

Abstract
Background and Purpose
Negative allosteric modulators (NAMs) that target the calcium‐sensing receptor (CaS receptor) were originally developed for the treatment of osteoporosis by stimulating the release of endogenous parathyroid hormone, but failed in human clinical trials. Several chemically and structurally distinct NAM scaffolds have been described, but it is not known how these different scaffolds interact with the CaS receptor to inhibit receptor signalling in response to agonists.
Experimental Approach
In the present study, we used a mutagenesis approach combined with analytical pharmacology and computational modelling to probe the binding sites of four distinct NAM scaffolds.
Key Results
Although all four scaffolds bind to the 7‐transmembrane and/or extracellular or intracellular loops, they occupy distinct regions, as previously shown for positive allosteric modulators of the CaS receptor. Furthermore, different NAM scaffolds mediate negative allosteric modulation via distinct amino acid networks.
Conclusion and Implications
These findings aid our understanding of how different NAMs bind to and inhibit the CaS receptor. Elucidation of allosteric binding sites in the CaS receptor has implications for the discovery of novel allosteric modulators.

Abbreviations
- 7TM
7‐transmembrane
- BMS compound 1
(S)‐6‐((1‐hydroxy‐3‐phenylpropan‐2‐yl)amino)‐N‐(2‐phenoxyethyl)‐2‐(3,4,5‐trimethoxyphenyl)nicotinamide
- BPMC
biased probability Monte Carlo
- Ca2+i
intracellular calcium
- Ca2+o
extracellular calcium
- ECL
extracellular loop
- ICL
intracellular loop
- JTT 305
2′‐((R)‐1‐((R)‐3‐((1‐(4‐chloro‐3‐fluorophenyl)‐2‐methylpropan‐2‐yl)amino)‐2‐hydroxypropoxy)ethyl)‐3‐methyl‐[1,1′‐biphenyl]‐4‐carboxylic acid
- NAM
negative allosteric modulator
- NPS2143
(R)‐2‐chloro‐6‐(2‐hydroxy‐3‐((2‐methyl‐1‐(naphthalen‐2‐yl)propan‐2‐yl)amino)propoxy)benzonitrile
- PAM
positive allosteric modulator
- Pfizer compound 1
(R)‐2‐(2‐hydroxyphenyl)‐3‐(1‐phenylpropan‐2‐yl)‐5‐(trifluoromethyl)pyrido[4,3‐d]pyrimidin‐4(3H)‐one
- Ronacaleret
(R)‐3‐(3‐(3‐((1‐(2,3‐dihydro‐1H‐inden‐2‐yl)‐2‐methylpropan‐2‐yl)amino)‐2‐hydroxypropoxy)‐4,5‐difluorophenyl)propanoic acid
- WT
wild‐type
What is already known
Negative allosteric modulators of calcium‐sensing receptors bind to the 7‐transmembrane domain of the receptor.
What this study adds
We identified overlapping but different binding sites for different negative allosteric modulators in this domain.
What is the clinical significance
Identification of allosteric binding sites in the calcium‐sensing receptor will facilitate structure‐based drug discovery.
1. INTRODUCTION
The human calcium‐sensing (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=12 (as designated by the International Union of Pharmacology (IUPHAR) (Bikle et al., 2019)) is a class C GPCR that regulates https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1785 secretion in response to subtle changes in extracellular calcium (Ca2+ o; Conigrave, 2012). Small molecule inhibitors of the CaS receptor, which stimulate increases in serum PTH by mimicking a drop in Ca2+ o, were of interest to a number of pharmaceutical companies for the treatment of osteoporosis (Leach & Gregory, 2017). https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=716 (Figure 1), the first reported CaS receptor inhibitor (Gowen et al., 2000), binds within the 7‐transmembrane domain (7TM) of the receptor, thus acting as a negative allosteric modulator (NAM) of Ca2+ o acting predominantly via the extracellular domain (Geng et al., 2016; Zhang et al., 2016). The structurally related arylalkylamine NAMs, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9473 (Figure 1) and JTT‐305, and the chemically distinct quinazolinone NAMs, AXT914 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9475 (Figure 1), proceeded into clinical trials as osteoporosis therapies, but all failed to stimulate the formation of new bone to levels comparable to those achieved with recombinant PTH injections (Cosman et al., 2016; Fitzpatrick, Dabrowski, et al., 2011; Fitzpatrick, Smith, et al., 2011; Fitzpatrick et al., 2012; Halse et al., 2014; John et al., 2014; Widler et al., 2008). A number of additional CaS receptor NAM scaffolds, including https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=10590 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=10589 (Figure 1), have also been developed but have not progressed to clinical trials.
Figure 1.
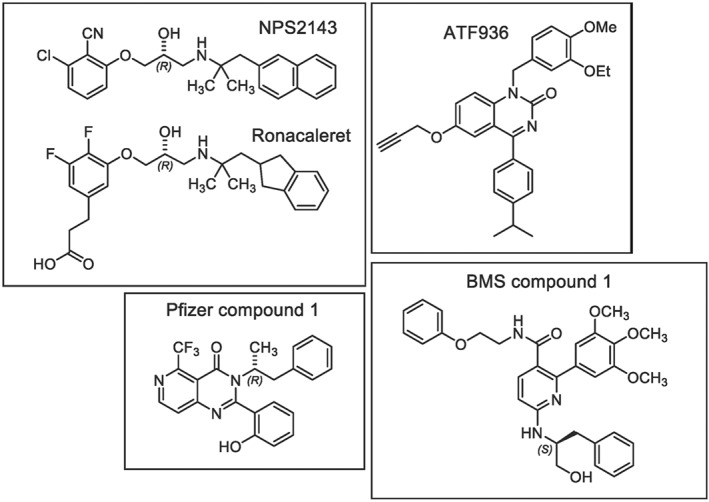
Chemical structures of CaS receptor NAMs
Although interest in developing NAMs of the CaS receptor for osteoporosis has diminished, there remains the possibility of repurposing these compounds for calcium metabolism disorders caused by naturally occurring gain‐of‐function CaS receptor mutations (Roberts et al., 2019) or CaS receptor‐mediated inflammation in asthmatic lung (Yarova et al., 2015). However, there is limited understanding of how different NAMs bind to and inhibit the CaS receptor, in part because the structure of the CaS receptor 7TM domain has not been determined. This information is necessary for understanding potential effects of naturally occurring mutations on the binding and allosteric actions of these drugs.
Although some allosteric modulators of the CaS receptor, including L‐amino acids, glutathione peptides (Conigrave & Hampson, 2010), and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8375 (Alexander et al., 2015), bind to the receptor's extracellular domain, all small molecule modulators identified to date appear to bind within the CaS receptor 7TM and/or extracellular loops (ECLs; Bu, Michino, Wolf, & Brooks, 2008; Leach et al., 2016; Miedlich, Gama, Seuwen, Wolf, & Breitwieser, 2004; Petrel et al., 2003; Petrel et al., 2004; Widler et al., 2010). We previously demonstrated that the CaS receptor 7TM/ECL domains contain an extended cavity that accommodates multiple allosteric binding sites (Leach et al., 2016). Similarly, previous studies predict that CaS receptor NAMs bind to distinct binding sites within the CaS receptor 7TM/ECL domains (Arey et al., 2005; Widler et al., 2010). For instance, NPS2143 and other arylalkylamine PAMs and NAMs possess a central secondary amino group that is charged at physiological pH and is predicted to form a salt bridge with Glu8377.32 (superscript residue numbers are based on those assigned in Dore et al., 2014) in TM7 (Bu et al., 2008; Leach et al., 2016; Miedlich et al., 2004; Petrel et al., 2003; Petrel et al., 2004). Accordingly, substitutions of Glu8377.32 abolish arylalkylamine modulator binding, and additional substitutions of residues that line the 7TM cavity abolish or significantly reduce arylalkylamine modulator affinity. In contrast, the activity of ATF936 and related compounds is in part altered by distinct mutations to those that reduce arylalkylamine activity, suggesting quinazolinone NAMs may bind in a different manner (Widler et al., 2010). Similarly, BMS compound 1 is predicted to bind to a site that is topographically distinct from the NPS2143 binding site, because it did not displace a radiolabelled NPS2143 analogue in radioligand binding studies (Arey et al., 2005).
