA and CPT treatment are within physiological range, we analyzed cell viability at each time point (Figure S4). The results show that the cell viability changes in these experiments (Figures 5A–5C) were <33%, 7%, and 30%, respectively. Therefore, cell death is not likely a significant factor in the DPC coefficient readings from the ARK assay. These data demonstrate that enzymatic DPCs can be effectively detected with a significant span of readout by the ARK assay.
Parallel Comparison between the ARK Assay and the RADAR Assay in the Detection of CPT-Induced DPCs
Background level and signal recovery are two key aspects for both direct and indirect DPC assays. We have shown that the ARK assay has substantially improved sensitivity as an indirect DPC assay. To compare the capability of the ARK assay to direct DPC assay approaches, we performed a parallel comparison between the ARK assay and the RADAR assay. HeLa, 293A, and TK6 cells were treated or mock-treated with 10 μM CPT for 1 h. Cells were harvested and subjected to the standard RADAR assay. Results from the TOP1 antibody blotting produced 6.4 ± 2.0-, 4.1 ± 0.9-, and 10.0 ± 3.6-fold TOP1cc induction for 293A, HeLa, and TK6 cells, respectively (Figures 6A and 6B).
Figure 6. Parallel Comparison of Assay Readout between the RADAR and ARK Assays.
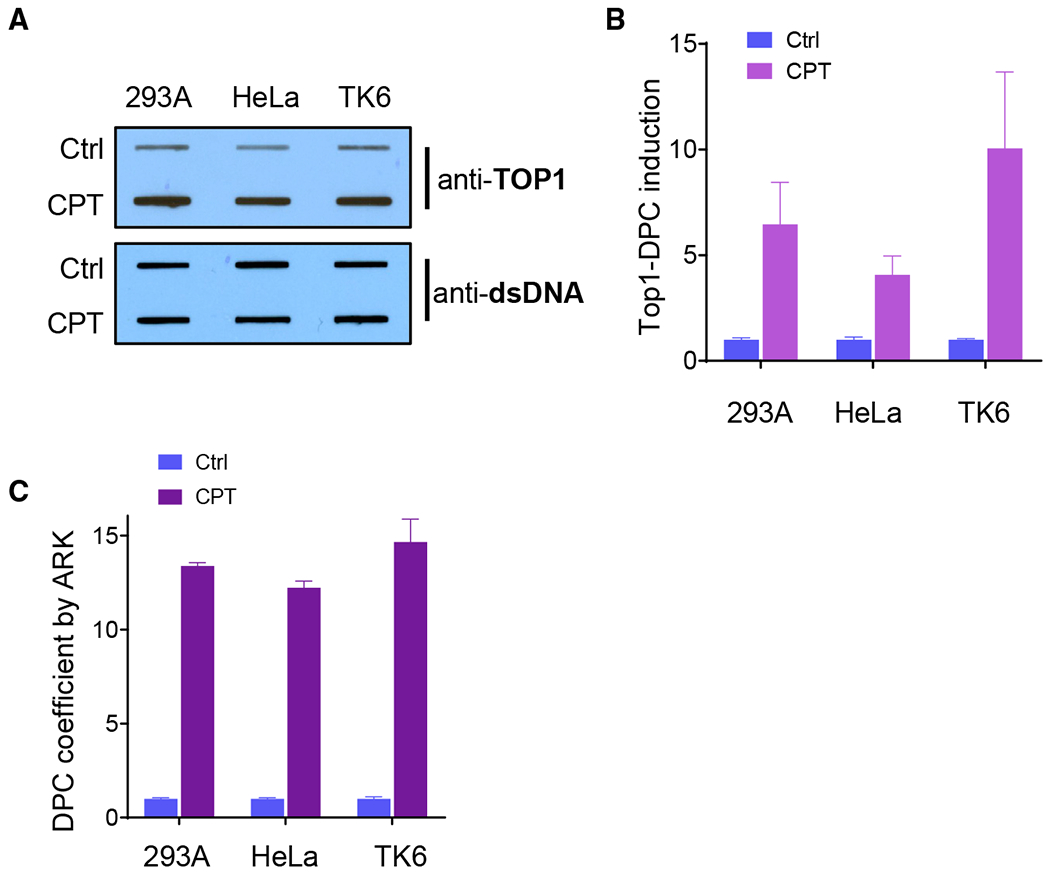
(A) RADAR assay of 293A, HeLa, and TK6 cells treated with 10 μM CPT for 1 h. Upper panel: representative slot blot of DPC samples visualized by an anti-TOP1 antibody as performed by the standard RADAR assay. Lower panel: slot blot (using anti-double-stranded DNA [dsDNA]) of DNA isolated from corresponding samples in the upper panel.
(B) Relative TOP1-DPC induction by normalizing the TOP1-DPC chemiluminescent signal to the corresponding DNA signal in (A). The background levels of the mock-treated sample (Ctrl) for each cell line were set to 1.
(C) ARK detection of TOP1-DPC from identical cell samples used in (A).
(D) DPC isolates were prepared by the ARK assay protocol from identical cell samples used in (A) and blotted with an anti-TOP1 antibody (upper panel). Lower panel: slot blotting of DNA isolated from corresponding samples in the upper panel.
The number of biological repeats ≥3. The error bars depict standard deviations.
In parallel, identical cell preparations were analyzed using the ARK assay. As shown in Figure 6C, the fold increases in TOP1cc induction are 13.4 ± 0.31, 12.2 ± 0.6, and 14.7 ± 2.1, for 293A, HeLa, and TK6 cells, respectively. This result suggests that the ARK assay can be more sensitive than the antibody-based RADAR assay in detecting TOP1cc DPCs.
