

Abstract
Introduction:
Systemic lupus erythematosus (SLE) is a multi-system inflammatory autoimmune disease of incompletely understood etiology. It is thought that environmental exposures “trigger” or accelerate the disease in genetically-predisposed individuals.
Areas covered:
Substantial epidemiological evidence exists to support the association between cigarette smoking and the risk of incident SLE. Recent evidence points to current smoking as the specific risk factor, with decreasing risk 5 years after smoking cessation, and the greatest risk for disease characterized by the presence of SLE- specific autoantibodies. Research has begun to search for possible explanations for the temporal nature of the relationship between current smoking and autoantibody positive-SLE. Here we review potential biologic mechanisms linking smoking and SLE risk, including effects upon T and B cells, inflammatory cytokines, oxidative stress, and the formation of short-lived DNA adducts.
Expert Commentary:
The directions for future research in this field include studies of gene-environment interactions, epigenetics, metabolomics and putative biologic mechanisms.
Keywords: SLE (systemic lupus erythematosus), smoking, pathogenesis, etiology, risk
1.0. Introduction
Systemic lupus erythematosus (SLE) is an inflammatory rheumatic disease of immunologic origin. It can present in any number of ways affecting multiple systems of the body: from autoantibody production, fever and rash to polyarthritis, pericarditis and nephritis [1]. Due to the heterogeneous nature of the disease, there is not one phenotype that encompasses all presentations of SLE. The lack of uniformity among SLE patients has made the study of the etiology of the disease complex.
2.0. Genes and Environment both play roles in SLE Pathogenesis
A widely-accepted etiologic model of SLE is that environmental exposures “trigger” the disease in genetically predisposed individuals [1,2]. That is, both genetics and environmental factors play pivotal roles in the development of the disease. Between 80–90% of SLE cases are in women, and disease severity is much greater in African-American and non-white populations [3,4]. Additionally, 5–12% of first-degree relatives of individuals with SLE will develop the disease [5]. Studies of twin concordance can be used in epidemiology to discern both the role of genetics and the influence of environmental factors on disease susceptibility. Twin studies in SLE have shown much higher disease concordance in monozygotic twins (24%–57%) than in dizygotic twins, indicating there is a genetic component to the disease [6–10]. However, even with monozygotic twins the disease concordance is not 100%, indicating genetics do not fully explain a person’s risk for developing SLE. Thus, we can look to the interaction of environmental factors with genetics, which may help us understand both SLE disease susceptibility and mechanistic pathways in its development.
Strong epidemiological evidence exists linking a number of environmental factors, including crystalline silica exposure [11–13], oral contraceptive use [14,15], and current cigarette smoking [16–18] to the development of SLE. Smoking has been causally linked to the development of other autoimmune diseases, including rheumatoid arthritis (RA) and Graves’ disease [19–23]. In RA, smoking is associated with seropositivity of the rheumatoid factor (RF) even in subjects who do not have rheumatoid arthritis [24], suggesting smoking is associated not only with autoimmune disease onset, but may contribute specifically to autoantibody formation in this disease. As in RA, significant epidemiological evidence also exists to support the association between cigarette smoking and the risk of incident SLE.
3.0. Epidemiologic Evidence linking Smoking to Increased SLE Risk
A number of past observational studies, both case-control and cohort designs, suggest cigarette smoking is related to increased risk of developing SLE, while a few others have not found a clear association [18,25–30]. The case-control studies span a wide range of geographic locations, and also vary widely in smoking exposure collection methods (i.e. definition of smoking status, specificity questions defining an exposure window) and SLE phenotype. Prospective cohort studies help alleviate some of the recall bias present in case-control studies, and are particularly useful in examining smoking exposure data as they are collected before the onset of disease. A meta-analysis of seven case-control and two cohort studies that assessed cigarette smoking as a risk factor for developing SLE found a significant risk for the development of SLE among current smokers compared to non-smokers (OR 1.50, 95% CI 1.09–2.08), but not past smokers (OR 0.98, 95% CI 0.75–1.27) [16]. A later meta-analysis with a couple of newer case-control studies confirmed these findings [31].
A growing body of evidence thus points to current smoking specifically as a risk factor for incident SLE, and research in the field has begun to search for possible explanations for the temporal nature of the relationship between smoking and the disease. A recent large cohort study involving more than 238,000 women followed in the prospective Nurses’ Health Study cohorts confirmed that the increased risk of developing SLE was primarily only among current smokers, or past smokers who had quit recently (within 4–5 years) [16]. After this window of time, a smoker’s risk of developing SLE returned to that of a lifelong non-smoker. That is, the effects of cigarette smoking as they relate to SLE development are potentially reversible. This impermanence implies a biological pathway that is potentially reversible.
3.1. Smoking and SLE-specific autoantibodies
Another important development in the study of environmental exposures and SLE risk has been the stratification of SLE subtypes by autoantibody. SLE is a heterogeneous disease characterized by both clinical manifestations and presence of autoantibodies. Just as RA is classified as seronegative or seropositive based on the presence of disease-specific antibodies (RF, anti-CCP), SLE can be classified by the presence of one or many different autoantibodies including many ENAs (anti-Sm, anti-SSA/SSB, anti-RNP) and anti-dsDNA. The anti-dsDNA/autoantibody positive subgroup of SLE is of particular interest because it represents a more severe phenotype of the disease [32]. Anti-dsDNA positive SLE patients are more likely to develop nephritis and vasculitis, two of the worse disease manifestations. Given the homogeneity in this more specific phenotype, and the fact that anti-dsDNA autoantibodies are very specific to SLE [33,34], it is hypothesized that this subtype of SLE could have a different pathogenesis than the more heterogeneous, generalized disease. In the recent work from the Nurses’ Health Study by Barbhaiya et al. [32], the association of smoking with incident SLE risk was examined stratified by anti-dsDNA status: current smoking was strongly associated with the risk of anti-dsDNA+ SLE (hazard ratio, HR 1.86) [Figure 1]. Risk of anti-dsDNA+ SLE was almost doubled among current smokers, and increased by 60% among people with >10 pack-years of smoking. However, a more recent study did not uncover associations between cigarette smoking and anti-dsDNA antibodies among healthy individuals and those at increased risk for developing SLE by virtue of a strong family history [35], perhaps because the development of these antibodies is highly specific for SLE.
