

This study defines a single-cell proteomic atlas of human OA cartilage to reveal rare subpopulations that stratify patients.
Abstract
Aging or injury leads to degradation of the cartilage matrix and the development of osteoarthritis (OA). Because of a paucity of single-cell studies of OA cartilage, little is known about the interpatient variability in its cellular composition and, more importantly, about the cell subpopulations that drive the disease. Here, we profiled healthy and OA cartilage samples using mass cytometry to establish a single-cell atlas, revealing distinct chondrocyte progenitor and inflammation-modulating subpopulations. These rare populations include an inflammation-amplifying (Inf-A) population, marked by interleukin-1 receptor 1 and tumor necrosis factor receptor II, whose inhibition decreased inflammation, and an inflammation-dampening (Inf-D) population, marked by CD24, which is resistant to inflammation. We devised a pharmacological strategy targeting Inf-A and Inf-D cells that significantly decreased inflammation in OA chondrocytes. Using our atlas, we stratified patients with OA in three groups that are distinguished by the relative proportions of inflammatory to regenerative cells, making it possible to devise precision therapeutic approaches.
INTRODUCTION
Osteoarthritis (OA) is a highly prevalent, age-related disease of the joints, characterized by cartilage degeneration, loss of mobility, and chronic pain. Much work has been done investigating several aspects of its complex etiology, including the contributions of metabolic, epigenetic, genetic, and cellular factors. However, no disease-modifying drugs exist to treat OA, with the current standard of care being limited to pain management, followed by eventual joint replacement. Recent and ongoing work has highlighted the important interplay between aging, inflammation, and loss of regenerative potential in multiple tissues. Although cartilage is a relatively simple tissue, with a single cell type being encapsulated in its secreted extracellular matrix, the variable degree of degeneration associated with each patient with OA suggests that understanding this tissue at a single-cell level can provide insights into the onset and progression of pathology.
Defining the precise subpopulations that constitute cartilage will also aid strategies for cartilage tissue engineering or for enhancing endogenous cartilage regeneration. Unlike other skeletal tissues, cartilage has a remarkably low regeneration potential. Even injuries sustained in youth remain unrepaired, giving rise to the fibrocartilaginous tissue that can lead to accelerated OA pathology. Multiple studies have explored the putative cartilage progenitor cells (CPCs) in articular cartilage by characterizing their cell surface markers and describing their function (1). Notably, the CPC populations were reported to be enriched in OA cartilage, having an increased migratory potential, the ability to form highly clonal populations, and multipotency (i.e., the ability to give rise to chondrocytes, osteoblasts, and adipocytes in culture) (2–4). Recently, the human skeletal stem cell (hSSC) was identified (5), further suggesting another fountain of cells for repair. However, despite the existence of these putative regenerative populations, overall cartilage repair remains low, both in healthy and diseased states. Cartilage repair is variable even in younger, non-OA patients who undergo cartilage related injuries, such as anterior cruciate ligament rupture or degenerative meniscal tears, with some patients having a good recovery while others develop OA over a decade or so. Collectively, this suggests that there are factors preventing effective repair and regeneration of the tissue and that these factors vary between patients.
One source of this limited repair might be the chronic inflammation experienced by the joint. The synovium is known to be infiltrated by a variety of immune cells (6), and several inflammatory cytokines have been detected in the synovial fluid of patients with OA (7). Further, several studies have characterized the actions of the hypoxia factors (HIFs), nitric oxide, reactive oxygen species, nuclear factor κB (NF-κB) signaling, and other pathways that maintain the proinflammatory environment. To understand how this milieu might affect the proregenerative populations, such as the CPCs, we used single-cell mass cytometry [cytometry by time-of-flight (cyTOF)] to map both the proregenerative cell populations and inflammatory populations. By simultaneously being able to map cell identity and signaling states, we could observe how cells interact and influence each other. Furthermore, these maps provide us with a cell population–based stratification of patients with OA, which may aid in targeted OA therapeutics in the future.
RESULTS
High-dimensional mass cytometry–based profiling of normal and OA cartilage
Toward our goal of profiling rare stem/progenitor-like populations within normal and OA cartilage, we used cyTOF, a mass spectrometry–based high-dimensional method for single-cell detection of isotope-labeled antibodies (Fig. 1A) (8). While cyTOF panels have to be preselected for each experiment, this technique provides the advantage that a large number of cells can be easily profiled in multiple samples without being cost prohibitive. This profiling at the protein level is complementary to single-cell transcriptomics and can provide a snapshot of the active signaling pathways in a specific subpopulation. After a detailed study of the literature and our preliminary data, a panel of 33 markers was labeled and optimized (see Methods and table S1) for profiling chondrocytes. This panel included cell surface receptors, adhesion molecules, signaling mediators, and cell cycle and transcription factors that are known to be important for cartilage homeostasis (table S1). Samples were collected from the surgical waste of patients with OA undergoing total knee arthroplasty [according to an Institutional Review Board (IRB) protocol approved by Stanford University], digested and expanded for a single passage in high-density culture as previously described (9). Each sample had an expression ratio of Col2a1/Col1a1 between 10 and 100 (fig. S1A), and the expression of MMP3, MMP9, and MMP13 was 10- to 10,000-fold higher in OA cartilage compared to normal, as expected (fig. S1, B to D). An average of 3 × 104 and 10 × 104 cells were assayed per OA or normal sample, respectively, and, to ensure chondrogenicity, only the SOX99/CD44 double-positive cells were further analyzed (fig. S1E). For visualization, the total population was downsampled to 9%, representing 9000 cells, and cells were projected onto a two-dimensional plane using t-distributed stochastic neighbor embedding (tSNE) (Fig. 1B). The spatial representations of OA and normal cells are distinct, although no single sample (patient) for either normal or OA samples was observed to dominate this representation (Fig. 1, C and D). Analysis of SOX9 and CD44 staining showed high levels of staining across all cells, ensuring the chondrogenic phenotype of the cells with no dedifferentiation observed during sample processing (Fig. 1, E and F). We proceeded to analyze the single-cell data from 20 OA and 5 normal samples. We observed known features of the OA landscape, for example, the expansion of NOTCH1-expressing chondrocytes in OA (Fig. 1, E and F). Phosphorylated NF-κB (pNF-κB), in contrast, could not readily distinguish between normal and OA samples, which both consisted of populations manifesting high, medium, and low levels of signaling (Fig. 1, E and F).
Fig. 1. High-dimensional profiling of normal and OA chondrocytes using mass cytometry.
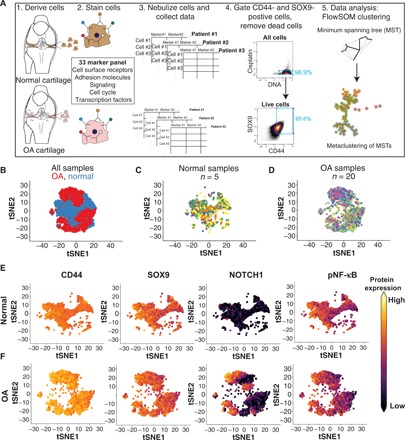
(A) Schematic outlining the procedures used to profile chondrocytes by mass cytometry. Briefly, cells are dissociated from cartilage tissue, stained with metal-conjugated antibodies, and analyzed using cyTOF. The resulting data are then gated for live, SOX9/CD44-positive chondrocytes that are used for downstream analyses, including identifying clusters with FlowSOM. (B) tSNE projections of the normal (blue) and OA (red) chondrocytes where each cell is represented by a dot. Each group was downsampled randomly to 9000 cells. (C) Normal chondrocytes colored by patient sample, downsampled to 9000 cells. (D) OA chondrocytes colored by patient sample, downsampled to 9000 cells. (E) tSNE plots of 9000 normal chondrocytes, colored by the expression of two chondrogenic markers (SOX9 and CD44), the cell surface receptor NOTCH1, and pNF-κB. Expression is set at the max of each channel and is comparable between (E) and (F). (F) tSNE plots of 9000 OA chondrocytes, colored by the expression of two chondrogenic markers (SOX9 and CD44), the cell surface receptor NOTCH1, and pNF-κB. Expression is set at the max of each channel and is comparable between (E) and (F).