To further test whether the ARK assay is capable of TOP1cc detection under an extended dose range, we treated 293A, HeLa, and TK6 cells with 2.5 μM CPT for 1 h and performed the ARK and RADAR assays in parallel on the same samples. The ARK assay detected an average DPC fold induction of 8.0 ± 0.15, 8.6 ± 0.74, and 8.5 ± 0.58 compared to 1.9 ± 0.44, 2.3 ± 0.20, and 2.0 ± 0.32 from the RADAR assay for 293A, HeLa, and TK6 cells, respectively (Figure S5). Results from these comparisons suggest that the ARK protocol for DPC detection is more sensitive in TOP1cc detection than the RADAR assay, with a lower dose range of CPT. These advantages render this new assay a versatile and reliable method in analyzing DPC repair activities under physiological conditions.
Removal of DPCs in Nucleotide Excision Repair and Fanconi Anemia Pathway-Deficient Cells
The nucleotide excision repair (NER) pathway has been shown to repair DPCs with small protein moieties (Minko et al., 2002; Nakano et al., 2009) or to act in the removal of DPC repair intermediates in which the bulkiness of the protein adducts is proteolytically reduced (Reardon and Sancar, 2006). The Fanconi anemia pathway has been implicated in DPC repair, as observed in Fanconi anemia pathway-deficient cells that accumulate DNA crosslinking damage arising from endogenous FA (Pontel et al., 2015). Furthermore, cells deficient in the Fanconi anemia pathway are sensitive to DPC-inducing agents (Rosado et al., 2011). To address the question of whether these distinct repair mechanisms contribute to DPC repair, we generated in HeLa cells an XPA−/− mutant to serve as an NER-deficient model, an FANCL−/− mutant to serve as a Fanconi anemia pathway-deficient model, and a double-knockout mutant for these genes to serve as a model deficient in both pathways (Figures S6 and S7). When HeLa cells were exposed to FA, the formation of FANCD2 foci was observed in the wild-type but not in the FANCL−/− mutant, reflecting the activation of the Fanconi anemia pathway (Figure S7C). Analyses of clonogenic survival (Figure 7A) showed that the loss of XPA leads to a slight increase in FA sensitivity, while the deletion of FANCL drastically decreases cell survival. The XPA−/−/FANCL−/− mutant exhibited a minute increase in sensitivity compared to the FANCL−/− cells. These observations indicate that the NER mechanism has a limited function in FA-induced DNA crosslinking repair, while the Fanconi anemia pathway plays a more significant role in countering the genotoxicity from FA exposure.
Figure 7. Removal of DPCs in Fanconi Anemia and NER Mutants.
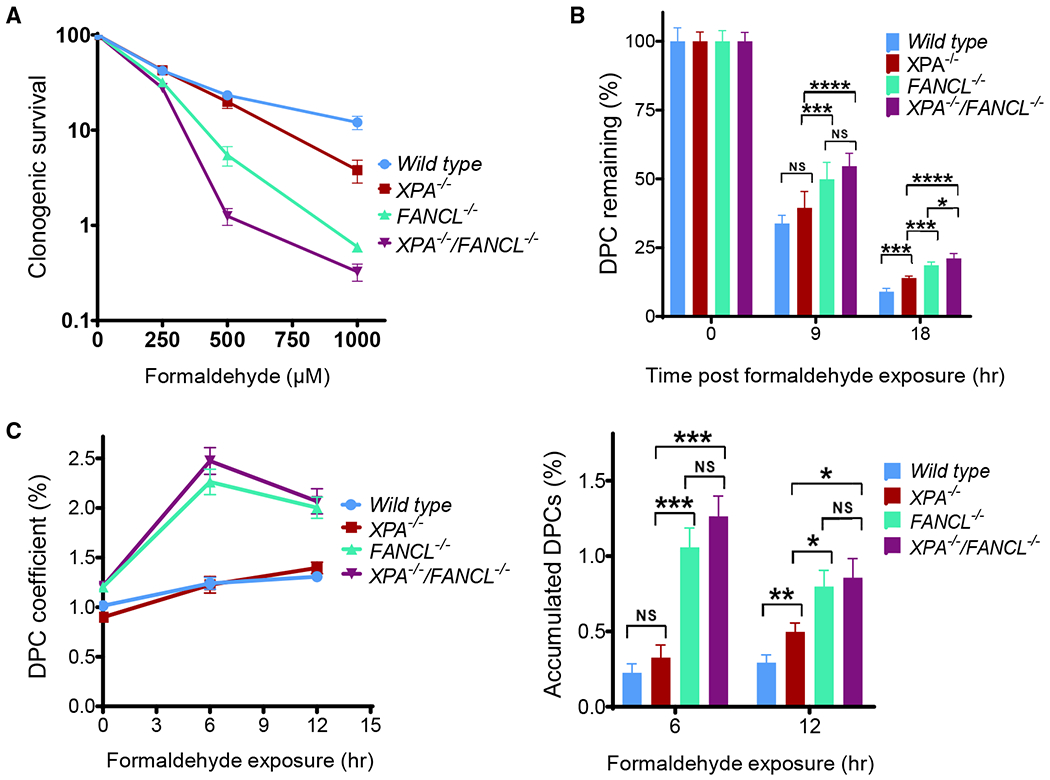
(A) Clonogenic survival of HeLa wild-type, XPA−/−, FANCL−/−, XPA−/−/FANCL−/− cells treated with FA.
(B) Removal of DPCs in wild-type HeLa cells and other indicated knockout derivatives. Cells were exposed to 500 μM FA for 2 h. DPC coefficients were determined for each cell line at indicated time points and normalized against the 0 time point to arrive at the percentages of DPC remaining.
