

Abstract
Many eukaryotic protein-coding genes are able to generate exonic circular RNAs. Most of these covalently linked transcripts are expressed at low levels, but some accumulate to higher levels than their associated linear mRNAs. Here, we highlight a number of methodologies that have been developed in recent years to identify and characterize these transcripts, which have revealed an increasingly detailed view of how circular RNAs can be generated and function. It is now clear that modulation of circular RNA levels can result in a variety of molecular and physiological phenotypes, including effects on the nervous system, innate immunity, microRNAs, and many disease relevant pathways.
Keywords: circRNA, backsplicing, pre-mRNA splicing, innate immunity, microRNA, translation
Circular RNAs are generated from many eukaryotic protein-coding genes
Most eukaryotic genes are disrupted by intronic sequences that must be removed from nascent precursor mRNA (pre-mRNAs) by the spliceosome [reviewed in 1]. It had long been thought that most introns are removed sequentially and rapidly once they have been transcribed, thereby allowing each exon to be covalently linked to the next and a functional linear mRNA to be produced (Figure 1A, top). Nevertheless, splicing decisions are highly regulated, and many genes generate a variety of mature spliced transcripts with unique combinations of exons [reviewed in 2, 3]. This includes circular RNAs that have covalently linked ends and are generated when a splice donor (5′ splice site) is joined to an upstream splice acceptor (3′ splice site) via a form of alternative splicing (see Glossary) termed backsplicing (Figure 1A, bottom) [4–6]. All internal exons of a gene (excluding the first and last exon) can theoretically be subjected to backsplicing, but RNA-seq has revealed that only a small subset of possible backsplicing events occur in cells [7–9]. It is important to keep in mind, however, that there are more than 200,000 exons in the human genome, so these relatively rare events still result in thousands or more unique circular RNAs being present in any given cell type [10]. Most of these circular RNAs are expressed at low levels, but these transcripts are naturally resistant to degradation by exonucleases and, in some cases, accumulate to levels that exceed that of their cognate linear mRNAs [7]. This suggests that the main function of some protein-coding genes may be to generate circular RNAs rather than linear mRNAs or their expected protein products.
Figure 1. Eukaryotic pre-mRNAs can be spliced to generate a linear or circular RNA.
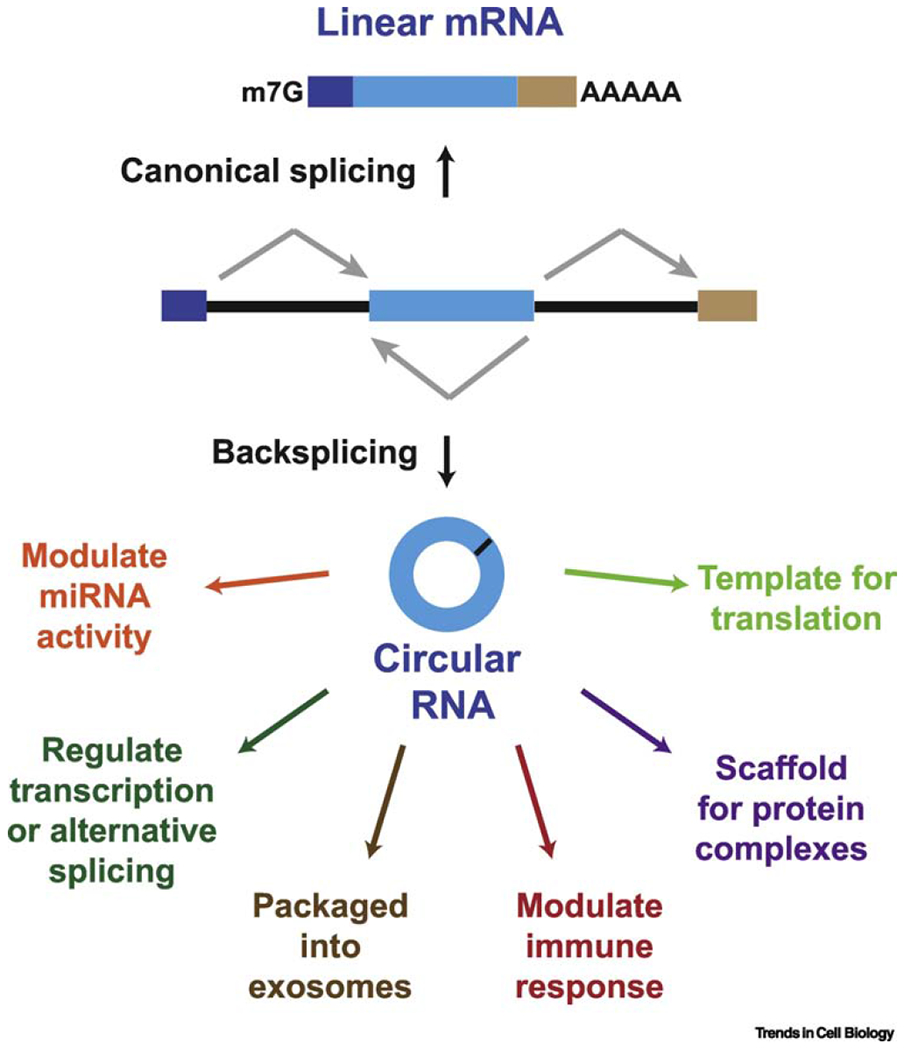
(Top) When splice sites (ss) are joined in a linear order by the pre-mRNA splicing machinery, a canonical linear mRNA is generated that is also capped and polyadenylated. (Bottom) Alternatively, backsplicing can join a 5′ ss to an upstream 3′ ss, resulting in production of a circular RNA whose ends are covalently linked by a 3′-5′ phosphodiester bond and can function via a number of distinct molecular mechanisms.
Key questions in the field have been: (i) how many circular RNAs are expressed in cells?; (ii) how is the choice between linear vs. circular RNA production made?; and (iii) how do circular RNAs function? In recent years, a number of new methodologies to identify and characterize circular RNAs have been developed, which have revealed important insights into each of these questions. Here, we highlight some of the latest findings on circular RNAs and refer readers to other reviews for more details on earlier studies [11–14]. An increasingly detailed picture has emerged for how the biogenesis of circular RNAs is tightly regulated in a combinatorial manner. Once these mature circular RNAs are generated, they can then have important molecular functions and their mis-regulation can result in phenotypes, including in animal models.
Improved methods for genome-wide detection of circular RNAs
Circular RNAs are most commonly detected using high-throughput RNA sequencing (RNA-seq) followed by identification of spliced reads that span backsplicing junctions [reviewed in 15]. A number of computational algorithms to identify such reads have been developed, including ones that can reconstruct full-length circular RNAs [16], but there are striking differences in their sensitivity and precision [17, 18]. For example, when the same RNA-seq dataset was inputted to five different algorithms, ~40% of putative circular RNAs were predicted by only a single algorithm. Some of these transcripts may be reverse transcriptase artifacts or trans-spliced RNAs and thus at least two algorithms should be used to identify circular RNAs [19], and transcripts of interest must be validated using independent approaches such as Northern blotting or RT-PCR [20]. Further complicating matters is that most backsplicing reactions are much less efficient than canonical splicing events [21]. This means that most circular RNAs are expressed at low levels, with only a handful of reads (if any) spanning their backsplicing junctions in a standard RNA-seq library. One way to overcome this problem is to use exon capture probes prior to RNA-seq, thereby enabling gene loci that generate circular RNAs of interest to be sequenced with higher depth [22].