Normal and OA cartilage landscape consists of both abundant and rare subpopulations
To find unique subpopulations in the normal and OA cartilage, we used the algorithm FlowSOM (10) to define clusters (see Methods) based on the similarity of expression of cell surface receptors and intracellular markers. FlowSOM identified 20 clusters or subpopulations in our data (Fig. 2, A and B ). Using an alternate algorithm, X-shift (11), we observed a similar number and composition of clusters (fig. S1F), providing an independent validation of the FlowSOM analyses. A standard scaled distribution matrix for all the surface receptors and intracellular markers used to define the 20 clusters with FlowSOM (Fig. 2C) demonstrates the molecular identity of these clusters. For example, clusters 1 and 2 are marked by high intercellular adhesion molecule (ICAM), clusters 12 and 16 have a high expression of NOTCH1, STRO1, and CD166, and clusters 15 and 20 have high interleukin-1 receptor 1 (IL1R1) and tumor necrosis factor receptor II (TNFRII). Using the 20 clusters, we observed that the patients with OA were highly anticorrelated with the normal samples (fig. S2A). On the basis of the known functions of the molecules that defined each subpopulation, we broadly defined clusters as CPC clusters or non-CPC clusters (Fig. 2D and fig. S2B).
Fig. 2. Normal and OA cartilage landscape consists of both abundant and rare subpopulations.
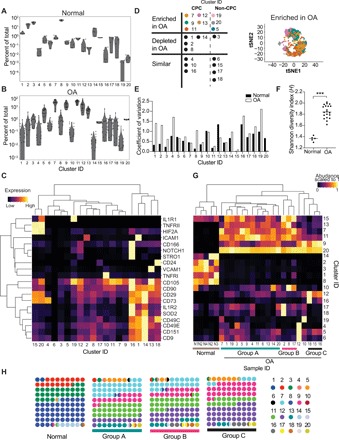
(A) Abundance of each of the 20 clusters called by FlowSOM analysis in normal samples. Each point represents a single sample. (n = 5). (B) Abundance of each of the 20 clusters called by FlowSOM analysis in OA samples (n = 20). Each point represents a single sample. (C) Expression of cell surface receptors used for delineating the 20 clusters. Expression is averaged between all cells of a given cluster ID. Color is scaled to 1 for each protein between all the clusters. Dendograms were drawn using complete-linkage hierarchical clustering. (D) Table of the cluster IDs that are enriched, depleted, or similar between OA and normal samples. Colors in the enriched section correspond to the tSNE projection on the right. The tSNE projection contains cells from clusters that are enriched in OA compared to normal, sampled to 9000 cells. Enrichment, depletion, or similarity between the ranked means of normal (n = 5) and OA (n = 20) cluster abundance was tested using an unpaired, two-tailed Mann-Whitney test with Bonferroni correction (α = 0.0025). Adjusted P values for all enriched or depleted clusters are 0.002. (E) Coefficient of variation (mean divided by SD) for each cluster in normal or OA samples. (F) Shannon’s diversity index (H) calculated for each normal and OA sample (see Methods). Theoretical max H value is 2.99. Equality between the means H values for OA (n = 20) and normal (n = 5) samples was tested using a two-tailed Mann-Whitney test. ***P = 0.001. (G) Hierarchical clustering of normal and OA samples by cluster abundances. Abundance is scaled to 1. Samples belonging to the three designated groups are labeled at the bottom. (H) Average cluster abundance in normal and group A, B, and C patients with OA. Each color designates a cluster ID.
We next wanted to investigate how the nature and frequency of the identified subpopulations varied between the normal and OA samples, specifically to determine whether populations were gained or lost with disease. On the basis of this idea, we categorized the clusters into three groups: (i) increased in OA, (ii) unchanged between OA and normal, and (iii) decreased in OA. Eight subpopulations (clusters 5, 7, 9, 11, 12, 13, 19, and 20) were enriched in the OA samples compared to normal; five subpopulations (clusters 1, 2, 3, 8, and 14) were depleted compared to normal, while seven subpopulations (clusters 4, 6, 10, 15, 16, 17, and 18) remained unchanged between the OA and normal samples (Fig. 2D and fig. S2B). Quantitation of the frequency of these populations revealed interpatient heterogeneity, which we quantified using the coefficient of variation (Fig. 2E). CPC clusters 4 and 16 along with the non-CPC cluster 19 were among the most variable between patients with OA, while clusters 15 and 20 were the least variable (Fig. 2E). As an alternative way to quantify this heterogeneity, we used a metric used in population ecology, known as Shannon’s diversity index, which describes how heterogeneous and evenly distributed populations are in an ecosystem. On the basis of the 20 populations identified by FlowSOM, we observed that (i) OA samples had a higher Shannon diversity index (H value) and, additionally, (ii) the range of H values for patients with OA was larger than for normal samples, indicating a loss of population evenness in OA (Fig. 2F). A direct comparison between the OA and normal samples is difficult, however, as the number of OA samples (n = 20) is much larger than normal samples (n = 5). Hence, this dataset may be missing some of the potentially higher variability clusters in normal cartilage.
Using these populations, whose unique identities are detailed in later sections, we performed hierarchical clustering of the 20 OA patient and 5 normal samples in our study. Our goal was to identify subsets of patients with unique compositions of these rare populations. Such characterization, common in the cancer field, can be helpful in designing targeted therapeutic strategies tailored to groups of patients with similar molecular underpinnings driving their disease. As expected, all the normal samples clustered together (Fig. 2G). We observed three major groups of patients, with some patients that clustered only with themselves. Group A, the largest of the three groups with 12 patients, was enriched in clusters 7 and 11, marked by CD105 expression (Fig. 2, G and H). Group B, consisting of three patients, was enriched in clusters 17 and 18, the CD24+ populations, and group C, also consisting of three patients, was characterized by a high abundance of clusters 9, 12, and 16 that were identified to be NOTCH1/VCAM-1 (vascular cell adhesion molecule–1)–positive CPC (Fig. 2, G and H). In the following sections, we will detail the unique characteristics of these populations and the etiology that they reveal about the underlying patients with OA.
Patients are differentially enriched in inflammatory and noninflammatory CPCs
Several studies (3, 4, 12–19) have found CPCs that have the ability to give rise to chondrocytes, show self-renewal in culture, and are able to migrate in OA cartilage. These CPCs are believed to be the origin of the highly clonal characteristic clusters (20, 21) found in OA cartilage. Their role in OA disease pathology, however, remains unclear, especially whether they contribute to disease onset and progression. To address these questions and better characterize the CPCs and their cross-talk with other cartilage-resident cells, we had designed our cyTOF panel to include 13 previously described markers for CPCs (Fig. 3A and table S1). Of the 20 clusters identified using FlowSOM, 12 clusters were found to be enriched for these CPC markers in a variety of combinations (Fig. 3A and fig. S3A). The rest of the clusters, designated non-CPC, are very low in their expression of the CPC markers as shown for clusters 3, 5, and 6 (fig. S2A). In contrast to previous observations, we found that there are three variants of CPC subpopulations that are depleted in OA (Fig. 3B), which we termed CPC I. Out of the rest, two clusters were unchanged between normal and OA cartilage, termed CPC II, and six clusters were enriched in OA cartilage, comprising some of the previously described CPC populations, which we termed CPC III (Fig. 3B).
Fig. 3. Patients with OA are differentially enriched in types of CPCs.
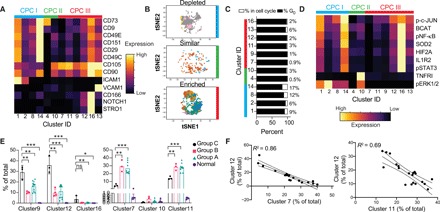
(A) Expression of the 13 CPC markers among the clusters that are enriched for them. Expression is scaled to 1 between all clusters. (B) tSNE projections of the type I (depleted), type II (similar), and type III (enriched) CPCs in OA, colored by cluster ID, where each cluster ID has a different color. Cells are sampled to 9000 when possible. (C) Cell cycle analysis for each cluster. Cell cycle stages were analyzed for each cell, and then, the proportion of the population in G0 and in the cell cycle was calculated for each cluster. The percentage in the cell cycle is given to the right of each bar graph. (D) Cell signaling and other intracellular and cell surface receptor markers for the CPC clusters. Expression is scaled to 1. (E) Cluster abundance for each sample in the OA groups and normal cells. Significance is tested with a multiple-test corrected Welch’s t test. ns, not significant. (F) Correlation between abundance of each cluster, labeled on each axis. Each point represents a patient with OA. The full matrix of correlations between clusters is plotted in fig. S3A. *P = 0.05, **P = 0.01, and ***P = 0.001.