(C) Left panel: DPC accumulation in wild-type HeLa cells and other indicated knockout derivatives continuously exposed to low-dose FA (75 μM) for 6 and 12 h; right panel: analysis of FA-induced DPC accumulation among HeLa cells examined in the left panel after formaldehyde treatment for 6 and 12 h, respectively. Accumulated DPCs are calculated from the DPC coefficient at the selected time point with a deduction of background level (time 0) for corresponding HeLa cell lines.
Number of biological repeats with duplication: n = 2 for (A), n = 4 for (B) and (C). The error bars depict standard deviation. One-way ANOVA analyses for the indicated time points (9 and 18 hr in B; 6 and 12 h in C) generated p < 0.0002 for the F-test, and interested pain/vise Tukey test results are displayed.
To determine whether the clonogenic survival outcomes are connected to the DPC repair capacities of these mutants, we measured the DPC coefficient in the XPA and FANCL mutants using the ARK assay. XPA−/−, FANCL−/−, and XPA−/−/FANCL−/− cells were subjected to an acute treatment of FA (500 μM) for 2 h, and the levels of protein-conjugated DNA were determined at 9 and 18 h after the treatment. As shown in Figure 7B, the XPA−/−/FANCL−/− and the FANCL−/− mutants retained significantly higher and indistinguishable levels of DPCs compared to wild-type HeLa cells and the XPA−/− mutant, suggesting that FANCL function contributes to DPC repair.
To validate the results derived from acute DPC induction, the mutant cells were treated with sustained exposure to low-dose formaldehyde (75 μM) for 6 and 12 h and followed by the ARK assay to determine the accumulation of DPCs. We found that the XPA−/−/FANCL−/− and the FANCL−/− mutants exhibited a markedly increased DPC level compared to the XPA−/− mutant and wild-type HeLa cells. The relatively lower levels of DPC accumulation in XPA−/−/FANCL−/− and FANCL−/− mutants at the 12-h time point are likely attributable to the diminishing potency of FA in the culture medium and the presence of FANCL-independent repair mechanism(s). These results, which are consistent with the clonogenic survival data, suggest that the Fanconi anemia mechanism is involved in the removal of FA-induced DPC lesions.
DISCUSSION
In this study, we sought to develop a sensitive and versatile assay for the quantitative analysis of DPCs. The new assay, ARK, features a stringent DPC-isolation scheme that effectively reduced the background level of existing assays without signal loss. We showed that the ARK assay can detect both enzymatic and non-enzymatic DPCs with high sensitivity and expanded readout range. Analyses using DPC repair genetic models indicate that the ARK assay is effective in detecting their DPC repair defects. Moreover, the ARK assay retains the simplicity of the indirect DPC assay, allowing a large number of samples to be analyzed within 7 h.
Among the two categories of DPC assays, a direct assay quantifies DNA-conjugated proteins and an indirect assay measures protein-linked DNA as a percentage of total DNA. However, the isolation of DPCs is critical to both direct and indirect assays in terms of sensitivity and range of readout. A key factor in DPC isolation is the contamination from the noncovalent association between DNA and proteins. The main feature of the DPC isolation method in the ARK assay is the introduction of dual denaturation steps to minimize the noncovalent DNA protein complex, which is a major source of false DPC signals. The guanidine-mediated chaotropic lysis provides, in general, a more complete homogeneous cell lysis, which is capable of stripping most hydrogen bond- and charge-based DNA-protein associations from the abundance of chromatin proteins such as histones and high mobility group proteins. The resulting DPC isolate is further subjected to SDS-mediated protein denaturation, providing an additional and irreversible disruption of noncovalent DNA-protein association. Most likely, the dual denaturation steps and dual ethanol-KCl precipitation substantially decreases the amount of noncovalent DNA-protein complexes, allowing increased assay sensitivity and an extended readout range in the ARK assay (Figure 2D).
Additional optimizations of DPC preparation contributed to the improved background reading and readout range of the ARK assay. We found that the background reading is reduced by ~2-fold when cells were lysed with heated guanidine buffer, presumably allowing more complete dissociation and denaturation of noncovalent protein binding to the DNA. Optimized shearing of DPC samples also decreases the background. As such, each step of the ARK assay has gone through optimization to achieve the ~10-fold improvement over the convention protocol.
From a technical standpoint, the ARK assay retains key advantages of the indirect assay, capable of global enzymatic and non-enzymatic DPC detection without relying on antibody detection and image-based quantifications. DPCs formed under physiological conditions can be readily and consistently detected from 100,000 cultured cells. The entire ARK protocol takes <7 h to complete. The short assay duration and the procedural simplicity render the ARK assay capable of processing a large number of samples. The procedure can also assay tissue samples and be adapted to high-throughput purposes.
As an indirect assay, ARK provides effective measurement of global DPCs. For protein-specific DPC analysis, this assay may have its limitations. We showed that for TOP1cc, our protocol yielded comparable or much higher fold induction compared to the conventional RADAR assay (Figures 6 and S5). When assaying 5-aza-2′-deoxycytidine-induced DNA methyltransferase 1 DPCs (Kiianitsa and Maizels, 2013), the induction fold from the ARK assay is 2.5 (data not shown), whereas the RADAR assay with the same samples can reach 3.0-fold (Kiianitsa and Maizels, 2013). When combined with ELISA detection, the RADAR assay provides enhanced measurement of low abundant and protein-specific DPCs with high-quality antibodies (Kiianitsa and Maizels, 2014).
