

Abstract
Cigarette smoking affects the oral microbiome, which is related to various systemic diseases. While studies that investigated the relationship between smoking and the oral microbiome by 16S rRNA amplicon sequencing have been performed, investigations involving metagenomic sequences are rare. We investigated the bacterial species composition in the tongue microbiome, as well as single-nucleotide variant (SNV) profiles and gene content of these species, in never and current smokers by utilizing metagenomic sequences. Among 234 never smokers and 52 current smokers, beta diversity, as assessed by weighted UniFrac measure, differed between never and current smokers (pseudo-F = 8.44, R2 = 0.028, p = 0.001). Among the 26 species that had sufficient coverage, the SNV profiles of Actinomyces graevenitzii, Megasphaera micronuciformis, Rothia mucilaginosa, Veillonella dispar, and one Veillonella sp. were significantly different between never and current smokers. Analysis of gene and pathway content revealed that genes related to the lipopolysaccharide biosynthesis pathway in Veillonella dispar were present more frequently in current smokers. We found that species-level tongue microbiome differed between never and current smokers, and 5 species from never and current smokers likely harbor different strains, as suggested by the difference in SNV frequency.
Subject terms: Microbial genetics, Plaque, Next-generation sequencing
Introduction
Cigarette smoking is associated with many oral diseases such as periodontitis and oral cancer1. The oral microbiome are reported to play a vital role in the pathogenesis of such oral diseases2, and are also important in systemic diseases such as diabetes mellitus3 and rheumatoid arthritis4. The change of oral microbiome induced by cigarette smoking could be associated with systemic diseases; thus, the relationship between cigarette smoking and the oral microbiome is of interest.
Recently, numerous studies investigating quantitative differences in oral microbiome across smoking status using 16S rRNA amplicon sequencing technology have been performed. An analysis of mouth rinse samples performed by Kato et al., revealed that Neisseria was less abundant, while members of the Veillonellaceae family were more abundant, in current smokers5. A large study, conducted by Wu et al., indicated that anaerobic bacteria favor the oral environment in smokers, and that aerobic bacteria were less abundant6. Moreover, they revealed that the functional metagenomic profile, which included the degradation of certain toxic compounds contained in cigarettes, also differed between current and never smokers. Mason et al., investigated differences in the subgingival community and also identified elevation of anaerobic bacteria and cariogenic species7. However, although species-level differences in diversity and abundance were investigated using 16S rRNA amplicon sequencing in some previous studies, investigations involving metagenomic sequences are rare, as previous studies suggested that species-level prediction based on 16S rRNA had low accuracy8. Besides, there are few studies that investigated differences in single-nucleotide variant (SNV) profiles and gene content per species in the oral environment across smoking status, which cannot be explored by amplicon sequencing.
Here, we conducted the metagenomic profiling of tongue plaque samples in a Japanese health-checkup cohort. Species-level differences of diversity and abundance, as well as SNV profiles and gene content of the bacterial species, were investigated across the smoking status.
Results
Characteristics and statistics of participant metagenomic sequences
A flowchart summarizing participant selection for this study is shown in Fig. 1. Table 1 details the background of the 286 participants included in the study. There were significant differences in sex, BMI category, and presence of caries between current and never smokers. After quality filtering and removal of human genome contamination, the average reads (SD) across participants was 9769630 (3920068) and the minimum and maximum numbers of reads were 3169504 and 24023935, respectively. The distribution of filtered reads across the groups were visualized in Fig. 2.
Fig. 1. Flowchart detailing participant selection.
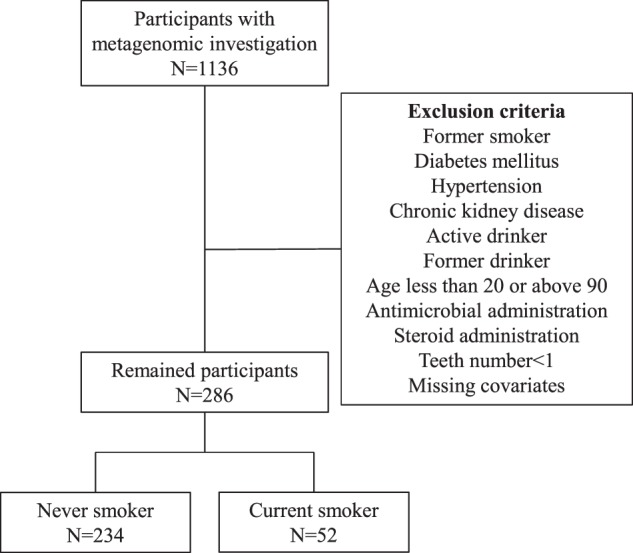
Flowchart detailing participant selection. Overall, 286 participants were selected.
Table 1.
