

Abstract
The genome of the allotetraploid species Coffea arabica L. was sequenced to assemble independently the two component subgenomes (putatively deriving from C. canephora and C. eugenioides) and to perform a genome-wide analysis of the genetic diversity in cultivated coffee germplasm and in wild populations growing in the center of origin of the species. We assembled a total length of 1.536 Gbp, 444 Mb and 527 Mb of which were assigned to the canephora and eugenioides subgenomes, respectively, and predicted 46,562 gene models, 21,254 and 22,888 of which were assigned to the canephora and to the eugeniodes subgenome, respectively. Through a genome-wide SNP genotyping of 736 C. arabica accessions, we analyzed the genetic diversity in the species and its relationship with geographic distribution and historical records. We observed a weak population structure due to low-frequency derived alleles and highly negative values of Taijma’s D, suggesting a recent and severe bottleneck, most likely resulting from a single event of polyploidization, not only for the cultivated germplasm but also for the entire species. This conclusion is strongly supported by forward simulations of mutation accumulation. However, PCA revealed a cline of genetic diversity reflecting a west-to-east geographical distribution from the center of origin in East Africa to the Arabian Peninsula. The extremely low levels of variation observed in the species, as a consequence of the polyploidization event, make the exploitation of diversity within the species for breeding purposes less interesting than in most crop species and stress the need for introgression of new variability from the diploid progenitors.
Subject terms: Computational biology and bioinformatics, Evolution, Genetics, Plant sciences
Introduction
Coffea arabica is an allopolyploid species (2n = 4x = 44) resulting from the hybridization between two species most closely related to C. eugenioides and C. canephora1. The allopolyploid speciation of C. arabica has a broad time interval estimate spanning 10,000 or 665,000 years BP2,3. Unlike many tropical tree crops, Arabica coffee is not clonally propagated. Cultivars and landraces are typically propagated by seed. The mating system is primarily based on self-fertilization, although pollinator-mediated outcrossing may occasionally occur. The predominant autogamy leads to high levels of inbreeding.
C. arabica is indigenous to Ethiopia and South Sudan which represents its primary center of diversity4 (FAO-1964, ORSTOM-1966). Yemen is a secondary dispersal center5. Several accounts of the early history of C. arabica germplasm usage and movements are available in the literature, but the most complete and best documented publication is the one of Haarer (1958). Sometimes during the 14th century, coffee seeds were brought out of the forests of South Western Ethiopia to Yemen, where coffee cultivation expanded to satisfy the demand of a growing number of coffee houses in Moccha and Cairo at the end of the 15th century. The center of diversity in South Western Ethiopia corresponded to the area that has been for centuries under the control of the Kingdom of Kafa. This area was described as an impenetrable “citadelle” until Menelik II conquered the Kingdom in 1897. Hence, early escapes of coffee seeds from South Western Ethiopia were likely occasional and the coffee cultivation in Yemen has started from a narrow genetic basis. C. arabica was then spread worldwide from Yemen rather than from its primary center of origin in Ethiopia. Significant out-of-Yemen movements were documented (i) in 1670, from Yemen to India with some seeds smuggled by Baba Budan and (ii) in 1715, from Yemen to Bourbon Island (today Ile de la Réunion) with very few seeds. The former gave rise to the Typica variety after the Dutch brought some seeds from India to today’s Indonesia in 1696 and 1699. From Indonesia through Europe, Typica reached the Americas in 1723. The latter gave rise to the Bourbon variety that reached the Americas and East Africa in the mid-19th century. In India, the early seeds introduced in 1670 were grown locally for centuries. In the early 20th century, East Africa (today Burundi, Rwanda, DR Congo, Kenya, Tanzania and Uganda) started coffee cultivation and introduced seeds from Yemen, Bourbon, Typica and Indian varieties and very few Ethiopian landraces, which leaked from Ethiopia in the pockets of travelers (Geisha and Rume Sudan for instance). Hence, East Africa could be considered as a melting pot of all different varieties descending from early coffee cultivation in Yemen6.
It is evident that molecular analyses of genetic diversity are needed to support this scenario that is based on geographical and historical data. In the past, microsatellites or RAPDs have been used for studying genetic diversity in C. arabica7–9. More recently, a molecular study of C. arabica germplasm was performed by Merot-L’anthoene et al.10 using an 8.5k SNP array, which represents the first genome-wide analysis of the genetic diversity in this species but it suffers from the ascertainment bias associated with the limited breadth of variation included in the SNP discovery panel. Unbiased approaches to investigate genetic diversity are supposed to rely on the availability of a C. arabica genome sequence. The genome of a modern accession of C. canephora, one of the progenitors of C. arabica, has been sequenced11, but the assembly of a diploid genome provides limited support for analyzing sequencing data from tetraploid coffee germplasm. Genome sequencing initiatives of tetraploid accessions have been launched by several research groups (https://coffeegenome.ucdavis.edu/12, among others) but an open-access genome assembly, with a reliable sorting of homoeologous sequences, is not yet available. Decoding the allotetraploid genome of C. arabica is therefore mandatory to have accurate genotyping-by-sequencing (GBS) studies in this species.
One of the challenges in short read sequencing of polyploid genomes is the difficulty in assembling a reference sequence for the haploid complement by disentangling its homoeologous components. Here we used short read sequencing of pooled BAC clones. Each DNA pool contained ~3% of the haploid genome and was sequenced and assembled separately. This strategy resulted into the first public draft genome of C. arabica L. that enabled us to undertake the first GBS approach for polymorphism detection on C. arabica accessions and its parental species. This allowed us to (i) confirm the geographical structure of the genetic diversity of C. arabica which originated after a single event of polyplodization that gave rise to the species and was partly shaped by early movements of planting material, (ii) detect sources of diversity for coffee breeding and (iii) understand the relatedness between the canephora subgenome of C. arabica and the modern diploid C. canephora, some of which are used for the production of Robusta coffee beans.
For this analysis we selected C. arabica varieties conserved in the CATIE International Coffee Collection in Costa Rica. This collection does not only contain elite cultivars, but also the most extensive sampling of C. arabica genetic diversity available outside of Ethiopia. We studied also 93 Yemeni genotypes collected from farmers’ fields in Yemen. Based on the GBS analysis of 736 C. arabica accessions we developed a description of the genetic diversity of the species in the context of their geographical distribution and historical records.