Results from our study and others show that Fanconi anemia pathway mutant mice and cells are deficient in repairing FA-mediated crosslinking damage (Ridpath et al., 2007; Rosado et al., 2011), which include DPCs and DNA interstrand crosslinks. Using the ARK assay, we show that DPC repair is impaired in Fanconi anemia mutant cells, supporting the notion that the Fanconi anemia pathway is involved in DPC removal and that the accumulation of DPC may account for, at least in part, the increased FA sensitivity of Fanconi anemia mutant cells. The ARK assay results also showed that the loss of XPA has a minimal impact on the accumulation of DPCs. This observation is consistent with the notion that the NER mechanism has a limited role in the overall DPC repair in mammalian cells (Ide et al., 2011). However, the Fanconi anemia pathway is unlikely the only mechanism for DPC removal, as reflected by our results, demonstrating that DPC removal is partially impaired in the XPA−/−/FANCL−/− and FANCL−/− mutants (Figures 7B and 7C). SPRTN-dependent and proteasome-mediated DPC repair mechanisms have been shown to be independent of the Fanconi anemia mechanism (Larsen et al., 2019). Given the highly variable nature of the protein adducts in DPCs, it is most likely that the repair of DPC lesions is carried out by multiple pathways with varying contributions in different cell types and conditions.
The ARK assay we describe here offers a novel and effective tool for quantitative DPC analyses. With markedly increased sensitivity, more questions regarding the formation, repair, and regulation of cellular DPCs can be experimentally addressed with this new methodology. Because the ARK provides a comprehensive measurement of cellular DPCs and does not rely on antibodies, its application on DPC repair biology extends to both enzymatic and non-enzymatic DPCs. Given the increasing interest in DPC repair biology and its direct connections to prevalent anti-cancer drugs, the ARK assay is expected to contribute to both basic and translational studies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lei Li (leili@mdanderson.org). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All cell lines used in this study were authenticated with STR marker using MD Anderson Cancer Center Cell Line Identify service (Institutional policy ACA #1044) and tested negative for mycoplasma contamination using the MycoAlert Kit (Lonza). Human cell lines HeLa and 293A were purchased from American Type Culture Collection (ATCC) and Invitrogen, respectively, and both cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 100 U/ml penicillin-streptomycin. TK6 and its isogenic knockout mutants SPRTN−/−, TDP1/2−/− (double knockout for both TDP1 and TDP2) were generated by the Takeda group and maintained in RPMI-1640 containing 10% horse serum, 2 mM glutamine, 100 U/ml penicillin-streptomycin, 1 mM sodium pyruvate, 4500 mg/L glucose, and 1500 mg/L sodium bicarbonate. Cells were cultured at 37°C in a humidified incubator infused with 5% CO2.
HeLa cells are of female origin. Other cell lines, such as 293A and TK6 are of unknown gender origin.
METHOD DETAILS
Generation of SPRTN−/− human TK6 cells
To disrupt SPRTN, we designed a guide RNA, 5′-TCAAAAGGGGTTCGCTGAGACGG-3′, targeting the second exon and gene-targeting constructs. The arrangement of the CRISPR target site and integration cassette is depicted in Figures S3A–S3C. We generated two gene-targeting constructs containing the NeoR or PuroR marker using the Seamless Cloning and Assembly Kit (Thermo Fisher Scientific, US). The genomic DNA was amplified from TK6 cells with primers F1 and R1 (primer information is available in Table S1). The resulting PCR product was further amplified using primers F2 and R2 and primers F3 and R3 to obtain the 5′- and 3′-arms, respectively. The 5′ ends of the F2 and R2 primers have 20-nt sequences identical to the upstream and downstream of the ApaI site in both the DT-A-pA/loxP/PGK- NeoR-pA/loxP and DT-A-pA/loxP/PGK-PuroR-pA/loxP vectors (provided from the Laboratory for Animal Resources and Genetic Engineering, Center for Developmental Biology, RIKEN Kobe, http://www2.clst.riken.jp/arg/cassette.html) (Ninagawa et al., 2014). The 5′ ends of the F3 and R3 primers have 20-nt sequences identical to the upstream and downstream of the AflII site in these vectors. The DT-A-pA/loxP/PGK-NeoR-pA/loxP or DT-A-pA/loxP/PGK-PuroR-pA/loxP vectors were digested with both ApaI and AflII, which cut at the 5′ and 3′ of the selection marker genes, respectively. We combined the digested DNAs containing either NeoR or PuroR marker with the 5′- and 3′-arms using the Seamless Cloning and Assembly Kit (Thermo Fisher Scientific, US), and generated the two gene-targeting constructs containing the NeoR or PuroR marker. 6 μg of CRISPR and 2 μg each of the two gene-targeting vectors were transfected into 4 × 106 TK6 cells using the Neon Transfection System (Life Technologies, US) with three times 1350 V pulse with a 10 ms pulse width. After electroporation, cells were released into 20 mL drug-free medium containing 10% horse serum. Forty-eight hours later, cells were seeded into 96-well plates for selection with both G418 (Neomycin) and puromycin antibiotics for three weeks. The gene disruption was confirmed by genotyping PCR (using F4/F5 and R4 primers) and RT-PCR (using F6 and R5 primers) (Figures S3A–S3C).
Generation of TDP1/2−/− TK6 Cells
To generate the double mutant of TDP1−/−/TDP2−/−, two targeting vectors and pX330-gRNA (the gRNA was inserted into the BbsI site of pX330 vector (Cat# 42230, Addgene, US) (Cong et al., 2013) of TDP2 were transfected into TDP1−/− mutant cells, which was described previously (Hoa et al., 2016). To delete the TDP2 gene, we generated a guide RNA, 5′-CCAAGAAGGTCCAAACTTCG-3′, targeting the catalytic site. The left and right arms were amplified using F7/R7 and F8/R8 primers, respectively (Table S1). The resulting fragments were assembled with either DT-ApA/NEOR or DT-ApA/PUROR having been digested with ApaI and AflII, using a GeneArt Seamless cloning kit (Invitrogen). Gene target events were confirmed by genomic PCR using two sets of primers (F4/R6 for NEOR and F5/R6 for PUROR) and western blotting against anti-Tdp2 antibody (A302-737A, Bethyl) (Figures S3D and S3E).