Figure 1:
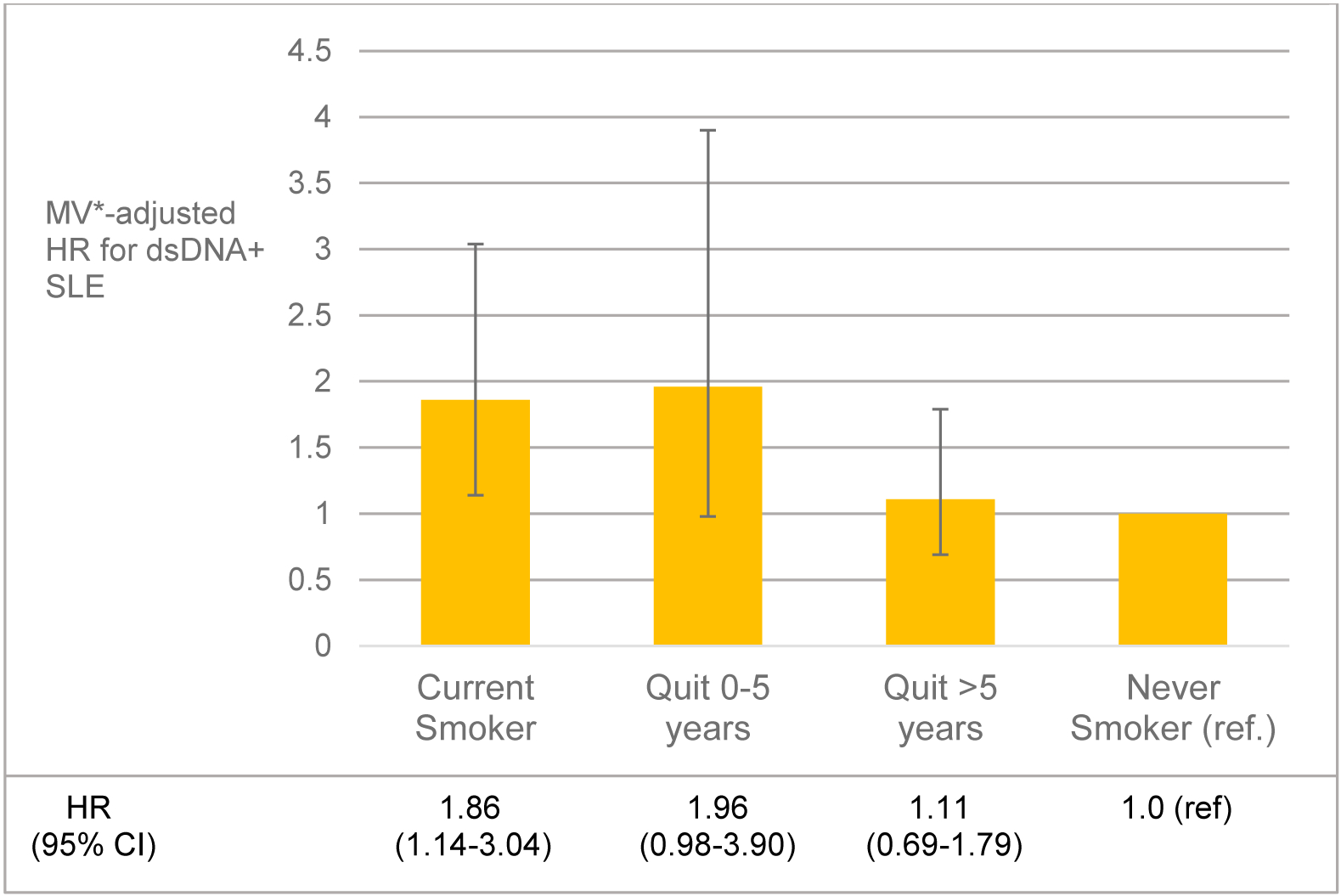
Association of smoking cessation and risk of anti-double stranded DNA positive (dsDNA+) SLE among participants in Nurses’ Health Study and Nurses’ Health Study II. p for heterogeneity between the cohorts >0.05 for all analyses. *Adjusted for age, race, questionnaire cycle, cohort, alcohol intake, body mass index, median family income from US census, oral contraceptive use and menopausal status/postmenopausal hormone use. CI, confidence interval; HR, hazard ratio; MV, multivariable; SLE, systemic lupus erythematosus. From Barbhaiya M, Tedeschi SK, Lu B, et al., Cigarette smoking and the risk of systemic lupus erythematosus, overall and by anti-double stranded DNA antibody subtype, in the Nurses’ Health Study cohorts. Ann Rheum Dis, 2018. 77(2): p. 196–202; with permission.
3.2. Potential gene-environment interactions
Interactions between genetic and environmental factors are critical in disease etiology, but have yet to be well examined in SLE. Genetic and environmental risk factors must be studied simultaneously to uncover the pathogenic mechanisms involved in SLE risk. In statistical terms, a gene-environment interaction is present when the effect of genotype on disease risk depends on the level of exposure to an environmental factor, or vice versa [36,37]. The multiple gene-environment interactions that could occur may explain why no single gene or single environmental factor has been found to be associated with SLE in most individuals. Some genes, such as PTPN22 and certain HLA types, have been associated with increased risk of several autoimmune diseases and may be general “autoimmune susceptibility genes,” responsible for common steps in the break of immune tolerance. Similarly, some environmental exposures may act to trigger several autoimmune diseases, acting non-specifically to cause systemic inflammation, oxidative stress and the up-regulation of many genetic pathways via epigenetic mechanisms. Other exposures may be related to risks of specific autoimmune diseases and subtypes, given their biologic effects and the underlying genetic susceptibility of the individual. Analyses of gene-environment interactions and their relationships to immune phenomena have been very informative in RA [38–42], are an essential next step in the study of SLE etiology, and may lead to new strategies for identification of those at highest risk and for prevention of this severe autoimmune disease.