Participants’ background.
| Clinical values | Never smokers (n = 234) | Current smokers (n = 52) | p-value |
|---|---|---|---|
| Age (years), mean (SD) | 47.99 (15.47) | 43.56 (10.53) | 0.05 |
| Sex: # female (%) | 192 (82.1) | 32 (61.5) | 0.002 |
| Teeth number, mean (SD) | 25.63 (5.55) | 26.15 (5.30) | 0.534 |
| eGFR (mL/min/1.73 m2), mean (SD) | 82.59 (15.15) | 83.76 (12.03) | 0.605 |
| HbA1c (%), mean (SD) | 5.66 (0.30) | 5.68 (0.29) | 0.675 |
| Systolic blood pressure (mmHg), mean (SD) | 115.09 (11.94) | 111.94 (12.04) | 0.087 |
| Diastolic blood pressure (mmHg), mean (SD) | 70.11 (8.53) | 68.81 (10.13) | 0.337 |
| Pack-year index, mean (SD) | NaN (NA) | 13.77 (15.13) | NA |
| FEV1.0%, mean (SD) | 83.28 (6.54) | 82.06 (6.73) | 0.228 |
| BMI category # (%) | 0.003 | ||
| 0–18.5 kg/m2 | 23 (9.8) | 10 (19.2) | |
| ≥18.5–25 kg/m2 | 173 (73.9) | 28 (53.8) | |
| ≥25–30 kg/m2 | 29 (12.4) | 14 (26.9) | |
| ≥30 | 9 (3.8) | 0 (0.0) | |
| Caries present # (%) | 66 (28.2) | 23 (44.2) | 0.036 |
| Suspected of having periodontitis # (%) | 117 (50.0) | 33 (63.5) | 0.109 |
BMI body mass index, HbA1c hemoglobin A1c, eGFR estimated glomerular filtration rate, FEV1.0% percent predicted forced expiratory volume in 1 s, SD standard deviation.
Fig. 2. The distribution of reads across groups.

The barplot and distribution of reads across groups of never and current smokers are shown. Blue indicates never smokers, while yellow indicates current smokers.
Differences in beta diversity and relative abundance
First, beta diversity and relative abundance were compared to characterize overall microbiome differences at the species-level. Beta diversity was compared between groups by weighted UniFrac measure, and a statistically significant difference were identified (pseudo-F = 8.44, R2 = 0.028, p = 0.001). The PCoA plot of weighted UniFrac distance is shown in Fig. 3. Next, the relative abundance of the species was compared. Overall, 1157 species were present across all samples. Among them, 313 species that have a relative abundance of at least 0.0001 in above 20% of all the samples were tested by MaAsLin2, and 38 species showed statistically significant differences in relative abundance. We visualized the violin and box plot of relative abundances for these species (Fig. 4). 29 species including Porphyromonas endodontalis (coef. = 0.023, standard error = 0.0037, q < 0.001), Streptococcus oralis (coef. = 0.020, standard error = 0.0041, q < 0.001), Streptococcus parasanguinis (coef. = 0.036, standard error = 0.0071, q < 0.001) were significantly present more abundant in current smokers, while nine species including Neisseria subflava (coef. = −0.062, standard error = 0.013, q = 0.0012), Lautropia mirabilis (coef. = −0.013, standard error = 0.0031, q = 0.0027), Neisseria flavescens (coef. = −0.050, standard error = 0.013, q = 0.0087) were present less abundant in current smokers. Species were listed according to the lowest 3 q-values. All the statistical results are presented in Supplementary Data Set 1. Additionally, species relative abundance table were deposited in Supplementary Data.
Fig. 3. The principal coordinate plot of beta diversity measured by weighted UniFrac.
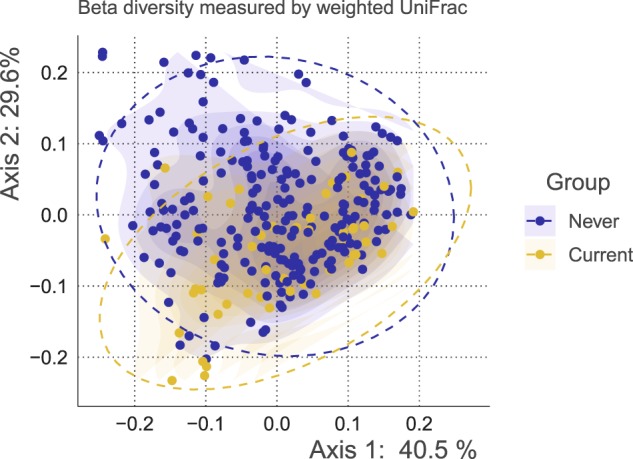
The x- and y-axes represent the first and second principal coordinates with the proportion of variance. The 95% confidence ellipse is shown for each group. The points in blue indicate never smokers, while the points in yellow indicate current smokers.
Fig. 4. The violin and boxplot demonstrating significant differences in relative species abundance.

The group of never or current smokers are shown on the x-axis and the relative abundances is shown on the y-axis (on a logarithmic scale). Blue indicates abundance in never smokers, while yellow indicates abundance in current smokers.