Generation of XPA−/− and XPA−/−FANCL−/− HeLa cells
The XPA−/− and XPA−/−FANCL−/− mutant HeLa cell lines were generated on the wild-type and FANCL−/− mutant background, respectively. The FANCL−/− mutant was generated before as previously reported (Tian et al., 2017). gRNA targeting the XPA locus (5′-GGCGGCTTTAGAGCAACCCG-3′) inserted into lentiGuide-Puro (Addgene #52963) and together with lentiCas9-Blast (Addgene #52962) was used to infect wild-type or FANCL−/− cells (Mali et al., 2013; Sanjana et al., 2014). Following selection, single cells were seeded in 96-well plates. Western blots were used to screen for XPA null clones (ThermoFisher Cat# MS-650-P0). Candidate clones negative for XPA expression were subjected to genotyping to identify indels for both alleles. Ten or more clones of each genomic PCR fragment were sequenced to confirm the knockout genotype. The genetic identities of all mutant cell lines were validated by Sequenom Pro SNP fingerprinting.
Drug treatment
293A and HeLa cells were grown to 60%–70% confluence in 6-well plates and TK6 cells were maintained at 0.5 × 106 cells/ml in 100-mm Petri dishes before treatment with formaldehyde for 2 hr and CPT or ETO for 1 hr. For dose-response studies, cells were collected immediately after the treatment. For time-course experiments of DPC repair, cells were treated with 400 μM of formaldehyde for 2 hr, washed, and replaced with fresh media. Cells were collected at different time points as needed. For DPC accumulation studies under low doses of formaldehyde and CPT, continuous treatment was carried out for either 12 hr or 24 hr before collection.
Quantification of DNA by the PicoGreen dsDNA assay
The assays were performed in a 96-well plate (Corning) following the manufacturer’s instructions. The final total volume for each reaction/well was 250 μl. DNA standards of concentrations 8 ng, 80 ng, 400 ng, 800 ng, 1400 ng, 2000 ng/mL were prepared in a volume of 125 μL TE buffer and placed in the wells. Samples of proper volume, which was determined by fitting the best linear range, were diluted with TE buffer in the wells and adjusted to a final volume of 125 μl. Equal volume (125 μl) of the PicoGreen reagent diluted (1:200) from the original stock was added to the wells and mixed. Fluorescence detection was carried out on a Synergy2 plate reader (BioTek). The DNA quantity for each sample was derived from the fluorescence comparison with the standard curve. A control sample from a mock experimental well was used as a blank. The excitation and emission wavelengths were 485 nm and 528 nm, respectively. The detection protocol setting for Optics position was top 50% and the Gain was set at 50.
Isolation of DPC-associated DNA by the ARK procedure
To harvest cells, culture media was removed through aspiration directly from 6-well plates for adherent cells or after centrifugation (300 × g for 5 min) on the Thermo IEC CentraCL3 centrifuge for suspension cells. For 293A and HeLa 0.8 × 106 cells and for TK6 2.5 × 106 cells were lysed (without cell wash) in 950 μL of M buffer (MB) prewarmed at 55°C for 5-10 min. MB consists of 5.6 M GTC, 10 mM Tris–HCl (pH 6.5), 20 mM EDTA, 4% Triton X-100, 1% Sarkosyl, and 1% dithiothreitol. Cell lysates were collected after scraping and mild shearing with a 1-mL pipettip (passing 6 times). After a brief spin to eliminate bubbles, the lysates were further sheared by passing through a 22-gauge needle 6 times and followed by DNA precipitation with an equal volume of pre-chilled ethanol (−20°C). Free DNA and DPCs were recovered as a pellet after a full speed centrifugation (micro-centrifuge) at 4°C for 20 min. The pellet was washed at 4° C one time in the buffer composed of 20 mM Tris-HCl pH 6.5, 150 mM NaCl and 50% ethanol. Samples (no air drying) can either be stored at −80°C or used directly in the next step of the protocol. To dissolve the DNA/DPC pellet 0.5 mL of pre-warmed 1% SDS, 20 mM Tris-HCl (pH 7.5) was added and incubated at 42°C for 6 min. The samples were briefly spun (3500 × g, 30 s, on micro-centrifuge) and shorn by passing through a 25-gauge needle 5 times. As soon as 3-4 samples were processed, a 45 s full-speed centrifugation was performed to reduce the bubbles and 0.5 mL of 200 mM KCl, 20 mM Tris-HCl (pH 7.5) buffer was added to precipitate SDS-bound DPCs. The samples were incubated on ice for 6 min to complete the DPC precipitation. The precipitate was pelleted after a full-speed centrifugation for 5 min in cold room. The supernatant was collected and set aside on ice for DNA measurement. The DPC pellet was washed in 1.5 mL of 100 mM KCl, 20 mM Tris-HCl (pH 7.5) by incubating at 55°C for 10 min, on ice for 6 min, followed by centrifugation at 20,000 × g speed in a micro-centrifuge at 4°C for 5 min. The supernatant was collected and combined with the previously collected supernatant for total DNA measurement. The wash procedure was repeated one more time before dissolving the DPC pellet in 1 mL of the proteinase K buffer (100 mM KCl, 20 mM Tris-HCl, pH 7.5, and 10 mM EDTA). Proteins were digested by adding proteinase K to a final concentration of 0.2 mg/ml and incubating at 55°C for 45 min. The digestion was chilled on ice for 6 min and centrifuged at 20,000 × g for 10 min at 4°C to remove any debris. The 1 mL supernatant in this tube contains the DPC-associated DNA.
To determine the DPC coefficient, 10 μL from the 4 mL of the recovered free DNA supernatant, and 62.5 μL from the 1 mL supernatant of the DPC resuspension were used for DNA quantification by the PicoGreen assay kit. The DPC coefficient is expressed as the percentage of DNA from DPCs over the total DNA in each sample.