In the present study, we probed how different CaS receptor NAM chemotypes bind to the CaS receptor 7TM domain. Our results support the existence of a second allosteric binding site utilised by BMS compound 1 and suggest other CaS receptor NAMs may bind to distinct regions of the extended 7TM cavity.
2. METHODS
2.1. Cell lines
FlpIn TRex HEK 293 cells were maintained in DMEM with 5% FBS. FlpIn TRex cells stably expressing the cmyc‐tagged WT and mutant CaS receptors have been described previously (Davey et al., 2012; Leach et al., 2016) and were maintained in DMEM supplemented with 5% FBS and antibiotic selection (200 μg·ml−1 hygromycin; 5 μg·ml−1 blasticidin). Cells were routinely tested for mycoplasma using the Lonza MycoAlert™ mycoplasma detection kit.
2.2. Intracellular calcium (Ca2+ i) mobilisation assays
Ca2+ i mobilisation assays were performed as previously described (Leach et al., 2016). Briefly, FlpIn HEK293 TRex stable cell lines were seeded at 80,000 cells per well in poly‐D‐lysine (50 μg·ml−1) coated clear 96‐well plates and incubated overnight at 37°C, 5% CO2, in the presence of 100 ng·ml−1 tetracycline to induce receptor expression. Cells were washed in assay buffer (150‐mM NaCl, 2.6‐mM KCl, 1.18‐mM MgCl2, 10‐mM D‐glucose, 10‐mM HEPES, 0.1‐mM CaCl2, 0.5% BSA, 4‐mM probenecid, pH 7.4) and loaded with 1‐μM Fluo‐4 or Fluo‐8 AM in assay buffer for 1 hr. Modulators (0, 0.03, 0.1, 0.3, 1, 3, or 10 μM) were pre‐incubated for 20 min prior to the addition of agonist (Ca2+ o). Agonist addition and measurement of Ca2+ i mobilisation was performed in duplicate at 37°C using a Flexstation 1 or 3 (Molecular Devices; Sunnyvale, CA, USA), using 485‐nm excitation and 525‐nm emission. Data were normalised to the maximum Ca2+ i mobilisation response produced by 1‐μM ionomycin.
2.3. Computational modelling
In silico docking studies were performed using ICM‐Pro (Molsoft L.L.C, USA), guided by the crystal structures of isolated 7TM domains of metabotropic glutamate receptor subtype 1 (Wu et al., 2014) and subtype 5 (Christopher et al., 2015), as previously described (Leach et al., 2016). Identification of NAM binding sites was guided by our mutagenesis data, where residues that reduced affinity (pK B) were assumed to contribute to the binding site. For docking studies, NAMs were initially placed in the centre of the presumed binding site before being extensively sampled by biased probability Monte Carlo sampling in internal coordinates. Based on previous predictions of a salt bridge between the charged secondary amine in NPS2143 and Glu8377.32 (Leach et al., 2016) and on the reduction in pK B values for ronacaleret caused by Ile substitution of Glu8377.32 (see Section 3), ronacaleret was docked by extensive conformational sampling of the ligand and the residue side‐chains lining the pocket, while maintaining adequate distance to allow a salt bridge interaction between the charged secondary amine in ronacaleret and the carboxylate of Glu8377.32. For ATF936, Pfizer compound 1, and BMS compound 1, there were no predicted anchor points that could be used to guide docking. Therefore, the hydrogen bonding, van der Waals, and hydrophobic and electrostatic potential of the CaS receptor 7TM binding cavity were calculated to create a “grid potential map,” which was used to score the binding potential of randomised conformations of the NAM. Binding scores were calculated as described previously (Totrov & Abagyan, 1999). Ten poses with the lowest calculated binding scores were retained for each NAM. For ATF936, Pfizer compound 1, and BMS compound 1, these poses were further refined using a “flexible receptor” approach, by undertaking biased probability Monte Carlo optimisation of receptor residue sidechains (Abagyan & Totrov, 1994), while simultaneously sampling NAM shape and position by Monte Carlo randomisation. Final results were assessed using a combination of ICM docking scores and calculated binding energies (Neves, Totrov, & Abagyan, 2012; Schapira, Totrov, & Abagyan, 1999), coupled with manual inspection for agreement with mutagenesis data and with published structure activity relationship (SAR) data (described below).
Initial attempts to dock ATF936 into our CaS receptor grid potential map produced poses with poor docking scores that were inconsistent with our mutagenesis data. However, previous SAR work indicated that only a propargyl moiety was well tolerated at the 6 position (Widler et al., 2010). In contrast, alkoxy, phenolic, or mono‐ or di‐substituted amino groups at this position resulted in reduced NAM potency, indicating that bulkier or hydrophilic substituents were not favourable (Widler et al., 2010). We therefore hypothesised that the propargyl substituent may occupy a defined hydrophobic pocket not captured in our grid potential map. An ATF936 derivative without the propargyl was consequently docked, and the propargyl was reintroduced during the flexible receptor refinement. Using this approach, we achieved compound binding poses consistent with our mutagenesis data.
As found with ATF936, initial docking attempts with Pfizer compound 1 into our grid potential maps produced poses that were inconsistent with our mutagenesis data. This was most likely due to Pfizer compound 1's steric bulk, which restricted its access to residues deeper in the 7TM cavity that were shown via mutagenesis to be important for its binding. Hence, we utilised the SCARE method (Bottegoni, Kufareva, Totrov, & Abagyan, 2008) to increase receptor sidechain mobility during the docking procedure. The SCARE method systematically changes each residue in the binding pocket to Ala when creating grid potential maps. While the docking of Pfizer compound 1 into the resultant maps produces poses with significant steric clashes with residue side chains in the pocket, the flexible receptor refinement determined whether sidechain movements could accommodate these poses.
It must be noted that the final proposed pose for each NAM is a prediction based on the current mutagenesis data and available SAR (see Section 3 for both). However, alternative poses that were also compatible with mutagenesis and SAR data are also possible, and we therefore show a representative alternative pose in the Supplementary File S1.
2.4. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Group sizes and number of independent experiments appropriately reflect the variability in the datasets and magnitude of the signal size. Outliers were included in data analysis and presentation.
Ca2+ i mobilisation data were normalised to the vehicle response (considered 0%) and the response to 1‐μM ionomycin (considered 100%) to reduce variability caused by experimental variations in cell density and Fluo‐4‐AM or Fluo‐8‐AM loading efficiency. Data describing the interaction between Ca2+ o and the NAMs were fitted to the following operational model of allosterism (Gregory, Giraldo, Daio, Christopoulos, & Leach, 2019):
| (1) |
where K A is the affinity of the orthosteric agonist; K B is the affinity of the allosteric ligand; τA and τB are the operational efficacies of the orthosteric agonist and allosteric ligand, respectively; α and β are the allosteric effects on orthosteric agonist affinity and efficacy, respectively; [A] and [B] are the orthosteric agonist and allosteric ligand concentrations, respectively; Em is the maximal system response; nT is the transducer slope linking agonist concentration to response; nB is the binding slope linking agonist concentration to occupancy; and [C] is the contaminating Ca2+ o concentration present in the assay buffer. In all instances, nT was constrained to unity.
Non‐linear regression analysis was performed in GraphPad Prism 7 or 8 (GraphPad Prism, RRID:SCR_002798). Affinity, cooperativity, and efficacy parameters were estimated as logarithms to generate Gaussian data (Christopoulos, 1998). Statistical analysis was undertaken only for studies where each group size was at least n = 5. The declared group size is the number of independent experiments, and statistical analysis was done using these independent experiment values. Post hoc tests were conducted when F in the ANOVA achieved the necessary level of statistical significance and there was no significant variance inhomogeneity. A one‐way ANOVA with Tukey's multiple comparisons was performed to determine whether NAM affinity or cooperativity at the WT differed significantly between NAMs, where P < .05 was considered significant and was marked on figures and tables accordingly. A one‐way ANOVA with Dunnett's multiple comparisons was performed to determine whether NAM affinity or cooperativity was significantly different at the mutant versus WT receptors, where P < .05 was considered significant and was marked on figures and tables accordingly. BMS compound 1 affinity and cooperativity estimates at two mutants (Val833ECL3Ala and Ser834ECL3Ala) were not subjected to statistical analysis owing to small group sizes of n < 5. Experiments relating to these two mutants were a preliminary screen to assess the validity of our molecular docking studies. As there was no obvious change in BMS compound 1 affinity or cooperativity values at these two mutants, further n numbers were not pursued.