The CPC I clusters were characterized by lower CD105 expression in contrast to the CPC III clusters (Fig. 3A). Cluster 1 and 2 cells were distinct in having a high expression of CD54 (ICAM) (Fig. 3A). Previous work exploring markers for stem or progenitor cells had noted that cells with high CD54 and CD55 expression had higher levels of aldehyde dehydrogenase activity, associated with stem cell function (22). Cluster 14 was distinguished by the expression of CD151, i.e., tetraspanin, a cell adhesion marker, which was described to mark chondrocytes with higher chondrogenic potential in an in vitro study (23). Cell cycle analysis showed that CPC I clusters had the highest percentage of cells that were cycling (Fig. 3C), although, overall, the number of cycling cells was low, as expected for postmitotic chondrocytes (<20%). The CPC I clusters are exclusively characterized by extracellular signal–regulated kinase 1/2 signaling, while the other clusters, with the exception of the CPC II cluster 10, are not (Fig. 3D). Out of the CPC II clusters, cluster 4 is characterized by a high CD73 expression and is not predominantly active in any of the tested signaling pathways (Fig. 3D). CD73 has recently been identified to be one of the critical markers on an adult hSSC population (5). The CPC III populations contained clusters that were enriched for many inflammatory signaling pathways. Clusters 12, 13, and 16 were high in the expression of pNF-κB, pSTAT3 (phosphorylated Signal transducer and activator of transcription 3), β-catenin, and HIF2A, associated with inflammation in OA. However, CPC III also had populations that were low in these pathways, namely, clusters 7, 9, and 11 (Fig. 3D). Cluster 16 appears to be the quintessential CD105/CD90-high, NOTCH1/STRO1–driven migratory CPC that has been previously identified in OA cartilage (15, 24). Group C patients had a significantly higher percentage of the proinflammatory clusters 9, 12, and 16 and a lower percentage of low-inflammation clusters 7 and 11 (Fig. 3E). This anticorrelation between clusters 9 and 11, clusters 12 and 7, and clusters 16 and 14 (Fig. 3F and fig. S3, A and B) held across the 20-patient cohort, suggesting that these patients might be particularly driven by this cellular subtype.
Identification of a rare inflammation-amplifying population in OA cartilage
We further analyzed the non-CPC populations that were identified by our panel, with a focus on putative inflammatory populations that might contribute to pathology. Among these were clusters 15 and 20, which are characterized by the coexpression of two cytokine receptors, IL1R1 (CD121A) and TNFRII (CD120B) (Fig. 4, A to C). Cluster 20 is significantly expanded in OA cartilage compared to the normal cartilage (Fig. 4D). Clusters 15 and 20 vary in the quantity IL1R1 expression, with cluster 20 having a higher level of IL1R1 (Fig. 4E). However, both clusters 15 and 20 have similarly high levels of TNFRII and HIF2A expression (Fig. 4E).
Fig. 4. Identification of a rare immune recruiting population in OA cartilage.
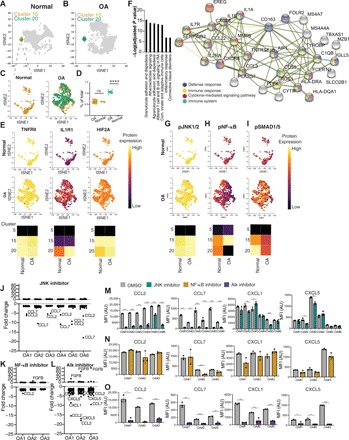
(A) tSNE projection of normal cells (gray) with clusters 15 and 20 colored, sampled at 9000 cells. (B) tSNE projection of OA cells (gray) with clusters 15 and 20 colored, sampled at 9000 cells. (C) A magnified projection of clusters 15 and 20 from normal and OA samples, ****P = 0.0001. (D) Quantification of the abundance of clusters 15 and 20 in normal and OA samples. Significance is tested using Welch’s t test. Each point represents a sample. (E) Magnified projection of clusters 15 and 20 depicting expression of the two cell surface receptors, TNFRII and IL1R1, and of intracellular HIF2A. Expression is scaled to max value in dataset for each protein and is comparable across normal and OA samples. Heatmap below the tSNE depicts quantification of average expression in representative chondrocytes (cluster 5) in comparison to clusters 15 and 20. (F) Single-cell RNA (scRNA) sequencing data from (25) reanalyzed. Cells expressing TNFRII and IL1R1 were sorted in silico, and their transcriptome was compared to the rest of the OA cells and used for Gene Ontology (GO) term and STRING analyses. (G) Same as in (E), for signaling markers pJNK1/2, pNF-κB (H), and pSMAD1/5 (I). (J) Fold change in cytokines from human 62-plex Luminex assay between dimethyl sulfoxide (DMSO) and JNK inhibitor treatment. (K) Fold change in cytokines from human 62-plex Luminex assay between DMSO and NF-κB inhibitor treatment. (L) Fold change in cytokines from human 62-plex Luminex assay between DMSO and Alk inhibitor treatment. (M) Raw mean fluorescence intensity (MFI) values for cytokines that were significantly altered between DMSO- and JNK-treated samples in at least five of six tested OA samples. Significance was first tested for using analysis of variance (ANOVA) with multiple corrections for the 62 comparisons, and then, t test with Tukey’s correction was applied for each comparison on a patient-by-patient sample. Each point represents an independent technical treatment and cytokine analyses for the same patient (n = 6 patients with OA). AU, arbitrary units. (N and O) Same as in (M) but with NF-κB and Alk inhibitors, respectively (n = 3 patients with OA). *P = 0.05, **P = 0.01, and ***P = 0.001.
To further understand the molecular underpinnings of these subpopulations, we used publicly available single-cell RNA (scRNA) sequencing data (25). We were able to successfully identify cells that expressed both IL1R1 and TNFRSF1B transcripts in the scRNA sequencing data. These cells represented about ~2% of sequenced cells, validating the frequency we observed by cyTOF. Chondrocytes that expressed both transcripts were sorted in silico, and the differentially expressed genes and pathways were analyzed. The IL1RI/TNFRII-expressing chondrocytes were found to be highly enriched in pathways related to innate and adaptive immune cells, inflammation, and altered T and B cells signaling in arthritis (Fig. 4F). These analyses suggest that the IL1RI/TNFRII cells might act to recruit immune cells to the joint space. We, therefore, termed clusters 15 and 20 inflammation-amplifying (Inf-A) chondrocytes. Upon analyzing their signaling status, the Inf-A clusters showed exclusive signaling through pJNK (phosphorylated c-Jun N-terminal kinase) and pSMAD1/5 compared to the rest of the chondrocyte clusters (Fig. 4, G and I). In contrast, pNF-κB levels in clusters 15 and 20 were similar to other clusters identified (Fig. 4H). Despite its rarity, cluster 20 was highly consistent among patients, with TNFRII expression and JNK and SMAD1/5 phosphorylation levels consistently high across all patients with OA in cluster 20, and more variable in cluster 15 (fig. S4A). Cluster 20 shows the lowest coefficient of variation in the OA samples (Fig. 2E).