K-SDS assay
The K-SDS assay was performed by following the previously described non-isotopic protocol (Costa et al., 1996; Liu et al., 2006; Ye et al., 2013; Zhitkovich and Costa, 1992). Briefly, 0.8 × 106 cells were collected through trypsinization, washed twice with ice-cold PBS, and resuspended in 100 μL PBS. The cells were lysed with 0.5 mL of a 2% SDS solution and the DNA was sheared by passing the lysates through a 22-gauge needle six times. The lysates were frozen at −80°C and thawed at 65°C for 10 min. An equal volume of 200 mM KCl, 20 mM Tris-HCl (pH 7.5) buffer was added to precipitate SDS-bound DPCs via incubation at 4°C for 6 min. After centrifugation at 6,000 × g for 5 min at 4°C, the supernatants were collected. The resulting pellets were washed three times at 65°C for 10 min with a solution consisting of 100 mM KCl, 20 mM Tris-HCl (pH 7.5), followed by incubation on ice for 6 min for precipitation each cycle. All supernatants were combined for free DNA measurement. The final pellets were digested with proteinase K, and 80 μl of BSA (5 mg/ml) was added to each sample. Quantification of both DPC-bound DNA and free DNA was performed by the PicoGreen assay kit.
RADAR assay
The RADAR assay for TOP1cc was performed following the published protocol (Kiianitsa and Maizels, 2013). Briefly, cells were seeded in 6-well plates and treated with 2.5-10 μM CPT. After treatment, cells were lysed in 0.8 mL MB (6M GTC, 10 mM Tris-HCl, pH 6.5, 20 mM EDTA, 4% Triton X-100, 1% Sarkosyl and 1% dithiothreitol). Nucleic acids were precipitated out of the lysate by adding half volume (0.4 mL) of 100% ethanol, incubating at −20 for 5 min, and centrifuging at maximum speed for 15 min. After 2 washes in 75% ethanol, pellets were resuspended in 0.2 mL of 8 mM NaOH without drying. For immunodetection, a small portion of the lysates were diluted in Tris-buffered saline [10mM Tris (pH 7.5), 150mM NaCl] for a final volume of 0.2 mL, which was then slot blotted onto a nitrocellulose membrane with the Bio-Rad BioDot SF. Band intensities were analyzed with the online ImageJ software and normalized to the amount of loaded DNA. Antibodies used in this assay were rabbit anti-TOP1 (Bethyl, 1:2000) and mouse anti-dsDNA (Abcam, 1:2000).
Clonogenic survival assay
Cells at 50% confluency were treated with formaldehyde for 2 hours in serum-free media and then seeded in duplicates in 6-well plates. After 10 to 14 days in regular medium, colonies were fixed with 6% glutaraldehyde (v/v) and stained with 0.5% crystal violet (w/v) for visualization.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using built-in functions in GraphPad Prism Version 8. For experiments involving three or more groups but only one variable, one-way ANOVA analysis was conducted to broadly determine if the means of all groups are all the same through an F-test with p < 0.05 being considered significant. If significant, further Tukey-Cramer test was performed for comparisons between any two groups. Analysis involving only two groups was carried out using Student t tests with p < 0.05 being considered statistically significant. Symbols for different significance levels are assigned as the following: * for p < 0.05; ** for p < 0.01; *** for p < 0.001; **** for p < 0.0001; NS for not significant. All data for measured variables were expressed as means ± SD as indicated. Sample size was n ≥ 3 containing both biological and experimental replicas, as indicated.
DATA AND CODE AVAILABILITY
No new datasets or code were generated in this paper.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Tdp2 antibody | Bethyl Laboratories | Cat# A302-737A; RRID:AB_10631698 |
| anti-TOP1 antibody | Bethyl Laboratories | Cat# A302-589A; RRID:AB_2034865 |
| anti-dsDNA antibody | Abcam | Cat# ab27156; RRID:AB_470907 |
| anti-XPA antibody | Lab Vision | Cat# MS-650-P0; RRID:AB_142101 |
| anti-vinculin antibody | Sigma-Aldrich | Cat# V9131; RRID:AB_477629 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Formaldehyde (FA) | Sigma-Aldrich | Cat# F8775 |
| Guanidine thiocyanate (GTC) | Sigma-Aldrich | Cat# G9277 |
| Camptothecin (CPT) | Sigma-Aldrich | Cat# 390238 |
| PicoGreen | Thermo Fisher Scientific | Cat# P7589 |
| MycoAlert | Lonza | Cat# LT07-218 |
| Etoposide (ETO) | Abcam | Cat# ab120227 |
| Critical Commercial Assays | ||
| GeneArt Seamless Cloning and Assembly Kit | Thermo Fisher Scientific | Cat# A13288 |
| Neon Transfection Kit | Thermo Fisher Scientific | Cat# MPK10025 |
| Experimental Models: Cell Lines | ||
| 293A | Invitrogen | Cat# R70507 |
| TK6 | Dr. Shunichi Takeda | N/A |
| TK6 SPRTN−/− | This paper | N/A |
| TK6 TDP1−/− | Hoa et al., 2016 | N/A |
| TK6 TDP1/2−/− | This paper | N/A |
| HeLa | ATCC | ATCC CCL-2; RRID:CVCL_0030 |
| HeLa XPA−/− | This paper | N/A |
| HeLa FANCL−/− | Tian et al., 2017 | N/A |
| HeLa XPA−/−;FANCL−/− | This paper | N/A |
| Oligonucleotides | ||
| SPRTN gRNA: TCAAAAGGGGTTCGCTGAGACGG | This paper | N/A |
| TDP2 gRNA: CCAAGAAGGTCCAAACTTCG | This paper | N/A |
| XPA gRNA: GGCGGCTTTAGAGCAACCCG | This paper | N/A |
| TK6 Primers F/R 1-8 see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| DT-A-pA/loxP/PGK- NeoR-pA/loxP | Laboratory for Animal Resources and Genetic Engineering, Center for Developmental Biology, RIKEN Kobe, http://www2.clst.riken.jp/arg/cassette.html | Cassette #16 |
| DT-A-pA/loxP/PGK-PuroR-pA/loxP | Ninagawa et al., 2014 | N/A |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Cong et al., 2013 | Addgene Cat# 42230; RRID:Addgene_42230 |
| lentiGuide-Puro | Sanjana et al., 2014 | Addgene Cat# 52963; RRID:Addgene_52963 |
| lentiCas9-Blast | Sanjana et al., 2014 | Addgene Cat# 52962; RRID:Addgene_52962 |
| Software and Algorithms | ||
| ImageJ | ImageJ | https://imagej.nih.gov/ij/; RRID:SCR_003070 |
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
Highlights.