It has nevertheless become most common to validate and enrich for circular RNAs using RNase R [8] (Figure 2A), a highly processive 3′-5′ exonuclease that can digest many linear RNAs [23] (Figure 2B, compare control RNA-seq library in red to RNA-seq libraries generated after RNase R treatment in brown and blue). Given that circular RNAs have no 3′ end, they are resistant to digestion by RNase R, become enriched in the remaining RNA population, and hence are more easily detected when RNA-seq libraries are subsequently generated (Figure 2C). However, it is now recognized that hundreds (or more) of linear transcripts fail to be efficiently digested by RNase R [24, 25]. This includes linear RNAs with highly structured 3′ ends, including histone mRNAs (Figure 2D) and many abundant small noncoding RNAs such as snRNAs, tRNAs, and the RNA component of RNase P (RPPH1) (Figure 2E). Such structured transcripts fail to be digested because they lack 3’ single-stranded overhangs of at least seven nucleotides, which are required for initial binding by RNase R [26]. These RNAs can, however, be digested if 3′ terminal poly(A) tails are added to their ends in vitro by E. coli poly(A) polymerase (E-PAP) prior to incubation with RNase R [24, 25] (Figure 2D–E). We recently found that RNase R also abruptly stalls at internal G-quadruplex structures, thereby only partially degrading hundreds of linear mRNAs that contain them [24] (Figure 2F). G-quadruplex structures are stabilized by the presence of K+ cations (which are present in the commonly used RNase R reaction buffer), but not by cations with smaller ionic radius such as Li+ [27]. Indeed, replacing K+ with Li+ in the reaction buffer (Figure 2A) was sufficient to enable RNase R to proceed through the G-quadruplexes and fully degrade these mRNAs (Figure 2F). Therefore, to obtain higher purity circular RNAs, we now recommend that one should first treat RNA samples with poly(A) polymerase followed by RNase R digestion in the presence of Li+ (Figure 2A, Right).
Figure 2. Enrichment of circular RNAs using RNase R.

(A) To simultaneously deplete linear RNAs and enrich for circular RNAs, the 3′-5′ exonuclease RNase R is commonly used. In standard protocols (left), total RNA is treated with RNase R in a K+-containing buffer, but a number of linear RNAs fail to be digested by this protocol. We thus recently developed an improved protocol (right) in which total RNA is first treated with E. coli poly(A) polymerase (E-PAP) followed by incubation with RNase R in a Li+-containing buffer. (B-F) HeLa total RNA was subjected to the indicated treatments and RNA-seq libraries were then prepared [data from 24]. Sequencing read coverage is shown for (B) LGALS3, (C) HIPK3, (D) HIST2H2AB, a member of the histone H2A family, (E) RPPH1, the RNA component of RNase P, and (F) PPP1R8. The HIPK3 circular RNA is designated in purple and the G-quadruplex in the 3′ UTR of the PPP1R8 mRNA is indicated in green. The standard RNase R protocol using KCl fails to digest HIST2H2AB mRNA and the RPPH1 noncoding RNA, and only digests the PPP1R8 mRNA up until the G-quadruplex structure. In contrast, the improved protocol allows efficient digestion of these RNAs coupled to increased enrichment of the HIPK3 circular RNA.
In total, RNA-seq experiments done by many groups have revealed thousands of exonic circular RNAs that are generated from protein-coding genes across the eukaryotic tree of life, including in metazoans, plants, fungi, and protists [7–10, 22, 24, 28–37]. These transcripts are often produced from more than 10% of expressed genes in a given cell type and consist of one or more exons, with any internal introns typically being fully spliced out (although there can be alternative splicing events within circular RNAs [38, 39]). Notably, there is seldom a clear correlation between the levels of linear and circular RNAs from a given gene. Most of these circular RNAs are instead expressed at low levels (e.g. in human mammary epithelial cells, the average circular RNA was found to be <3% as abundant as linear transcripts from the same gene [40]). However, some circular RNAs accumulate to much higher levels than their cognate linear mRNAs. This is especially true in the nervous system, where the vast majority of circular RNAs have been detected [29, 31,41,42]. Circular RNAs have further been found to be globally up-regulated in the brain as multiple species age [29, 43–45], but the functional significance (if any) of this observation remains unknown. Like other classes of RNAs, mature circular RNAs can contain modified nucleotides, including N6-methyladenosine (m6A) [46, 47], and can fold into secondary structures that can be important for their function [48].
Both sequence features and trans-acting factors dictate the efficiency of circular RNA biogenesis
Early studies of the mouse Sry circular RNA revealed that ~50 kb of near perfectly complementary intronic sequences flank this exon [6], and that inclusion of ~400 nt of the flanking repeats was sufficient for production of this circular RNA from the intervening exon [49]. It is now recognized that most circular RNAs are similarly flanked by longer than average introns that have complementary repeats, often Alu elements in human cells [8, 32, 41, 50]. Base pairing between these intronic repeats is required for biogenesis of many circular RNAs (likely by bringing the intervening splice sites into close proximity) (Figure 3A), with 30–40 nt repeats being sufficient for exon circularization in some cases [21, 51–53]. Notably, the presence of multiple repetitive elements in a gene can enable distinct circular RNAs to be produced, and these transcripts can have differing expression levels depending on how efficiently each pair of sequences binds to one another (Figure 3B) [32, 38]. Indeed, one can accurately predict many circular RNAs by simply searching for complementary sequences in the flanking introns [50]. This indicates that intronic repeat sequences are important players in the biogenesis of many circular RNAs [54, 55], but it must be stressed that the presence of repeats is not sufficient for exon circularization. There are now clear examples of circular RNAs generated from readthrough transcription events [22, 56] or in a manner independent of repeat sequences, including in S. pombe where a circular RNA from the mrps16 gene is generated only after an exon skipping event and re-splicing of the lariat (Figure 3C) [57]. A correlation between exon skipping and circular RNA production has been noted at other genes [58, 59], including at the Arabidopsis SEPALLATA3 gene where a circular RNA forms an R-loop with its cognate gene locus to promote further skipping of that particular exon [60].
Figure 3. Circular RNA biogenesis can be controlled by intronic repeat sequences, exon skipping, and the levels of core spliceosome components.

(A) Backsplicing can be induced when inverted repeat sequences (orange arrows) in the flanking introns base pair to one another, thereby bringing the intervening splice sites into close proximity. (B) There are often multiple intronic repeat sequences (labeled as i, ii, iii, and iv) present in a pre-mRNA, which allows distinct mature RNAs to be produced depending on which repeats base pair to one another. When repeats in different introns base pair, backsplicing can be induced to yield the indicated circular RNAs. In contrast, a linear mRNA is produced if repeats in a single intron (ii and iii) base pair to one another. (C) Exon skipping events result in a mature linear mRNA and an intron lariat, which can be re-spliced to generate a stable circular RNA and a double lariat structure that is then debranched and degraded. (D) In pre-mRNAs with long introns, spliceosome components, including the U1 and U2 snRNPs, first assemble on exons to form exon junction complexes. These cross-exon interactions must then be replaced with cross-intron interactions to allow generation of a mature linear mRNA (left). When spliceosome activity is limiting and the exon is sufficiently long, the full spliceosome tends to instead assemble across an exon, resulting in backsplicing and generation of a circular RNA (right).