To date, there have been only a few investigations into gene-environment interactions in SLE susceptibility and these have been limited by relatively small sample sizes. In a study of 152 SLE cases and 427 controls in Japan, smoking and two candidate SLE risk genes, STAT4 and TNFRSF1B, were studied [43]. An interaction between smoking (as ever/never) and TNFRSF1B risk genotypes GG or GT was found, with an OR of 5.42 (95%CI 2.48, 11.84) for SLE in the presence of both factors and a significant attributable proportion of 0.49 (49% of the excess risk of SLE among smokers with a G allele was due to an interaction). No interaction between smoking and STAT4 SLE risk alleles was found. These findings suggest that smoking and the TNFRSF1B risk genotype act together along the same biologic pathway to increase SLE risk (although TNFRSF1B has not been confirmed as a SLE risk gene in other populations [44]). This research group has also studied interactions between smoking and detoxification genes CYP1A1, GSTM1, N-acetyltransferase-2 (NAT2) in SLE risk in the same population. An interaction between smoking (ever/never) and the NAT2 slow acetylator genotype was found with OR 6.44, 95%CI 3.07, 13.52 in the presence of both risk factors and a significant attributable proportion due to interaction of 0.50[45]. The NAT2 enzyme is responsible for detoxification of arylamines produced by cigarette smoking (and reduces the oxidation of these compounds) [46]. Those who possess the slow-acetylator genotype may be particularly susceptible to smoking’s effects. Similarly, ever smokers with the CYP1A1/GSTM1 risk genotype had significantly elevated SLE risk (OR 17.5, 95%CI 3.20, 95.9), again pointing to genes involved in handling of toxic exposures, such as the many contained in cigarette smoke [47]. Gene-environment interaction between a different exposure, ultraviolet light, and GST risk polymorphisms was also reported in 43 cases and 298 controls in the Carolina Lupus Study, with an elevated risk among those with both factors [48]. Additionally, the Roxbury Lupus study studied 93 SLE cases and found a gene-environment interaction between residential proximity to toxic waste sites and GST risk genotypes in influencing earlier age at SLE onset [45,49].
4.0. Potential Biologic Mechanisms for the Association of Smoking with Increased Risk of SLE [Figure 2]
Figure 2:
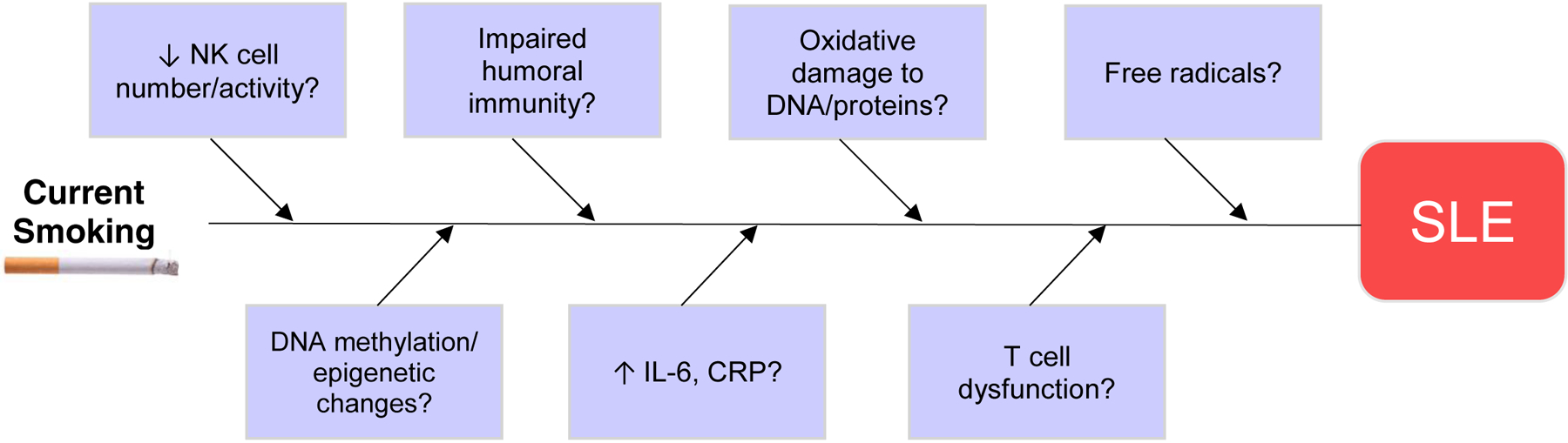
Schematic showing the potential biologic mechanisms linking smoking and SLE risk. It is thought that environmental exposures “trigger” or accelerate the disease in genetically-predisposed individuals. Substantial epidemiological evidence exists to support the association between cigarette smoking and the risk of incident SLE.