Results
Genome sequencing and assembly
A BAC library was constructed from a C. arabica plant of the variety ‘Bourbon Vermelho’. BAC clones amounting to a ~2.8X genome coverage were arranged into 96 pools of 384 clones (Table S1). DNA pools were sequenced independently, generating 488 Gbp that were assembled using ABySS v1.3.713. We also produced for the same individual 42.7 Gbp from genome-wide mate pairs spanning 2 kbp DNA fragments (Table S1). Such mate pairs were used for scaffolding BAC contigs, resulting in 164,254 scaffolds, with an N50 of 19,010 and an L50 of 22.3 kbp, amounting to a total length of 1.536 Gbp. Based on k-mer analysis of paired-end whole genome shotgun (WGS) sequences, we estimated a ~1.3 Gbp genome size. For low values of k, e.g. k = 16, we observed a bimodal distribution of coverage in C. arabica (Fig. S1). One peak pointed to a value of ~54X, corresponding to the expected mean genome coverage after read trimming and filtering. Another peak pointed to a 2-fold higher coverage that is expected when sequences are identical between subgenomes. We did not observe any peak corresponding to half of the diploid genome coverage that would have been expected in the presence of substantial levels of heterozygosity, compatible with the self-fertilizing nature of C. arabica. Conversely, for higher values of k, e.g. k = 51, the k-mer analysis produced a single peak exactly at the expected genome coverage, indicating a relatively high level of diversity between the two subgenomes. We then generated 38 Gbp of short reads from a C. eugenioides accession, corresponding to a coverage of approximately 54X (Table S1), while raw reads from a C. canephora doubled-haploid accession DH200-9411 corresponding to a 66X coverage were downloaded from the NCBI Sequence Read Archive (SRA). After masking repetitive sequences from C. arabica scaffolds we compared 51-mers generated from each scaffold with 51-mers obtained from C. eugenioides and C. canephora WGS reads. A total of 25,315 scaffolds, amounting to a total length of 444 Mbp, shared more 51-mers with C. canephora than with C. eugenioides and were therefore assigned to the canephora subgenome. A total of 26,627 scaffolds, amounting to a total length of 527 Mbp, shared more 51-mers with C. eugenioides than with C. canephora and were therefore assigned to the eugenioides subgenome. The remaining 112,312 scaffolds, amounting to a total length of 565 Mbp, could not be assigned with high confidence to either subgenome due to either their short size or the presence of repetitive sequences or the high similarity between homoeologous sequences. We sequenced 560 million reads, corresponding to 70 Gb of RNA-seq from 8 different tissues of C. arabica, ‘Bourbon Vermelho’ variety (Table S1 and section methods). We used these sequences to predicted 46,562 non-redundant gene models in the C. arabica genome that include 92.4% of the plant orthologs set of BUSCO14, see Supplementary Material for details. Of these, 21,254 and 22,888 genes were located on scaffolds assigned to the canephora and eugenioides subgenomes, respectively, while 2,420 genes were located on unassigned scaffolds. Gene predictions are downloadable from the website https://worldcoffeeresearch.org/work/coffea-arabica-genome/.
The tetraploid genome in the sequenced accession of ‘Bourbon’ did not show major chromosomal deletions compared to a reference diploid genome of one of the progenitor species (C. canephora). We detected a single large chromosomal rearrangement, corresponding to an event of homoeologous replacement of canephora DNA with eugeniodes DNA in the terminal 1.2-Mbp of chromosome 7, involving 179 predicted genes (Fig. 1), in agreement with previous findings based on the lack of inter-homeologue polymorphisms (hemi-SNP) in that region15. ‘Bourbon’ is therefore an autopolyploid across this gene-rich region, carrying four copies of eugenioides DNA that likely originated through a homology-directed repair of a double-strand break in the canephora chromosome. This event likely occurred immediately after hybridization because we did not detect hemi-SNP across that region in any accession of C. arabica analyzed in this study. We selected approximatively 1 Mbp of shared nucleotide sequence between the canephora and eugeniodes subgenomes in ‘Bourbon Vermelho’, using the largest scaffolds that contained single-copy genes (BUSCO). Shared regions within each scaffold were identified using (B)LastZ and realigned using MUSCLE. Nucleotide diversity (π) between C. arabica subgenomes based on hemi-SNPs amounted to 3.1 × 10−2.
Figure 1.

Homoeologous replacement on chromosome 7. The brown rectangle indicates a large event of homoeologous replacement. The upper plot indicates the fractions of nucleotides in 100-kb non-overlapping windows classified as gene space (yellow), repetitive DNA (red), and intergenic low-copy DNA (white background). The lower plots illustrate the average read coverage in 2-kb non-overlapping windows (red dots) and a cubic smoothing spline of the data (black line), the alignment of C. arabica scaffolds, sorted by their subgenome assignment (canephora in green, eugenioides in blue) against the genome reference of the diploid C. canephora.
Coffea arabica genetic diversity
We used GBS data to estimate genetic diversity in a sample of 736 accessions of C. arabica (Dataset S2), presumably representing a large fraction of the diversity available in the species. The nucleotide diversity estimate obtained is low (π = 2.3 × 10−4, based on 652 SNPs across 193,873 informative nucleotides), a value that is one order of magnitude lower than that estimated in the present-day germplasm of the two progenitor diploid species (C. canephora π = 2.6 × 10−3, C. eugenioides π = 1.1 × 10−3). We observed a very peculiar distribution of private mutations (i.e. variant sites detected in a single individual only), with such mutations corresponding to 74.4% of the variant sites. In order to exclude an underestimation of π due to any alignment bias between the native and derived subgenomes used as a reference genome in our procedure of read mapping (see Materials and Methods for more details) or due to the fragmentation of our reference, we repeated this analysis by aligning dRAD reads against a chromosome-scale assembly of C. arabica ‘Caturra Vermelho’ that became recently available (GenBank assembly accession: GCA_003713225.1). We confirmed the levels of π both in the progenitor diploid species (C. canephora π = 2.3 × 10−3, C. eugenioides π = 1.1 × 10−3) and C. arabica (π = 2.0 × 10−4) across a larger sample of 339,526 nucleotides.
The value of Taijma’s D in C. arabica was highly negative (−2.51), as expected for a population undergoing expansion after a recent severe bottleneck. A large fraction (87.6%) of variant sites in C. arabica showed a minor allele frequency lower than 0.05. Among the variant sites that were present in more than one individual of C. arabica, 28.7% did not violate Hardy-Weinberg equilibrium, 6% showed an excess of heterozygotes and the majority (65.3%) showed a deficiency of heterozygotes, as expected for an autogamous species.
The severity of the bottleneck effect and the unsubstantial carry-over of ancestral diversity from the Coffea genus, represented in this study by the most likely parental species, into the population of C. arabica suggest that the tetraploid species has originated from a single event of hybridization. This conclusion is supported also by the distribution of private alleles among the three species (C. arabica, C. canephora, C. eugenioides) that shows that the vast majority of SNPs identified in C. arabica (Fig. 2A) is not shared with either of the parental species, confirming that most of the variation present today in C. arabica has arisen after the polyploidization event and also indicating that there have not been major introgression events from the two parental species into C. arabica.
Figure 2.

Genetic diversity in Coffea. Number of variant sites and their distribution among the three species (panel A), number of individuals n = 53 in C. canephora, n = 10 in C. eugenioides, n = 736 in C. arabica. Principal Component Analysis within C. arabica using all variant sites (panel B) or only variant sites on the canephora (panel C) or on the eugeniodes subgenome (panel D), separately. Individuals were grouped a priori into geographical classes. Principal Component Analysis of the Arabica populations (panel E) using all variant sites, as in panel B, in relation to ancestry assignment provided by the software STRUCTURE. Map of Ethiopia and Yemen showing the locations of coffee accessions (panel F), as a function of their STRUCTURE population, as in panel E.
We performed in silico simulations with msprime16 to assess whether diversity levels and distribution are compatible with a recent origin of all current C. arabica accessions from a single polyploid individual. We simulated mutation accumulation with mutation rates ranging from 6.5 × 10−10 to 3.25 × 10−8 per base per generation, and under four alternative hypothetical demographic models (Dataset S1) of a current population of effective size Ne = 10,000 or 50,000 initiated with a single hybrid individual 10,000 or 20,000 years ago, which attained the final size in 200 generations of exponential growth followed by a constant size or recovering to the final size from a bottleneck that occurred 1,000 years ago. The other parameters in the model were kept constant (recombination rate 1 × 10−8, generation per year = 0.2, sampling size = 700 individuals). These models predicted a nucleotide diversity π ranging between 1.2 × 10−4 and 2.5 × 10−4 for the highest mutation rate and between 2.4 × 10−6 and 5 × 10−6 for the lowest mutation rate and a proportion of private SNPs ranging from 23% (polyploidization 10,000 years ago, Ne =10,000) to 35% (polyploidization event 10,000 years ago, Ne =50,000, Dataset S1). Considering a generation time of 5 years, the highest mutation rate per generation of 3.25 × 10−8, that provides diversity estimates that are very similar to those observed, corresponds, on a per year basis, to that estimated in Arabidopsis17. Since coffee varieties and landraces are propagated by seed for establishing new plantations, we also simulated a recent expansion since the beginning of massive coffee cultivation (approx. 0.4 kya) to a current size of Ne = 500,000. This model did not predict substantial changes in the expected values of π but it generated higher expected values of private SNPs (42%) that are closer to the observed values.