Regardless if repeat sequences are involved in their biogenesis, the expression level of likely all circular RNAs is controlled by RNA binding proteins. The RNA helicase DHX9 (DExH-Box Helicase 9) binds double-stranded RNAs and can inhibit many backsplicing events, likely by directly unwinding the double-stranded regions in the flanking introns or recruiting ADAR (adenosine deaminase acting on RNA) enzymes that convert adenosines to inosines [50, 61]. On the other hand, the alternative splicing factor Quaking (QKI) promotes production of many circular RNAs during human epithelial-mesenchymal transition (EMT) [30] and the Drosophila Muscleblind (Mbl) protein binds to its own pre-mRNA to promote production of a circular RNA, thereby dampening expression of the canonical linear Mbl mRNA [62]. At the Drosophila laccase2 gene, it has been shown that base pairing between intronic repeat sequences promotes backsplicing, whereas multiple hnRNPs (heterogeneous nuclear ribonucleoproteins) and SR proteins inhibit production of this circular RNA [52]. It thus appears that circular RNA levels are controlled in a combinatorial manner with various RNA binding proteins fine-tuning the efficiency of backsplicing. Considering that each gene has its own unique set of protein-binding sites, one can imagine how this code is used to generate a variety of distinct circular RNA expression patterns and how it can enable changes in these patterns as cells change their state.
Canonical 5′ and 3′ splice sites are required for backsplicing and >99% of annotated human circular RNAs have been found to be flanked by the canonical splicing motif AG-GT [22]. However, recent work has now revealed that most circular RNAs may be generated post-transcriptionally [21, 51] and that circular RNA levels increase when core spliceosome factors are depleted or pharmacologically inhibited [56, 63, 64]. The spliceosome is formed in a stepwise and dynamic manner [reviewed in 1], and our group showed in Drosophila that individually depleting many spliceosome components, including proteins that are part of the U1 or U2 snRNPs (e.g. snRNP-U1–70K or the SF3b complex, respectively) or are required for the catalytic steps in splicing (e.g. Slu7), resulted in decreased linear mRNA levels coupled to increased circular RNA expression [56]. Likewise, circular RNAs were up-regulated when Drosophila cells were treated with Pladienolide B, a pharmacological inhibitor of the SF3b complex [56]. Similar increases in circular RNA expression patterns were observed when rat neurons were treated with the splicing inhibitor Isoginkgetin [64]. We thus propose that remodeling of exon definition complexes [65] to cross-intron complexes may not occur as efficiently when core spliceosome complexes are depleted or inhibited (Figure 3D) [56]. This limits the production of canonically spliced linear mRNAs and instead allows single exon circular RNAs to become the preferred splicing outcome. Indeed, recent biochemical and structural work using S. cerevisiae has now shown that exon definition complexes can be directly converted to catalytically competent backsplicng complexes if the exon is sufficiently long to allow binding of the U4/U6.U5 tri-snRNP [66].
Once generated, most circular RNAs are stable and traffic to the cytoplasm
Due to their covalently closed structure, circular RNAs are naturally resistant to digestion by RNA exonucleases and generally have much longer half-lives than linear RNAs (often 24 hours or longer) [40]. The CDR1as/ciRS-7 circular RNA contains a near perfect target site for the miR-671 microRNA and can be cleaved by Argonaute-2 (Ago2) to trigger transcript degradation [53, 67], but this regulatory strategy appears to be unique to this locus. It is thus still largely unclear how circular RNAs are degraded in normal, unstressed cells. Some circular RNAs may be eliminated via packaging into extracellular vesicles, such as exosomes [68–70]. However, it seems most likely that yet to be identified RNA endonucleases (besides RNase L [48], discussed below) can trigger degradation of specific circular RNAs when appropriate, e.g. when these transcripts are modified with m6A [71].
A subset of circular RNAs remain in the nucleus and have been proposed to control transcription or alternative splicing patterns [41, 60, 72], but the vast majority of circular RNAs accumulate in the cytoplasm. The mechanism of how circular RNAs are exported from the nucleus remains incompletely understood, but it is now clear that the lengths of these transcripts are measured to dictate the mode of nuclear export (Figure 4) [73]. In Drosophila, long (>800 nt) circular RNAs accumulate in the nucleus upon depletion of the DExH/D-box helicase Hel25E. The two human homologs of Hel25E have likewise been implicated in control of circular RNA localization: UAP56 (DDX39B) is required for the export of long (>1300 nt) circular RNAs, whereas URH49 (DDX39A) is required for export of short (<400 nt) circular RNAs [73]. Hel25E homologs are well-established players in the export of many linear mRNAs [74], but other known export factors appear to be dispensable for circular RNA export in Drosophila [73]. In neurons, circular RNAs can be enriched in synapses [42], suggesting the possible existence of directed transport mechanisms for these transcripts but the details remain unknown.
Figure 4. Circular RNAs are exported to the cytoplasm in a length-dependent manner.
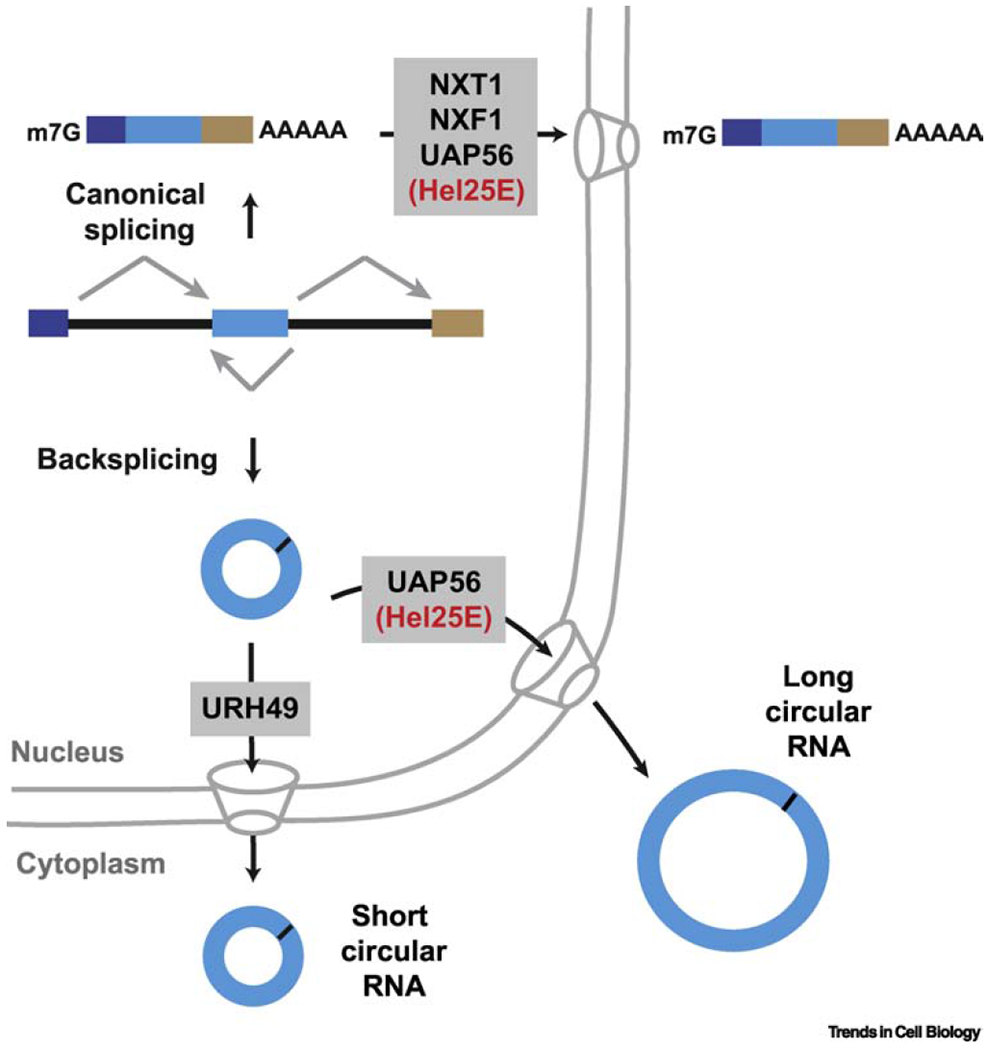
The export of many linear mRNAs from the nucleus requires binding of a set of proteins, including UAP56 (Hel25E in Drosophila), which enable recruitment of the NXT1/NXF1 heterodimeric export receptor and trafficking through the nuclear pore complex. RNAi screening has now revealed roles for Drosophila Hel25E and its human homologs UAP56/URH49 in nuclear export of circular RNAs. The length of the mature circular RNA dictates which of these factors is required.