A sensitive assay for the global measurement of DNA-protein crosslinks (DPCs)
Stringent DPC purification scheme improves DPC detection and minimizes background signals
Assay is capable of both enzymatic and nonenzymatic DPC detections
DPC repair is impaired in Fanconi anemia pathway-deficient cells
ACKNOWLEDGMENTS
This work is supported in part by NIH R01 CA190635 and CA193124-Project 3 to L.L.; the Cancer Prevention and Research Institute of Texas (RP150538 to J.C., T.P., and L.L.); and the Olive Stringer endowed professorship (L.L.). S.T. and L.K.S. are recipients of the JSPS Core-to-Core international collaboration program.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.067.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Barker S, Weinfeld M, Zheng J, Li L, and Murray D (2005). Identification of mammalian proteins cross-linked to DNA by ionizing radiation. J. Biol. Chem 280, 33826–33838. [DOI] [PubMed] [Google Scholar]
- Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, and Zou L (2010). CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chválová K, Brabec V, and Kaspárková J (2007). Mechanism of the formation of DNA-protein cross-links by antitumor cisplatin. Nucleic Acids Res. 85, 1812–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Zhitkovich A, Gargas M, Paustenbach D, Finley B, Kuykendall J, Billings R, Carlson TJ, Wetterhahn K, Xu J, et al. (1996). Interlaboratory validation of a new assay for DNA-protein crosslinks. Mutat. Res 369, 13–21. [DOI] [PubMed] [Google Scholar]
- de Graaf B, Clore A, and McCullough AK (2009). Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair (Amst.) 8, 1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, and Walter JC (2014). Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell 159, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielden J, Ruggiano A, Popovic M, and Ramadan K (2018). DNA protein crosslink proteolysis repair: from yeast to premature ageing and cancer in humans. DNA Repair 71, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, and Patel KJ (2012). Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489, 571–575. [DOI] [PubMed] [Google Scholar]
- Heck HD, Casanova M, and Starr TB (1990). Formaldehyde toxicity-new understanding. Crit. Rev. Toxicol 20, 397–426. [DOI] [PubMed] [Google Scholar]
- Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsutsui K, et al. (2016). Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 64, 1010. [DOI] [PubMed] [Google Scholar]
- Ide H, Shoulkamy MI, Nakano T, Miyamoto-Matsubara M, and Salem AM (2011). Repair and biochemical effects of DNA-protein crosslinks. Mutat. Res 711, 113–122. [DOI] [PubMed] [Google Scholar]
- Ide H, Nakano T, Salem AMH, and Shoulkamy MI (2018). DNA-protein cross-links: Formidable challenges to maintaining genome integrity. DNA Repair 71, 190–197. [DOI] [PubMed] [Google Scholar]
- Kiianitsa K, and Maizels N (2013). A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Res. 41, e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiianitsa K, and Maizels N (2014). Ultrasensitive isolation, identification and quantification of DNA-protein adducts by ELISA-based RADAR assay. Nucleic Acids Res. 42, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Yu R, Hartwell HJ, Moeller BC, Bodnar WM, and Swenberg JA (2016). Measurement of Endogenous versus Exogenous Formaldehyde-Induced DNA-Protein Crosslinks in Animal Tissues by Stable Isotope Labeling and Ultrasensitive Mass Spectrometry. Cancer Res. 76, 2652–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, and Patel KJ (2011). Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58. [DOI] [PubMed] [Google Scholar]
- Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Räschle M, Walter JC, and Duxin JP (2019). Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell 73, 574–588. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, Philipp M, Sim JCH, Smith KR, Oehler J, et al. (2014). Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat. Genet 46, 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Li CM, Lu Z, Ding S, Yang X, and Mo J (2006). Studies on formation and repair of formaldehyde-damaged DNA by detection of DNA-protein crosslinks and DNA breaks. Front. Biosci 11, 991–997. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, and Machida YJ (2014). Spartan deficiency causes genomic instability and progeroid phenotypes. Nat. Commun 5, 5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, Flatten KS, Sieben CJ, Peterson KL, Baker DJ, Nam HJ, Kim MS, Smyrk TC, Kojima Y, Machida Y, et al. (2017). Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 45, 4564–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X, Groehler A 4th, Michaelson-Richie ED, Villalta PW, Campbell C, and Tretyakova NY (2017). Mass Spectrometry Based Proteomics Study of Cisplatin-Induced DNA-Protein Cross-Linking in Human Fibrosarcoma (HT1080) Cells. Chem. Res. Toxicol 30, 980–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko IG, Zou Y, and Lloyd RS (2002). Incision of DNA-protein crosslinks by UvrABC nuclease suggests a potential repair pathway involving nucleotide excision repair. Proc. Natl. Acad. Sci. USA 99, 1905–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mórocz M, Zsigmond E, Toth R, Enyedi MZ, Pintér L, and Haracska L (2017). DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA. Nucleic Acids Res. 45, 3172–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Katafuchi A, Matsubara M, Terato H, Tsuboi T, Masuda T, Tatsumoto T, Pack SP, Makino K, Croteau DL, et al. (2009). Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem 284, 27065–27076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Xu X, Salem AMH, Shoulkamy MI, and Ide H (2017). Radiation-induced DNA-protein cross-links: mechanisms and biological significance. Free Radic. Biol. Med 107, 136–145. [DOI] [PubMed] [Google Scholar]