Differences in SNV frequency
We next analyzed whether there are differences in bacterial genome SNV frequency across smoking status. For SNV profiling, 26 species that have sufficient coverage in at least 10% of the samples were analyzed. The number of participants with sufficient read depth for the species is summarized in Supplementary Data Set 1. Among these 26 species, a distance matrix calculated from the SNV frequency table was compared by permutational multivariate analysis of variance (PERMANOVA). Overall, 5 species; Actinomyces graevenitzii (profiled in 38 never and 13 current smokers, pseudo-F = 2.60, R2 = 0.051, q = 0.013, Actinomyces_graevenitzii_58300 in the MIDAS database), Megasphaera micronuciformis (profiled in 37 never and 13 current smokers, pseudo-F = 1.56, R2 = 0.032, q = 0.021, Megasphaera_micronuciformis_62167 in the MIDAS database), Rothia mucilaginosa (profiled in 25 never and 14 current smokers, pseudo-F = 6.47, R2 = 0.124, q = 0.009, Rothia_mucilaginosa_62109 in the MIDAS database), Veillonella dispar (profiled in 81 never and 31 current smokers, pseudo-F = 20.98, R2 = 0.16, q = 0.009, Veillonella_dispar_61763 in the MIDAS database), one Veillonella sp (profiled in 116 participants, 100 never and 16 current smokers, pseudo-F = 2.71, R2 = 0.023, q = 0.009, Veillonella_sp_62404 in the MIDAS database) showed statistically significant differences in SNV frequency between current and never smokers. The PCoA plots and dendrograms based on SNV frequency are shown in Figs 5 and 6. Those 5 species were selected for the downstream analysis. The exact number of SNVs and the genome length were summarized in Table 2. Participants’ background with average sufficient read depth of species included in the downstream analysis are summarized in Supplementary Data Set 1. The SNV location and SNV frequency of the samples for all the species are deposited in Supplementary Data. As the proportion of participants with “suspected of periodontitis” was higher in current smokers, we performed the comparison between those with healthy periodontal status and those with suspected of periodontitis controlling for age, sex, BMI, teeth number, caries status, and smoking. There were no species that showed significant differences between them (Supplementary Data Set 1). We performed the analysis with the newly created custom database for the 5 species that significantly differed, and the result was similar (Supplementary Data Set 1). We additionally performed multiple sequence alignment of the core-genome of these 5 species, and visualize the tree in Supplementary Fig. 1. Additionally, the dendrogram based on the distance matrix calculated from SNV frequency table, with RefSeq deposited sequences are visualized in Supplementary Fig. 2. Further, the proportion of within- and between-group strain sharing are described in Supplementary Data Set 1.
Fig. 5. The principal coordinate plots based on the distance matrix of species differed significantly between never and current smokers in terms of SNV frequency.

The x- and y-axes represent the first and second principal coordinates, respectively, with the proportion of variance. The points in blue indicate never smokers, while the points in yellow indicate current smokers, and the green points indicate the reference in the database.
Fig. 6. The dendrogram based on the distance matrix of species that differed significantly between never and current smokers in terms of SNV frequency.

The circular dendrogram is shown for each species significantly differed in terms of SNV frequency. Red branches indicate the leaf of current smokers, and blue branches indicate the leaf of never smokers.
Table 2.
The detailed number of SNVs and the genome length.
| Species_id | Number_of_snv | Number_of_snv_in_cds | m-equal-r(count) | m-equal-r(%) | Genome_length |
|---|---|---|---|---|---|
| Actinomyces_graevenitzii_58300 | 97,158 | 90,456 | 80,295 | 0.82643735 | 2,196,917 |
| Megasphaera_micronuciformis_62167 | 87,204 | 80,271 | 77,932 | 0.8936746 | 1,765,528 |
| Rothia_mucilaginosa_62109 | 299 | 274 | 283 | 0.94648829 | 2,264,603 |
| Veillonella_dispar_61763 | 5930 | 4714 | 4983 | 0.84030354 | 2,118,767 |
| Veillonella_sp_62404 | 28,233 | 25,005 | 25,319 | 0.89678745 | 2,176,752 |
m-equal-r the number of SNV that the major allele are as same as the reference allele.
Differences in gene and pathway content of species
We next investigated differences in gene and pathway content across species present in current or never smokers. The gene presence table was analyzed by logistic regression models. The gene differential results for 5 species were summarized in Table 3. There were no significant differences in terms of gene presence frequency between current and never smokers in Megasphaera micronuciformis. Those genes yielded up-regulated and down-regulated KO identifiers for each species and the KEGG pathways were inferred by MinPath. Up-regulated pathways were as follows; purine and pyrimidine metabolism, and lipopolysaccharide biosynthesis in Veillonella dispar, pentose phosphate pathway, glycine, serine and threonine metabolism, and phenylalanine, tyrosine and tryptophan biosynthesis in Rothia mucilaginosa. Down-regulated pathways were as follows; galactose metabolism in Actinomyces graevenitzii, pyrimidine metabolism and ubiquinone and other terpenoid-quinone biosynthesis in Rothia mucilaginosa, glycolysis/gluconeogenesis, cysteine and methionine metabolism, and pyrimidine metabolism in Veillonella dispar, and pentose phosphate pathway, purine metabolism, and cysteine and methionine metabolism in Veillonella sp. Note that the pathway with the highest three total number of involved KO identifiers was listed, and genes that were differentially presented between current and never smokers are listed in Supplementary Data Set 1, and the inferred pathways are summarized in Supplementary Data Set 1. In addition, a gene status heatmap displaying presence or absence of four species that have significantly differed genes is shown in Fig. 7. Further, the gene presence table for all the species were deposited in Supplementary Data.
Table 3.