2.5. Materials
Pfizer compound 1 (>95% purity via HPLC), ATF936 (98.6% purity via HPLC), and ronacaleret (>95.6% purity via HPLC) were synthesised by Institut de Recherches Servier (France) using published methods (Li, et al., 2013; Smithkline Beecham Corporation, 2002; Widler et al., 2010). BMS compound 1 (>95% purity via HPLC) was synthesised by SYNthesis Med Chem Research PTY (Parkville, Australia) or in house as described previously (Yang et al., 2009). The Flp‐In™ TREX™ HEK 293 cells (RRID:CVCL_U427) were purchased from Invitrogen (Carlsbad, USA) and are not listed as commonly misidentified cell lines by ICLAC. Fluo‐4‐AM and Fluo‐8‐AM (acetoxymethyl ester) were purchased from Molecular Devices (San Jose, USA) and Abcam (Cambridge, USA), respectively, and ionomycin from Cayman Chemicals (Michigan, USA). DMEM (high glucose), poly‐D‐lysine, hygromycin B, blasticidin HCl, tetracycline, and all other reagents were purchased from Sigma‐Aldrich (St. Louis, USA).
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Rationale for NAMs and mutations studied
Four chemically and structurally distinct NAMs were selected for inclusion in the study (Figure 1). Ronacaleret is an indanylpropyl amine with a two‐carbon linker separating the indanyl motif and the secondary amine that is charged at physiological pH. Ronacaleret is structurally related to the prototypical NAM, NPS2143, the latter being a naphthylpropyl amine with a two‐carbon linker separating the naphthyl motif and the protonated secondary amine. Due to their structural similarities, we hypothesised that ronacaleret would bind in a similar manner to our previously predicted NPS2143 binding pose (Leach et al., 2016). ATF936 belongs to the quinazolinone chemical class, is uncharged at physiological pH, and was previously proposed to bind in a distinct manner to NPS2143 (Widler et al., 2010). BMS compound 1 is a decorated nicotinamide analogue, which has the largest predicted topographical polar surface area due to the presence of multiple oxygen and nitrogen atoms, and is predicted to be uncharged at physiological pH. BMS compound 1 has the greatest steric bulk of the selected CaS receptor NAMs, and it has previously been hypothesised to bind to a site distinct from that for NPS2143 (Hu et al., 2006). Pfizer compound 1 is a pyridopyrimidinone and contains a phenolic moiety that is predicted to be predominantly negatively charged at physiological pH due to extensive resonance stabilisation. Pfizer compound 1 has not previously been studied in terms of the effect of 7TM mutations. Mutations were selected based on those previously shown to alter CaS receptor NAM binding and/or activity and predicted to line the extended 7TM cavity (Bu et al., 2008; Leach et al., 2016; Miedlich et al., 2004; Petrel et al., 2003; Petrel et al., 2004; Widler et al., 2010). All mutants studied here are expressed at the cell surface and are functional with respect to their ability to respond to Ca2+ o or PAMs (Table S1; Leach et al., 2016). Thus, these mutations do not alter CaS receptor structure, expression, or stability such that the receptor is deemed non‐functional.
3.2. CaS receptor NAMs bind with different affinities and cooperativities at the CaS receptor
With the exception of our study that identified the NPS2143 binding site (Leach et al., 2016), previous mutagenesis‐based structure–function studies and prior CaS receptor NAM discovery have almost exclusively relied on measurements of NAM inhibition of a single Ca2+ o concentration to determine NAM potency (IC50). This is because no commercial radioligands are available for the CaS receptor, preventing estimation of affinity via radioligand binding assays. Given that potency is a composite value of allosteric modulator affinity, cooperativity (magnitude and direction of modulation between the modulator and agonist), and efficacy (modulator agonism or inverse agonism), potency alone cannot distinguish mutation effects or SAR on modulator affinity, cooperativity, or efficacy and is therefore a misleading parameter when quantifying NAM pharmacology. Therefore, we first sought to quantify the actions of each NAM (ronacaleret, ATF936, Pfizer compound 1, and BMS compound 1) at the WT CaS receptor. To do so, we tested CaS receptor‐mediated Ca2+ i mobilisation and analysed allosteric interactions between NAMs and Ca2+ o with an operational model of allosterism (Equation 1) to quantify the modulator functional affinity (referred to here as pK B) and cooperativity (αβ) (Table 1). We did not detect NAM‐mediated agonism or inverse agonism in the Ca2+ i mobilisation assay, indicating that the NAMs had no intrinsic efficacy (denoted τB in the operational model of allosterism and constrained to 0 in our analysis).
Table 1.
Functional affinity (pK B) and cooperativity (Logαβ) parameters for CaS receptor modulators determined in Ca2+ i mobilisation assays, as described
| Ronacaleret | ATF936 | Pfizer compound 1 | BMS compound 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pK B | Logαβ (αβ) | n | pK B | Logαβ (αβ) | n | pK B | Logαβ (αβ) | n | pK B | Logαβ (αβ) | n | |
| WT | 6.35 ± 0.09 | −1.56 ± 0.16 (0.03) | 6 | 7.59 ± 0.10 | −2.30 ± 0.15 (0.005) | 6 | 7.82 ± 0.14 | −2.03 ± 0.13 (0.009) | 5 | 6.97 ± 0.07 | −1.47 ± 0.05 (0.03) | 12 |
| F6682.56A | NA | NA | 4 | 7.76 ± 0.09 | −2.37 ± 0.16 (0.004) | 5 | 7.35 ± 0.14 | −1.34 ± 0.13* (0.05) | 5 | 7.26 ± 0.12 | −2.08 ± 0.13* (0.008) | 5 |
| G670ECL1A | 6.90 ± 0.08* | −1.29 ± 0.05 (0.05) | 5 | 8.21 ± 0.05* | −2.61 ± 0.08 (0.002) | 5 | 8.27 ± 0.05 | −2.17 ± 0.05 (0.007) | 5 | 6.87 ± 0.09 | −1.58 ± 0.08 (0.03) | 5 |
| R6803.32A | NA | NA | 3 | 7.56 ± 0.10 | −1.90 ± 0.13 (0.01) | 5 | 6.67 ± 0.19* | −0.97 ± 0.10* (0.11) | 5 | 6.73 ± 0.10 | −1.12 ± 0.07 (0.08) | 5 |
| F6843.36A | NA | NA | 3 | NA | NA | 3 | NA | NA | 4 | 6.70 ± 0.10 | −2.02 ± 0.32* (0.009) | 5 |
| F6883.40A | NA | NA | 3 | NA | NA | 3 | NA | NA | 4 | 6.70 ± 0.10 | −1.92 ± 0.27 (0.01) | 5 |
| E767ECL2A | 6.43 ± 0.12 | −1.17 ± 0.11 (0.07) | 5 | 8.24 ± 0.09* | −3.35 ± 0.27* (0.0004) | 5 | 8.33 ± 0.09* | −2.55 ± 0.10* (0.003) | 5 | 7.88 ± 0.11* | −2.32 ± 0.12* (0.005) | 5 |
| W8186.50A | 7.94 ± 0.07* | −1.96 ± 0.06 (0.01) | 5 | 5.78 ± 0.26* | −0.52 ± 0.12* (0.30) | 5 | 7.39 ± 0.07 | −2.29 ± 0.13 (0.005) | 5 | 7.32 ± 0.12 | −1.12 ± 0.07 (0.08) | 5 |
| F8216.53A | NA | NA | 4 | NA | NA | 4 | NA | NA | 4 | NA | NA | 4 |
| Y8256.56A | NA | NA | 3 | 7.18 ± 0.10 | −1.90 ± 0.14 (0.01) | 5 | 7.91 ± 0.07 | ~ − 100 | 5 | 6.82 ± 0.09 | −1.93 ± 0.15 (0.01) | 5 |
| E8377.32I | NA | NA | 3 | 7.97 ± 0.09 | −2.18 ± 0.12 (0.007) | 5 | 6.44 ± 0.12* | −1.17 ± 0.09* (0.07) | 5 | 6.06 ± 0.11* | −1.31 ± 0.20 (0.05) | 5 |
| I8417.36A | NA | NA | 4 | 6.64 ± 0.11* | −1.24 ± 0.11* (0.06) | 5 | 6.62 ± 0.15* | −0.88 ± 0.07* (0.13) | 5 | 6.50 ± 0.13* | −1.37 ± 0.11 (0.04) | 6 |
Note. Data are mean ± SEM from the indicated number of independent experiments (n). NA denotes not determined due to no or weak modulator activity.