- Ninagawa S, Okada T, Sumitomo Y, Kamiya Y, Kato K, Horimoto S, Ishikawa T, Takeda S, Sakuma T, Yamamoto T, and Mori K (2014). EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol 206, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi C, Grothusen G, Anandarajan V, Martinez-Lage Garcia M, Terlecky D, Corzo K, Tanaka K, Nakagawa H, and Noguchi E (2017). Genetic controls of DNA damage avoidance in response to acetaldehyde in fission yeast. Cell Cycle 16, 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olin KL, Cherr GN, Rifkin E, and Keen CL (1996). The effects of some redox-active metals and reactive aldehydes on DNA-protein cross-links in vitro. Toxicology 110, 1–8. [DOI] [PubMed] [Google Scholar]
- Pommier Y, and Marchand C (2011). Interfacial inhibitors: targeting macro-molecular complexes. Nat. Rev. Drug Discov 11, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Huang SY, Gao R, Das BB, Murai J, and Marchand C (2014). Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 19, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, Chandrasekaran G, Broecker V, Wei W, Liu L, et al. (2015). Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol. Cell 60, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quievryn G, and Zhitkovich A (2000). Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 21, 1573–1580. [PubMed] [Google Scholar]
- Quiñones JL, Thapar U, Yu K, Fang Q, Sobol RW, and Demple B (2015). Enzyme mechanism-based, oxidative DNA-protein cross-links formed with DNA polymerase β in vivo. Proc. Natl. Acad. Sci. USA 112, 8602–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, and Sancar A (2006). Repair of DNA-polypeptide crosslinks by human excision nuclease. Proc. Natl. Acad. Sci. USA 103, 4056–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, Buerstedde JM, Gillespie DA, Sale JE, Yamazoe M, et al. (2007). Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 67, 11117–11122. [DOI] [PubMed] [Google Scholar]
- Rosado IV, Langevin F, Crossan GP, Takata M, and Patel KJ (2011). Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol 18, 1432–1434. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulkamy MI, Nakano T, Ohshima M, Hirayama R, Uzawa A, Furusawa Y, and Ide H (2012). Detection of DNA-protein crosslinks (DPCs) by novel direct fluorescence labeling methods: distinct stabilities of aldehyde and radiation-induced DPCs. Nucleic Acids Res. 40, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, and Jentsch S (2015). DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol 16, 455–460. [DOI] [PubMed] [Google Scholar]
- Stingele J, Schwarz MS, Bloemeke N, Wolf PG, and Jentsch S (2014). A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 158, 327–338. [DOI] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjar S, Tainer JA, et al. (2016). Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Mol. Cell 64, 688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenberg JA, Kerns WD, Mitchell RI, Gralla EJ, and Pavkov KL (1980). Induction of squamous cell carcinomas of the rat nasal cavity by inhalation exposure to formaldehyde vapor. Cancer Res. 40, 3398–3402. [PubMed] [Google Scholar]
- Tian Y, Shen X, Wang R, Klages-Mundt NL, Lynn EJ, Martin SK, Ye Y, Gao M, Chen J, Schlacher K, and Li L (2017). Constitutive role of the Fanconi anemia D2 gene in the replication stress response. J. Biol. Chem 292, 20184–20195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trask DK, DiDonato JA, and Muller MT (1984). Rapid detection and isolation of covalent DNA/protein complexes: application to topoisomerase I and II. EMBOJ. 3, 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretyakova NY, Michaelson-Richie ED, Gherezghiher TB, Kurtz J, Ming X, Wickramaratne S, Campion M, Kanugula S, Pegg AE, and Campbell C (2013). DNA-reactive protein monoepoxides induce cell death and mutagenesis in mammalian cells. Biochemistry 52, 3171–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, and Sedgwick B (2002). Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419, 174–178. [DOI] [PubMed] [Google Scholar]
- Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I, et al. (2016). Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Mol. Cell 64, 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walport LJ, Hopkinson RJ, and Schofield CJ (2012). Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol 16, 525–534. [DOI] [PubMed] [Google Scholar]
- Ye X, Ji Z, Wei C, McHale CM, Ding S, Thomas R, Yang X, and Zhang L (2013). Inhaled formaldehyde induces DNA-protein crosslinks and oxidative stress in bone marrow and other distant organs of exposed mice. Environ. Mol. Mutagen 54, 705–718. [DOI] [PubMed] [Google Scholar]
- Yu R, Lai Y, Hartwell HJ, Moeller BC, Doyle-Eisele M, Kracko D, Bodnar WM, Starr TB, and Swenberg JA (2015). Formation, Accumulation, and Hydrolysis of Endogenous and Exogenous Formaldehyde-Induced DNA Damage. Toxicol. Sci 146, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Sharma A, Ju L, Murai J, Umans L, Vermeire L, Pommier Y, Takeda S, Huylebroeck D, Caldecott KW, and El-Khamisy SF (2012). TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic Acids Res. 40, 8371–8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A, and Costa M (1992). A simple, sensitive assay to detect DNA-protein crosslinks in intact cells and in vivo. Carcinogenesis 13, 1485–1489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new datasets or code were generated in this paper.