There are many models of how environmental exposures may putatively contribute to SLE pathogenesis, among them, oxidative stress involving intracellular organelles such as mitochondria and endoplasmic reticulum [50–52], elevated cytokine-driven systemic inflammation accelerating autoimmune disease onset [53–55], and impaired T- and B-cell function [56–60]. Given the well-documented in vivo effects of cigarette smoke, there are many different biologic pathways in which it may be causally related to SLE. Cigarette smoke contains at least 50 known carcinogens, including tars, nicotine, carbon monoxide, and polycyclic aromatic hydrocarbons [28]. It exists in two phases: a tar/particulate phase and a gaseous phase, both of which are highly carcinogenic [50]. Cigarette smoke increases expression of the Fas (CD95) membrane receptor on B and CD4 T lymphocyte cell surfaces [61]. CD95 is an essential membrane receptor; it transmits signals for apoptosis (programmed cell death) in lymphocytes [62], thereby playing a large role in immune homeostasis. With increased expression of CD95 on their surfaces, these cells are more sensitive to apoptotic signals and thus likely undergo apoptosis unnecessarily. This potentially induces autoimmunity by overburdening the clearance mechanisms for apoptotic material, which are not intended to handle excessive clearance [63]. Cigarette smoke has been demonstrated to impair T-cell function, reducing natural killer cells and impairing both humoral and cell-mediated immunity [64–66]. Moreover, it is possible that cigarette smoke could influence the function of T cells in SLE pathogenesis, including Th17 and Th22 cell functions, via the aryl hydrocarbon receptor (AhR), which has been shown to be activated by benzopyrenes present in cigarette smoke [67,68].
4.1. Oxidative stress and increased concentrations of inflammatory markers from cigarette smoke may be associated with incident SLE
Another likely mechanism by which cigarette smoke may be associated with incident SLE is via oxidative stress. Both the tar and particulate phases of cigarette smoke contain high concentrations of free radicals and are capable of activating endogenous sources of them as well [69,70]. Oxidative stress and abnormal mitochondrial function, as well as endoplasmic reticulum stress, are strongly implicated in SLE pathogenesis [51,52]. Free radicals also interact with DNA and cause epigenetic changes as well as mutations [50]. It is also possible that the reactive oxygen species produced by tobacco smoke metabolism damage DNA, leading to the formation of DNA adducts [71]. Freemer et. al [71] previously reported an association of smoking with anti-dsDNA+ SLE among current smokers. They found that SLE patients who were current smokers at the time of evaluation (for anti-dsDNA) were more likely to be seropositive than nonsmokers. This suggests that smoking may play a role in anti-dsDNA autoantibody formation. While anti-dsDNA autoantibodies are known markers of SLE disease activity, there is also evidence they may play a role in disease pathogenesis [72]. There are few animal models of SLE and cigarette smoke exposure. In one murine model of SLE, animals exposed to both low- and high-dose cigarette smoke showed an increased IgG anti-chromatin activity, which continued up to 4 months after smoke exposure cessation [73]. Thus, it is possible that smoking may induce anti-dsDNA+ SLE by inducing production of anti-dsDNA autoantibodies [71]. They propose an explanation for smoking-induced anti-dsDNA autoantibody formation: metabolism of cigarette smoke produces reactive oxygen species (ROS), which are known to damage both DNA and endogenous proteins [74,75]. The damage of DNA causes DNA adducts to form. These DNA adducts are recognized as foreign, and thus the body produces autoantibodies to them. It has been shown that SLE patients have higher levels of DNA adducts than do controls [76]. These DNA adducts have a half-life of 9–13 weeks [77], which provides a possible explanation for why an increased risk of developing anti-dsDNA+ SLE is only seen in current smokers.
By non-specifically activating cytokine-driven pathways of systemic inflammation, cigarette smoking may also play a role in lowering the threshold for SLE onset. Smoking is known to affect the concentration and activation of some WBCs, including leukocytes, which are associated with increased concentrations of the inflammatory markers CRP and IL-6 [78–81]. As SLE is a multisystem inflammatory disease, the effect of cigarette smoke on generalized markers of inflammation may non-specifically accelerate the onset and/or severity of disease.
4.2. Epigenetic modifications from exposure to cigarette smoke may yield autoimmune-inducing effects
Epigenetic modifications, heritable from one cell cycle to the next, regulate DNA transcription, but do not alter nucleotide sequence and are potentially reversible [82]. As only a small, specific subset of genes need to be expressed in any given human cell, there must be some sort of regulatory system in place to control which genes are exposed and open to transcription, and when. Two of the major mechanisms of epigenetic regulation, methylation and acetylation, are known to be influenced by environmental factors [83] and many epigenetic changes have been described as a result of exposure to cigarette smoke [84]. Thus, it is quite possible autoimmune-inducing effects of cigarette smoke act via epigenetic modifications, although this has not been well studied to our knowledge.
In a murine model, injecting epigenetically modified, demethylated DNA into CD4+ T cells was shown to yield a lupus-like syndrome in the mice [85,86]. Other studies have shown differential DNA methylation patterns with twin discordance in SLE specifically [87,88]. The evidence potentially linking epigenetic modifications to SLE is growing, and represents a field that warrants significantly more research.
5.0. Expert Commentary
Although large strides in understanding SLE’s complex pathogenesis have been made in recent years, ongoing investigation is warranted.
Research examining gene-environment interactions in the context of a genome-wide association study (GWAS) has the potential to be informative. GWAS evaluates hundreds of thousands of genetic loci for association with disease [89]. We can compare the genetic profiles of SLE cases to those of healthy controls by selecting a subset of single nucleotide polymorphisms (SNPs) to look at, either on genes thought to be related to SLE, or genes that are located near SLE gene candidates (by high linkage disequilibrium). By controlling for other factors that can contribute to significant genetic differences (age, sex, race), we can use GWAS to help narrow down which genes are more likely to be related to SLE, based on statistical associations with the likelihood of SLE development [41]. Future research could investigate gene-smoking interactions in determining SLE risk. Synergy between a genotype and smoking would indicate that smoking acts through a specific gene or set of genes to increase the risk of SLE. There is still much work to be done in this field.
Studying how environmental exposures could lead to epigenetic modifications and thus influence autoimmune disease risk is complex and a nascent field. There are little human data in SLE; in cancer and other diseases there is more. A study of twins discordant for SLE has pointed to epigenetic differences playing a role in SLE pathogenesis [88], but epigenetic modifications are difficult to study in humans given the long-lasting cumulative effects of a lifetime of many different potential exposures. Well-designed, in vitro, animal and human studies may soon shed light on how smoking and other environmental exposures influence gene expression and then downstream risk of complex autoimmune diseases, including SLE.