Next, we sought to explore the functional effects of inhibiting these Inf-A cells in OA cartilage by capitalizing on their distinct signaling through JNK. Chondrocytes derived from six patients were cultured for 48 hours in the presence of JNK inhibitor II, and the secretome was analyzed via 62 antibody human Luminex panels. Across all six patients, a variety of cytokines were altered (fig. S4B), many trending toward significance. Restricting our analysis to only those cytokines that were altered in five or more patients (>83% response rate), we observed a significant decrease in C-C motif chemokine ligand 2 (CCL2) and CCL7 after JNK inhibition (Fig. 4, J and M). CCL2 and CCL7 are well-established chemoattractants for monocytes and are known to be altered during OA progression (26). Genetic deletions of CCL2 and its receptor CCR2 prevent the development of surgical OA, further underscoring the importance of CCL2 as a key modulator in pathology (27). In contrast, inhibition of NF-κB activity with BMS-345541 (28) did not affect CCL2 or CCL7 secretion in OA chondrocytes (Fig. 4, K and N), suggesting that the effect is specific to the Inf-A population. As a complementary approach, we inhibited SMAD1/5, the other exclusive signaling pathway of the Inf-A cells, using an ALK (activin receptor-like kinase) inhibitor. ALK receptors are the most common upstream target of SMAD1/5 signaling in OA (29). As hypothesized, ALK inhibitor treatment resulted in a decrease in the same cytokines affected by the JNK inhibitor, CCL2, and CCL7, and, additionally, C-X-C motif chemokine ligand 1 (CXCL1) and CXCL5 (Fig. 4, L and O), two other leukocyte attracting factors. Collectively, these data are consistent with the transcriptional data suggesting that the IFNR1 (interferon receptor 1)/TNFRII–coexpressing cells mark a rare OA subpopulation that is potentially responsible for immune recruitment to the joint. We demonstrated that inhibition of this rare population can significantly affect the overall secretome of the end-stage OA chondrocytes.
A CD24+ chondrocyte population mitigates inflammation in OA cartilage
Our previous work established a role for the cell surface receptor CD24 in mitigating inflammation in healthy and induced pluripotent stem cell (iPSC)-derived chondrocytes (30). Although CD24 is highly expressed in juvenile and iPSC-derived chondrocytes, its expression is decreased with age (30), potentially underscoring the age-related etiology of OA. We included CD24 in our cyTOF panel to understand the interplay of CD24+ cells with the other regenerative and inflammatory subpopulations in the OA joint. FlowSOM-derived clusters 17 and 18 were found to be most enriched in CD24 expression (Fig. 5, A and C). Both clusters 17 and 18 were found in equal numbers in normal and OA cartilage; however, there was a high variability in their abundance between patients (Fig. 5B). In agreement with our previous report, CD24 cells decreased with age (fig. S5A) and were among the least reactive groups to undergo stimulation by the proinflammatory cytokine IL1B (fig. S5B). Therefore, we termed clusters 17 and 18 inflammation-dampening (Inf-D) I and II cells, respectively. Inf-D II cells had the highest levels of CD24 expression and also had higher levels of Sox9 and CD44, although expression in Inf-D I cells was comparable with normal cells (Fig. 5C). To further characterize the function of these CD24+ cells, we used the same previously published scRNA sequencing dataset and sorted out CD24+ cells. Consistent with our hypothesis that the CD24+ cells are capable of immune modulation, we observed an enrichment for pathways related to inflammation and immune cell trafficking and cross-talk (Fig. 5D and fig. S5C). In addition, the CD24+ cells showed an enrichment of oxidative phosphorylation pathways, suggesting that these cells could have different metabolic processes compared to other chondrocytes (Fig. 5D and fig. S5C).
Fig. 5. A CD24+ subpopulation mitigates inflammation in OA cartilage.
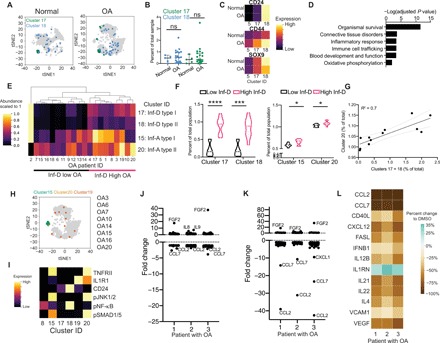
(A) tSNE projection of normal and OA cells (gray) with clusters 17 and 18 colored, sampled at 9000 cells each. (B) Abundance of each cluster per sample. Differences between the means were tested using Welch’s t test. (C) Heatmaps of chondrogenic markers SOX9 and CD44, as well as CD24. Expression is scaled to the highest expressing cell in the group. (D) scRNA sequencing data from (25), reanalyzed. Cells expressing CD24 with a high Col2a1/Col1a1 ratio were sorted in silico, and their transcriptome was compared to the rest of the OA cells and used for GO term and STRING analyses. (E) Hierarchical clustering of OA samples based on clusters 15, 17, 18, and 20. Abundance is scaled to one for each cluster. Groups are labeled along the x axis. (F) Violin plots of abundance of clusters 17, 18, 15, and 20 in low and high Inf-D groups. Each sample is represented as a point. (G) Correlation between the abundance of cluster 20 with clusters 17 + 18. Ninety-five percent confidence interval is shown in gray dashed line. Slope of line tested is significantly nonzero. (H) tSNE projection of OA cells, with clusters 15, 20, and 19 labeled, sampled at 9000 cells. (I) Heatmaps of the average expression of each marker in the given cluster. (J) Fold change in cytokines from human 62-plex Luminex assay between control and 3-isobutyl-1-methylxanthine (IBMX) treatment. (K) Fold change in cytokines from human 62-plex Luminex assay between control and a combined IBMX and JNK inhibitor treatment. (L) Percent change in cytokine MFI between control and the combined IBMX/JNK inhibitor treatment.
To understand the interplay between Inf-A and Inf-D cells in the OA cartilage, we analyzed their abundance in the cohort of 20 patients and used hierarchical clustering to order patients by the content of their Inf-A and Inf-D cells. The patients were clearly stratified into two large categories of patients: Inf-D–low and Inf-D–high patients with OA (Fig. 5E). The Inf-D–high group had concomitantly high levels of the Inf-A clusters than the Inf-D–low group (Fig. 5F). In addition, a positive correlation was observed between the percentage of Inf-A and Inf-D cells in patients (Fig. 5G). This led us to hypothesize that a combination strategy of enhancing Inf-D while inhibiting Inf-A populations could be effective in mitigating inflammation in OA cartilage. We also noted a small and highly variable population, cluster 19, which had a mixed character. Cluster 19 showed IL1R1 expression without the inflammatory signature that we observed in the Inf-A I and Inf-A II cells (pJNK1/2 and pSMAD1/5) (Fig. 5, H and I ) and curiously also expressed CD24. These cells were only present in 8 of the 20 patients (Fig. 5I) but further suggested that CD24 expression in the Inf-D cells can dampen inflammation.
To test this hypothesis, we first induced mild CD24 overexpression by treating cells with 3-isobutyl-1-methylxanthine (IBMX), an adenosine 3′,5′-monophosphate inhibitor that has been shown to increase CD24 expression in adipocytes (31). Treatment with 0.5 mM IBMX for 48 hours up-regulated CD24 expression by two- to fourfold in OA chondrocytes (fig. S5D). IBMX increased the gene expression of the mitochondrial genes Tfam, and Pgc1a (fig. S5E), although no consistent effect was, however, observed on MMP13 expression (fig. S5E). Using the 62-plex Luminex assay, we observed a modest down-regulation of CCL2 and CCL7; however, these effects were milder than the direct inhibition of the Inf-A signaling (Fig. 5J).
We then tested a combination treatment of JNK inhibitor with IBMX for 48 hours. We observed a greater magnitude decreased in CCL2 and CCL7 with the combination treatment (Fig. 5K) as compared to the single treatment with JNK inhibitor (Fig. 4, J and M). In addition, the combination therapy further mitigated inflammation by reducing the secretion of targets such as IL21, IL22, VCAM, and IFNB1 (Fig. 5L). Similar to JNK inhibitor treatment, matrix metalloproteinase (MMP) gene expression remained unaffected by the combination treatment (fig. S5F). These data, however, suggest that targeting multiple combinations of rare cell types in OA cartilage may be beneficial in mitigating inflammation.