Under the assumption of a recent origin of all C. arabica accessions from a single individual that underwent polyploidization, that seems to be fully supported by both the available data and the in silico simulations, expectations can be derived on the patterns and distribution of genetic diversity within the C. arabica population itself and in comparison to diversity within the ancestral species.
We analyzed diversity within C. arabica in relation to diversity in 35 C. canephora accessions using only the variants identified in the canephora subgenome by performing a Principal Component Analysis (PCA) where the first two PC’s explained 16.9 and 8.2% of the variance (Fig. 3).
Figure 3.
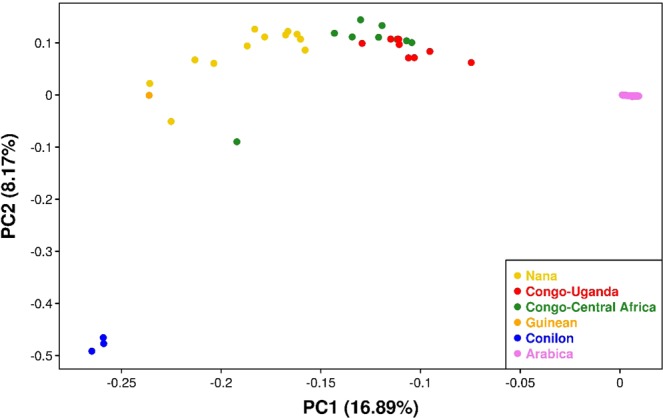
Principal Component Analysis for Arabica and canephora individuals using only the variants found in the C. canephora subgenome. Analysis of the first two axes with the percent variation explained (Nana group, Robusta Congo-Uganda group, Robusta Congo Central Africa group, Guinean group and Conilon group are represented in yellow, red, green, orange and blue, respectively).
The C. canephora accessions are representative of the diversity within the species including what is commercially identified as “Conilon”, e.g. C. canephora accessions used for coffee production in Brazil, and “Robusta”, e.g. C. canephora accessions cultivated elsewhere in the world18. The genetic diversity of the modern C. canephora is much wider than the one of the canephora subgenome of C. arabica, as expected under the single polyploidy event scenario. The Robusta groups of modern C. canephora from Congo-Central Africa and Congo-Uganda appear to be the closest to the lineage that has donated the canephora subgenome to C. arabica, confirming the observations based on a SNP chip array analysis10. Interestingly, in this PCA, the “Conilon” group is clearly separated from other C. canephora groups that instead form a continuum.
A principal component analysis was then conducted solely in C. arabica individuals using 698 variant sites identified across 129,638 informative positions over the two subgenomes (Fig. 2B) or using separately variant sites on either subgenome (Fig. 2C,D). The structure observed was very similar whether or not subgenome-specific SNPs were merged; nevertheless, the percentages explained by the first two components, that are always very low, were slightly higher when using the eugenioides subgenome, meaning that using the genetic variation across this subgenome enables a better description of the structure of the population. As expected under a scenario where the polymorphisms are extremely recent and mostly private, the substructuring of the population appears very weak, even though still detectable. The first axis (PC1) separates the majority of the Ethiopian population (Landrace cultivated, Survey Ethiopia) from the Yemeni population (other geographical classes, Dataset S2) but some Ethiopian genotypes were located close to the Yemeni category and vice versa (Fig. 2), as expected based on a recent origin (dating to the 14th century) of the Yemeni population from Ethiopian accessions (Chevalier 1929). Indian old varieties and East African old varieties, as well as Bourbon/Typica varieties, overlapped with the area of the PCA plane populated by Yemeni germplasm (Fig. 2). The group ‘Landrace cultivated’ covered entirely the area of the PCA plane populated by Yemeni germplasm and only partially the area of the PCA plane populated by Ethiopian germplasm, suggesting that part of the genetic diversity present in Ethiopia is currently used for cultivation only locally and another part has not been exploited yet. The separation between widely cultivated varieties and part of the Ethiopian wild germplasm was corroborated by ancestry assignment performed with the software STRUCTURE. At K = 2, one ancestry group (G1) included the vast majority of C. arabica accessions that were either surveyed in Ethiopia or were recently brought out of Ethiopia (Fig. 2E,F, Table S2,: Fig. S2) and the other group (G2) included all the Yemeni varieties, their descendants and a few Ethiopian accessions grown in the easternmost part of the country. By running STRUCTURE only on G1 a clear structuration of the Ethiopian germplasm was revealed (Additional text).
Discussion
Whole genome sequencing is revolutionizing our understanding of biology and the field is developing rapidly due to advances in DNA sequencing technologies. The accuracy of genome sequencing and assembly of complex genomes is affected by different factors ranging from plant material availability to genome size, GC content, repeat content, ploidy level, sequencing technology, and bioinformatic software19–22. Long-read sequencing technologies, such as those of Pacific Biosciences or Oxford Nanopore Technologies, are currently used for reconstructing Mb-long chromosome sequences or both haplotypes of highly heterozygous genomes23–27. Short-read sequencing is less expensive, provides more coverage with the same investment and thus more sequence accuracy, but it leads to more fragmented assemblies. Several applications such as the analysis of gene content, the discovery and application of genetic markers and studies of genetic diversity can be undertaken using draft whole genome shotgun assemblies, which are relatively quick to produce. They allow to reconstruct efficiently the non-repetitive fraction of the genome, which is the most informative for those applications. In our study we adopted a strategy to maximize the accuracy of sequence assembly, with the aim of reconstructing separately homoeologous sequences and provide a reference for studying allelic variation in C. arabica. Our genome assembly is relatively fragmented with an L50 of 22.3 kbp but it contains 92.4% of the conserved set of plant single-copy orthologs (BUSCO) and allowed us to assign 94.8% of the predicted genes to their parental genome. We predicted 46,562 protein‐coding genes in C. arabica, which is nearly double the number (25,574) of one of its ancestor C. canephora11. This high number is fully compatible with the hypothesis of a recent genome doubling and limited gene loss or pseudogenization since the event of polyploidisation to the present time.
Given the extremely low level of heterozygosity of C. arabica, the main issue we had to face was the ploidy level28. To tackle this, we could have opted to isolate chromosomes through flow cytometry29 but we could not consider this technology due to lack of appropriate genetic stocks that would allow for easy chromosome isolation. We thus opted for a hierarchical sequencing approach of BAC pooling where the likelihood of two homoeologous fragments occurring in the same pool was extremely low30,31, leading to dramatic reduction of the genome complexity in the assemblies of each pool. In this paper, we show a successful application of this genome assembly for studying Arabica diversity.