Metabolomics (the study of metabolites and their relationship to disease) may also help us understand how environmental factors interact with genetics and influence disease risk [41]. Metabolic shifts and abnormal metabolites may be mediators on the pathway between current smoking and the onset of SLE. Basic science research into the myriad biologic effects of cigarette smoke can certainly help us understand how this carcinogen may have an effect on SLE pathogenesis. Studies with particularly susceptible groups, such as family members of people with SLE, or people with a high genetic risk profile, are also helping to investigate genetic causes of SLE and the immunologic events preceding its onset [90].
6.0. Five-Year View
It is hoped that within five years research will have identified specific gene-environment interactions, leading to a greater understanding of SLE pathogenesis. The study of smoking and other environmental exposures in relation to epigenetic modifications, intracellular oxidative stress and autoantibody production will likely uncover new targets and means of preventing SLE.
7.0. Conclusion
Ultimately, we are seeking to solve a very complicated puzzle that is the specific pathogenesis of SLE. We have a wealth of evidence to support the fact that SLE arises from a combination of genetic factors and environmental exposures and likely stochastic factors as well. Identifying the environmental causes of SLE and determining how they act holds great potential. Not only are such studies giving us clues to the biologic mechanisms of disease that could be targeted and interrupted, but also, exposure to environmental factors can be modified, presenting an opportunity for disease prevention.
Table 1:
Summary of current epidemiological data on smoking and lupuss
| Author [ref.] | Year | Study Type | Finding |
|---|---|---|---|
| Costenbader et al. [16] | 2004 | Meta-analysis 9 case-control + 2 cohort studies | Current smoking was associated with an increased risk of SLE. Current smoking OR: 1.50 Past smoking OR: 0.98 Never smoked OR: 1.0 (ref) |
| Jiang et al. [31] | 2015 | Meta-analysis 11 case-control + 2 cohort studies | Smoking was associated with an increased risk of SLE. Current smoking OR: 1.56 Past smoking OR: 1.23 Never smoked OR: 1.0 (ref.) |
| Freemer et al. [71] | 2006 | Case-only | Current smoking was associated with a higher risk of dsDNA seropositivity among SLE patients. Current smoking OR: 4.0 Never smoked OR: 1.0 (ref.) |
| Young et al. [35] | 2014 | Cohort | Smoking was not associated with increased risk of autoantibody production in SLE patients or in healthy controls. |
| Barbhaiya et al. [32] | 2018 | Cohort | Current smoking was associated with anmti-dsDNA+ SLE. Current smoking HR: 1.86 Past smoking HR: 1.31 Never smoked HR: 1.0 (ref.) |
8.0. Key Issues.
The interaction of environmental and genetics factors may help us to understand both SLE disease susceptibility and mechanistic pathways in its development.
A growing body of evidence suggests current smoking specifically is a risk factor for incident SLE, with risk decreasing significantly 4–5 years after smoking cessation.
Some genes, such as PTPN22 and certain HLA types, may be general “autoimmune susceptibility genes,” just as some environmental exposures may act non-specifically to trigger several autoimmune diseases.
There are many models of how environmental exposures may putatively contribute to SLE pathogenesis, including oxidative stress, elevated systemic inflammation, and impaired T- and B-cell function.
Future research in this field should include examining gene-environment interactions in the context of a genome-wide association study (GWAS), epigenetic differences and their potential role in SLE pathogenesis, and metabolomics, the study of metabolites and their relationship to disease.
Declaration of Interests/Acknowledgements:
Dr. Costenbader’s research is supported by NIAMS R01 AR057327 and K24 AR066109.
References
- 1.Costenbader KH, Karlson EW. Cigarette smoking and systemic lupus erythematosus: a smoking gun? Autoimmunity, 38(7), 541–547 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Barbhaiya M, Costenbader KH. Environmental exposures and the development of systemic lupus erythematosus. Curr Opin Rheumatol, 28(5), 497–505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fessel WJ. Systemic lupus erythematosus in the community. Incidence, prevalence, outcome, and first symptoms; the high prevalence in black women. Arch Intern Med, 134(6), 1027–1035 (1974). [PubMed] [Google Scholar]
- 4.McCarty DJ, Manzi S, Medsger TA Jr., Ramsey-Goldman R, LaPorte RE, Kwoh CK. Incidence of systemic lupus erythematosus. Race and gender differences. Arthritis Rheum, 38(9), 1260–1270 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Walport MJ. Complement and systemic lupus erythematosus. Arthritis Res, 4 Suppl 3, S279–293 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballestar E. Epigenetics lessons from twins: prospects for autoimmune disease. Clin Rev Allergy Immunol, 39(1), 30–41 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Deapen D, Escalante A, Weinrib L et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum, 35(3), 311–318 (1992). [DOI] [PubMed] [Google Scholar]
- 8.Grennan DM, Parfitt A, Manolios N et al. Family and twin studies in systemic lupus erythematosus. Dis Markers, 13(2), 93–98 (1997). [PubMed] [Google Scholar]
- 9.Jarvinen P, Aho K. Twin studies in rheumatic diseases. Semin Arthritis Rheum, 24(1), 19–28 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Perdriger A, Werner-Leyval S, Rollot-Elamrani K. The genetic basis for systemic lupus erythematosus. Joint Bone Spine, 70(2), 103–108 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Parks CG, Cooper GS, Nylander-French LA et al. Occupational exposure to crystalline silica and risk of systemic lupus erythematosus: a population-based, case-control study in the southeastern United States. Arthritis Rheum, 46(7), 1840–1850 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Parks CG, Cooper GS, Nylander-French LA, Storm JF, Archer JD. Assessing exposure to crystalline silica from farm work: a population-based study in the Southeastern United States. Ann Epidemiol, 13(5), 385–392 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Parks CG, Cooper GS. Occupational exposures and risk of systemic lupus erythematosus. Autoimmunity, 38(7), 497–506 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Sanchez-Guerrero J, Karlson EW, Liang MH, Hunter DJ, Speizer FE, Colditz GA. Past use of oral contraceptives and the risk of developing systemic lupus erythematosus. Arthritis Rheum, 40(5), 804–808 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Costenbader KH, Feskanich D, Stampfer MJ, Karlson EW. Reproductive and menopausal factors and risk of systemic lupus erythematosus in women. Arthritis Rheum, 56(4), 1251–1262 (2007). [DOI] [PubMed] [Google Scholar]
- 16.**.Costenbader KH, Kim DJ, Peerzada J et al. Cigarette smoking and the risk of systemic lupus erythematosus: a meta-analysis. Arthritis Rheum, 50(3), 849–857 (2004). [DOI] [PubMed] [Google Scholar]; This is the first meta-analysis to demonstrate a statistically significant association between current smoking, specifically, and development of SLE.