P < .05, significantly different from WT; one‐way ANOVA with Dunnett's multiple comparisons post test.
NAM affinities at the WT CaS receptor varied significantly (one‐way ANOVA with Tukey's multiple comparisons test) by up to 30‐fold, with a rank pK B order of Pfizer compound 1 = ATF936 > BMS compound 1 > ronacaleret (Table 1). Similarly, there was up to a sixfold significant difference in NAM cooperativity, with a rank Logαβ order of ATF936 = Pfizer compound 1 > ronacaleret = BMS compound 1 (Table 1). Therefore, even at equal receptor occupancy, individual NAMs would have different capacities to inhibit the responsiveness of the CaS receptor to Ca2+ o.
3.3. 7TM residues differentially contribute to NAM affinity in a chemotype‐specific manner
To identify residues essential for the binding of the four different NAM chemotypes, we next investigated the effects of amino acid substitutions on NAM functional affinity. Substitutions that altered modulator affinity were defined as those that caused a significant pK B change in comparison to the WT receptor (Table 1). NAM effects on Ca2+ o concentration response curves are shown in Figure 2. Changes in NAM affinity and cooperativity are summarised in Figure 3. Based on these criteria, we identified two key amino acid residues that, when mutated, either completely abolished NAM activity or reduced the affinity of all four NAM chemotypes: Phe8216.53 and Ile8417.36. Our previous work has shown that Ala substitution of these residues is unlikely to have a deleterious effect on overall receptor stability and structure, as Phe8216.53Ala and Ile8417.36Ala mutant receptors are expressed at the cell surface at levels comparable to the WT CaS receptor and they retain sensitivity to Ca2+ o (Table S1) and the CaS receptor PAM, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=3947 (Leach et al., 2016). This demonstrates that all four NAM chemotypes are sensitive to mutations in the 7TM cavity.
Figure 2.
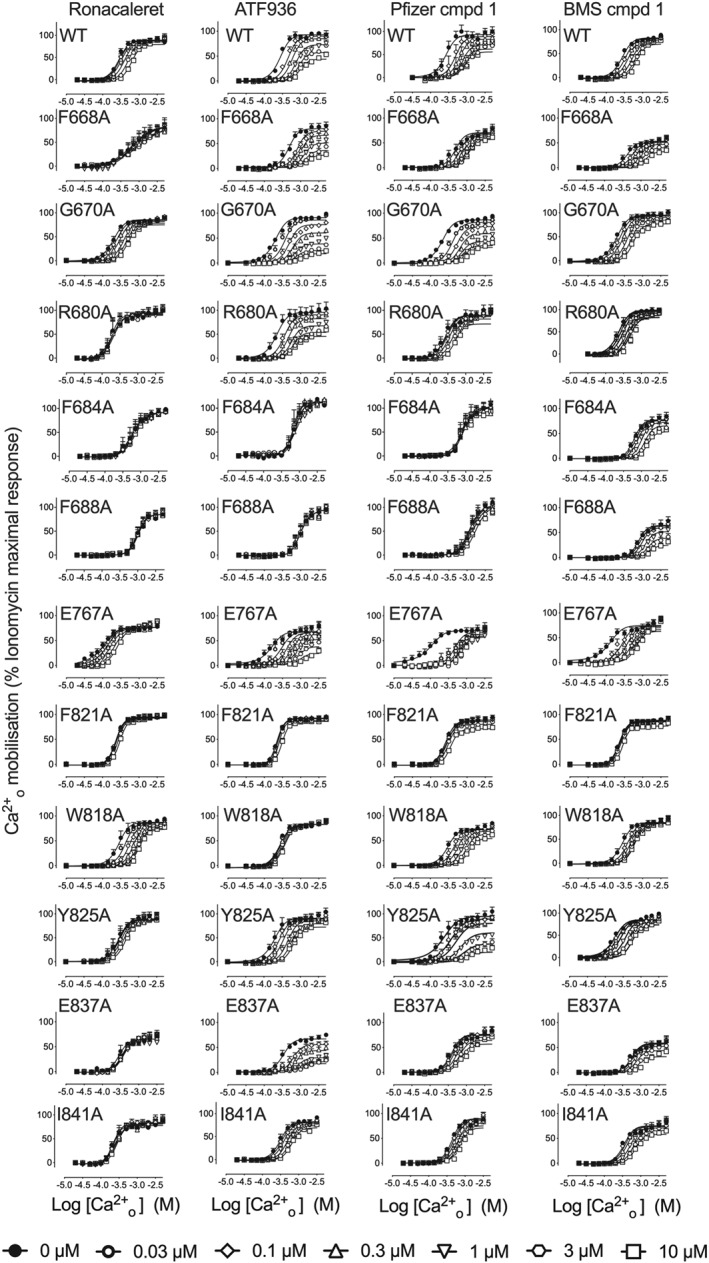
7TM mutations alter the activity of CaS receptor NAMs. Concentration‐response curves to Ca2+ o in the absence and the presence of CaS receptor NAMs were determined in Ca2+ i mobilisation assays to identify mutations that altered NAM activity. Data are mean + SEM pooled from the number of independent experiments stated in Table 1 performed in duplicate. Only data where an n of five independent experiments were performed were used for statistical analysis. Curves are the best fit of Equation (1) to the data. Vehicle (buffer) has been plotted as the lowest [Ca2+ o] in accordance with the logarithmic scale
Figure 3.
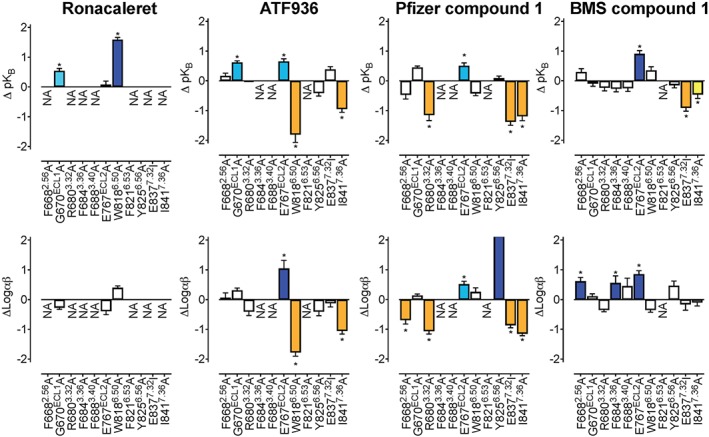
7TM and ECL mutations alter the affinity and cooperativity of CaS receptor NAMs. An operational model of allosterism (Equation 1) was fitted to concentration–response curves to Ca2+ o in the absence and the presence of each NAM determined in Ca2+ i mobilisation assays 1 to determine the change in NAM affinity (ΔpK B) and cooperativity (ΔLogαβ) at mutant CaS receptors in comparison to the WT receptor. White bars represent no significant change in pK B or Logαβ. Coloured bars that sit above and below zero represent a greater than twofold increase (light teal), greater than fivefold increase (dark blue), greater than twofold decrease (yellow), or greater than fivefold decrease (orange) significant change in pK B or Logαβ, respectively. Data are the ΔpK B or ΔLogαβ + SEM calculated from WT and mutant pK B or Logαβ values and experiment numbers shown in Table 1. *P < .05, significantly different from WT; one way ANOVA with Dunnett's multiple comparisons post test. NA refers to values that are not applicable or could not be accurately determined. For Y825A treated with Pfizer compound 1, the increase in Logαβ was too great to be accurately determined
We also identified a number of residues that when mutated differentially perturbed the binding of NAMs. For instance, Ile substitution of Glu8377.32, which is predicted to form a key salt bridge with the prototypical NAM, NPS2143, abolished the activity of ronacaleret and reduced the affinity of Pfizer compound 1 and BMS compound 1. However, this mutation had no effect on ATF936 affinity (Table 1). Ala substitution of Phe6843.36 and Phe6883.40 abolished the activity of ronacaleret, Pfizer compound 1, and ATF936, but had no effect on BMS compound 1 affinity. Phe6682.56Ala abolished the binding of ronacaleret, but had no effect on ATF936, Pfizer compound 1, or BMS compound 1. Trp8186.50Ala decreased ATF936 affinity, increased ronacaleret affinity, and had no effect on Pfizer compound 1 or BMS compound 1 affinity, whereas Glu767ECL2 increased ATF936, Pfizer compound 1 and BMS compound 1 affinity but was without effect on ronacaleret. Other examples of differential effects of mutations can be seen in Table 1 and Figure 2. Together, these findings suggest that the four NAM chemotypes form distinct contacts or bind to different binding sites in the 7TM cavity of the CaS receptor.