The summary of the differential analysis of genes.
| Species_id | Never | Current | Total-gene | All-zero | All-one | Tested | Down | Up |
|---|---|---|---|---|---|---|---|---|
| Actinomyces_graevenitzii_58300 | 38 | 13 | 2471 | 46 | 918 | 1507 | 10 | 7 |
| Megasphaera_micronuciformis_62167 | 37 | 13 | 1724 | 87 | 1210 | 427 | 0 | 0 |
| Rothia_mucilaginosa_62109 | 107 | 30 | 1765 | 2 | 187 | 1576 | 24 | 37 |
| Veillonella_dispar_61763 | 85 | 31 | 1890 | 10 | 349 | 1531 | 24 | 271 |
| Veillonella_sp_62404 | 115 | 23 | 1991 | 19 | 303 | 1669 | 197 | 3 |
Never the number of never smokers profiled for genes, Current the number of current smokers profiled for genes, Total-gene the number of total genes, All-zero the number of genes that were absent in all samples, All-one the number of genes that were present in all samples, Tested the number of genes tested, Down the number of genes down-regulated, Up the number of genes up-regulated.
Fig. 7. Heatmap depicting gene presence or absence status.

Rows indicate sample (red: current smokers, blue: never smokers), and columns indicate statistical significance (gray: not significant, black: significant) along with q-values (on a logarithmic scale). Heatmap cells indicate gene presence or absence (red: absent, yellow: present).
Random forest classification
We performed binary classification of current and never smokers by random forest algorithm to characterize how these differences can discriminate current and never smokers. The performance was highest when the input was gene presence table of Veillonella dispar (0.930 [0.060], mean [SD]). The performance was 0.913 [0.077], 0.824 [0.098], 0.817 [0.107], and 0.801 [0.128] respectively (mean [SD]), when the input was SNV frequency table of Veillonella dispar, SNV frequency table of Veillonella sp, gene presence table of Veillonella sp, and SNV frequency table of Actionmyces graevenitzii. Only the model with AUROC above 0.8 were listed, and other performances regarding 5 species profiled were summarized in Table 4. When the input was the relative abundance of all the species presented in samples, the performance was 0.745 [0.097]. These results indicated that current and never smokers can be discriminated by the SNV frequency or gene presence table of Veillonella dispar, one of Veillonella sp, and Actinomyces graevenitzii, which suggested these species within the oral microbiome of current and never smokers have distinct SNV frequency or gene presence frequency characteristics.
Table 4.
The performance of random forest classifier measured by AUROC.
| Input | Species | Mean | Stdev |
|---|---|---|---|
| relative_abundances | – | 0.7453 | 0.0971 |
| snv | Actinomyces_graevenitzii_58300 | 0.7396 | 0.2034 |
| snv | Megasphaera_micronuciformis_62167 | 0.6631 | 0.1182 |
| snv | Rothia_mucilaginosa_62109 | 0.7467 | 0.204 |
| snv | Veillonella_dispar_61763 | 0.9132 | 0.0773 |
| snv | Veillonella_sp_62404 | 0.8238 | 0.0984 |
| Gene | Actinomyces_graevenitzii_58300 | 0.8012 | 0.1283 |
| Gene | Megasphaera_micronuciformis_62167 | 0.6554 | 0.1977 |
| Gene | Rothia_mucilaginosa_62109 | 0.6842 | 0.0939 |
| Gene | Veillonella_dispar_61763 | 0.9298 | 0.0596 |
| Gene | Veillonella_sp_62404 | 0.8174 | 0.1069 |
Mean the mean value of 5-fold cross validation, Stdev the standard deviation value of 5-fold cross validation.
Discussion
This study explored the species-level bacterial composition, SNV profile, and gene content of species in the tongue microbiome, with particular focus on the relationship with cigarette smoking. The tongue is exposed to many chemical compounds during cigarette smoking and many of these compounds are considered harmful9. In the present study, we found 5 species that differed in terms of SNV frequency. The relative abundance of Veillonella dispar was high in current smokers, and the SNV profile differed in current smokers compared to never smokers, which suggests that current smokers have different strains of Veillonella dispar. It seems likely that Veillonella genus is related with cigarette smoking, considering the results of previous studies which reported quantitative changes of Veillonella5,6.
Veillonella are strict-anaerobic, Gram-negative diplococci from the phylum Firmicutes that are commensal oral bacteria, and are reported to be nitrate and nitrite reducing bacteria10. They were also reported in some clinical case reports such as prosthetic joint infection11. In various studies using 16S rRNA amplicon sequencing technology, Veillonella were reported to be related to smoking. Wu et al. reported that Veillonella were more abundant in mouthwash samples of smokers, and suggested that the anaerobic characteristics of these bacteria make them tolerant to the smoking environment5. Kato et al. also found that the Veillonellaceae family were more abundant in smoker’s saliva samples4. Veillonellaceae were also reported to be related to certain cancers, such as lung cancer, and are reported to be a good marker of squamous cell carcinoma in the salivary microbiome12. In addition, Veillonella parvula were reportedly found frequently in the lower air tracts of lung cancer patients13. Furthermore, a study comparing microbiome in bronchoalveolar lavage fluid of lung cancer patients and benign mass patients revealed that Megasphaera and Veillonella were more abundant in lung cancer patients14.
Additionally, Veillonella dispar gene and pathway content analysis revealed that some genes in Veillonella dispar were present with high frequency in current smokers compared to never smokers. Interestingly, the lipopolysaccharide (LPS) biosynthesis pathway was up-regulated in the current smoker group, from the pathway reconstruction based on the significantly different genes. LPS are located in outer membrane of Gram-negative bacteria like Veillonella, and consist of lipid A, core oligosaccharide, and a distal polysaccharide15. In the review by Delwiche et al., they postulated that Veillonella produces significant endotoxic LPS by sugar assimilation of not only ribose, but also fructose being incorporated into LPS16. Additionally, Veillonella LPS were reported to have relatively high endotoxicity17. Our results suggested that Veillonella dispar have a potentially different activities related to LPS production in current and never smokers, judging from the KEGG pathway analysis.