- 17.Hardy CJ, Palmer BP, Muir KR, Sutton AJ, Powell RJ. Smoking history, alcohol consumption, and systemic lupus erythematosus: a case-control study. Ann Rheum Dis, 57(8), 451–455 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghaussy NO, Sibbitt WL Jr., Qualls CR. Cigarette smoking, alcohol consumption, and the risk of systemic lupus erythematosus: a case-control study. J Rheumatol, 28(11), 2449–2453 (2001). [PubMed] [Google Scholar]
- 19.Costenbader KH, Feskanich D, Mandl LA, Karlson EW. Smoking intensity, duration, and cessation, and the risk of rheumatoid arthritis in women. Am J Med, 119(6), 503 e501–509 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Criswell LA, Merlino LA, Cerhan JR et al. Cigarette smoking and the risk of rheumatoid arthritis among postmenopausal women: results from the Iowa Women’s Health Study. Am J Med, 112(6), 465–471 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Hutchinson D, Shepstone L, Moots R, Lear JT, Lynch MP. Heavy cigarette smoking is strongly associated with rheumatoid arthritis (RA), particularly in patients without a family history of RA. Ann Rheum Dis, 60(3), 223–227 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karlson EW, Lee IM, Cook NR, Manson JE, Buring JE, Hennekens CH. A retrospective cohort study of cigarette smoking and risk of rheumatoid arthritis in female health professionals. Arthritis Rheum, 42(5), 910–917 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Prummel MF, Wiersinga WM. Smoking and risk of Graves’ disease. JAMA, 269(4), 479–482 (1993). [PubMed] [Google Scholar]
- 24.Jonsson T, Thorsteinsson J, Valdimarsson H. Does smoking stimulate rheumatoid factor production in non-rheumatic individuals? APMIS, 106(10), 970–974 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Benoni C, Nilsson A, Nived O. Smoking and inflammatory bowel disease: comparison with systemic lupus erythematosus. A case-control study. Scand J Gastroenterol, 25(7), 751–755 (1990). [DOI] [PubMed] [Google Scholar]
- 26.Ekblom-Kullberg S, Kautiainen H, Alha P, Leirisalo-Repo M, Julkunen H. Smoking and the risk of systemic lupus erythematosus. Clin Rheumatol, 32(8), 1219–1222 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Formica MK, Palmer JR, Rosenberg L, McAlindon TE. Smoking, alcohol consumption, and risk of systemic lupus erythematosus in the Black Women’s Health Study. J Rheumatol, 30(6), 1222–1226 (2003). [PubMed] [Google Scholar]
- 28.Kiyohara C, Washio M, Horiuchi T et al. Cigarette smoking, alcohol consumption, and risk of systemic lupus erythematosus: a case-control study in a Japanese population. J Rheumatol, 39(7), 1363–1370 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Nagata C, Fujita S, Iwata H et al. Systemic lupus erythematosus: a case-control epidemiologic study in Japan. Int J Dermatol, 34(5), 333–337 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Washio M, Horiuchi T, Kiyohara C et al. Smoking, drinking, sleeping habits, and other lifestyle factors and the risk of systemic lupus erythematosus in Japanese females: findings from the KYSS study. Mod Rheumatol, 16(3), 143–150 (2006). [DOI] [PubMed] [Google Scholar]
- 31.*.Jiang F, Li S, Jia C. Smoking and the risk of systemic lupus erythematosus: an updated systematic review and cumulative meta-analysis. Clin Rheumatol, 34(11), 1885–1892 (2015). [DOI] [PubMed] [Google Scholar]; This 2015 meta-analysis updates and builds upon the findings of Costenbader et al., 2004 and finds an increased risk of SLE among current and past smokers.
- 32.*.Barbhaiya M, Tedeschi SK, Lu B et al. Cigarette smoking and the risk of systemic lupus erythematosus, overall and by anti-double stranded DNA antibody subtype, in the Nurses’ Health Study cohorts. Ann Rheum Dis, 77(2), 196–202 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study found a strong association between current smoking and dsDNA+ SLE, suggesting smoking is involved in dsDNA+ SLE pathogenesis.
- 33.Sun KH, Yu CL, Tang SJ, Sun GH. Monoclonal anti-double-stranded DNA autoantibody stimulates the expression and release of IL-1beta, IL-6, IL-8, IL-10 and TNF-alpha from normal human mononuclear cells involving in the lupus pathogenesis. Immunology, 99(3), 352–360 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yung S, Cheung KF, Zhang Q, Chan TM. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J Am Soc Nephrol, 21(11), 1912–1927 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.*.Young KA, Terrell DR, Guthridge JM et al. Smoking is not associated with autoantibody production in systemic lupus erythematosus patients, unaffected first-degree relatives, nor healthy controls. Lupus, 23(4), 360–369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study found no clear associations between smoking and autoantibody production in subjects with SLE or in healthy controls.