DISCUSSION
In this study, we built the first single-cell, proteomic atlas for healthy and osteoarthritic adult articular cartilage. Cartilage regeneration and OA remain unmet medical needs. Therefore, a high-resolution cellular atlas of articular cartilage tissue lays the foundation for insight into disease pathology, drug strategies, and tissue engineering. Using a panel of 33 markers, we identified multiple populations that constitute the articular cartilage landscape, including rare populations that contribute to disease pathology and interpatient heterogeneity.
Recently, an scRNA sequencing map of knee cartilage was reported from a cohort of 10 patients with OA and outlined several known and cell populations (25). Our study complements this single-cell transcriptomic data, with the additional advantage that the proteomic snapshot provides insight into the status of signaling pathways in the identified subpopulations. The single-cell proteomic approach is especially pertinent in robustly identifying rare cell populations that are difficult to discern from RNA sequencing data, where only 1600 cells were studied from all the patients with OA. In contrast, the ability to map 30,000 to 100,000 cells per patient in a 20-patient cohort by the cyTOF method provided us a robust dataset to find and validate statistically significant rare subpopulations. A recent study on rare senescent cell populations in OA cartilage has shown the influence of these small populations in OA pathology (32). Removal of senescent cells significantly impaired OA progression in a mouse model and modulated end-stage human OA chondrocytes, underscoring the need for further studies on other rare populations that might contribute to OA pathology. In addition, frequent discrepancies between gene and protein expression have been reported in OA, further signifying the need for complementary proteomic and transcriptomic studies.
The ability to measure a large number of cells with high precision allowed us to identify two, rare chondrocyte subpopulations (Inf-A and Inf-D), which constitute only 0.5 to 1.5% of all chondrocytes. However, pharmacologically targeting these small populations led to a dampening of inflammatory cytokines at the population level. The Inf-A cells express both the TNFRII and IL1R1 receptors, are consistently expanded in OA compared to normal cartilage, and are characterized by activated JNK1/2 and SMAD1/5 pathways. An analysis of their transcriptomes from the published scRNA sequencing dataset suggests that these cells may function to recruit immune cells. Inhibition of these cells using a JNK inhibitor led to an overall reduction of secreted CCL2 and CCL7, cytokines implicated in immune cell recruitment (33, 34). Genetic knockout of JNK1 or JNK2 ameliorates disease symptoms in a collagenase-induced model of rheumatoid arthritis (RA) (35), and inhibition of JNK protects joints from characteristic degeneration (36). These mouse models can be used in future studies to test a putative immune cell recruitment function of the Inf-A population. However, unlike in RA models, JNK inhibitors have not been systematically studied as a therapy in animal models of OA. TNFRII antibodies also have a strong therapeutic index in RA (37). Our work suggests that some of these therapies may also be successful in targeting OA.
The other novel population identified in our study is the Inf-D chondrocytes, which are characterized by the expression of CD24, a cell surface receptor we had previously reported to be enriched in juvenile cartilage and associated with resistance to inflammatory cues (30). Expression of CD24 in Inf-A cells, a subpopulation observed in some patients, led to complete inhibition of JNK activation. In addition, the positive correlation between Inf-A and Inf-D populations in a subset of patients led us to hypothesize an interplay between these two populations. Combinatorial treatment with JNK inhibitor (lowering Inf-D) and IBMX, a small molecular activator of CD24 (increasing Inf-D), showed a greater decrease in CCL2, CCL7, CXCL1, CXCL5, and other inflammatory cytokines than JNK inhibition alone. Our data, therefore, provide insights into the interplay between multiple cellular populations that likely contribute to the chronic inflammatory environment that is observed in end-stage OA cartilage. A deeper understanding of these populations, their cross-talk, and relative influence can help devise single or combinatorial biologic candidates that can tilt the inflammatory balance in a way that can be beneficial in the later or early stages of OA progression.
Our data also served to redefine the cartilage stem and progenitor-like populations that reside in adult cartilage. We validated the existence of CD105/CD90-, NOTCH1-, and STRO1-expressing CPCs that have been previously described in OA and are highly inflammatory. In addition, we described other CPC populations in OA cartilage that express CD90 and CD105 but are low in inflammation. It will be interesting to compare the regenerative potential of these different subpopulations of CPCs, especially in a low-inflammation microenvironment. Since CD24 is a marker for younger chondrocytes with a higher regenerative potential, it is possible that our combinatorial treatment can boost regenerative populations in addition to mitigating inflammation. Our data also reveal that CD24 expression is associated with mitochondrial biogenesis, another characteristic associated with younger healthy chondrocytes. The data also reveal CPC I as progenitor populations that are lost in OA. Future studies are needed to determine how these CPCs are lost during OA progression and whether reintroduction of these CPCs can benefit cartilage regeneration. A particularly interesting subgroup to follow is the CD73-expressing cells, as CD73 has recently been identified to characterize the hSSCs in bone marrow, which can self-renew and give rise to cartilage, bone, and fat progenitor cells (5).
By characterizing chondrocyte populations in patients with OA, we stratified patients by the abundance of each population. This practice is well established in the cancer field, where patient heterogeneity and tumor subtyping play an ever-increasing role in precision medicine. Identification of the 20 different subpopulations in cartilage allowed revealed three major categories of patients with OA. Group A represents 60% of the patients, while groups B and C represent 15% each. Group C patients were distinguished from group A and B patients by an expansion of the inflammatory NOTCH1/STRO1-expressing CPCs, which are also highly active in proinflammatory pathways such as NF-κB and HIF2A. Group B patients had an expansion of the Inf-D population. A subset of patients driven by inflammation has been suggested previously as well on the basis of RNA sequencing (38) and DNA methylation patterns (39, 40) in cartilage. Future work will reveal the molecular mechanism(s) that drive this heterogeneity, which may be related to the multifactorial etiology of OA that is affected by the genetic, epigenetic, metabolic, as well as lifestyle factors of the patient populations. Such work will also benefit from studying the interactions of the CPCs and Inf-A and Inf-D cells in multifactorial systems that take into account all the other cell types present in the joint.
In summary, this study provides the first high-dimensional cyTOF map for adult cartilage, revealing multiple, rare subpopulations that coexist in health and disease. Collectively, our data highlight the complex interplay between Inf-A and Inf-D populations and regenerative populations in cartilage and suggest that altering the balance between these populations could provide novel therapeutic strategies for OA. In future studies, refined panels and larger cohort sizes can provide a powerful platform for the stratification of patients with OA based on the underlying cellular drivers of their disease. Ultimately, these stratification efforts would allow for targeted testing of drugs for each patient subset, to establish personalized medicine strategies for OA.
METHODS
Study design
Research objectives
Our objective was to profile rare populations of CPCs in OA patient samples and determine their interactions. We designed a curated panel of antibodies (see below) and tested a cohort of 20 OA patient and 5 normal samples. Observations from this dataset were then more thoroughly tested.
Research subjects
Chondrocytes were derived from OA cartilage or healthy cartilage samples. All experiments were performed on primary cells.
Experimental design
We collected a cohort of 20 patients, which passed several quality control parameters (see below) and included a variety of ages and a balanced pool of male/female patients. Samples that did not pass quality control metrics were not used for downstream analysis. Patient samples were selected on the basis of previously established quality control criteria, namely, the expression ratio of Col2a1/Col1a1 (see methods below) and the expression of MMP genes. Follow-up analysis was conducted on a separate panel of OA chondrocytes to ensure that we could independently see the same results.
Blinding
Researchers were not blind to disease status or treatment when analyzing the data.
Data inclusion/exclusion criteria
All collected data points were used for assays performed after drug treatment. All datasets were quality controlled, and wells or data points that did not pass quality control metrics were not used. This includes (i) Luminex wells that did not give acceptable standard bead readings, (ii) quantitative polymerase chain reaction (qPCR) wells that did not give suitable Ct values for Actin, and (iii) cells analyzed by cyTOF that did not have high SOX9 or CD44 expression. Quality control exclusions were performed before analysis of data. After exclusion of points for these reasons, no additional points were excluded.
Replicates
All drug treatments were performed in independent technical replicates for each patient (i.e., cells derived from the same patient were treated three times with drug versus control). All drug treatments were performed in three to six patient samples.