As we only identified one amino acid mutation that abolished BMS compound 1 activity (Phe8216.53Ala, which globally abolished all NAM activity) and only one other mutation (Glu8377.32Ile) that reduced BMS compound 1 affinity more than threefold, we examined the activity of BMS compound 1 at an N terminally truncated CaS receptor composed of amino acids 599–903 to confirm that the BMS compound 1 binding site was not located in the CaS receptor N terminal domain. Because Ca2+ o has reduced activity at this N terminally truncated receptor (Gregory et al., 2018), we performed interaction studies between the ago‐PAM, AC265347, and the NAM, BMS compound 1. These experiments demonstrated that BMS compound 1 retained activity at the N terminally truncated CaS receptor, where it caused a rightward shift in the AC265347 concentration–response curves (Figure S1). Thus, the 7TM domain accommodates the binding of BMS compound 1 to the CaS receptor.
3.4. Different NAM chemotypes utilise distinct residues to transmit negative cooperativity
We next evaluated the effect of mutations in the 7TM or ECL domains of the CaS receptor on cooperativity between each NAM chemotype and Ca2+ o. Similar to our earlier findings with NPS2143, we did not identify any mutations that altered the cooperativity between Ca2+ o and ronacaleret (Table 1), although seven of the 10 mutations abolished ronacaleret activity, preventing quantification of ronacaleret cooperativity with Ca2+ o at these mutants. In contrast, several residues that reduced or increased affinity for ATF936, Pfizer compound 1, or BMS compound 1 also reduced or increased their cooperativity. Thus, Glu767ECL2Ala increased negative cooperativity of ATF936, Pfizer compound 1, and BMS compound 1 with Ca2+ o, whereas cooperativity with Ca2+ o was reduced for ATF936 at Trp8186.50Ala, Pfizer compound 1 at Glu8377.32Ile, and was reduced for ATF936 and Pfizer compound 1 at Ile8417.36Ala (Table 1). In contrast, several mutations reduced or increased NAM cooperativity without altering NAM affinity, such as Phe6683.40Ala, which increased Pfizer compound 1 and decreased BMS compound 1 cooperativities with Ca2+ o. Collectively, these findings suggest that cooperativity is not exclusively linked to residues that contribute to the binding of NAMs and that both shared and distinct residues transmit cooperativity mediated by different NAM chemotypes.
3.5. Delineation of NAM binding sites by combining structure–function insights with structure activity relationships (SAR)
To better understand the effects of mutations on NAM affinity and cooperativity, we docked the four NAM chemotypes into a homology model of the 7TM domain of the CaS receptor, using results from our mutagenesis as a guide to predict the likely binding pocket and pose (see Section 2, Figures 4 and 5). Poses shown in Figures 4 and 5 are those that, upon manual inspection, were the most compatible with mutagenesis and SAR data. However, a representative alternative pose for each NAM is shown in Figure S2. Although less favourable based on lower docking scores and SAR data, alternative poses shown in Figure S2 are representative of the most distinct pose to our proposed pose, while still being compatible with mutagenesis data based on predicted binding proximity to residues that contribute to NAM affinity.
Figure 4.
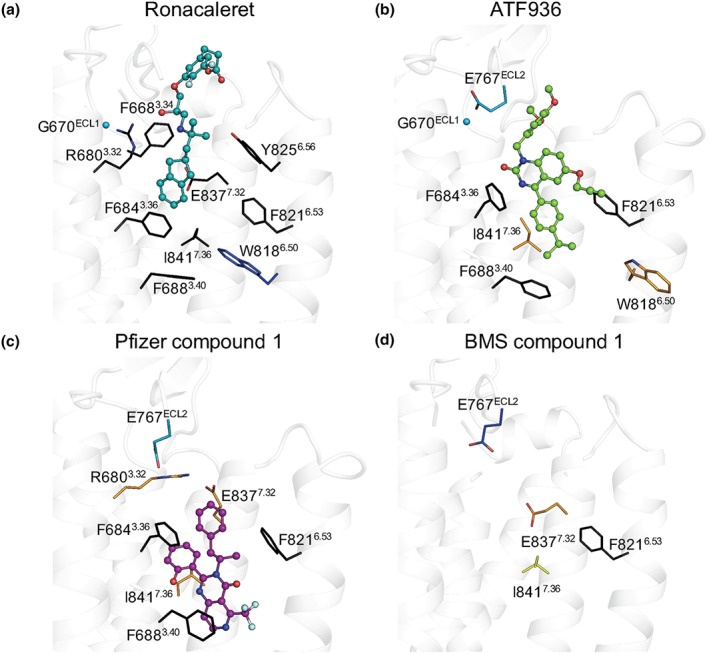
Mutagenesis and computational modelling predict CaS receptor NAMs bind in overlapping but disparate binding orientations within the 7TM domain. (a) Ronacaleret, (b) ATF936, and (c) Pfizer compound 1 are shown in sticks and balls docked in the 7TM domain (white helical ribbons). Mutated residues are shown as sticks and are coloured according to a greater than twofold increase (light teal), greater than fivefold increase (dark blue), greater than twofold decrease (yellow), or greater than fivefold decrease (orange) in binding affinity. When affinity could not be determined (i.e. a complete abolishment of NAM activity), residues are shown in black. Salt bridges are shown as red dashes, and Gly670ECL1 is shown as a sphere. While residues that contribute to BMS compound 1 affinity are shown (d), BMS compound 1 is not docked in the 7TM because predicted docking poses were not compatible with the mutagenesis or SAR data
Figure 5.
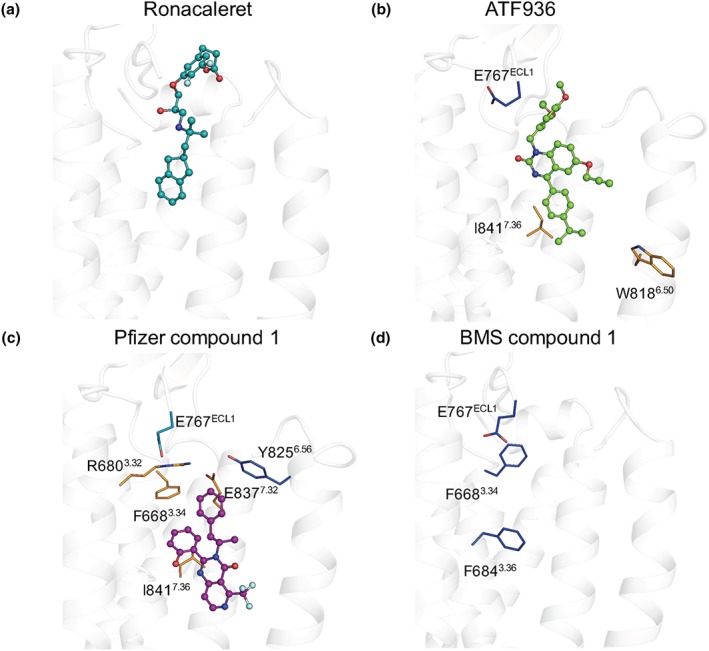
Mutagenesis and computational modelling reveal different residues contribute to NAM cooperativity. (a) Ronacaleret, (b) ATF936, and (c) Pfizer compound 1 are shown in sticks and balls docked in the 7TM domain (white helical ribbons). Mutated residues are shown as sticks and are coloured according to a greater than twofold increase (light teal), greater than fivefold increase (dark blue), or greater than fivefold decrease (orange) in cooperativity. While residues that contribute to BMS compound 1 affinity are shown (d), BMS compound 1 is not docked in the 7TM because predicted docking poses were not compatible with the mutagenesis or SAR data
For ronacaleret, all 10 top scoring poses were located in a similar position due to the docking constraint applied to maintain ronacaleret proximity to Glu8377.32. Only the substituted phenylpropanoate adopted significantly different positions, as shown in Figure S2. Due to the substituted phenylpropanoate, ronacaleret has a more extended conformation. Thus, the most likely pose was one where the phenylpropanoate extends upwards towards the ECLs, much like the substituted phenyl in NPS2143. Thus, unsurprisingly, the predicted pose for ronacaleret (Figures 4a and 5a) was similar to that predicted for NPS2143 (Leach et al., 2016). The hydrophobic indanyl extends into a hydrophobic–aromatic pocket formed by Phe6883.40, Phe8216.53, Phe6843.36, and Ile8417.36 in a manner similar to the position occupied by the naphthyl group of NPS2143 (Leach et al., 2016).