- 36.Botto LD, Khoury MJ. Commentary: facing the challenge of gene-environment interaction: the two-by-four table and beyond. Am J Epidemiol, 153(10), 1016–1020 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Clayton D, McKeigue PM. Epidemiological methods for studying genes and environmental factors in complex diseases. Lancet, 358(9290), 1356–1360 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Costenbader KH, Chang SC, De Vivo I, Plenge R, Karlson EW. Genetic polymorphisms in PTPN22, PADI-4, and CTLA-4 and risk for rheumatoid arthritis in two longitudinal cohort studies: evidence of gene-environment interactions with heavy cigarette smoking. Arthritis research & therapy, 10(3), R52 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karlson EW, Chang SC, Cui J et al. Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Annals of the rheumatic diseases, 69(1), 54–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arkema EV, Goldstein BL, Robinson W et al. Anti-citrullinated peptide autoantibodies, human leukocyte antigen shared epitope and risk of future rheumatoid arthritis: a nested case-control study. Arthritis research & therapy, 15(5), R159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sparks JA, Costenbader KH. Genetics, environment, and gene-environment interactions in the development of systemic rheumatic diseases. Rheum Dis Clin North Am, 40(4), 637–657 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim K, Jiang X, Cui J et al. Interactions between amino acid-defined major histocompatibility complex class II variants and smoking in seropositive rheumatoid arthritis. Arthritis & rheumatology (Hoboken, N.J, 67(10), 2611–2623 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.*.Kiyohara C, Washio M, Horiuchi T et al. Cigarette smoking, STAT4 and TNFRSF1B polymorphisms, and systemic lupus erythematosus in a Japanese population. J Rheumatol, 36(10), 2195–2203 (2009). [DOI] [PubMed] [Google Scholar]; This group found an association between cigarette smoking and SLE, differentiated by a specific tumor necrosis factor receptor allele, demonstrating a potential gene-environment interaction.
- 44.Chadha S, Miller K, Farwell L et al. Haplotype analysis of tumour necrosis factor receptor genes in 1p36: no evidence for association with systemic lupus erythematosus. Eur J Hum Genet, 14(1), 69–78 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Kiyohara C, Washio M, Horiuchi T et al. Cigarette smoking, N-acetyltransferase 2 polymorphisms and systemic lupus erythematosus in a Japanese population. Lupus, 18(7), 630–638 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Hein DW. Acetylator genotype and arylamine-induced carcinogenesis. Biochim Biophys Acta, 948(1), 37–66 (1988). [DOI] [PubMed] [Google Scholar]
- 47.Kiyohara C, Washio M, Horiuchi T et al. Risk modification by CYP1A1 and GSTM1 polymorphisms in the association of cigarette smoking and systemic lupus erythematosus in a Japanese population. Scand J Rheumatol, 41(2), 103–109 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Fraser PA, Ding WZ, Mohseni M et al. Glutathione S-transferase M null homozygosity and risk of systemic lupus erythematosus associated with sun exposure: a possible gene-environment interaction for autoimmunity. J Rheumatol, 30(2), 276–282 (2003). [PubMed] [Google Scholar]
- 49.Karlson EW, Watts J, Signorovitch J et al. Effect of glutathione S-transferase polymorphisms and proximity to hazardous waste sites on time to systemic lupus erythematosus diagnosis: results from the Roxbury lupus project. Arthritis Rheum, 56(1), 244–254 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Pryor WA, Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci, 686, 12–27; discussion 27–18 (1993). [DOI] [PubMed] [Google Scholar]
- 51.Lee WS, Sung MS, Lee EG et al. A pathogenic role for ER stress-induced autophagy and ER chaperone GRP78/BiP in T lymphocyte systemic lupus erythematosus. J Leukoc Biol, 97(2), 425–433 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol, 9(11), 674–686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Churg A, Zay K, Shay S et al. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am J Respir Cell Mol Biol, 27(3), 368–374 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Garey KW, Neuhauser MM, Robbins RA, Danziger LH, Rubinstein I. Markers of inflammation in exhaled breath condensate of young healthy smokers. Chest, 125(1), 22–26 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Seagrave J, Barr EB, March TH, Nikula KJ. Effects of cigarette smoke exposure and cessation on inflammatory cells and matrix metalloproteinase activity in mice. Exp Lung Res, 30(1), 1–15 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Hughes DA, Haslam PL, Townsend PJ, Turner-Warwick M. Numerical and functional alterations in circulatory lymphocytes in cigarette smokers. Clin Exp Immunol, 61(2), 459–466 (1985). [PMC free article] [PubMed] [Google Scholar]
- 57.Kalra R, Singh SP, Savage SM, Finch GL, Sopori ML. Effects of cigarette smoke on immune response: chronic exposure to cigarette smoke impairs antigen-mediated signaling in T cells and depletes IP3-sensitive Ca(2+) stores. J Pharmacol Exp Ther, 293(1), 166–171 (2000). [PubMed] [Google Scholar]
- 58.Robbins CS, Dawe DE, Goncharova SI et al. Cigarette smoke decreases pulmonary dendritic cells and impacts antiviral immune responsiveness. Am J Respir Cell Mol Biol, 30(2), 202–211 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Savage SM, Donaldson LA, Cherian S, Chilukuri R, White VA, Sopori ML. Effects of cigarette smoke on the immune response. II. Chronic exposure to cigarette smoke inhibits surface immunoglobulin-mediated responses in B cells. Toxicol Appl Pharmacol, 111(3), 523–529 (1991). [DOI] [PubMed] [Google Scholar]