We identified quantitative differences between bacteria in current and never smokers at the species-level. In accordance with other studies, anaerobic bacteria, including those in the genus Streptococcus or Veillonella, as well as Fusobacterium nucleatum, were confirmed to be more abundant in current smokers7. In the genus Veillonella, Veillonella dispar was significantly more abundant in terms of relative abundance. In addition to quantitative differences measured by 16S rRNA sequence, which has also been described in some previous studies, the present study provided more specific information regarding differences in the tongue microbiome between current and never smokers.
Our results suggested that Veillonella dispar gene presence table was able to distinguish current smokers and never smokers with the highest performance using the random forest classifier, which indicated that Veillonella dispar from current and never smokers have distinct characteristics compared to other species investigated. Additionally, the sequences of strains deposited in RefSeq under Veillonella dispar were placed in different groups in the dendrogram, suggesting that strains deposited in RefSeq could be grouped according to other criteria, such as SNV frequency and functional differences.
Further, our study demonstrated that Actinomyces graevenitzii, Megasphaera micronuciformis, Rothia mucilaginosa, and one Veillonella sp also displayed the potential of having different strains at the species-level based on the SNV frequency difference, and some genes from those species were significantly differed. Actinomyces graevenitzii has not been reported in association with cigarette smoking. Megasphaera micronuciformis were reported to be found more commonly in cigarette smoker’s upper gastrointestinal tract, as detected using the microarray18, and they are under the family Veillonellaceae. The genus Rothia is an oral commensal, and Rothia mucilaginosa was recently reported to be increased in current smoker’s buccal microbiome19, although our analysis in tongue microbiome did not find significant difference in terms of relative abundance. We performed strain sharing pattern analysis to further elucidate the meaning of these differences, and two Veillonella species have relatively high strain sharing within groups compared to between groups. However, we cannot extrapolate functional consequences based on these SNV findings. Additionally, there is currently no strict definition of strain20, and the definition is given by the tool basis.
The current study has several strengths. The study explored differences in SNV profiles of tongue microbiome across smoking status using community-wide metagenomics data, and it revealed that smokers may harbor different strains in some species compared to never smokers. Further, we profiled the differences in gene content for these species, which had not been thoroughly investigated previously. As well, we minimized the effect of confounders by various parameters which are said to affect oral microbiome.
The present study also contained limitations. First, due to the cross-sectional nature of the study, it is unknown whether these results indicate that species present originally in the tongue flora underwent nucleotides alteration due to cigarette smoking, or whether some species favor the habitat created by smoking. In addition, we did not include dietary information, which may also influence the tongue microbiome. The detailed functional consequences, as well as the clinical implication, of differences in SNV or gene profiles remain unclear and further investigations will be necessary. Online tools other than MIDAS, such as StrainPhlAn21 or metaSNV22, are available to investigate species or strain-level microbiome profiles. These tools utilize different databases, and differences in databases may influence the profile results. The default database of MIDAS utilized clustering with a cutoff of 95% genome-wide ANI, which correspond to the standard definition of species. Thus, the present analysis could detect variations under the species-level, and we validated the result using a custom-built database based on RefSeq. We cannot determine which species got sufficient read depth to profile SNV beforehand. Additionally, we could not dissect the effect of smoking and periodontitis on the tongue microbiome thoroughly as cigarette smoking is a risk factor for periodontitis, and the prevalence of periodontitis in current smokers in our cohort was high. In addition, the oral examination was based on one-time measurement when they admitted to the health-checkup, so the detailed treatment history of oral diseases, or how often they got maintenance treatment regarding oral diseases were unknown.
In summary, we conclude that tongue microbiome of cigarette smokers differed from that of never smokers at a species-level resolution. Moreover, Veillonella dispar was suggested to have a different SNV profile and gene content in smokers, thus implying different functionality. In addition to previous studies that mainly investigated quantitative differences in relative abundances of the oral microbiome, we show that differences related to SNV profile and gene content may also be present between current and never smokers. However, the clinical implication of these differences requires further investigation.
Methods
Study population and covariate assessment
This study was approved by the Ethical Committee of Hirosaki University (approval number: 2016-028) and all participants provided written informed consent. The participants were drawn from the Iwaki health-checkup cohort conducted in 2016. The participants’ covariate information was obtained from questionnaires administered upon induction into the study. Medical history, current medication use, and smoking and drinking history was provided by the participants. Body mass index (BMI) was calculated from participants’ height and weight, and was classified into four categories according to the World Health Organization criteria. Smoking status was classified as current, former, and never. Drinking status was classified as non-drinker, current drinker, and former drinker. Natural tooth number, caries number, and periodontal status were directly examined by the dentists, and caries number was categorized as having caries or not. Periodontal status was classified as suspected of having periodontitis or not. Specifically, if participants had the a depth of gingivitis pocket ≥4 mm and/or gum bleeding, they were categorized as suspected of having periodontitis, according to the definition of the Community Periodontal Index23. Patients were excluded based on the following criteria: patients younger than 20 years or older than or equal to 90 years, use of antihypertensive drugs, selection of “currently under treatment”, “currently followed up” or “previously under treatment” of hypertension in the questionnaire, systolic blood pressure of 140 mmHg or above, diastolic blood pressure of 90 mmHg or above, use of antidiabetic drugs, selection of “currently under treatment”, “currently followed up” or “previously under treatment” of diabetes mellitus in the questionnaire, HbA1c of 6.5 or above, estimated glomerular filtration rate below 30, use of antimicrobials or steroids, current or former drinkers, former smokers, those who have no teeth, and those with missing information regarding any of their covariates. Overall, 52 current smokers and 234 never smokers were included in the study. We measured the predicted percentage of forced expiratory volume in 1 s and pack-year index (the number of cigarettes smoked per day divided by 20, multiplied by the number of years of smoking).