Coffee is a recent beverage relative to the time scale of mankind. Ethiopian nomadic mountain people were probably the first to recognize coffee’s stimulating effect. However, it is estimated that the use of coffee brewing as we know it today would have begun in the Middle Ages in Yemen. The first archaeological evidence of beverage coffee consumption was found in Zabid (Yemen) a city at the south end of the Arabian Peninsula32. Yemen would have become the first coffee market. In Ethiopia, drinking coffee was formally prohibited for Christians until the early 20th century and the Ethiopian Coffee ceremony is considered a recent invention33. The prevailing assumption is that coffee seeds were introduced from Ethiopia to Yemen. From southwestern Ethiopia, wild coffee genotypes should have been acclimated to the other side of the gulf. However, domestication of C. arabica over a long period of time (end of 17th century) has also been reported in Ethiopia by scientific observers or travelers in the Harar region, Zeghie peninsula, Sidamo, and Welega province34. During the 15th and 16th centuries, coffee cultivation was developed within Yemen to meet local needs. In Yemen, people transformed the mountainsides into terraced hillsides, built irrigation networks, and invented farming techniques including growing coffee without shade. In our study we compared some ‘Typica’ and ‘Bourbon’–derived cultivars with the Yemeni and Ethiopian accessions. In Latin America, breeders exploited these two narrow genetic bases, which resulted in ‘Typica’ and ‘Bourbon’–derived cultivars, all showing similar agronomic traits and high susceptibility to the major coffee diseases and pests35. Today, over 80% of Arabica coffee is produced in Latin America and Arabica coffee production is still based to a large extent on cultivars developed long ago by line selection within the ‘Typica’ and ‘Bourbon’ varieties or among seedlings originating from crosses between these varieties36.
We have chosen to analyze genetic diversity and population divergence within the C. arabica species and in comparison to the ancestral donor species using an unbiased resequencing-based method such as GBS rather than using a SNP chip such as the one recently developed for coffee10. Even though we have analyzed a total number of SNPs that is lower than that present on the above mentioned chip, the unbiased view of sequence diversity provided by the GBS approach has allowed us, on one hand, to obtain robust estimates of nucleotide diversity, allele frequency spectra and other population parameters that could be used to infer past population demographic history and, on the other hand, to avoid the ascertainment bias in estimating population divergence that is intrinsic to SNP chip-based approaches caused by the limited number of individuals from specific populations present within the initial SNP discovery panels37,38.
We found in C. arabica the lowest level of genetic diversity reported so far in crop species (Table S4), only comparable to levels observed for bread wheat, another recent allopolyploid species, and an exceedingly large fraction of private alleles, both to individual accessions as well as to the species in comparison to the progenitor species. All evidence collected, including that from forward computer simulations of mutation accumulation, are compatible with the origin of C. arabica from a single polyploidization event that occurred in very recent evolutionary times. This is the simplest model for allopolyploid evolution (Doyle and Egan 2010) and predicts that all variation observed in the new species is due to new mutations arisen after the polyploidization event. During the early stages following polyploid formation, the occurrence of a ‘genomic shock’ leading to gene loss and/or homeologous recombination has been observed in some species39. In C. arabica we observed a single instance of homeologous replacement at the tip of chromosome 7 and thus we do not have evidence of a significant contribution of major structural rearrangements occurring after polyploidization.
Despite the hypothesized very recent origin of all accessions from a single allopolyploid and the consequent very recent origin of all polymorphisms surveyed, our results revealed genetic differentiation between Ethiopian accessions, still present in rainforest areas at the Southwesternmost range of distribution of the species, and the germplasm used for intensive plantation in Eastern Ethiopia and in Yemen that is also genetically similar to all cultivated varieties worldwide, from East Africa and India as well as belonging to the Bourbon/Typica lineages. These results are in agreement with historical information on a worldwide spreading of C. arabica out of Yemen through the Bourbon/Typica lineages and point to the existence of wild Ethiopian germplasm that could represent a yet untapped, even though very limited, reservoir of novel genetic variation for improvement of C. arabica cultivated varieties (see supplementary materials for a more detailed analysis of variation patterns within the Ethiopian population).
In the study of Lashermes et al.40, the authors used RAPD markers to genotype 20 accessions, observed two groups, consisting of either cultivated coffee or wild Ethiopian accessions. A similar study7 with 119 accessions and 16 markers showed similar results. Finally, a study by Silvestrini et al.41 based on 73 accessions and 15 SSR markers found only two groups, the cultivated group from Yemen and a second group representing accessions from Ethiopia.
Genetic diversity is the foundation of the genetic improvement of crops and has become a strategic economic and cultural identity issue. The challenges of C. arabica germplasm conservation in Ethiopia have been reviewed by Labouisse et al.42 and regional human overpopulation appears to be the main cause of accelerated destruction of the biodiversity in the montane forests of southwestern Ethiopia43. Knowledge of coffee genetic diversity at the molecular level is essential for effective strategic conservation in ex situ collections and for protection of in situ populations, as well as the use of these resources to meet both current and future breeding needs. The vast germplasm collection of CATIE is easily accessible but its use for crop improvement is still very limited. Molecular markers used in the present study helped to clarify the structure of the genetic diversity of the species. The wild germplasm collected from the forest areas in Ethiopia is already considered a valuable source of new diversity for breeding programs. For example, controlled crosses between wild Ethiopian progenitors and American cultivars (i.e. descendants from the Yemeni population) have produced high yielding F1 hybrids44, suggesting heterotic groups that can be further differentiated and optimized through targeted selection45. The genetic diversity of C. arabica available within the collections outside of Ethiopia is currently exploited for breeding programs to cope with the challenges of climate change around the world. While a global coffee genetic resource conservation consortium is certainly needed to ensure that existing coffee genetic resources are preserved in Ethiopia, the low level of genetic diversity in C. arabica compared to the much higher diversity in the present-day populations of its progenitors suggests that introgression events into the allotetraploid species from the diploid species such as those exploited to achieve coffee rust resistance in derivatives of the Timor hybrid46,47 are of paramount importance to broaden substantially the genetic diversity in the cultivated germplasm and increase environmental, economic and social sustainability of coffee cultivation.
Conclusions
In our study we adopted a hierarchical sequencing approach to reduce the genome complexity and consequently to maximize the accuracy of sequence assembly of Coffea arabica, a tetraploid species, with the aim of reconstructing separately homoeologous sequences and provide a reference for studying allelic variation in Arabica. Our genome assembly is relatively fragmented, but it contains most of the conserved set of plant single-copy orthologs and allowed us to assign most of the predicted genes to their parental genome and to perform an in-depth unbiased analysis of Arabica population history and diversity. We found in C. arabica a very low level of genetic diversity and an extremely large fraction of private alleles, both when considering individual accessions as well as when comparing the polyploid species with the two progenitor species. All evidence collected are compatible with the origin of C. arabica from a single polyploidization event that occurred in very recent evolutionary times, as attested also by in silico forward simulations under different demographic scenarios.
Our results still reveal genetic differentiation between a group of ‘wild’ Ethiopian accessions and another group including other ‘wild’ Ethiopian accessions and most of cultivated accessions studied, including commercial germplasm belonging to the ‘Typica’ and/or ‘Bourbon’ lineages. The Ethiopian reservoir may be exploited for improvement of C. arabica cultivated varieties but, due to its limited divergence from the commercially grown materials, we suggest exploiting introgression events from the diploid parental species into the allotetraploid species, as in the case of the famous Timor hybrid. The extremely recent origin and low genetic diversity of Coffea arabica make many of the traditionally used approaches in plant breeding, trait mapping and gene isolation less efficient and the development of novel alternative approaches a definitive must.