- 60.Anolik JH. B cell biology: implications for treatment of systemic lupus erythematosus. Lupus, 22(4), 342–349 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Bijl M, Horst G, Limburg PC, Kallenberg CG. Effects of smoking on activation markers, Fas expression and apoptosis of peripheral blood lymphocytes. Eur J Clin Invest, 31(6), 550–553 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Walczak H, Krammer PH. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res, 256(1), 58–66 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun, 34(3), J258–265 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Burton RC. Smoking, immunity, and cancer. Med J Aust, 2(9), 411–412 (1983). [DOI] [PubMed] [Google Scholar]
- 65.Hersey P, Prendergast D, Edwards A. Effects of cigarette smoking on the immune system. Follow-up studies in normal subjects after cessation of smoking. Med J Aust, 2(9), 425–429 (1983). [PubMed] [Google Scholar]
- 66.Moszczynski P, Zabinski Z, Moszczynski P Jr., Rutowski J, Slowinski S, Tabarowski Z. Immunological findings in cigarette smokers. Toxicol Lett, 118(3), 121–127 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Hong CH, Lee CH, Yu HS, Huang SK. Benzopyrene, a major polyaromatic hydrocarbon in smoke fume, mobilizes Langerhans cells and polarizes Th2/17 responses in epicutaneous protein sensitization through the aryl hydrocarbon receptor. Int Immunopharmacol, 36, 111–117 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Baricza E, Tamasi V, Marton N, Buzas EI, Nagy G. The emerging role of aryl hydrocarbon receptor in the activation and differentiation of Th17 cells. Cell Mol Life Sci, 73(1), 95–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pryor WA, Stone K, Zang LY, Bermudez E. Fractionation of aqueous cigarette tar extracts: fractions that contain the tar radical cause DNA damage. Chem Res Toxicol, 11(5), 441–448 (1998). [DOI] [PubMed] [Google Scholar]
- 70.Kunchithapautham K, Atkinson C, Rohrer B. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J Biol Chem, 289(21), 14534–14546 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.*.Freemer MM, King TE Jr., Criswell LA. Association of smoking with dsDNA autoantibody production in systemic lupus erythematosus. Ann Rheum Dis, 65(5), 581–584 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study found an association of smoking with anti-dsDNA seropositivity among SLE patients, potentially providing insight into the mechanisms of autoantibody formation.
- 72.Arbuckle MR, James JA, Kohlhase KF, Rubertone MV, Dennis GJ, Harley JB. Development of anti-dsDNA autoantibodies prior to clinical diagnosis of systemic lupus erythematosus. Scand J Immunol, 54(1–2), 211–219 (2001). [DOI] [PubMed] [Google Scholar]
- 73.Rubin RL, Hermanson TM, Bedrick EJ et al. Effect of cigarette smoke on autoimmunity in murine and human systemic lupus erythematosus. Toxicol Sci, 87(1), 86–96 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Hung RJ, Boffetta P, Brennan P et al. Genetic polymorphisms of MPO, COMT, MnSOD, NQO1, interactions with environmental exposures and bladder cancer risk. Carcinogenesis, 25(6), 973–978 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Wu X, Zhao H, Suk R, Christiani DC. Genetic susceptibility to tobacco-related cancer. Oncogene, 23(38), 6500–6523 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Lunec J, Herbert K, Blount S, Griffiths HR, Emery P. 8-Hydroxydeoxyguanosine. A marker of oxidative DNA damage in systemic lupus erythematosus. FEBS Lett, 348(2), 131–138 (1994). [DOI] [PubMed] [Google Scholar]
- 77.Mooney LA, Santella RM, Covey L et al. Decline of DNA damage and other biomarkers in peripheral blood following smoking cessation. Cancer Epidemiol Biomarkers Prev, 4(6), 627–634 (1995). [PubMed] [Google Scholar]
- 78.Bermudez EA, Rifai N, Buring JE, Manson JE, Ridker PM. Relation between markers of systemic vascular inflammation and smoking in women. Am J Cardiol, 89(9), 1117–1119 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Petitti DB, Kipp H. The leukocyte count: associations with intensity of smoking and persistence of effect after quitting. Am J Epidemiol, 123(1), 89–95 (1986). [DOI] [PubMed] [Google Scholar]
- 80.Smith CJ, Fischer TH. Particulate and vapor phase constituents of cigarette mainstream smoke and risk of myocardial infarction. Atherosclerosis, 158(2), 257–267 (2001). [DOI] [PubMed] [Google Scholar]
- 81.Tracy RP, Psaty BM, Macy E et al. Lifetime smoking exposure affects the association of C-reactive protein with cardiovascular disease risk factors and subclinical disease in healthy elderly subjects. Arterioscler Thromb Vasc Biol, 17(10), 2167–2176 (1997). [DOI] [PubMed] [Google Scholar]
- 82.Costenbader KH, Gay S, Alarcon-Riquelme ME, Iaccarino L, Doria A. Genes, epigenetic regulation and environmental factors: which is the most relevant in developing autoimmune diseases? Autoimmun Rev, 11(8), 604–609 (2012). [DOI] [PubMed] [Google Scholar]
- 83.Ballestar E, Esteller M, Richardson BC. The epigenetic face of systemic lupus erythematosus. J Immunol, 176(12), 7143–7147 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics, 7, 113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lu Q, Kaplan M, Ray D et al. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum, 46(5), 1282–1291 (2002). [DOI] [PubMed] [Google Scholar]
- 86.Lu Q, Wu A, Richardson BC. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol, 174(10), 6212–6219 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol, 179(9), 6352–6358 (2007). [DOI] [PubMed] [Google Scholar]
- 88.Javierre BM, Fernandez AF, Richter J et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res, 20(2), 170–179 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Neale BM, Purcell S. The positives, protocols, and perils of genome-wide association. Am J Med Genet B Neuropsychiatr Genet, 147B(7), 1288–1294 (2008). [DOI] [PubMed] [Google Scholar]
- 90.Parks CG, de Souza Espindola Santos A, Barbhaiya M, Costenbader KH. Understanding the role of environmental factors in the development of systemic lupus erythematosus. Best Pract Res Clin Rheumatol, 31(3), 306–320 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]