Sample collection, DNA extraction, and Illumina shotgun sequencing
For each of the participants, in the morning of admission and before breakfast and tooth brushing, tongue plaque samples were obtained by brushing the dorsal surface of tongue 4–5 times with a swab. The swab head was then placed in a collection tube containing 4 M guanidium thiocyanate, 100 mM Tris-HCl (pH 8.0), 40 mM EDTA and 0.001% bromothymol blue. The samples were mixed with zirconia beads using a FastPrep 24 instrument (MP Biomedicals, Santa Ana, California, USA). DNA was extracted from the bead-treated suspensions using an automatic nucleic acid extractor and MagDEA DNA 200 (GC) (Precision System Science, Chiba, Japan). Quantification and quality assessment of extracted DNA was performed using the 2200 TapeStation System (Agilent, Santa Clara, California, USA). DNA samples were fragmented using LE220 (Covaris, Woburn, Massachusetts, USA), and subjected to library preparation using the TruSeq ChIP Library Preparation Kit (Illumina, San Diego, California, USA) following the manufacturer’s instructions. Prepared libraries were evaluated using the 2200 TapeStation System, and quantified by quantitative PCR using KAPA Library Quantification Kits (KAPA Biosystems, Wilmington, Massachusetts, USA). Sequencing was performed on the HiSeq2500 instrument (Illumina, San Diego, California, USA) with 101 bp paired-end reads.
Metagenomic sequence processing
Raw paired-end reads were quality filtered via sickle version 1.3324 using the sliding window approach and the following parameters: average quality threshold of 30 and the minimum length of 20. Reads were then mapped to the human reference genome (GRCh38) by Burrows-Wheeler Aligner MEM (BWA-MEM) version 0.7.1725 with the default parameter set and mapped reads with alignment length greater than 80 were subsequently discarded.
Relative abundance and SNV profiling
Metagenomic Intra-Species Diversity Analysis System (MIDAS) was used to profile species abundance, SNV frequency, and gene content in each species26. First, we aligned quality-filtered paired-end sequence reads to the reference database, containing 31,007 sequences comprising 5952 species provided by default in MIDAS, by HS-BLASTN27 to obtain species-level abundance per sample (run_midas.py species in default parameter, and merge_midas.py species with default parameter). The resulting alignment files were processed by MIDAS to profile SNVs against the bacterial representative genome (run_midas.py snps with the default parameter). Specifically, species with ≥3.0X coverage (total base pair aligned to the representative sequence of the species divided by total genome length of the species) were profiled SNV. Subsequently, the SNV profile was merged across samples, and only bi-allelic positions were chosen. Participants with average read depth across reference sites with at least 1 mapped read of ≥5.0 were merged SNV per species, and species with a number of profiled participants ≥10% of the total number of participants were included in the SNV analysis. Other parameters were in accordance with the preset option ‘–core-snps’ (merge_midas.py snps –core-snps). We additionally performed a validation analysis using the updated custom database for species that differed significantly in terms of SNV frequency. The complete genome sequences for species significantly differed were downloaded from the NCBI RefSeq28 based on the assembly summary on 17 December 2019. The average nucleotide identity (ANI) of the sequences was calculated by FastANI29, and the sequences were hierarchically clustered based on the ANI. The representative sequence was chosen based on the medoid of the clusters, and the build_db.py script was deployed. We again analyzed the SNV frequency differences for significant species using the newly created database and computed the result. Additionally, the SNV profile of metagenomics in our cohort was compared with that of sequences available in the NCBI RefSeq. In order to compare the reference sequences with our metagenomic sequences, we simulated 9,000,000 metagenomic sequence reads from reference sequences using wgsim, as MIDAS expect metagenomic sequences to profile SNV, with parameters set to simulate no errors, insertions, or deletions. Subsequently, these reads were processed by the MIDAS pipeline via the same method used to profile metagenomic samples from the cohort. Hierarchical clustering based on the distance matrix calculated from SNV frequency was performed and a dendrogram was constructed for visualization of the result.
Gene content and pathway analysis
We also performed pan-genome gene content analysis to infer the gene content of species per sample by MIDAS using the default parameters (run_midas.py genes with the default parameter), and results were merged across samples (merge_midas.py genes with the default parameter). Gene presence status was visualized by a heatmap, produced by R library ComplexHeatmap30. Additionally, we inferred functional pathways using significantly up-regulated or down-regulated genes to compare the functional characteristic of species between groups. Specifically, the nucleotide sequences of significantly differed genes were aligned to the Kyoto Encyclopedia of Genes and Genomes (KEGG) prokaryotic protein database31 by DIAMOND version 0.8.2232 with the default parameter. One gene was matched to one KEGG prokaryotic protein with the lowest e-value. Subsequently, the protein was matched to KEGG Orthologue (KO) database. The KEGG pathways were inferred by MinPath version 1.4 from the resulting KO identifier. MinPath infers the conservative estimate of pathways by a parsimony approach rather than a naïve mapping approach using a set of genes33.