Methods
Plant material for genome sequencing and annotation
Sequencing for genome assembly was performed using an individual of C. arabica ‘Bourbon Vermelho’. Seedlings were obtained by somatoclonal embryogenesis from cherries imported from a production area called Ahuachapan in El Salvador. Nine tissues/organs (young leaves, leaves, stems, roots, red drupes, green drupes, multiple drupes, meristems, buds) were sampled from the same variety. Sequencing for subgenome assignment was performed using an accession of C. eugenioides derived from an illycaffè greenhouse in Rivignano, Udine, Italy.
Library construction for genome sequencing
A BAC library of 175,872 BAC clones was constructed from genomic DNA by Lucigen Corporation (Lucigen Corporation). 36,864 BACs were randomly selected, inoculated into 96 384-well plates and grown at 37 °C for 22 hours in 2x LB medium supplemented with chloramphenicol antibiotic. The 384 bacterial cultures of each plate were mixed in a single tube. Each pool was subjected to alkaline lysis of bacterial cells and BAC vector was amplified in vitro with Bacteriophage Phi29 polymerase (IllustraTM TempliPhiTM Large Construct V2 kit, Resnova) with an isothermic reaction at 20 °C for 16 hours. BAC DNA was then purified with ethanol and sodium-acetate precipitation and resuspended in distilled water. BAC pools were quantified on a fluorometer (Qubit, Invitrogen) and visualized on a 0.8% agarose gel in 1x TBE buffer. Illumina libraries were constructed with the NexteraTM DNA Sample Preparation kit (New England Biolabs), following the manufacturer’s protocol.
Libraries were then purified with magnetic beads AMPure XP (Agencourt), quantified on a Caliper GX (Perkin Elmer), and sequenced using an Illumina HiSeq2000 (Illumina), generating 100-bp paired ends. Libraries from 12 pools were also sequenced with an Illumina MiSeq sequencer, generating 250-bp paired ends.
Whole-Genome Shotgun libraries were constructed from genomic DNA of the same individual of C. arabica and from one accession of C. eugenioides using the Illumina TruSeq DNA Sample prep kit, according to the manufacturer’s protocol. C. arabica WGS was performed using an Illumina HiSeq2000 and generated 100-bp paired ends. C. eugenioides WGS was performed using an Illumina HiSeq2000 and generated 125-bp paired ends.
For the same individual of C. arabica a 2-3 kbp mate-pair library was constructed using a Mate Pair Library v2 Sample Preparation kit following the Illumina protocol without gel electrophoresis size selection. The libraries were validated using a Bioanalyzer 2100 (Agilent), quantified using Qubit (Invitrogen) and then sequenced on Illumina HiSeq2000.
Reads trimming and filtering
Reads were quality trimmed with erne-filter v1.4.348 using default parameters and minimum read length of 50 bp. Adapters were removed with cutadapt49 using default parameters but -O 5 -n 2 -m 35. Cloning vector, Escherichia coli and chloroplast reads were filtered with erne-filter v1.4.348. Mate pairs were trimmed and filtered using the same procedure as above and then sorted into genuine mate pairs or paired-ends with internally developed Perl scripts based on the presence/absence of the Biotine signature.
De novo assembly, k-mer analysis and subgenome identification
Each BAC pool was assembled independently with ABySS v1.3.713 with default parameters but k = 71, aligner=map, b = 1000000, p = 0.95, s = 500, n = 10. WGS mate pairs were used for scaffolding contigs within each BAC pool with SSPACE v3.050 with a minimum of 10 mate pair links to join adjacent contigs. Repetitive DNA was masked with RepeatMasker with the following parameters: -qq -nolow -norna -no_is -gff with repeat library derived from the C. canephora genome11. k-mer analysis was carried out with Jellyfish51 with parameters -c 3 -s 10 G and -m either 16 or 51. Effective sequencing depth (N) was estimated with k-mer analysis based on modal k-mer frequency (M), read length (L), k-mer length (K) and on the formula N = M*L/(L-K + 1)52; genome size was derived from sequencing yield divided by N. For C. canephora k-mer analysis, reads were downloaded from NCBI Sequence Read Archive experiments ERX294808, ERX294809, ERX294819, ERX294831, ERX294847, ERX294857, ERX294862, ERX294873, ERX294881, and ERX294885. C. eugenioides and C. canephora 51-mers were generated from Illumina reads using Jellyfish51 with parameters -m 51 -c 3 -s 10 G. Scaffolds were classified with internally developed Perl scripts as belonging to either the canephora or eugenioides subgenome if they either shared more 51-mers with C. canephora or shared more 51-mers with C. eugenioides, respectively. Scaffolds with less than 1000 available 51-mers, i.e. either very short or containing mostly repetitive DNA, or with similar numbers of shared 51-mers for both parental species (difference < 10%) remained unclassified.
RNA-Seq and transcript analysis
RNAs were extracted using Spectrum Plant Total RNA Kit (SIGMA) following the manufacturer’s protocol (www.sigmaaldrich.com/). 1.5 µg of good quality RNA (R.I.N. > 7) was used as starting material for library preparation with the Illumina mRNA-Seq Sample Prep kit v2.0 following the manufacturer’s instructions (www.illumina.com/). The poly-A mRNA was fragmented for 1.5 minutes at 94 °C and all purification steps were performed using 1X Agencourt AMPure XP beads. Library quality and quantity were assessed using the Agilent Bioanalyzer 2100 High Sensitivity and Qubit DNA High Sensitivity (Invitrogen) as described more in detail in53. Libraries were pooled together, and the obtained pool checked on an Agilent Bioanalyzer 2100 in order to determine the molarity. Paired-end sequencing was performed on the Illumina HiSeq2500 (www.illumina.com/systems/sequencing-platforms/hiseq-2500.html) generating 125-base reads.
Trimmomatic54 was used for adapter clipping and quality trimming. The minimum read length was set to 35 bp and a minimum quality score of 20 within a sliding window of 5. RNA-seq reads were aligned on the reference genome using hisat255 using default parameters and setting the maximum intron length to 50 kbp. Genome-guided transcript reconstruction was performed independently for each RNA-library using stringtie56, setting the minimum junction coverage to 5 (option –j). The transcripts were further assembled using PASA57, a eukaryotic genome annotation tool that exploits spliced alignments of expressed transcript sequences to automatically model gene structures.
Gene prediction
Gene prediction resulted integrating several sources of evidence: (i) RNA-seq data; (ii) nucleotide and protein alignments; (iii) de novo gene training and prediction. Five different programs were used for ab initio gene prediction: Augustus58,59, Snap60, Glimmer61 and GeneMark62. Intron coordinates derived from RNA-Seq read alignments were provided to GeneMark. Gene models generated by PASA were used to train Snap, Glimmer and Augustus. Briefly, the PASA alignment assemblies were used to automatically extract protein coding regions in order to generate a high-quality data set for training ab initio gene predictors. To discard the lowest quality gene models generated by PASA, from the training dataset, only complete genes and validated through a similarity search with blast against a dataset of C. canephora were considered. From the blast search, only proteins with a match with an e-value lower that 1e-30 and an alignment coverage higher than 90% were used to train the ab initio predictors.
Nucleotide and protein sequences ranging from close to distantly related organisms belonging to eudycotyledon taxonomic rank, Gentianales order, Coffea genus and C. canephora species were downloaded from NCBI and aligned to the reference genome using exonerate (https://www.ebi.ac.uk/about/vertebrate-genomics/software/exonerate). Only high-quality alignments were retained applying the following stringent criteria: 30% identity and 70% alignment coverage at the protein level, 50% identity and 70% alignment coverage at the nucleotide level.