Methods for modulating circular RNA levels have enabled the first phenotypes to be revealed in animal models
The molecular and physiological functions of most circular RNAs remain unknown, but there has been increasing progress in this area thanks to the development of methods to overexpress and knock-down/out specific circular RNAs. Circular RNAs can be generated in vitro using self-splicing introns [75–78] or splint ligation approaches [79] and subsequently added to cells. There are also now plasmid and viral-based methods for expressing circular RNAs of interest using the endogenous spliceosome machinery, in some cases with minimal production of linear RNAs [32, 52, 53, 80–82]. Conversely, specific circular RNAs can be depleted using short hairpin RNAs (shRNAs)/small interfering RNAs (siRNAs) that target the backsplicing junction or via removal of the flanking repeat elements (or the entire locus in the case of CDR1as/ciRS-7 [83]) using CRISPR/Cas9 genome editing [21, 84–86]. These technologies have now enabled high-throughput screening projects, including a recent shRNA screen that revealed 171 circular RNAs (representing 11.3% of screened circular RNAs) that are essential for cell proliferation in human prostate cancer cell lines [36]. Notably, in >90% of cases where the circular RNA was found to be essential in that screen, its linear mRNA counterpart was not, suggesting that circular RNAs can drive phenotypes independent of their linear counterparts.
Going one step further, the first circular RNA knockout animals have now been generated: knockout of CDR1as/ciRS-7 resulted in mice with impaired sensorimotor gating, a phenotype associated with neuropsychiatric disorders [83] and knockout of cia-cGAS in mice led to decreased numbers of long-term hematopoietic stem cells, which was often associated with severe anemia and death [86]. It should be noted that knockout of CDR1as/ciRS-7 was accomplished by removing the entire exon that circularizes, thereby also removing the protein-coding CDR1 gene that is present on the opposite strand [83]. Nevertheless, expression of CDR1 mRNA was not detected in any examined wild type mouse tissue, suggesting the observed phenotypes are most likely due to loss of the circular RNA. To knock out cia-cGAS, one of the complementary sequences in the flanking introns was removed, thus providing more confidence that the phenotypes are due to alterations in this circular RNA [86]. Ultimately, the gold standard will be to show that phenotypes observed upon knockout of a circular RNA can be rescued by re-expressing the circular RNA from a plasmid or viral vector. For example, adeno-associated virus (AAV) mediated over-expression of a circular RNA from the mouse Fndc3b gene resulted in improved cardiac repair in a myocardial infarction model [82].
Molecular functions for circular RNAs: Moving beyond simple microRNA sponging models
As of now, the CDR1as/ciRS-7 circular RNA has been studied in most detail, in part because it harbors more than 60 evolutionarily conserved miR-7 binding sites [9, 53]. This circular RNA is highly expressed in the brain, especially in excitatory neurons [83], and localizes to the cytoplasm and neuronal cell extensions [87]. Given its many binding sites for miR-7, it has long been assumed that CDR1as/ciRS-7 sequesters this microRNA and prevents it from regulating expression of its target mRNAs (Figure 5A) [9, 53]. However, recent work in mice revealed that knocking out this circular RNA did not lead to decreased levels of most miR-7 target genes in the brain as predicted by the microRNA sponging model [83]. Instead, the levels of mature miR-7 were slightly reduced (~2-fold), which resulted in increased expression of some miR-7 target genes, including immediate early genes that are markers of neuronal activity. It thus appears that the main molecular function of CDR1as/ciRS-7 may be in protecting miR-7 from degradation and/or controlling its temporal and spatial localization. In doing so, this circular RNA may ensure that appropriate amounts of this microRNA are delivered to neuronal cell extensions and synapses to control expression of miR-7 target genes (Figure 5A) [67, 83]. Indeed, it is now recognized that CDR1as/ciRS-7 is part of a larger post-transcriptional regulatory network in the brain that involves several other noncoding RNAs, including the long noncoding RNA Cyrano that has a site with unusually high complementarity to miR-7 which triggers miR-7 destruction [67]. Additional circular RNAs have been proposed to sequester or alter the expression of other microRNAs [29, 36, 88], including the mouse testis-specific Sry circular RNA that harbors 16 putative binding sites for miR-138 [53]. However, most circular RNAs are expressed at low levels and contain few microRNA binding sites [40, 89], making them highly unlikely to function as microRNA sponges or competing endogenous RNAs (ceRNAs) [90].
Figure 5. Circular RNAs can modulate microRNA activity or be translated.