Our top 10 predicted docking poses for ATF936 were similar in location and orientation (see Figure S2). Our chosen docking pose for ATF936 (Figures 4 and 5) suggests this compound may sit slightly lower than ronacaleret, with the isopropylphenyl motif oriented towards the hydrophobic–aromatic pocket formed by Phe6883.40, Phe8216.53, Phe6843.36, or Ile8417.36 (Figure 4b). Even the pose that was most distinct from that shown in Figures 4 and 5, which sat slightly higher in the binding pocket, did not extend towards the ECLs as far as ronacaleret, and reached deeper into the 7TM (Figure S2). Our proposed ATF936 pose is consistent with our mutagenesis data demonstrating a loss of ATF936 activity or a reduction in ATF936 affinity upon mutation of Phe6883.40, Phe8216.53, Phe6843.36, or Ile8417.36. The predicted ATF936 pose is also consistent with SAR data demonstrating that ATF936 NAM activity is dependent upon the presence of a number of refined substituents at pivotal positions around a highly functionalised quinazolinone, with large variability only allowable at the N1 position. For example, replacement of the isopropyl substituent attached to the phenyl group with hydrocarbon isosteres such as ethyl, cyclopropyl, and tert‐butyl were tolerated. However, the incorporation of an additional phenyl ring exhibited a modest reduction in compound potency. Introduction of functional groups able to participate in hydrogen bonding (ether and ester) and electrostatic interactions (carboxylic acid) resulted in 21‐fold or greater potency reduction (Widler et al., 2010). These findings are consistent with these substituents being less favourable for binding into the hydrophobic–aromatic pocket. The 3‐ethoxy‐4‐methoxylphenyl motif is hence positioned towards the ECLs (Figure 4b), consistent with published SAR data demonstrating a diverse range of truncated and substituted alkyl and benzylic substituents are well tolerated at this position (Widler et al., 2010). The minimal change in NAM potency upon removal or extension of substituents at the N1 position suggest that this portion of ATF936 does not form major binding interactions with the receptor. Further, our predicted ATF936 pose suggests Glu767ECL2 faces away from the ethoxy–methoxyphenyl motif. This is consistent with the ethoxy‐methoxyphenyl motif being ring activated and possessing a slight negative charge that would repel the γ‐carboxylate of Glu767ECL2. Finally, the ATF936 propargyl group, which increased potency, is predicted to be located in a hydrophobic pocket containing Phe8216.53, consistent with SAR data suggesting a preference for smaller hydrophobic substituents at this position that are less likely to impede access to the pocket.
Pfizer compound 1 docking studies produced multiple distinct poses that were consistent with our mutagenesis characterisation of the 7TM binding site. However, one pose produced significantly better docking and energy scores and was consistent with published SAR data (Figure 4c). The phenol moiety was positioned between Ile8417.36 and Phe6843.36, consistent with a reduction in affinity or complete loss of Pfizer compound 1 activity upon substitution of these residues (Table 1, Figure 5c). The phenolic group could facilitate a possible hydrogen bond interaction with the receptor backbone, consistent with published SAR that showed a fivefold reduction in modulator potency when the phenol moiety was substituted for a tolyl group. The pyrimidin‐4(3H)‐one core sat between Phe6843.36, Phe8216.53, and Ile8417.36 (Figure 4c), residues shown to be important for activity or affinity (Table 1, Figure 5c). This places the trifluoromethyl group attached to the fused pyrido ring in close proximity to TM6, consistent with limited tolerance for replacement of this moiety with larger substituents, which would prevent the access of Pfizer compound 1 to the well‐recognised cavity formed by TM 3, 5, and 6. Finally, the phenylpropanyl moiety is predicted to sit adjacent to Glu8377.32 and Arg6803.32, two residues important for the affinity of Pfizer compound 1. Although a number of alternative docking poses for Pfizer compound 1 were predicted, which were higher in the 7TM binding cavity and extended closer towards the ECLs (Figure S2), these poses were less compatible with mutagenesis data because they positioned Pfizer compound 1 further away from residues that contribute to its affinity.
Our modelling attempts for BMS compound 1 based on our initial mutagenesis predicted a potential binding mode towards the top of the 7TM bundle, similar to the position occupied by ronacaleret (Figure S2). However, preliminary data indicated that substitution of ECL3 residues (Val833 and Ser834), which from this initial pose were predicted to interact with BMS compound 1, were not important for activity (Table S2). Indeed, residues such as Tyr8256.56, implicated by this pose to form direct polar interactions, were also not important for the affinity or cooperativity of BMS compound 1 (Table 1, Figure 5d). Only Phe8216.53 abolished BMS compound 1 activity, but this residue was not predicted to form a direct contact with the compound in our model and was shown to be important for NAM activity generally (Figure S2). Collectively, the mutational data do not support the pose predicted for BMS compound 1, which is consistent with previous work indicating that BMS compound 1 and an NPS2143 analogue allosterically interact when occupying the CaS receptor (Arey et al., 2005). Therefore, our data confirms that BMS compound 1 is likely to use an alternative allosteric binding site for its activity as a NAM for the CaS receptor (Figure 6).
Figure 6.
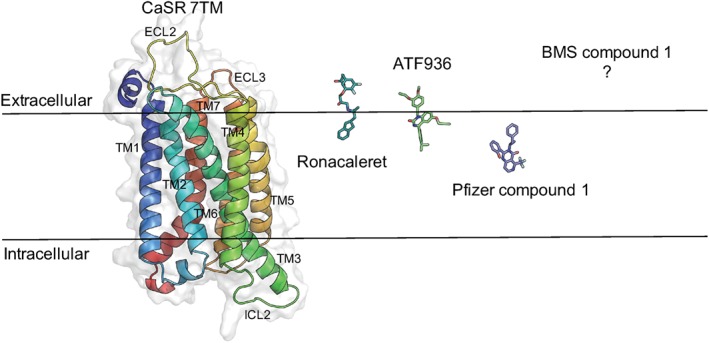
Different NAM binding sites within the 7TM domain of the CaS receptor. Ronacaleret, ATF936, and Pfizer compound 1 predicted binding poses in our homology model. The black line represents the boundary of the lipid bilayer, and the 7TM domain is shown as ribbons, and NAMs are represented as stick structures
4. DISCUSSION AND CONCLUSIONS
The current study has probed the function and binding of four structurally distinct NAMs within the 7TM and/or ECL domains of the CaS receptor. Quantitation of NAM effects at the WT CaS receptor reveals substantial differences in the affinity of the different NAMs and their ability to modulate CaS receptor activity. Further, mutagenesis, combined with analytical pharmacology and computational modelling, predicts that CaS receptor NAMs from diverse chemotypes occupy distinct binding sites (Figure 6), which may in part reflect differences in the ability of the NAMs to inhibit CaS receptor signalling.
We first fully characterised NAM affinity and cooperativity at the WT CaS receptor and demonstrated disparate affinities and cooperativities between the four NAMs. For instance, ronacaleret affinity is 20‐ to 30‐fold lower than that of the highest affinity NAMs, ATF936 and Pfizer compound 1, and ronacaleret also had a sixfold weaker cooperativity than ATF936. Similarly, ronacaleret and BMS compound 1 cooperativities are threefold to sixfold lower than Pfizer compound 1 and ATF936, respectively. Although the failure of ronacaleret in the clinic has been attributed to its poor pharmacokinetic profile, these data suggest that its affinity and cooperativity also distinguish ronacaleret (and indeed BMS compound 1) from other CaS receptor NAMs. These findings highlight the importance of quantifying affinity and cooperativity, rather than relying on modulator potency values, as these parameters may have important implications for putative NAM clinical efficacy.