Statistical analysis
Participant backgrounds were compared via one-way ANOVA and chi-squared test. Beta diversity was assessed by the weighted UniFrac measure, and compared between current and never smokers by PERMANOVA using adonis function in R package vegan34, adjusted for age, sex, BMI category, teeth number, presence of caries, and periodontal status, and was subsequently visualized by principal coordinate analysis (PCoA) plot. Default phylogenetic trees provided by MIDAS were used. Beta diversity calculation was performed by the function UniFrac in R library phyloseq35. Species abundance was compared between current and never smokers using Multivariate Association with Linear Models 2 (MaAsLin2)36 adjusted for age, sex, BMI category, teeth number, presence of caries, and periodontal status. Relative abundances were arc sine square root transformed, and taxa that had a minimum abundance of 0.0001 in at least 20% of all samples were tested (“transform = AST”, “min_abundance = 0.0001”, and “min_prevalence = 0.2”). The distance between samples was calculated based on the Manhattan distance, which accounted for the sum of the differences of non-reference allele frequency per SNV positions divided by the number of SNV positions. These distances were calculated based on the sub-set of participants that have sufficient average read depth for the candidate species. Subsequently, these distances were tested for association with smoking status by PERMANOVA adjusted for age, sex, BMI category, teeth number, presence of caries, and periodontal status using adonis function. Permutations were set to 999. The distance matrix of significant taxa was subsequently visualized by PCoA plot. In addition, hierarchical clustering, based on the distance matrix calculated from SNV frequency, was performed, and the resulting dendrogram was visualized. Additionally, we built a strain-level phylogenetic tree by identifying core-genome regions that have high coverage across multiple samples. We performed the call_consensus.py script for the species significantly differed in the SNV frequency comparison with the following parameters; –site_maf 0.01, –site_depth 5, –site_prev 0.90, –sample_depth 10, –sample_cov 0.40, –size_ratio 5.0 as suggested by the MIDAS manual. Subsequently, FastTree37 was used to build a phylogenetic tree for the species and we visualized the resulting tree. Within groups strain sharing were measured by the MIDAS script strain_tracking.py, with the parameter of –min_freq 0.90 and –min_reads 10 in the strain_tracking.py id_markers, which identifies SNV that rarely occur in different unrelated samples, and with the default parameter in the strain_tracking.py track_markers, which identifies marker SNV sharing between samples. We defined the SNV sharing cutoff level of 5%, and described the proportion of SNV sharing within groups. Genes contained in representative genomes included in the database were identified and annotated using prokka38 version 1.14.0, in addition to the default annotation provided by MIDAS. Specifically, the exact matching of scaffold identifiers, start position, and end position was performed for the MIDAS default database and prokka annotation. Prokka was used with the default parameter. Gene content of each species was compared between current and never smokers by logistic regression models implemented in brglm using the binary table of gene presence of each species adjusted for age, sex, BMI category, teeth number, presence of caries, and periodontal status39. Genes present or absent in all the samples, or genes that were present or absent in all the samples in each group were excluded before testing. All statistical tests were two-sided, and a p-value, or false discovery rate adjusted q-value, of <0.05 was considered statistically significant40. All analysis was performed using R or Python. The plots were organized and generated using ape, DECIPHER, dendextend, firatheme, and ggplot241–45.
Random forest classification
Classification of current and never smokers was achieved using the random forest classifier using the species abundance table, SNV frequency, and gene presence table of each species. The performance was evaluated by area under receiver operator characteristic curve (AUROC). Five fold cross-validation was performed, and the mean and standard deviation of AUROC was calculated. Random forest classification was performed by scikit-learn RandomForestClassifier function, default parameters provided for the function, and a fixed seed number was used46.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary.
Supplementary information
Acknowledgements
This work was supported by JST COI Grant Number JPMJCE1302. The super-computing resource was provided by the Human Genome Center, the Institute of Medical Science, and the University of Tokyo. The funders of the study had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. Y.O. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Author contributions
N.S. designed the study, analyzed the data, interpreted the data, and wrote the manuscript. M.K., T.H., R.Y., E.U., S.I., S.N., M.Y., and Y.O. analyzed the data, interpreted the data, helped write the manuscript, and commented on drafts. W.K., K.S., Y.T., I.T., S.N., and K.M. performed the medical checkups, and gathered and summarized the data. All authors read and confirmed the final manuscript. Y.O. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Data availability
All the data supporting the conclusion of this analysis are available on Supplementary Information, the public repository (Supplementary Data, 10.6084/m9.figshare.11611089), or on reasonable request to the corresponding author.
Code availability
The scripts are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41522-020-0121-6.