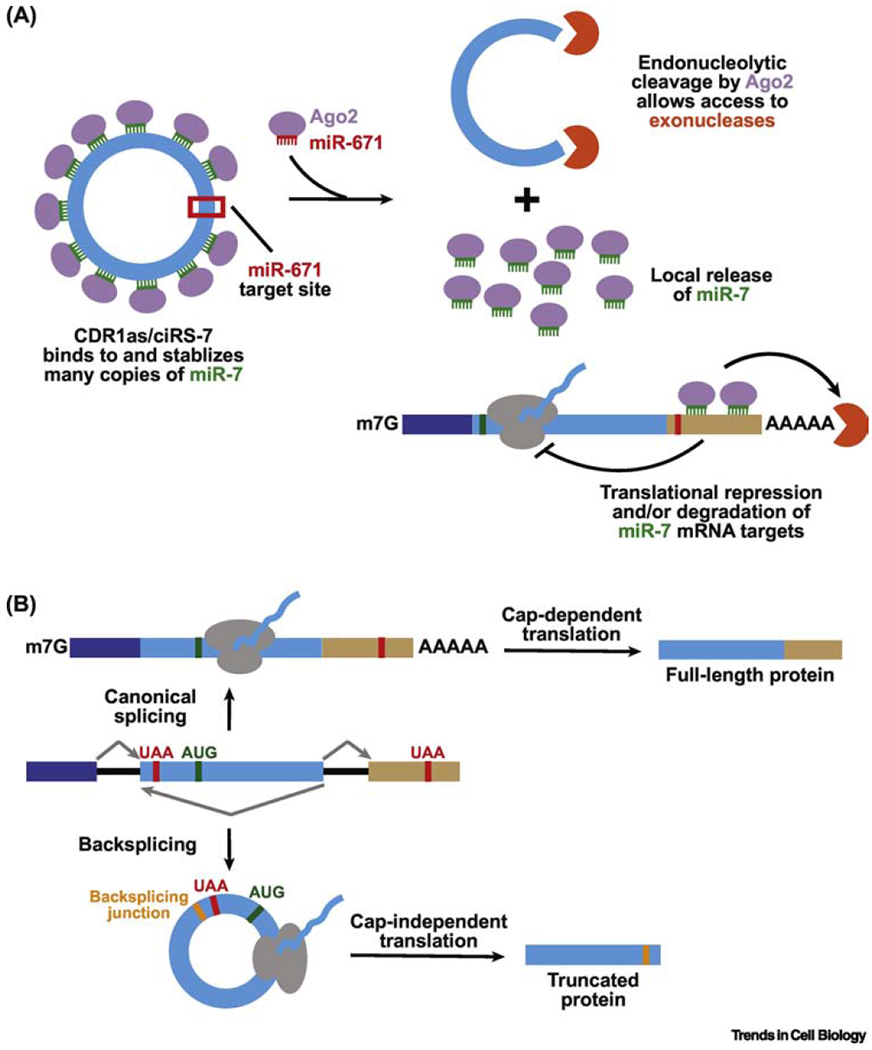
(A) The CDR1as/ciRS-7 circular RNA contains many sites complementary to the seed region of the miR-7 microRNA along with a single, near perfect target site for miR-671. Binding of miR-671 triggers endonucleolytic cleavage of the circular RNA by Argonaute-2 (Ago2), which likely leads to recruitment of exonucleases and full degradation of CDR1as/ciRS-7. This allows release of the miR-7 transcripts that can then bind and down-regulate the expression of specific mRNAs. (B) Linear and circular RNAs generated from a protein-coding gene locus may be translated to yield distinct protein products. Here, the linear mRNA can be translated in a cap-dependent manner to yield a full-length protein, while the circular RNA is translated in a cap-independent manner to generate a truncated protein that terminates at a stop codon encountered after the backsplicing junction.
How else might circular RNAs function in cells? It has long been known that synthetic circular RNAs can be translated if they contain an internal ribosome entry site (IRES) [75, 91, 92], and a number of endogenous circular RNAs contain the start codon that is used for initiation from the cognate linear mRNA [reviewed in 93]. Several groups have now used ribosome profiling, mass spectrometry, and/or expression plasmids to suggest that a subset of endogenous circular RNAs may indeed generate proteins (Figure 5B) [84, 94–96]. In some cases, the presence of m6A in the circular RNA may help recruit the ribosome [95], but this conclusion has been questioned [97] and the underlying mechanisms are still largely unclear. Further work is now needed to determine how many of these translated products have functions and/or accumulate to high levels, especially given that other studies have failed to find any evidence for circular RNA translation [33, 42, 89]. It is nevertheless tempting to speculate that significant amounts of protein could be made over the long lifetimes of circular RNAs or that translation of these transcripts could be induced in a localized manner or under specific conditions, e.g. times of stress [94, 95]. Besides encoding a protein, circular RNAs bind a variety of RNA binding proteins and, in some cases, may serve as a scaffold or alter the activity of the proteins [98]. For example, a circular RNA from the PABN1 gene has been reported to sequester HuR, thereby preventing it from activating translation of the linear PABN1 mRNA [99]. In other cases, the biogenesis of the circular RNA may itself be the critical regulatory event as the choice to backsplice can block production of the canonical linear mRNA [62].
Emerging connections between circular RNAs and the immune system
The first identified circular RNAs were pathogenic plant viroids [100] and recent years have revealed an increasing number of DNA viruses that generate circular RNAs [101–105]. It thus makes sense that eukaryotic immune systems may have evolved mechanisms to sense foreign circular RNAs. However, there is currently conflicting data on whether these transcripts are, in fact, immunogenic. Chen and colleagues have shown that addition of non-self circular RNAs (e.g. transcripts generated in vitro by group I catalytic introns) to mammalian cell lines or mouse models activates a potent innate immune response via the pattern recognition receptor RIG-I (Figure 6) [47, 78]. This is at least in part due to the lack of m6A modifications on the non-self circular RNAs, as addition of this modification and/or tethering the m6A reader protein YTHDF2 to the transcript was sufficient to suppress the immune responses [47]. On the other hand, Wesselhoeft and colleagues have argued that pure circular RNAs are not immunogenic even when they lack modified nucleotides, and that the discordant results between the studies are due to impurities in circular RNA preparations (Figure 6) [75, 97]. Further work is now required to reconcile the two sets of observations, especially by using the exact same RNA sequences and directly comparing the purification methods.
Figure 6. Circular RNAs and the immune system.

(Top) The biogenesis of circular RNAs can be modulated by factors that stabilize (NF90/NF110) or de-stabilize (DHX9 or ADAR) base pairing between intronic repeat sequences. After backsplicing, circular RNAs are exported to the cytoplasm where they are recognized as self and can bind a number of protein factors, including immune regulators such as PKR and NF90/NF110. (Bottom Left) It has been proposed that foreign circular RNAs may trigger an immune response in a RIG-I dependent manner, although this may be due to contaminants (e.g. short triphosphorylated RNAs) in the circular RNA preparation. (Bottom Right) Furthermore, viral infection can result in activation of RNase L, resulting in cleavage of many endogenous transcripts, including circular RNAs. Recent work suggests that digestion of the circular RNAs releases key immune regulators, including PKR, which can then bind the pathogenic dsRNAs and become activated to inhibit viral infection.
Besides potentially recognizing foreign circular RNAs, it is now clear that immune regulators can control the levels of endogenous circular RNAs. In particular, a recent genome-wide screen that aimed to identify circular RNA biogenesis factors in human HeLa cells revealed a number of double-stranded RNA (dsRNA) binding proteins, including NF90/NF110, that are involved in innate immunity [106]. In uninfected cells, NF90/NF110 can bind to nascent transcripts and promote backsplicing events by stabilizing base pairing interactions between intronic repeats (Figure 6). However, upon viral infection, NF90/NF110 re-localize to the cytoplasm, limiting the production of additional circular RNAs [106]. The steady-state levels of these transcripts can be further reduced during infections by the cytoplasmic endonuclease RNase L, which cleaves many viral and cellular transcripts (including circular RNAs) after UN dinucleotides [48]. It has recently been proposed that a rapid fall in circular RNA levels can play a critical role in the innate immune response, as it allows proteins bound to circular RNAs (e.g. NF90/NF110 or the dsRNA-dependent kinase PKR) to be released and activated upon recognition of the pathogenic RNAs (Figure 6). Indeed, over-expression of circular RNAs resulted in reduced PKR activity and increased viral mRNA expression in HeLa cells infected with EMCV (encephalomyocarditis virus) [48]. Therefore, circular RNAs can function as a group to modulate immune responses. These data further suggest that RNase L may act upstream of PKR, but other studies using RNase L knockout cells have found no requirement for RNase L in PKR activation [107, 108]. Furthermore, little long dsRNA is thought to be present in the cytoplasm of uninfected normal cells, so it is currently unclear why PKR would need to be subjected to such active suppression by circular RNAs or why robust transcriptional up-regulation of PKR following infection would be insufficient to dilute the effects of circular RNAs. Besides PKR, recent work suggests that a circular RNA may similarly inhibit the activity of cyclic GMP-AMP synthase (cGAS), a cytosolic DNA sensor [86]. Further work is now needed to clarify the exact interplay of circular RNAs and the various pattern recognition receptors, especially in well-characterized viral infection models and in autoimmune conditions. The latter is particularly relevant as it was recently suggested that circular RNAs are reduced in patients with the autoimmune disease systemic lupus erythematosus (SLE) [48].