Previously collected evidence was combined for gene prediction using EVidenceModeler57 in order to obtain a single gene model. EVidenceModeler (EVM) combines ab initio gene predictions and protein and transcript alignments into weighted consensus gene structures. To reduce false positive prediction and improve the overall gene prediction quality, several filters were applied:
Genes predicted only by ab initio programs were considered only if they were confirmed by at least two different ab initio programs, if they were complete (with a start and a stop codon) and longer than 300 base pairs.
Gene supported by external evidence (e.g. proteins/RNA-seq) were considered if confirmed by at least two different types of evidence or by one external evidence and one ab initio gene predictor.
Predicted genes with a low ab initio support (as per step 1) were further processed. Those supported by only one ab initio program were retained only if found in a database of Coffea protein sequences. While proteins with a sequence coverage match higher than 50% for both query and subject and an e-value lower than 1e-6 were recovered.
Gene models passing these filters were further processed using PASA to add UTR regions and predict alternative splicing.
To remove multiple isoforms of the same gene and sequence redundancy, DNA coding sequences were clustered using CD-HIT63 with option -g set to 1 and for each gene locus we selected the longest transcripts isoform. CD-HIT was initially run setting the clustering percentage identity to 0.9. A Perl script was developed to parse the output in order to empirically identify a threshold that clustered as many genes as possible while keeping subgenome k-mer classified genes in proper clusters. The optimal clustering percentage identity was obtained at 0.9961 (Fig. S5).
Gene Annotation
BLASTp similarity searches (e-value threshold of 1e−5) of C. arabica predicted genes were performed against the non-redundant protein database (NCBI). InterProscan564 was used to obtain the conserved protein domains and functional annotation. The databases used included PROSITE patterns, PRINTS, PFAM, PRODOM, SMART, TIGRFAM, and PANTHER. Gene Ontology and KEGG classifications were predicted running BLAST2GO 2.6.065 on the BLASTp and InterProscan outputs.
Plant material for genotyping
An extensive number of accessions from C. arabica (781), C. canephora (35) and C. eugenioides (10) were collected for genotypic analysis for this study. Because 45 of the Coffea arabica accessions failed to produce sufficient reads following GBS analysis they were removed from the study leaving a total of 736 Arabica accessions. The final Coffea arabica genotypes included in this study were represented as follows: 648 C. arabica accessions provided by CATIE and 88 C. arabica accessions collected in Yemen provided by Sana’a University. The C. canephora and C. eugenioides accessions were collected by IRD (Institut de Recherche pour le Développement, France)66 and provided by CATIE or CIRAD. The list of all 781 accessions is provided in Dataset S2.
In relation to this history, the Arabica accessions are coded using the following categories:
Survey Ethiopia: 441 accessions from the FAO survey in Ethiopia (FAO 1964), 84 from the ORSTOM survey (1966) in Ethiopia (Guillaumet 1967) and 16 accessions from the ‘Lejeune survey’ or other surveys collected in Ethiopia before 1957. In Dataset S2 geographic coordinates and altitude for 359 accessions provided by botanists during the FAO and ORSTOM surveys is provided.
Landrace cultivated: 49 accessions of Ethiopian cultivated populations that have been collected in Ethiopian farms in addition to the FAO and ORSTOM surveys. These surveys are less documented than the FAO and ORSTOM surveys.
Survey Yemen: 93 accessions representing subspontaneous-derived accessions cultivated in Yemen and collected by Sana’a University (88 accessions) or by FAO (5 accessions planted in the CATIE collection). Those accessions can be considered as domesticated. For 28 accessions provided by Sana’a University, the altitude and the geographical coordinates are reported in Dataset S2.
East Africa and Indian Old varieties: 45 accessions of varieties selected in the 30’s in India and East Africa.
Typica/Bourbon cultivars: Seven accessions from the CATIE field GeneBank that conformed to the botanical varieties described by Krug and Carvalho (1951). In this study they represent the two widely cultivated varieties in Asia and Latin America for more than two centuries.
DNA extraction and genotyping
Leaves were collected and lyophilized, and genomic DNA was extracted using the ADNid method (http://www.adnid.fr/index-2-4A.html) at ADNid (Montpellier, France).
Genotyping by sequencing (GBS) was conducted at the Cornell University Institute for Genomic Diversity (http://www.igd.cornell.edu/index.cfm/page/GBS.htm). Illumina template libraries were produced using the restriction enzyme PstI followed by single-end sequencing on a HiSeq2000 (Illumina) as previously described67.
GBS analysis
787 C. arabica varieties along with 10 C. eugenioides and 35 C. canephora accessions were subjected to GBS using the restriction enzyme PstI67. Ninety-six barcoded accessions were pooled, and each pool was run in one lane of a flow cell on an Illumina HiSeq2000 using a 91 bp single-end sequencing mode at the Institute for Genomic Diversity (IGD) at Cornell University. In total ~172 GB of DNA sequence data was obtained. Reads were initially processed to remove the individual barcodes corresponding to each accession and separated into individual fastq files for each accession using a custom python script. To avoid false positive SNP detection due to the allopolyploid nature of the C. arabica genome (i.e. polymorphism between the two subgenomes), two in silico reference sequences were generated, one for each of the two subgenomes. The canephora in silico reference was composed by (i) the assembled canephora subgenome, (ii) a full homoeologous complement of the assembled eugenioides subgenome, and (iii) the unassigned scaffolds. Similarly, the eugenioides in silico reference was composed by (i) the assembled eugenioides subgenome, (ii) a full homoeologous complement of the assembled canephora subgenome, and (iii) the unassigned scaffolds. In order to produce the full homoeologous complement of the assembled canephora subgenome, WGS reads of C. arabica ‘Bourbon Vermelho’ were aligned using BWA68 against the assembled canephora subgenome and the unassigned scaffolds. Then, homoeologous SNPs were called using GATK69 with default parameters and the alternative homoeologous reference was generated using GATK FastaAlternateReferenceMaker for sites with minimum depth 50 and allele frequency between 0.25 and 0.75. The same procedure was applied to obtain the full homoeologous complement of the assembled eugenioides subgenome.
GBS reads were aligned using BWA-MEM v0.7.1068 against the canephora and eugenioides in silico references in order to call allelic SNPs in the two subgenomes, respectively. For both artificial references, SNP calling was performed using Stacks v2.170. Only variant sites covered by at least 10 reads were retained. Heterozygous SNPs with an allele frequency lower than 0.25 or higher than 0.75 were discarded. Polymorphisms detected using the two artificial references were merged and only variant sites called in native scaffolds of each subgenome were retained for subsequent analyses. To obtain a dataset of SNPs for STRUCTURE and PC analyses, Stacks was run with the option –r (minimum percentage of individuals in a population required to process a locus for that population) set to 0.75 and the option–max-clipped (maximum soft-clipping level, in fraction of read length) set to the default value of 0.20, corresponding to a minimum stack length of 73 bp. Variants sites with seven or eight missing genotype calls out of the eight Bourbon/Typica accessions were interpreted as misalignments and filtered out.
Principal Component Analysis was performed using the R package ade471. A hierarchical study of the diversity has been conducted using a model-based clustering procedure with admixture as implemented in STRUCTURE v2.3.472. Runs were performed at values of k ranging from 1 to 13 with 10 replicates per K and a burn-in period of 75,000 and 75,000 MCMC repetitions. Plotting k vs ΔK indicated that the highest value was for 2 groups followed by 3 groups (Fig. S2). Hence, after applying a threshold of 0.80 of membership, STRUCTURE was performed again on each of the 2 populations. According to ΔK, the second population was divided in 2 groups (Fig. S2). A Principal Coordinates Analysis (PCoA) was performed in R (version 3.5.1, 64 bit; www.r-project.org/) with ape73, using provesti distance matrix calculated using poppr package74. The PCoA was plotted with ggplot2 (Fig. S3).