References
- 1.Chaturvedi P, Singh A, Chien C-Y, Warnakulasuriya S. Tobacco related oral cancer. BMJ. 2019;365:l2142. doi: 10.1136/bmj.l2142. [DOI] [PubMed] [Google Scholar]
- 2.Meurman JH. Oral microbiota and cancer. J. Oral Microbiol. 2010;2:5195. doi: 10.3402/jom.v2i0.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao E, et al. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe. 2017;22:120–128.e4. doi: 10.1016/j.chom.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015;21:895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 5.Kato I, et al. Oral microbiome and history of smoking and colorectal cancer. J. Epidemiol. Res. 2016;2:92–101. doi: 10.5430/jer.v2n2p92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu J, et al. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J. 2016;10:2435–2446. doi: 10.1038/ismej.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mason MR, et al. The subgingival microbiome of clinically healthy current and never smokers. ISME J. 2015;9:268–272. doi: 10.1038/ismej.2014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edgar RC. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ. 2018;6:e4652. doi: 10.7717/peerj.4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elders MJ, Perry CL, Eriksen MP, Giovino GA. The report of the Surgeon General: preventing tobacco use among young people. Am. J. Public Health. 1994;84:543–547. doi: 10.2105/AJPH.84.4.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hyde ER, et al. Metagenomic analysis of nitrate-reducing bacteria in the oral cavity: implications for nitric oxide homeostasis. PLoS ONE. 2014;9:e88645. doi: 10.1371/journal.pone.0088645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchandin H, et al. Prosthetic joint infection due to Veillonella dispar. Eur. J. Clin. Microbiol. Infect. Dis. 2001;20:340–342. doi: 10.1007/PL00011273. [DOI] [PubMed] [Google Scholar]
- 12.Yan X, et al. Discovery and validation of potential bacterial biomarkers for lung cancer. Am. J. Cancer Res. 2015;5:3111–3122. [PMC free article] [PubMed] [Google Scholar]
- 13.Rybojad P, Los R, Sawicki M, Tabarkiewicz J, Malm A. Anaerobic bacteria colonizing the lower airways in lung cancer patients. Folia Histochem. Cytobiol. 2011;49:263–266. doi: 10.5603/FHC.2011.0036. [DOI] [PubMed] [Google Scholar]
- 14.Lee SH, et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer. 2016;102:89–95. doi: 10.1016/j.lungcan.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delwiche EA, Pestka JJ, Tortorello ML. The veillonellae: gram-negative cocci with a unique physiology. Annu. Rev. Microbiol. 1985;39:175–193. doi: 10.1146/annurev.mi.39.100185.001135. [DOI] [PubMed] [Google Scholar]
- 17.Sveen K. The capacity of lipopolysaccharides from bacteroides, fusobacterium and veillonella to produce skin inflammation and the local and generalized Shwartzman reaction in rabbits. J. Periodontal Res. 1977;12:340–350. doi: 10.1111/j.1600-0765.1977.tb01525.x. [DOI] [PubMed] [Google Scholar]
- 18.Vogtmann E, et al. Association between tobacco use and the upper gastrointestinal microbiome among Chinese men. Cancer Causes Control. 2015;26:581–588. doi: 10.1007/s10552-015-0535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karabudak S, et al. Analysis of the effect of smoking on the buccal microbiome using next-generation sequencing technology. J. Med. Microbiol. 2019;68:1148–1158. doi: 10.1099/jmm.0.001003. [DOI] [PubMed] [Google Scholar]
- 20.Segata N. On the Road to Strain-Resolved Comparative Metagenomics. mSystems. 2018;3:190–217. doi: 10.1128/mSystems.00190-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017;27:626–638. doi: 10.1101/gr.216242.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costea PI, et al. metaSNV: A tool for metagenomic strain level analysis. PLoS ONE. 2017;12:e0182392. doi: 10.1371/journal.pone.0182392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.World Health Organization. Oral Health Surveys: Basic Methods. (World Health Organization, 2013).
- 24.Joshi, N. A., Fass, J. N. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. https://github.com/najoshi/sickle (2011).
- 25.Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
- 26.Nayfach S, Rodriguez-Mueller B, Garud N, Pollard KS. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 2016;26:1612–1625. doi: 10.1101/gr.201863.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Ye W, Zhang Y, Xu Y. High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 2015;43:7762–7768. doi: 10.1093/nar/gkv784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tatusova T, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 31.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 33.Ye Y, Doak TG. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 2009;5:e1000465. doi: 10.1371/journal.pcbi.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jari, O. et al. vegan: Community Ecology Package. R package version 2.5-6.https://CRAN.R-project.org/package=vegan (2019).
- 35.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mallick, H., Rahnavard, A., McIver, L. Maaslin2: Maaslin2. R package version 1.0.0.http://huttenhower.sph.harvard.edu/maaslin2 (2019).
- 37.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 39.Kosmidis, I. brglm: Bias Reduction in Binary-Response Generalized Linear Models.http://www.ucl.ac.uk/~ucakiko/software.html (2017).
- 40.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995;57:289–300. [Google Scholar]
- 41.Paradis E, Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 42.Wright ES. Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. R. J. 2016;8:352–359. doi: 10.32614/RJ-2016-025. [DOI] [Google Scholar]
- 43.Galili T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015;31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Kesteren, E-J. vankesteren/firatheme: firatheme version 0.2.1. 10.5281/zenodo.3604681 (2020).
- 45.Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, New York, 2016). https://ggplot2.tidyverse.org.
- 46.Pedregosa F, et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data supporting the conclusion of this analysis are available on Supplementary Information, the public repository (Supplementary Data, 10.6084/m9.figshare.11611089), or on reasonable request to the corresponding author.
The scripts are available from the corresponding author upon reasonable request.