The distinction between affinity and cooperativity facilitates identification of residues important for modulator binding. This is invaluable when interpreting SAR or molecular modelling studies, whereby CaS receptor residues important for modulator cooperativity are not necessarily the same as those that contribute to modulator affinity (Gregory et al., 2018; Keller et al., 2018; Leach et al., 2016). Thus, we probed the effects of 7TM and ECL amino acid mutations on NAM affinity and cooperativity. Our mutagenesis data suggest that all NAMs tested are sensitive to mutations in these receptor regions. For instance, Ala substitution of Phe6843.36, Phe6883.40, Phe8216.53, and Ile8417.36, which are predicted to contribute to a hydrophobic–aromatic network of residues towards the base of an extended 7TM cavity (Leach et al., 2016), either abolished the activity or reduced the affinity of ronacaleret, ATF936, and Pfizer compound 1. Similarly, Ala substitution of Phe8216.53 abolished BMS compound 1 activity and Ala substitution of Ile8417.36 reduced affinity. However, there were a number of mutations that differentially altered the activity or binding of diverse NAMs, suggesting that structurally distinct NAMs bind, at the very least, in a disparate manner, and potentially interact with different regions of the 7TM, ECL, or even ICL regions of the CaS receptor. This may be particularly true for BMS compound 1, whose activity was only abolished by Phe8216.53Ala, a mutation that abolished the activity of all the NAMs tested herein and increased the affinity of PAMs (Keller et al., 2018; Leach et al., 2016). The opposing sensitivities of CaS receptor PAMs and NAMs to the Phe8216.53Ala mutation, together with the finding that the Phe8216.53Ala mutant retains sensitivity to Ca2+ o (Table S1; Leach et al., 2016), suggest Phe8216.53 acts as an important mediator of CaS receptor allostery per se, rather than directly interacting with PAMs and NAMs. Thus, although the NAMs may not directly contact Phe8216.53, Ala substitution of Phe8216.53 is likely to stabilise a receptor conformation that is highly unfavourable for NAM binding. Similarly, Ile8417.36Ala may stabilise a conformation that is unfavourable for binding of small molecule compounds, either via disruption of direct ligand interactions or via indirect changes in receptor regions important for modulator binding. The latter is supported by findings that BMS compound 1 affinity was only reduced more than threefold at the Phe8216.53Ala and Glu8377.32Ile mutants. Thus, it is unlikely that BMS compound 1 makes major contacts with the 7TM or ECL residues tested. While surprising, this finding is not without precedence. Our homology modelling is based on a single CaS receptor 7TM domain and, while outside the scope of this study, dimer formation is important for CaS receptor function, harbouring potential additional modulator binding sites (Gregory et al., 2018; Jacobsen, Gether, & Bräuner‐Osborne, 2017). Further, alternative allosteric sites have been reported for other class C GPCRs (Chen, Goudet, Pin, & Conn, 2008; Noetzel et al., 2013), and the interaction between BMS compound 1 and NPS2143 appears to be allosteric in nature. The latter is supported by findings that although NPS2143 and BMS compound 1 have comparable affinity (BMS compound 1 pKB 6.97 [Table 1], NPS2143 pKB 6.66; Leach et al., 2016), concentrations of NPS2143 equal to and greater than its affinity inhibited the binding of a radiolabelled NPS2143 analogue, while equivalent BMS compound 1 concentrations had no effect (Arey et al., 2005) pointing towards a neutral allosteric interaction between these two NAMs.
Docking studies have previously been employed to predict the binding site of ATF936 and BMS compound 1 (Hu et al., 2006; Widler et al., 2010). In our model, ATF936 sits within the 7TM bundle in the same orientation as in previous docking attempts, with the isopropylphenyl group pointing into the bundle and towards the hydrophobic–aromatic pocket. However, there are differences in the overall position of the previously reported pose that are likely to be the result of using the class A rhodopsin structure for homology building, rather than the class C metabotropic glutamate receptors used in this study (Widler et al., 2010). Similarly, BMS compound 1 was previously docked into a rhodopsin‐based homology model, guided by mutation‐induced changes in modulator potency (Hu et al., 2006). However, through delineation of mutation‐induced changes in NAM affinity versus cooperativity, our work shows that mutation of Ile8417.36 to Ala, previously implicated as central for BMS compound 1 activity, caused only a mild drop in modulator affinity. Thus, prior approaches based on homology modelling to the class A rhodopsin structure have been unable to predict key docking interactions. Nonetheless, our CaS receptor homology model based on metabotropic glutamate receptors is not without its own limitations. Sequence identity between the CaS receptor and metabotropic glutamate receptors 7TM is low (29% and 33% homology with https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=289 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=293, respectively). Further, although the solved mGlu structures were bound to NAMs, each structure is representative of only one possible conformation, which is not necessarily similar to the conformation stabilised by different CaS receptor NAMs.
In contrast to BMS compound 1, the activities or affinities of ronacaleret, ATF936, and Pfizer compound 1 were abolished or reduced more than 10‐fold, respectively, by between five and eight mutations that line the predicted 7TM cavity, suggesting more contacts between these NAMs and the 7TM domain. Computational docking studies with ronacaleret, ATF936, and Pfizer compound 1 predicted binding poses for these NAMs within the 7TM cavity that were consistent with our mutagenesis data as well as previous SAR. In contrast, docking studies with BMS compound 1 indicated that the lowest energy predicted binding pose was inconsistent with our mutagenesis data, suggesting once again that BMS compound 1 may not bind to the binding site predicted by computational techniques. It is interesting to note, however, that Glu767ECL2Ala increased the affinity of BMS compound 1, in addition to ATF936 and Pfizer compound 1. In our CaSR homology model, Glu767ECL2 faces Glu8377.32 at the entrance of the predicted 7TM binding cavity. Ca2+ o and the naphthylethylamine PAM, cincacalcet, and naphthylpropylamine and indanylpropylamine NAMs, NPS2143 and ronacaleret, respectively, are predicted to provide a counter‐ion that offsets the electrostatic repulsion between Glu767ECL2 and Glu8377.32. We previously speculated that the Glu767 ECL2Ala mutation stabilises an active receptor state that preferentially binds agonists and PAMs, because cinacalcet and AC265347 affinity, and Ca2+ o potency, are increased at this mutant (Leach et al., 2016). However, given that ATF936, Pfizer compound 1 and BMS compound 1 also bind with higher affinity at the E767 ECL2A mutant, an active receptor conformation is unlikely to be responsible for their increased affinity at this mutant. Rather, mutating this residue may open up the top of the pocket, making the binding cavity more accessible to PAMs and NAMs. Thus, the increase in BMS compound 1 affinity at Glu767ECL2Ala means we cannot rule out its binding at the top of the 7TM/ECL cavity.
Our previous structure–function study identified only one mutation that significantly altered negative allosteric modulation mediated by NPS2143 (Leach et al., 2016). The cooperativity of the structurally related NAM, ronacaleret, was similarly unaffected by most mutations. This suggests that unlike PAMs, which appear to favour the receptor's transition to an active receptor state via the movement of numerous amino acid side‐chains, NPS2143 and ronacaleret may obstruct this transition, by adopting a rigid binding pose that restricts movement of the 7TM domains in the CaS receptor, thus holding the receptor in an inactive state (Leach et al., 2016). In contrast, the cooperativities of ATF936, Pfizer compound 1, and BMS compound 1 with Ca2+ o were sensitive to several mutations, suggesting that these NAMs stabilise different conformations.
Drugs that target distinct binding sites in the CaS receptor may offer unique pharmacological profiles. For instance, in comparison to the arylalkylamine PAMs, cinacalcet, NPSR568 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=719, which are weakly biased towards potentiation of CaS receptor ‐mediated mobilisation of Ca2+ i compared with the phosphorylation of ERK1/2 (pERK1/2), AC265347 preferentially modulates CaS receptor‐mediated pERK1/2 compared with Ca2+ i mobilisation (Cook et al., 2015) and has greater efficacy as an agonist. We have speculated that the differences in the binding of these two classes of compounds likely enables them to stabilise distinct CaS receptor conformations, resulting in the different biased signalling profiles and in turn distinct pharmacological effects. Our current data indicating that structurally different NAMs that bind to distinct regions of the CaS receptor have variations in affinity and cooperativity, support the notion that NAMs targeted towards different binding sites may also have unique pharmacological profiles. Importantly, naturally occurring mutations located in the extended 7TM cavity could render some NAMs inactive. An example is Phe8216.53, shown here to be globally important for NAM activity, and for which a naturally occurring gain‐of‐function Leu mutation is known (Leach et al., 2012). Hence, the availability of multiple NAMs that bind to different CaS receptor binding sites could provide additional treatment options for patients with gain‐of‐function mutations that reduce the affinity and/or cooperativity of one NAM.