Isolation and culture of primary chondrocytes from human cartilage
OA samples were procured from the discarded tissues of patients with radiographic OA undergoing total joint replacement, in accordance with the IRB protocol approved by Stanford University, as previously described (9). The age range for OA patient samples was 54 to 72 years old. Cartilage was shaved from the underlying bone, allowed to recover overnight at 37°C in complete media [HyClone Dulbecco's modified Eagle's medium:F12 (GE Healthcare, SH3002302) supplemented with 2 mM l-glutamine (Gibco, 25-030-149), 10% fetal bovine serum (FBS) (Corning, 35-016-CV), 1× antibiotic-antimycotic (Gibco, 15-240-062), and ascorbic acid (12.5 μg/ml; Eastman)] and then treated with collagenase (Collagenase II and IV, 2.5 mg/ml each; Worthington Biochem) in complete media overnight at 37°C. The next day, cells were strained, centrifuged, and plated at a high density of 2.6 × 104 cells/cm in complete media. Cells were allowed to become confluent on the plates and were passaged once using collagenase, before cyTOF experiments or drug treatments. Samples were checked for Col2a1/Col1a1 ratios and MMP3, MMP9, and MMP13 expression, before experimentation. Normal samples were either derived from expired cartilage allograft samples or shipped from the manufacturer (samples 1 to 4) or from the surgical waste of a notchplasty (sample 5) under an approved IRB and processed as described above.
RNA isolation and complementary DNA synthesis
Cells for RNA extraction were collected in RNA lysis buffer (Zymo Research) and processed according to the manufacturer’s specifications for the Quick-RNA MicroPrep Kit (Zymo Research, R1051), including the optional deoxyribonuclease I digestion. RNA quality and quantity were measured using the NanoDrop 1000 Spectrophotometer. All samples had A260/280 (absorbance at 260 and 280 nm) scores between 1.6 and 1.8.
Gene expression analyses
One milligram of RNA from each sample was reversed transcribed into complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368813). qPCR was performed using TaqMan gene-specific expression assays, FAM (carboxyfluorescein–labeled, for metalloproteinases 3, 9, and 13 (Hs00233962_m1, Hs00957562_m1, and Hs00233992_m1), with a universal master mix (Applied Biosystems, 4369016). Gene expression levels were normalized with FAM-labeled β-actin (Hs01060665_g1).
For Tfam, CD24, and PGC1a, we used the SybrGreen mastermix (Applied Biosystems, A25742) according to the manufacturer’s specifications. Primer sequences were as follows: Tfam_F: 5′-GCTCAGAACCCAGATGCA AAA-3′, Tfam_R: 5′-AGGAAGTTCCCTCCAACGC-3′; PGC1a_F: 5′-CCATGGATGAAGGGTACTTTTCTG-3′, PGC1a_R: 5′-CTTTTACCAAAGCAGCAGCC-3′; CD24_F: 5′-TACCCACGCAGATTTATT-3′, CD24_R: 5′-AGA GTGAGACCACGAAGA-3′; Actin_F: 5′-CACCAACTGGGACGACAT-3′, Actin_R: 5′-ACAGCCTGGATAGCAACG-3′. qPCR reactions included a 2-min incubation at 50°C to inactivate previous amplicons with uracil-DNA glycosylase, followed by a 10-min incubation at 95°C to activate the Taq polymerase. The amplification cycle, consisting of 15 s at 95°C and 1 min at 60°C, was repeated 40 times. The relative expression levels were determined using the ΔΔCt method (Ct gene of interest − Ct internal control), and relative gene expression is calculated using 2 − ΔΔCt method and plotted.
Drug treatment of OA cells
OA cells were seeded at high density in 12-well plates and treated with control [dimethyl sulfoxide (DMSO)] or drug the next day for 48 hours. Drug doses were determined on the basis of prior literature and validation: 0.5 mM IBMX (Sigma-Aldrich, I5879) (31), 50 μM JNK inhibitor II (Calbiochem, 420119), 25 μM NF-κB inhibitor BMS-345541 (Sigma-Aldrich, B9935) (28, 41), and 50 μM Alk inhibitor SB 431542 hydrate (Sigma-Aldrich, S4317) (42, 43) were used with appropriate dilution in DMSO.
Multiplex autoantibody assay
Cell culture supernatants were collected and spun down at 10,000g for 10 min at 4°C to remove any cells or cell debris and then snap-frozen in liquid nitrogen before performing the assay. This assay was performed in the Human Immune Monitoring Center at Stanford University. Human 62-plex kits were purchased from eBiosciences/Affymetrix and used according to the manufacturer’s recommendations with modifications as described below. Briefly, beads were added to a 96-well plate and washed in a BioTek ELx405 washer. Undiluted samples were added to the plate containing the mixed antibody-linked beads and incubated at room temperature (RT) for 1 hour, followed by overnight incubation at 4°C with shaking. Cold temperature and RT incubation steps were performed on an orbital shaker at 500 to 600 rpm. Following the overnight incubation, plates were washed in a BioTek ELx405 washer, and then, biotinylated detection antibody was added for 75 min at RT with shaking. The plate was washed as above, and streptavidin-phycoerythrin was added. After incubation for 30 min at RT, wash was performed as above, and reading buffer was added to the wells. Each sample was measured in duplicate. Plates were read using a Luminex 200 instrument with a lower bound of 50 beads per sample per cytokine. Custom assay control beads by Radix Biosolutions were added to all wells.
Conjugation of antibodies to metal isotopes
Antibodies were labeled according to the manufacturer’s specifications using the MAXPAR X8 Polymer labeling kit (Fluidigm). One tube was used per 100 μg of antibody. Antibodies were purchased labeling ready, without additives, whenever possible. Antibodies with carrier components such as albumin or glycerol were cleaned with Melon Gel IgG Purification columns (Thermo Fisher Scientific) after buffer exchange with Zeba Desalt Spin Columns (Thermo Fisher Scientific) as per the manufacturer’s specifications. Final antibody concentration was measured using a NanoDrop 1000 Spectrophotometer, set to IgG (immunoglobulin G) mode, diluted to the highest round value in W buffer with sodium azide, and stored at 4°C for later use. The complete list of conjugated antibodies, metal isotope, clone information, and the manufacturer can be found in table S1.
Titration of antibodies for cyTOF
Metal conjugated antibodies were tested in a three-point dilution curve, centered on their recommended or optimized fluorescence-activated cell sorting (FACS) concentration, with a 10-fold increase and decrease from this center value. Signal-to-noise ratio was compared by staining known negative samples, such as 293 T cells. The lowest concentration that had no increase in signal upon a 10-fold increase in concentration was used for the final staining concentration (see table S1).
Cell staining and cyTOF
OA cells were cultured to confluence in 10-cm dishes. On the collection day, cells were stained with 25 μM Idu (5-Iodo-2′-deoxyuridine) for 15 min at 37°C in the cell incubator and then with 0.5 μM cisplatin for 5 min at RT. Cells were then lifted with 0.25% trypsin-EDTA (Gibco) for 15 min at 37°C. Trypsin was quenched using media containing 10% FBS, and cell were washed three times with phosphate-buffered saline to remove any trace amounts of trypsin. Cells were fixed after straining through a 35 μM strainer in 1.6% paraformaldehyde (PFA) for 10 min at RT. Cells were washed four times with cells staining media, counted, and frozen in 1 million–cell aliquots in a small amount of cell staining media at −80°C. To stain, cells were thawed on ice and barcoded using the Cell-ID 20-plex Pd Barcoding Kit (Fluidigm) according to the manufacturer’s specifications. After barcoding, cells were labeled as previously described (8). Briefly, all barcoded samples were combined into one FACS tube and washed 3 times with cell staining media and stained with the cell surface antibodies for 30 min at RT according to the concentrations in table S1. Cells were then washed 2 times with cell staining media and permeabilized with 1 ml of cold methanol added dropwise with continuous gentle vortexing. Cells were incubated for 10 min on ice, with gentle vortexing every 2 to 3 min to avoid cell clumping, then washed in cell staining media, and stained with the intracellular antibodies for 30 min at RT. After 2 times washed with cell staining media, cells were resuspended in 1.6% PFA with Cell-ID Intercalator-Ir (Fluidigm) used at 1:2000. Cells were measured using the cyTOF 2 (Fluidigm) and injected using the supersampler. EQ (Four Element Calibration) beads (Fluidigm) were added just before runtime (1:10 dilution) to normalize signal over runtime.