Functions for circular RNAs in cancer
Compared to normal cells, there is a general decrease in circular RNA levels in most cancer types, suggesting a connection between cellular proliferation and the steady-state levels of these transcripts [22, 109]. Indeed, treating LNCaP prostate cancer cells with dinaciclib, a kinase inhibitor that decreases cellular proliferation, led to an overall increase (−50%) in circular RNA levels that was independent of changes in parent gene expression [22]. This suggests that circular RNAs may be diluted by cell division. It is nevertheless now clear that circular RNA levels can be associated with clinical outcomes in cancer patients and that some circular RNAs play functional roles in tumor cells [36, 110–112]. For example, a circular RNA encoded by the Zbtb7a (also known as POKEMON) gene can function as a proto-oncogenic RNA in mesenchymal tumors, whereas its linear counterpart functions as a tumor suppressor by encoding the Pokemon transcription factor [113]. On the other hand, a circular RNA from the Foxo3 gene promotes apoptosis and thus limits tumor growth [98, 114]. Additional functions will certainly be revealed for these transcripts in cancer and other diseases, such as Alzheimer’s disease [115], in the coming years. Furthermore, because circular RNAs are stable and present in exosomes and bodily fluids [68–70], they may represent promising new disease biomarkers [112].
Concluding Remarks
In summary, recent work has continued to make it clear that circular RNAs are not simply rare oddities or errors of pre-mRNA splicing, but instead tightly regulated transcripts that, at least in some cases, can perform important biological functions. Mechanisms that regulate the levels of many circular RNAs have been discovered, but many of the molecular details are still poorly understood and there are a number of key questions that remain unanswered (see Outstanding Questions). It is important to keep in mind that only a very small subset of circular RNAs identified by RNA-seq experiments has been studied (or validated by independent techniques), and thus we still know rather little about how most circular RNAs function. It thus appears that the field is at an important inflection point: it has become clear that many circular RNAs exist, especially in the nervous system, but it is now critical that we determine how important they are (or are not) for cellular physiology. High-throughput screening and animal models have begun to reveal phenotypes when the expression of individual circular RNAs are modulated, and many more studies are now required. Notably, many individual circular RNAs are expressed at very low levels which may make one question their significance, but recent work suggests the exciting idea that circular RNAs can work together as a group [48], thereby allowing even rare transcripts to contribute to key biological processes. Through continued improvements to methodologies and further exploration of circular RNAs in normal and disease states, it is certain that many more insights into these transcripts will be revealed in the coming years.
OUTSTANDING QUESTIONS.
Why is backsplicing generally less efficient than canonical splicing reactions?
What is the complete set of factors that control circular RNA expression and localization, and how do they collectively function to enable circular RNAs to have unique regulatory patterns?
Are there factors that are specifically required for backsplicing that are dispensable for canonical splicing reactions?
How are circular RNAs degraded in normal, unstressed cells?
How many circular RNAs truly function as competing endogenous RNAs to sponge microRNAs?
Is the translation of certain circular RNAs critical for their function and, if so, how?
How many circular RNAs result in an obvious phenotype when over-expressed or knocked out in vivo?
There is conflicting data on how in vitro synthesized circular RNAs interface with the immune system, so what features allow these transcripts to be recognized (or not recognized) by immune sensors?
The expression of circular RNAs globally increases in the brain as multiple species age, but are these changes beneficial, detrimental, or inconsequential?
Given their long half-lives, can circular RNAs serve as novel biomarkers or therapeutic modalities?
HIGHLIGHTS.
Circular RNAs are generated from many eukaryotic protein-coding genes when the pre-mRNA splicing machinery “backsplices” and joins a downstream 3′ splice site to an upstream 5′ splice site.
Circular RNA biogenesis is often facilitated by base pairing between intronic repeat elements, and the expression of these transcripts is further controlled by the combinatorial action of RNA binding proteins, the levels of core spliceosome components, and exon skipping events.
Once generated, most circular RNAs are highly stable and accumulate in the cytoplasm after being exported from the nucleus in a length-dependent manner.
The biological functions of most circular RNAs remain unknown, but it is becoming increasingly clear that specific circular RNAs may modulate the activity of microRNAs or RNA binding proteins, be translated to yield protein products, or regulate innate immune responses.
Circular RNAs are most abundant in neuronal tissues, accumulate with aging, and have functional roles in human diseases, including cancer.
ACKNOWLEDGEMENTS
We thank all members of the Wilusz laboratory for helpful discussions and suggestions. Research on circular RNAs in our laboratory is supported by NIH grants R35-GM119735 and R01-NS099371. J.E.W. is a Rita Allen Foundation Scholar.
GLOSSARY
- Alternative splicing
the process by which exons and introns are selectively included or excluded from the mature mRNA, thereby allowing genes to generate multiple distinct transcript isoforms that may have unique functions.
- Backsplicing
the joining of a 5′ splice site to an upstream 3′ splice site by the spliceosome, thereby producing a mature circular RNA that has covalently linked ends.
- Competing endogenous RNA (ceRNA)
an RNA that binds a microRNA, thereby competitively limiting the ability of that microRNA to bind and repress other transcripts.
- Exon definition complex
initial splicing complexes in which U1 and U2 snRNPs are bound at opposite ends of a single exon and are stabilized by a network of protein-protein interactions. These cross-exon interactions must then be converted to cross-intron interactions in order to yield a canonically spliced linear RNA [65].
- Exosome
extracellular vesicles generated from endosomal compartments of most eukaryotic cells that can be detected in biological fluids (e.g. blood, urine, and cerebrospinal fluid) and have been proposed to function in intercellular communication [116].
- G-quadruplex
a highly stable RNA structure built upon sets of four guanines that interact with each other in a planar manner via non-canonical Hoogsteen base pairing interactions.
- Internal Ribosome Entry Site (IRES)
an RNA element that enables translation initiation to occur in a cap-independent manner in the middle of a mRNA [117].
- Ribonuclease R (RNase R)
a 3′-to-5′ exonuclease that digests most linear RNAs, thus allowing circular RNAs to become enriched.