Given the low level of diversity of the species and the high frequency of the PstI restriction sites, we increased genome sampling for a better estimation of π, Tajima’s D and private or shared SNPs in C. arabica, C. canephora, and C. eugenioides by running Stacks with the option –r (minimum percentage of individuals in a population required to process a locus for that population) set to 0.50 and the option–max-clipped (maximum soft-clipping level, in fraction of read length) set to the value of 0.68, corresponding to a minimum stack length of 30 bp. For these analyses, sites in which all C. eugenioides accessions were homozygous reference and all C. canephora accessions were homozygous alternative, or vice versa, were interpreted as residual homoeologous SNPs and filtered out, as well as sites within missing data in C. eugenioides and C. canephora and >95% heterozygous calls in C. arabica. Nucleotide diversity and Tajima’s D were calculated for each group using the diversity.stats method included in the R package PopGenome75.
Supplementary information
Acknowledgements
We would like to thank Claudine Campa for helpful discussions on Coffea genomics. The research has been financed by Regione Friuli Venezia Giulia (L.R: 47/1978, Decreto n.893 dd.17/03/2014), illycaffè SpA (Trieste, Italy), Lavazza SpA (Torino, Italy), World Coffee Research and by USAID Grant award AID-OAA-A-14-00018.
Author contributions
G.D.G., G.G., T.S., C.M., M.M., B.B. conceived the work. S.S., L.T., G.D.G., C.M., M.M., B.B. designed the work. M.C., F.S.L., L.N., L.D.T., G.P., M.R.R., A.P., G.G., W.S., A.A.H., B.B. acquired the material. F.C., F.M., I.J. performed the experiments. S.S., L.T., D.S., G.M., M.V., S.P., N.V., P.K., N.B. run the bioinformatic analysis. S.S., L.T., G.D.G., D.S., G.V., S.M., C.M., M.M., B.B. interpreted the data. S.S., L.T., G.D.G., P.E.K., C.M., M.M., B.B. drafted the work and substantively revised it. All authors read and approved the final manuscript.
Data availability
The data have been deposited with links to BioProject accession number PRJNA554647 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/). The genome sequence is available at https://worldcoffeeresearch.org/work/coffea-arabica-genome/.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Simone Scalabrin and Lucile Toniutti.
Supplementary information
is available for this paper at 10.1038/s41598-020-61216-7.
References
- 1.Lashermes P, et al. Molecular characterisation and origin of the Coffea arabica L. genome. Mol. Gen. Genet. MGG. Springer. 1999;261:259–66. doi: 10.1007/s004380050965. [DOI] [PubMed] [Google Scholar]
- 2.Cenci A, Combes M-C, Lashermes P. Genome evolution in diploid and tetraploid Coffea species as revealed by comparative analysis of orthologous genome segments. Plant. Mol. Biol. 2012;78:135–45. doi: 10.1007/s11103-011-9852-3. [DOI] [PubMed] [Google Scholar]
- 3.Yu Q, Guyot R, de Kochko A, Rafael N-P. Micro-collinearity and genome evolution in the vicinity of an ethylene receptor gene of cultivated diploid and allopolyploid coffee species (Coffea) Plant. J. 2011;67:305–17. doi: 10.1111/j.1365-313X.2011.04590.x. [DOI] [PubMed] [Google Scholar]
- 4.Sylvain PG. Some observations on Coffea arabica L. in Ethiopia. Turrialba. 1955;5:37–53. [Google Scholar]
- 5.Fernie, L., Greathead, D., Meyer, F. & Monaco, L., Narasimhaswamy, R. FAO coffee mission to Ethiopia, 1964–65. FAO (1968).
- 6.Haarer, A. E. Modern Coffee production. Leonard Hill. (1958).
- 7.Anthony, F. et al. The origin of cultivated Coffea arabica L. varieties revealed by AFLP and SSR markers; 894–900 (2002). [DOI] [PubMed]
- 8.Aga E, Bryngelsson T, Bekele E, Salomon B. Genetic diversity of forest arabica coffee (Coffea arabica L.) in Ethiopia as revealed by random amplified polymorphic DNA (RAPD) Hereditas. 2003;138:36–46. doi: 10.1034/j.1601-5223.2003.01636.x. [DOI] [PubMed] [Google Scholar]
- 9.Tesfaye, K., Borsch, T., Govers, K. & Bekele, E. Characterization of Coffea chloroplast microsatellites and evidence for the recent divergence of C. arabica and C. eugenioides chloroplast genomes. Genome (2007). [DOI] [PubMed]
- 10.Merot-L’anthoene, V. et al. Development and evaluation of a genome-wide Coffee 8.5K SNP array and its application for high-density genetic mapping and for investigating the origin of Coffea arabica L. Plant Biotechnol J. (2019). [DOI] [PMC free article] [PubMed]
- 11.Denoeud F, et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science (80-) Am. Assoc. Advancement Sci. 2014;345:1181–4. doi: 10.1126/science.1255274. [DOI] [PubMed] [Google Scholar]
- 12.Tran, H. T. M. et al. SNP in the Coffea arabica genome associated with coffee quality. Tree Genet Genomes (2018).
- 13.Simpson J. T., Wong K., Jackman S. D., Schein J. E., Jones S. J.M., Birol I. ABySS: A parallel assembler for short read sequence data. Genome Research. 2009;19(6):1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. Genome analysis BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinforma. 2015;31:3210–2. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 15.Lashermes P, et al. Exchanges and Homeologous Gene Silencing Shaped the Nascent Allopolyploid Coffee Genome (Coffea arabica L.) Genes|Genomes|Genetics. 2016;6:2937–48. doi: 10.1534/g3.116.030858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelleher J, Etheridge AM, McVean G. Efficient Coalescent Simulation and Genealogical Analysis for Large Sample Sizes. Song YS, editor. PLOS Comput Biol. Public Library of Science. 2016;12:e1004842. doi: 10.1371/journal.pcbi.1004842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ossowski S et al. The rate and molecular spectrum of spontaneous mutations in arabidopsis thaliana. Science (80-) 2010. [DOI] [PMC free article] [PubMed]
- 18.Garavito A., Montagnon C., Guyot R., Bertrand B. Identification by the DArTseq method of the genetic origin of the Coffea canephora cultivated in Vietnam and Mexico. BMC Plant Biol. BMC Plant Biology 1–12 (2016). [DOI] [PMC free article] [PubMed]
- 19.Lander, E. S. & Waterman, M. S. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics (1988). [DOI] [PubMed]
- 20.Churchill, G. A. & Waterman, M. S. The accuracy of DNA sequences: Estimating sequence quality. Genomics (1992). [DOI] [PubMed]
- 21.Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA (2011). [DOI] [PMC free article] [PubMed]
- 22.Myers, E. W. Jr. A history of DNA sequence assembly. it - Inf Technol. (2016).