Quality control and data cleaning
Normalization over run time was performed using the EU beads using the previously published bead normalized (v0.3) available here: https://github.com/nolanlab/bead-normalization/releases with the default parameters. Samples were then debarcoded using the single-cell debarcoder available here: https://github.com/nolanlab/single-cell-debarcoder using the default parameters. Channel values were arcsine transformed and normalized between the two independent runs using two patients with OA that were loaded in both runs. The tower-independent runs were normalized to each other. Next, we selected for live cells by gating for cisplatin-negative, DNA (Ir195)–positive cells. Last, from live cells, we gated for SOX9/CD44 double-positive cells, which were included in the final analysis. On average, 98% of the OA and normal cells were live, and 95 and 64%, respectively, were in the SOX9/CD44 gate. Gating was performed using Cytobank.
FlowSOM analysis and tSNE projections
Clusters were called using FlowSOM (10). Analysis was performed using Cytobank’s online implementation using the standard settings. Clustering was performed using the cell surface receptors, HIF2A and SOD2 (superoxide dismutase 2); no signaling markers were included. The self-organizing map (SOM) was constructed using the 20 OA and 5 normal samples, and then, the same SOM was applied to the treated samples. tSNE projection was also performed using Cytobank’s online platform. All results, including FlowSOM clusters and tSNE coordinates, were exported as text files and manipulated for plotting in Python. We compared the results from our FlowSOM clusters to other clustering algorithms, including X-shift (24), and obtained similar numbers of clusters and patterns of expression within each cluster.
Data visualization and analysis
Data were visualized using Python and the NumPy (www.numpy.org/), pandas (https://pandas.pydata.org/pandas-docs/stable/), and seaborn (https://seaborn.pydata.org/) packages. Hierarchical clustering of samples or cell populations was performed using the seaborn clustermap function, using a complete-linkage algorithm, also known as the farthest neighbor clustering, in which clusters are decided on the basis of the two most dissimilar points. Complete linkage clustering avoids the chaining phenomenon that can occur with single-linkage methods, where clusters that may be very distant from each other are forced together because of a single element being close. Complete linkage tends to find compact clusters of equal diameters.
Reanalysis of scRNA sequencing data from GSE104782
Gene counts were downloaded from Gene Expression Omnibus and reanalyzed using custom Python scripts. Gene expression networks and pathway analyses were performed using Ingenuity Pathway Analysis (QIAGEN), Enrichr, and STRING.
Statistical analysis
Planned comparisons were performed with the GraphPad Prism software. We used (i) one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test to identify specific differences between drug treatment groups or between selected OA patient groups (for treatments, groups were only compared against DMSO controls, not against each other) and (ii) nonparametric, two-tailed Welch’s t test for comparisons between only two groups. P values were corrected for multiple hypothesis testing, such that the family-wise error was capped at 0.05, using the Bonferroni correction method. The exact method and specific P values for significant comparisons are stated in the appropriate results section. For cyTOF plots, although only 9000 cells were visualized on the tSNE plots in the figures, average values and other calculations or statistics were performed with all cells that met the required criteria.
Supplementary Material
Acknowledgments
We would like to thank Y. Rosenberg-Hasson at the Stanford Human Immune Profiling Center for help with the Luminex analysis, E. Migliore for help with sample acquisition, and N. Sahu for helpful feedback on the manuscript. Funding: P.S. and N.B. are supported by NIH/NIAMS grants R01 AR070865 and R01 AR070864, F.C.G. is supported by the NSF GRFP award. This work was also supported by a gift to the Department of Orthopedic Surgery from K. Thiery and D. O’Leary. Author contributions: F.C.G. conceptualized and designed the experiments, executed the study, analyzed and interpreted the data, and wrote the manuscript. R.B. and S.B. helped with the technical details of acquiring the data. P.S. assisted with data interpretation. S.M. assisted with data collection of the initial IBMX data. S.G., D.F.A., P.F.I., and C.C. provided OA or normal cartilage samples. N.B. conceptualized and designed the study, oversaw data collection, and wrote the manuscript. Competing interests: Patent “Methods and Compositions for the Treatment of Osteoarthritis” is provisionally filed. The organization issuing the patent is The Board of Trustees of the Leland Stanford Junior University, and the applicants are N.B. and F.C.G. (filed 2 October 2019; serial number 62/909,547). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaay5352/DC1
Fig. S1. Quality control of normal and OA chondrocyte samples used for cyTOF analysis.
Fig. S2. OA and normal samples are characterized by different populations of cells.
Fig. S3. CPC clusters differentiate OA patient subtypes.
Fig. S4. Inf-A population is marked by TNFRII and IL1R1.
Fig. S5. Inf-D population is marked by CD24 expression and low reactivity to inflammatory cues.
Table S1. Table of antibodies used in cyTOF panel.
REFERENCES AND NOTES
- 1.Jiang Y., Tuan R. S., Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat. Rev. Rheumatol. 11, 206–212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandes A. M., Herlofsen S. R., Karlsen T. A., Küchler A. M., Fløisand Y., Brinchmann J. E., Similar properties of chondrocytes from osteoarthritis joints and mesenchymal stem cells from healthy donors for tissue engineering of articular cartilage. PLOS ONE 8, e62994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koelling S., Kruegel J., Irmer M., Path J. R., Sadowski B., Miro X., Miosge N., Migratory chondrogenic progenitor cells from repair tissue during the later stages of human osteoarthritis. Cell Stem Cell 4, 324–335 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Fickert S., Fiedler J., Brenner R. E., Identification of subpopulations with characteristics of mesenchymal progenitor cells from human osteoarthritic cartilage using triple staining for cell surface markers. Arthritis Res. Ther. 6, R422–R432 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan C. K. F., Gulati G. S., Sinha R., Tompkins J. V., Lopez M., Carter A. C., Ransom R. C., Reinisch A., Wearda T., Murphy M., Brewer R. E., Koepke L. S., Marecic O., Manjunath A., Seo E. Y., Leavitt T., Lu W.-J., Nguyen A., Conley S. D., Salhotra A., Ambrosi T. H., Borrelli M. R., Siebel T., Chan K., Schallmoser K., Seita J., Sahoo D., Goodnough H., Bishop J., Gardner M., Majeti R., Wan D. C., Goodman S., Weissman I. L., Chang H. Y., Longaker M. T., Identification of the human skeletal stem cell. Cell 175, 43–56.e21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathiessen A., Conaghan P. G., Synovitis in osteoarthritis: Current understanding with therapeutic implications. Arthritis Res. Ther. 19, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mabey T., Honsawek S., Cytokines as biochemical markers for knee osteoarthritis. World J. Orthop. 6, 95–105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bendall S. C., Simonds E. F., Qiu P., Amir E. D., Krutzik P. O., Finck R., Bruggner R. V., Melamed R., Trejo A., Ornatsky O. I., Balderas R. S., Plevritis S. K., Sachs K., Pe’er D., Tanner S. D., Nolan G. P., Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor S. E. B., Smeriglio P., Dhulipala L., Rath M., Bhutani N., A global increase in 5-hydroxymethylcytosine levels marks osteoarthritic chondrocytes. Arthritis Rheumatol. 66, 90–100 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Van Gassen S., Callebaut B., Van Helden M. J., Lambrecht B. N., Demeester P., Dhaene T., Saeys Y., FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part J. Int. Soc. Anal. Cytol. 87, 636–645 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Samusik N., Good Z., Spitzer M. H., Davis K. L., Nolan G. P., Automated mapping of phenotype space with single-cell data. Nat. Methods 13, 493–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alsalameh S., Amin R., Gemba T., Lotz M., Identification of mesenchymal progenitor cells in normal and osteoarthritic human articular cartilage. Arthritis Rheum. 