- R-loop
a three-stranded nucleic acid structure that forms naturally during transcription and is comprised of a DNA:RNA hybrid, with the nontemplate DNA strand being single-stranded [118].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Wahl MC et al. (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136 (4), 701–18. [DOI] [PubMed] [Google Scholar]
- 2.Braunschweig U et al. (2013) Dynamic integration of splicing within gene regulatory pathways. Cell 152 (6), 1252–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nilsen TW and Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463 (7280), 457–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nigro JM et al. (1991) Scrambled exons. Cell 64 (3), 607–13. [DOI] [PubMed] [Google Scholar]
- 5.Cocquerelle C et al. (1993) Mis-splicing yields circular RNA molecules. FASEB J 7 (1), 155–60. [DOI] [PubMed] [Google Scholar]
- 6.Capel B et al. (1993) Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 73 (5), 1019–30. [DOI] [PubMed] [Google Scholar]
- 7.Salzman J et al. (2012) Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7 (2), e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeck WR et al. (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19 (2), 141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Memczak S et al. (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495 (7441), 333–8. [DOI] [PubMed] [Google Scholar]
- 10.Glazar P et al. (2014) circBase: a database for circular RNAs. RNA 20 (11), 1666–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilusz JE (2018) A 360 degrees view of circular RNAs: From biogenesis to functions. Wiley Interdiscip Rev RNA 9 (4), e1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X et al. (2018) The Biogenesis, Functions, and Challenges of Circular RNAs. Mol Cell 71 (3), 428–442. [DOI] [PubMed] [Google Scholar]
- 13.Kristensen LS et al. (2019) The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20 (11), 675–691. [DOI] [PubMed] [Google Scholar]
- 14.Patop IL et al. (2019) Past, present, and future of circRNAs. EMBO J 38 (16), e100836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo L and Salzman J (2016) Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17 (11), 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y et al. (2019) Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med 11 (1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen TB et al. (2016) Comparison of circular RNA prediction tools. Nucleic Acids Res 44 (6), e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng X et al. (2017) A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol 13 (6), e1005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen TB (2018) Improved circRNA Identification by Combining Prediction Algorithms. Front Cell Dev Biol 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tatomer DC et al. (2017) Inducible Expression of Eukaryotic Circular RNAs from Plasmids. Methods Mol Biol 1648, 143–154. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y et al. (2016) The Biogenesis of Nascent Circular RNAs. Cell Rep 15 (3), 611–624. [DOI] [PubMed] [Google Scholar]
- 22.Vo JN et al. (2019) The Landscape of Circular RNA in Cancer. Cell 176 (4), 869–881 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hossain ST et al. (2016) How RNase R Degrades Structured RNA: ROLE OF THE HELICASE ACTIVITY AND THE S1 DOMAIN. J Biol Chem 291 (15), 7877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao MS and Wilusz JE (2019) An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3’ ends. Nucleic Acids Res 47 (16), 8755–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panda AC et al. (2017) High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res 45 (12), e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vincent HA and Deutscher MP (2006) Substrate recognition and catalysis by the exoribonuclease RNase R. J Biol Chem 281 (40), 29769–75. [DOI] [PubMed] [Google Scholar]
- 27.Kwok CK et al. (2018) Detecting RNA G-Quadruplexes (rG4s) in the Transcriptome. Cold Spring Harb Perspect Biol 10 (7) pii: a032284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang PL et al. (2014) Circular RNA is expressed across the eukaryotic tree of life. PLoS One 9 (6), e90859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westholm JO et al. (2014) Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 9 (5), 1966–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conn SJ et al. (2015) The RNA binding protein quaking regulates formation of circRNAs. Cell 160 (6), 1125–34. [DOI] [PubMed] [Google Scholar]
- 31.Rybak-Wolf A et al. (2015) Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol Cell 58 (5), 870–85. [DOI] [PubMed] [Google Scholar]
- 32.Zhang XO et al. (2014) Complementary sequence-mediated exon circularization. Cell 159 (1), 134–147. [DOI] [PubMed] [Google Scholar]
- 33.Stagsted LV et al. (2019) Noncoding AUG circRNAs constitute an abundant and conserved subclass of circles. Life Sci Alliance 2 (3) pii: e201900398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maass PG et al. (2017) A map of human circular RNAs in clinically relevant tissues. J Mol Med (Berl) 95 (11), 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu Q et al. (2017) PlantcircBase: A Database for Plant Circular RNAs. Mol Plant 10 (8), 1126–1128. [DOI] [PubMed] [Google Scholar]
- 36.Chen S et al. (2019) Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell 176 (4), 831–843 e22. [DOI] [PubMed] [Google Scholar]
- 37.Ye CY et al. (2015) Widespread noncoding circular RNAs in plants. New Phytol 208 (1), 88–95. [DOI] [PubMed] [Google Scholar]
- 38.Zhang XO et al. (2016) Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res 26 (9), 1277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao Y et al. (2016) Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun 7, 12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enuka Y et al. (2016) Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res 44 (3), 1370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veno MT et al. (2015) Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol 16, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.You X et al. (2015) Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 18 (4), 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cortes-Lopez M et al. (2018) Global accumulation of circRNAs during aging in Caenorhabditis elegans. BMC Genomics 19 (1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gruner H et al. (2016) CircRNA accumulation in the aging mouse brain. Sci Rep 6, 38907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall H et al. (2017) Transcriptome profiling of aging Drosophila photoreceptors reveals gene expression trends that correlate with visual senescence. BMC Genomics 18 (1), 894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou C et al. (2017) Genome-Wide Maps of m6A circRNAs Identify Widespread and Cell-Type-Specific Methylation Patterns that Are Distinct from mRNAs. Cell Rep 20 (9), 2262–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen YG et al. (2019) N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol Cell 76 (1), 96–109 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu CX et al. (2019) Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell 177 (4), 865–880 e21. [DOI] [PubMed] [Google Scholar]
- 49.Dubin RA et al. (1995) Inverted repeats are necessary for circularization of the mouse testis Sry transcript. Gene 167 (1-2), 245–8. [DOI] [PubMed] [Google Scholar]
- 50.Ivanov A et al. (2015) Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 10 (2), 170–7. [DOI] [PubMed] [Google Scholar]
- 51.Liang D and Wilusz JE (2014) Short intronic repeat sequences facilitate circular RNA production. Genes Dev 28 (20), 2233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kramer MC et al. (2015) Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev 29 (20), 2168–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hansen TB et al. (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495 (7441), 384–8. [DOI] [PubMed] [Google Scholar]
- 54.Wilusz JE (2015) Repetitive elements regulate circular RNA biogenesis. Mob Genet Elements 5 (3), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong R et al. (2017) Increased complexity of circRNA expression during species evolution. RNA Biol 14 (8), 1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang D et al. (2017) The Output of Protein-Coding Genes Shifts to Circular RNAs When the Pre-mRNA Processing Machinery Is Limiting. Mol Cell 68 (5), 940–954 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barrett SP et al. (2015) Circular RNA biogenesis can proceed through an exon-containing lariat precursor. Elife 4, e07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zaphiropoulos PG (1996) Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: correlation with exon skipping. Proc Natl Acad Sci U S A 93 (13), 6536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kelly S et al. (2015) Exon Skipping Is Correlated with Exon Circularization. J Mol Biol 427 (15), 2414–2417. [DOI] [PubMed] [Google Scholar]
- 60.Conn VM et al. (2017) A circRNA from SEPALLATA3 regulates splicing of its cognate mRNA through R-loop formation. Nat Plants 3, 17053. [DOI] [PubMed] [Google Scholar]