- 23.Li, C., Lin, F., An, D., Wang, W. & Huang, R. Genome Sequencing and Assembly by Long Reads in Plants. Genes (Basel) 9 (2018). [DOI] [PMC free article] [PubMed]
- 24.Shimizu T, et al. Draft Sequencing of the Heterozygous Diploid Genome of Satsuma (Citrus unshiu Marc.) Using a Hybrid Assembly Approach. Front Genet. 2017;8:1–19. doi: 10.3389/fgene.2017.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koren Sergey, Rhie Arang, Walenz Brian P, Dilthey Alexander T, Bickhart Derek M, Kingan Sarah B, Hiendleder Stefan, Williams John L, Smith Timothy P L, Phillippy Adam M. De novo assembly of haplotype-resolved genomes with trio binning. Nature Biotechnology. 2018;36(12):1174–1182. doi: 10.1038/nbt.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pryszcz, L. P. & Gabaldon, T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 1–10 (2016). [DOI] [PMC free article] [PubMed]
- 27.Kajitani R, et al. Platanus-allee is a de novo haplotype assembler enabling a comprehensive access to divergent heterozygous regions. Nat Commun. 2019;10:1–15. doi: 10.1038/s41467-019-09575-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kyriakidou M, Tai HH, Anglin NL, Ellis D, Strömvik MV. Current Strategies of Polyploid Plant Genome Sequence Assembly. Front Plant Sci. 2018;9:1–15. doi: 10.3389/fpls.2018.01660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doležel J, Kubaláková M, Cihalikova J, Suchánková P, Šimková H. Chromosome Analysis and Sorting Using Flow Cytometry. Methods Mol Biol. 2011;701:221–38. doi: 10.1007/978-1-61737-957-4_12. [DOI] [PubMed] [Google Scholar]
- 30.Haiminen N, Feltus FA, Parida L. Assessing pooled BAC and whole genome shotgun strategies for assembly of complex genomes. BMC Genomics. 2011;12:1–13. doi: 10.1186/1471-2164-12-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Visendi P, et al. An efficient approach to BAC based assembly of complex genomes. Plant Methods. BioMed Central. 2016;12:1–9. doi: 10.1186/s13007-016-0107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brosh, N. Coffee Culture. Jerusalem: Israel Museum, editor (2002).
- 33.Pankhurst, R. The coffee ceremony and the history of coffee consumption in Ethiopia. Ethiop broader Perspect Pap XIIIth 18 Int Conf Ethiop Stud Kyoto, 12–17 December 1997. M. Shigeta. p. 516–39 (1997).
- 34.Sylvain Pierre G. Ethiopian coffee—its significance to world coffee problems. Economic Botany. 1958;12(2):111–139. doi: 10.1007/BF02862767. [DOI] [Google Scholar]
- 35.Bertrand, B., Aguilar, G., Santacreo, R. & Anzueto, F. El Mejoramiento Genetico En America Central. Desafios la caficultura en Centroam. B. Bertran. p. 407–56 (1999).
- 36.Van Der Vossen H, et al. Next generation variety development for sustainable production of arabica coffee (Coffea arabica L.): a review. Euphytica. 2015;204:243–56. doi: 10.1007/s10681-015-1398-z. [DOI] [Google Scholar]
- 37.Albrechtsen, A., Nielsen, F. C. & Nielsen, R. Ascertainment biases in SNP chips affect measures of population divergence. Mol Biol Evol. (2010). [DOI] [PMC free article] [PubMed]
- 38.Lachance, J. & Tishkoff, S. A. SNP ascertainment bias in population genetic analyses: Why it is important, and how to correct it. BioEssays (2013). [DOI] [PMC free article] [PubMed]
- 39.Gaeta, R. T., Pires, J. C., Iniguez-Luy, F., Leon, E. & Osborn, T. C. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell. (2007). [DOI] [PMC free article] [PubMed]
- 40.Lashermes P, Trouslot P, Anthony F, Combes MC, Charrier A. Genetic diversity for RAPD markers between cultivated and wild accessions of Coffea arabica. Euphytica. 1996;87:59–64. doi: 10.1007/BF00022965. [DOI] [Google Scholar]
- 41.Silvestrini M, et al. Genetic diversity of a Coffea Germplasm Collection assessed by RAPD markers. Genet Resour Crop Evol. 2008;55:901–10. doi: 10.1007/s10722-007-9295-5. [DOI] [Google Scholar]
- 42.Labouisse JP, Bellachew B, Kotecha S, Bertrand B. Current status of coffee (Coffea arabica L.) genetic resources in Ethiopia: Implications for conservation. Genet Resour Crop Evol. 2008;55:1079–93. doi: 10.1007/s10722-008-9361-7. [DOI] [Google Scholar]
- 43.Davis, A. P. et al. High extinction risk for wild coffee species and implications for coffee sector sustainability. Sci Adv. 1–9 (2019). [DOI] [PMC free article] [PubMed]
- 44.Bertrand B, et al. Comparison of bean biochemical composition and beverage quality of Arabica hybrids involving Sudanese-Ethiopian origins with traditional varieties at various elevations in Central America. Tree Physiol. 2006;26:1239–48. doi: 10.1093/treephys/26.9.1239. [DOI] [PubMed] [Google Scholar]
- 45.Hinze LL, Kresovich S, Nason JD, Lamkey KR. Population Genetic Diversity in a Maize Reciprocal Recurrent Selection Program Population Genetic Diversity in a Maize Reciprocal Recurrent Selection. Crop Sci. 2005;45:2435–42. doi: 10.2135/cropsci2004.0662. [DOI] [Google Scholar]
- 46.Clarindo, W. R., Carvalho, C. R., Caixeta, E. T. & Koehler, A. D. Following the track of “Híbrido de Timor” origin by cytogenetic and flow cytometry approaches. Genet Resour Crop Evol. (2013).
- 47.Herrera, J. C. et al. Genomic relationships among different Timor hybrid (Coffea L.) accessions as revealed by SNP identification and RNA-seq analysis. Adv Intell Syst Comput. (2014).
- 48.Del Fabbro C, et al. Evaluation of Read Trimming Effects on Illumina NGS Data Analysis. PLoS One. 2013;8:1–13. doi: 10.1371/journal.pone.0085024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 50.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE Summary. Bioinformatics. 2011;27:578–9. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 51.Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li R, et al. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463:311–8. doi: 10.1038/nature08696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wildhagen H, et al. Genes and gene clusters related to genotype and drought-induced variation in saccharification potential, lignin content and wood anatomical traits in Populus nigra. Tree Physiol. 2018;38:320–39. doi: 10.1093/treephys/tpx054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolger AM, Lohse M, Usadel B. Genome analysis Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim D, Langmead B, Salzberg S. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pertea Mihaela, Pertea Geo M, Antonescu Corina M, Chang Tsung-Cheng, Mendell Joshua T, Salzberg Steven L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haas, B. J. et al. Open Access Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced. Genome Biol. 9 (2008). [DOI] [PMC free article] [PubMed]
- 58.Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:465–7. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;11:1–11. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:1–9. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–9. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 62.Lomsadze A, Burns PD, Borodovsky M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014;42:1–8. doi: 10.1093/nar/gku557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 64.Jones P, et al. Sequence analysis InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–40. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 66.Anthony F, Berthaud J, Guillaumet JL, Lourd M. Collecting wild coffea species in Kenya and Tanzania. Plant Genet Ressources Newsl. 1987;69:23–9. [Google Scholar]
- 67.Elshire RJ, et al. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS One. 2011;6:1–10. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–40. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dray, S. & Dufour, A. The ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat Softw. 22 (2007).
- 72.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genet Soc Am. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paradis E, Claude J, Strimmer K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 74.Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ. 2 (2014). [DOI] [PMC free article] [PubMed]
- 75.Pfeifer B, Wittelsbu U, Ramos-onsins SE, Lercher MJ. PopGenome: An Efficient Swiss Army Knife for Population Genomic Analyses in R. Mol Biol Evol. 2014;31:1929–36. doi: 10.1093/molbev/msu136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data have been deposited with links to BioProject accession number PRJNA554647 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/). The genome sequence is available at https://worldcoffeeresearch.org/work/coffea-arabica-genome/.