50, 1522–1532 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Bernstein P., Sperling I., Corbeil D., Hempel U., Fickert S., Progenitor cells from cartilage—No osteoarthritis-grade-specific differences in stem cell marker expression. Biotechnol. Prog. 29, 206–212 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Dowthwaite G. P., Bishop J. C., Redman S. N., Khan I. M., Rooney P., Evans D. J. R., Haughton L., Bayram Z., Boyer S., Thomson B., Wolfe M. S., Archer C. W., The surface of articular cartilage contains a progenitor cell population. J. Cell Sci. 117, 889–897 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Grogan S. P., Miyaki S., Asahara H., D’Lima D. D., Lotz M. K., Mesenchymal progenitor cell markers in human articular cartilage: Normal distribution and changes in osteoarthritis. Arthritis Res. Ther. 11, R85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hattori S., Oxford C., Reddi A. H., Identification of superficial zone articular chondrocyte stem/progenitor cells. Biochem. Biophys. Res. Commun. 358, 99–103 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y., Hu C., Yu S., Yan J., Peng H., Ouyang H. W., Tuan R. S., Cartilage stem/progenitor cells are activated in osteoarthritis via interleukin-1β/nerve growth factor signaling. Arthritis Res. Ther. 17, 327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiraoka K., Grogan S., Olee T., Lotz M., Mesenchymal progenitor cells in adult human articular cartilage. Biorheology 43, 447–454 (2006). [PubMed] [Google Scholar]
- 19.McCarthy H. E., Bara J. J., Brakspear K., Singhrao S. K., Archer C. W., The comparison of equine articular cartilage progenitor cells and bone marrow-derived stromal cells as potential cell sources for cartilage repair in the horse. Vet. J. 192, 345–351 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Barbero A., Ploegert S., Heberer M., Martin I., Plasticity of clonal populations of dedifferentiated adult human articular chondrocytes. Arthritis Rheum. 48, 1315–1325 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Williams R., Khan I. M., Richardson K., Nelson L., McCarthy H. E., Analbelsi T., Singhrao S. K., Dowthwaite G. P., Jones R. E., Baird D. M., Lewis H., Roberts S., Shaw H. M., Dudhia J., Fairclough J., Briggs T., Archer C. W., Identification and clonal characterisation of a progenitor cell sub-population in normal human articular cartilage. PLOS ONE 5, e13246 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Unguryte A., Bernotiene E., Bagdonas E., Garberyte S., Porvaneckas N., Jorgensen C., Human articular chondrocytes with higher aldehyde dehydrogenase activity have stronger expression of COL2A1 and SOX9. Osteoarthr. Cartil. 24, 873–882 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Grogan S. P., Barbero A., Diaz-Romero J., Cleton-Jansen A.-M., Soeder S., Whiteside R., Hogendoorn P. C. W., Farhadi J., Aigner T., Martin I., Mainil-Varlet P., Identification of markers to characterize and sort human articular chondrocytes with enhanced in vitro chondrogenic capacity. Arthritis Rheum. 56, 586–595 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Karlsson C., Lindahl A., Articular cartilage stem cell signalling. Arthritis Res. Ther. 11, 121 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji Q., Zheng Y., Zhang G., Hu Y., Fan X., Hou Y., Wen L., Li L., Xu Y., Wang Y., Tang F., Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 78, 100–110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Longobardi L., Jordan J. M., Shi X. A., Renner J. B., Schwartz T. A., Nelson A. E., Barrow D. A., Kraus V. B., Spagnoli A., Associations between the chemokine biomarker CCL2 and knee osteoarthritis outcomes: The Johnston County Osteoarthritis Project. Osteoarthr. Cartil. 26, 1257–1261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raghu H., Lepus C. M., Wang Q., Wong H. H., Lingampalli N., Oliviero F., Punzi L., Giori N. J., Goodman S. B., Chu C. R., Sokolove J. B., Robinson W. H., CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann. Rheum. Dis. 76, 914–922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burke J. R., Pattoli M. A., Gregor K. R., Brassil P. J., MacMaster J. F., McIntyre K. W., Yang X., Iotzova V. S., Clarke W., Strnad J., Qiu Y., Zusi F. C., BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J. Biol. Chem. 278, 1450–1456 (2003). [DOI] [PubMed] [Google Scholar]
- 29.van Caam A., Madej W., Garcia de Vinuesa A., Goumans M.-J., ten Dijke P., Blaney Davidson E., van der Kraan P., TGFβ1-induced SMAD2/3 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthritis Res. Ther. 19, 112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J., Smeriglio P., Dragoo J., Maloney W. J., Bhutani N., CD24 enrichment protects while its loss increases susceptibility of juvenile chondrocytes towards inflammation. Arthritis Res. Ther. 18, 292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith N. C., Fairbridge N. A., Pallegar N. K., Christian S. L., Dynamic upregulation of CD24 in pre-adipocytes promotes adipogenesis. Adipocyte 4, 89–100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeon O. H., Kim C., Laberge R.-M., Demaria M., Rathod S., Vasserot A. P., Chung J. W., Kim D. H., Poon Y., David N., Baker D. J., van Deursen J. M., Campisi J., Elisseeff J. H., Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deshmane S. L., Kremlev S., Amini S., Sawaya B. E., Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 29, 313–326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsou C.-L., Peters W., Si Y., Slaymaker S., Aslanian A. M., Weisberg S. P., Mack M., Charo I. F., Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Invest. 117, 902–909 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han Z., Boyle D. L., Chang L., Bennett B., Karin M., Yang L., Manning A. M., Firestein G. S., c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Invest. 108, 73–81 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schepetkin I. A., Kirpotina L. N., Hammaker D., Kochetkova I., Khlebnikov A. I., Lyakhov S. A., Firestein G. S., Quinn M. T., Anti-inflammatory effects and joint protection in collagen-induced arthritis after treatment with IQ-1S, a selective c-Jun N-terminal kinase inhibitor. J. Pharmacol. Exp. Ther. 353, 505–516 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blüml S., Scheinecker C., Smolen J. S., Redlich K., Targeting TNF receptors in rheumatoid arthritis. Int. Immunol. 24, 275–281 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Soul J., Dunn S. L., Anand S., Serracino-Inglott F., Schwartz J.-M., Boot-Handford R. P., Hardingham T. E., Stratification of knee osteoarthritis: Two major patient subgroups identified by genome-wide expression analysis of articular cartilage. Ann. Rheum. Dis. 77, 423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rushton M. D., Reynard L. N., Barter M. J., Refaie R., Rankin K. S., Young D. A., Loughlin J., Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol. 66, 2450–2460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernández-Tajes J., Soto-Hermida A., Vázquez-Mosquera M. E., Cortés-Pereira E., Mosquera A., Fernández-Moreno M., Oreiro N., Fernández-López C., Fernández J. L., Rego-Pérez I., Blanco F. J., Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann. Rheum. Dis. 73, 668–677 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Murahashi Y., Yano F., Kobayashi H., Makii Y., Iba K., Yamashita T., Tanaka S., Saito T., Intra-articular administration of IκBα kinase inhibitor suppresses mouse knee osteoarthritis via downregulation of the NF-κB/HIF-2α axis. Sci. Rep. 8, 16475 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inman G. J., Nicolás F. J., Callahan J. F., Harling J. D., Gaster L. M., Reith A. D., Laping N. J., Hill C. S., SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 62, 65–74 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Laping N. J., Grygielko E., Mathur A., Butter S., Bomberger J., Tweed C., Martin W., Fornwald J., Lehr R., Harling J., Gaster L., Callahan J. F., Olson B. A., Inhibition of transforming growth factor (TGF)-β1–induced extracellular matrix with a novel inhibitor of the TGF-β type I receptor kinase activity: SB-431542. Mol. Pharmacol. 62, 58–64 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaay5352/DC1
Fig. S1. Quality control of normal and OA chondrocyte samples used for cyTOF analysis.
Fig. S2. OA and normal samples are characterized by different populations of cells.
Fig. S3. CPC clusters differentiate OA patient subtypes.
Fig. S4. Inf-A population is marked by TNFRII and IL1R1.
Fig. S5. Inf-D population is marked by CD24 expression and low reactivity to inflammatory cues.
Table S1. Table of antibodies used in cyTOF panel.