- 61.Aktas T et al. (2017) DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 544 (7648), 115–119. [DOI] [PubMed] [Google Scholar]
- 62.Ashwal-Fluss R et al. (2014) circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56 (1), 55–66. [DOI] [PubMed] [Google Scholar]
- 63.Stegeman R et al. (2018) Proper splicing contributes to visual function in the aging Drosophila eye. Aging Cell 17 (5), e12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang M et al. (2019) Long and Repeat-Rich Intronic Sequences Favor Circular RNA Formation under Conditions of Reduced Spliceosome Activity. iScience 20, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berget SM (1995) Exon recognition in vertebrate splicing. J Biol Chem 270 (6), 2411–4. [DOI] [PubMed] [Google Scholar]
- 66.Li X et al. (2019) A unified mechanism for intron and exon definition and back-splicing. Nature 573 (7774), 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kleaveland B et al. (2018) A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 174 (2), 350–362 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y et al. (2015) Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res 25 (8), 981–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lasda E and Parker R (2016) Circular RNAs Co-Precipitate with Extracellular Vesicles: A Possible Mechanism for circRNA Clearance. PLoS One 11 (2), e0148407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Preusser C et al. (2018) Selective release of circRNAs in platelet-derived extracellular vesicles. J Extracell Vesicles 7 (1), 1424473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Park OH et al. (2019) Endoribonucleolytic Cleavage of m(6)A-Containing RNAs by RNase P/MRP Complex. Mol Cell 74 (3), 494–507 e8. [DOI] [PubMed] [Google Scholar]
- 72.Li Z et al. (2015) Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22 (3), 256–64. [DOI] [PubMed] [Google Scholar]
- 73.Huang C et al. (2018) A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes Dev 32 (9–10), 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gatfield D et al. (2001) The DExH/D box protein HEL/UAP56 is essential for mRNA nuclear export in Drosophila. Curr Biol 11 (21), 1716–21. [DOI] [PubMed] [Google Scholar]
- 75.Wesselhoeft RA et al. (2018) Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat Commun 9 (1), 2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Puttaraju M and Been MD (1992) Group I permuted intron-exon (PIE) sequences self-splice to produce circular exons. Nucleic Acids Res 20 (20), 5357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ford E and Ares M Jr. (1994) Synthesis of circular RNA in bacteria and yeast using RNA cyclase ribozymes derived from a group I intron of phage T4. Proc Natl Acad Sci U S A 91 (8), 3117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen YG et al. (2017) Sensing Self and Foreign Circular RNAs by Intron Identity. Mol Cell 67 (2), 228–238 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Petkovic S and Muller S (2015) RNA circularization strategies in vivo and in vitro. Nucleic Acids Res 43 (4), 2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meganck RM et al. (2018) Tissue-Dependent Expression and Translation of Circular RNAs with Recombinant AAV Vectors In Vivo. Mol Ther Nucleic Acids 13, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Litke JL and Jaffrey SR (2019) Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat Biotechnol 37 (6), 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garikipati VNS et al. (2019) Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat Commun 10 (1), 4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Piwecka M et al. (2017) Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 357 (6357), eaam8526. [DOI] [PubMed] [Google Scholar]
- 84.Legnini I et al. (2017) Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol Cell 66 (1), 22–37 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng Q et al. (2016) Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 7, 11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xia P et al. (2018) A Circular RNA Protects Dormant Hematopoietic Stem Cells from DNA Sensor cGAS-Mediated Exhaustion. Immunity 48 (4), 688–701 e7. [DOI] [PubMed] [Google Scholar]
- 87.Kocks C et al. (2018) Single-Molecule Fluorescence In Situ Hybridization (FISH) of Circular RNA CDR1as. Methods Mol Biol 1724, 77–96. [DOI] [PubMed] [Google Scholar]
- 88.Kristensen LS et al. (2018) Circular RNAs are abundantly expressed and upregulated during human epidermal stem cell differentiation. RNA Biol 15 (2), 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo JU et al. (2014) Expanded identification and characterization of mammalian circular RNAs. Genome Biol 15 (7), 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bosson AD et al. (2014) Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 56 (3), 347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen CY and Sarnow P (1995) Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268 (5209), 415–7. [DOI] [PubMed] [Google Scholar]
- 92.Wang Y and Wang Z (2015) Efficient backsplicing produces translatable circular mRNAs. RNA 21 (2), 172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tatomer DC and Wilusz JE (2017) An Unchartered Journey for Ribosomes: Circumnavigating Circular RNAs to Produce Proteins. Mol Cell 66 (1), 1–2. [DOI] [PubMed] [Google Scholar]
- 94.Pamudurti NR et al. (2017) Translation of CircRNAs. Mol Cell 66 (1), 9–21 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang Y et al. (2017) Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res 27 (5), 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liang WC et al. (2019) Translation of the circular RNA circbeta-catenin promotes liver cancer cell growth through activation of the Wnt pathway. Genome Biol 20 (1), 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wesselhoeft RA et al. (2019) RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In Vivo. Mol Cell 74 (3), 508–520 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Du WW et al. (2016) Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 44 (6), 2846–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abdelmohsen K et al. (2017) Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol 14 (3), 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sanger HL et al. (1976) Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A 73 (11), 3852–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Toptan T et al. (2018) Circular DNA tumor viruses make circular RNAs. Proc Natl Acad Sci U S A 115 (37), E8737–E8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang JT et al. (2019) Identification of virus-encoded circular RNA. Virology 529, 144–151. [DOI] [PubMed] [Google Scholar]
- 103.Tagawa T et al. (2018) Discovery of Kaposi’s sarcoma herpesvirus-encoded circular RNAs and a human antiviral circular RNA. Proc Natl Acad Sci U S A 115 (50), 12805–12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ungerleider N et al. (2018) The Epstein Barr virus circRNAome. PLoS Pathog 14 (8), e1007206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao J et al. (2019) Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat Commun 10 (1), 2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li X et al. (2017) Coordinated circRNA Biogenesis and Function with NF90/NF110 in Viral Infection. Mol Cell 67 (2), 214–227 e7. [DOI] [PubMed] [Google Scholar]
- 107.Kindler E et al. (2017) Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog 13 (2), e1006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Khabar KS et al. (2003) RNase L mediates transient control of the interferon response through modulation of the double-stranded RNA-dependent protein kinase PKR. J Biol Chem 278 (22), 20124–32. [DOI] [PubMed] [Google Scholar]
- 109.Bachmayr-Heyda A et al. (2015) Correlation of circular RNA abundance with proliferation--exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 5, 8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yu J et al. (2018) Circular RNA cSMARCA5 inhibits growth and metastasis in hepatocellular carcinoma. J Hepatol 68 (6), 1214–1227. [DOI] [PubMed] [Google Scholar]
- 111.Guarnerio J et al. (2016) Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 165 (2), 289–302. [DOI] [PubMed] [Google Scholar]
- 112.Kristensen LS et al. (2018) Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 37 (5), 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guarnerio J et al. (2019) Intragenic antagonistic roles of protein and circRNA in tumorigenesis. Cell Res 29 (8), 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang W et al. (2016) Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene 35 (30), 3919–31. [DOI] [PubMed] [Google Scholar]
- 115.Dube U et al. (2019) An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat Neurosci 22 (11), 1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Raposo G and Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200 (4), 373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shatsky IN et al. (2018) Cap-Independent Translation: What’s in a Name? Trends Biochem Sci 43 (11), 882–895. [DOI] [PubMed] [Google Scholar]
- 118.Skourti-Stathaki K and Proudfoot NJ (2014) A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev 28 (13), 1384–96. [DOI] [PMC free article] [PubMed] [Google Scholar]