In conclusion, the current study provides new insights into how structurally distinct NAMs bind to the CaS receptor and mediate allosteric modulation of the endogenous agonist, Ca2+ o. This information may be used to inform the design and use of CaS receptor NAMs for the treatment of disorders of calcium metabolism and other disorders for which CaS receptor function is an important modifier.
AUTHOR CONTRIBUTIONS
T.J.M., A.N.K., E.K., A.D. and I.K. performed all experimental work. K.L., T.J.M., A.N.K. and E.K. analysed data. K.L., K.J.G., T.J.M., A.N.K., and B.C. wrote the manuscript. K.L., K.J.G., C.J.L., I.K. and A.C. conceived the studies and reviewed the manuscript draft(s).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 BMS compound 1 retains activity at an N terminally‐truncated CaS receptor . Ca2 + i mobilisation measured in response to the PAM‐agonist, AC265347, in the absence and presence of BMS compound 1. Curves are the best fit of a 3 parameter Hill equation to the data. Vehicle (buffer) has been plotted as the smallest [AC265347] in accordance with a logarithmic scale. Data are mean + SEM pooled from an n of 3 independent experiments performed in duplicate.
Figure S2 Alternative NAM docking poses. Alternative poses and associated docking scores for ronacaleret (A), ATF936 (B), and Pfizer compound 1 (C) in comparison to our proposed poses (ghosted) shown in Figures 4 and 5. Alternative poses are representative of those that are compatible with the mutagenesis and SAR data while being the most distinct alternative to our proposed pose. Also shown are two poses for BMS compound 1 (D) that were not supported by mutagenesis or SAR data. Scores for each pose were calculated as described in Totrov & Abagyan (1999), where lower scores represent lower ligand binding energies and therefore a more favourable interaction. Residues are those that alter NAM affinity as shown in Figure 4.
Table S1 WT and mutant Ca2 + o potency (pEC50), maximal response (Emax) and Hillslope determined in Ca2 + i mobilisation assays, and previously published mutant cell surface expression levels determined in FACS experiments. Data are mean ± s.e.m from the indicated number of experiments (n).
Table S2 Functional affinity (pKB) and cooperativity (Logαβ) parameters for BMS compound 1 at WT and ECL3 mutants determined in Ca2 + i mobilisation assays as described in the methods section. Data are mean ± s.e.m from the indicated number of independent experiments (n).
ACKNOWLEDGEMENTS
This work was supported by National Health and Medical Research Council of Australia (NHMRC) Project Grants APP1085143 and APP1138891, Australian Research Council Discovery Project Grant DP170104228, the National Institute of Health General Medical Sciences Grant GM117424, and the National Institute of Health Allergy and Infectious Diseases Grant AI118985.
Josephs TM, Keller AN, Khajehali E, et al. Negative allosteric modulators of the human calcium‐sensing receptor bind to overlapping and distinct sites within the 7‐transmembrane domain. Br J Pharmacol. 2020;177:1917–1930. 10.1111/bph.14961
Tracy M. Josephs and Andrew N. Keller contributed equally to this work.
REFERENCES
- Abagyan, R. , & Totrov, M. (1994). Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. Journal of Molecular Biology, 235, 983–1002. 10.1006/jmbi.1994.1052 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , … CGTP Collaborators (2019). The concise guide to pharmacology 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 174, S21–S141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. T. , Hunter, T. , Walter, S. , Dong, J. , Maclean, D. , Baruch, A. , … Tomlinson, J. E. (2015). Critical cysteine residues in both the calcium‐sensing receptor and the allosteric activator AMG 416 underlie the mechanism of action. Molecular Pharmacology, 88, 853–865. 10.1124/mol.115.098392 [DOI] [PubMed] [Google Scholar]
- Arey, B. J. , Seethala, R. , Ma, Z. , Fura, A. , Morin, J. , Swartz, J. , … Feyen, J. H. (2005). A novel calcium‐sensing receptor antagonist transiently stimulates parathyroid hormone secretion in vivo. Endocrinology, 146, 2015–2022. 10.1210/en.2004-1318 [DOI] [PubMed] [Google Scholar]
- Bikle D, Bräuner‐Osborne H, Brown EM, Chang W, Conigrave A, Hannan F, … Yarova, P. (2019). Calcium‐sensing receptor (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide to Pharmacology CITE 2019(4).
- Bottegoni, G. , Kufareva, I. , Totrov, M. , & Abagyan, R. (2008). A new method for ligand docking to flexible receptors by dual alanine scanning and refinement (SCARE). Journal of Computer‐Aided Molecular Design, 22, 311–325. 10.1007/s10822-008-9188-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu, L. , Michino, M. , Wolf, R. M. , & Brooks, C. L. 3rd (2008). Improved model building and assessment of the calcium‐sensing receptor transmembrane domain. Proteins, 71, 215–226. 10.1002/prot.21685 [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Goudet, C. , Pin, J. P. , & Conn, P. J. (2008). N‐{4‐Chloro‐2‐[(1,3‐dioxo‐1,3‐dihydro‐2H‐isoindol‐2‐yl)methyl]phenyl}‐2‐hydroxybenzamide (CPPHA) acts through a novel site as a positive allosteric modulator of group 1 metabotropic glutamate receptors. Molecular Pharmacology, 73, 909–918. 10.1124/mol.107.040097 [DOI] [PubMed] [Google Scholar]
- Christopher, J. A. , Aves, S. J. , Bennett, K. A. , Dore, A. S. , Errey, J. C. , Jazayeri, A. , … Congreve, M. (2015). Fragment and structure‐based drug discovery for a class C GPCR: Discovery of the mGlu5 negative allosteric modulator HTL14242 (3‐chloro‐5‐[6‐(5‐fluoropyridin‐2‐yl)pyrimidin‐4‐yl]benzonitrile). Journal of Medicinal Chemistry, 58, 6653–6664. 10.1021/acs.jmedchem.5b00892 [DOI] [PubMed] [Google Scholar]
- Christopoulos, A. (1998). Assessing the distribution of parameters in models of ligand‐receptor interaction: To log or not to log. Trends in Pharmacological Sciences, 19, 351–357. 10.1016/s0165-6147(98)01240-1 [DOI] [PubMed] [Google Scholar]
- Conigrave, A. D. (2012). Regulation of calcium and phosphate metabolism In Diseases of the parathyroid glands (pp. 13–51). New York, New York, Dordrecht, Heidelberg: Springer. [Google Scholar]
- Conigrave, A. D. , & Hampson, D. R. (2010). Broad‐spectrum amino acid‐sensing class C G‐protein coupled receptors: Molecular mechanisms, physiological significance and options for drug development. Pharmacology and Therapeutics, 127, 252–260. 10.1016/j.pharmthera.2010.04.007 [DOI] [PubMed] [Google Scholar]
- Cook, A. E. , Mistry, S. N. , Gregory, K. J. , Furness, S. G. , Sexton, P. M. , Scammells, P. J. , … Leach, K. (2015). Biased allosteric modulation at the CaSR engendered by structurally diverse calcimimetics. British Journal of Pharmacology, 172, 185–200. 10.1111/bph.12937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosman, F. , Gilchrist, N. , McClung, M. , Foldes, J. , de Villiers, T. , Santora, A. , … Denker, A. E. (2016). A phase 2 study of MK‐5442, a calcium‐sensing receptor antagonist, in postmenopausal women with osteoporosis after long‐term use of oral bisphosphonates. Osteoporosis International, 27, 377–386. 10.1007/s00198-015-3392-7 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey, A. E. , Leach, K. , Valant, C. , Conigrave, A. D. , Sexton, P. M. , & Christopoulos, A. (2012). Positive and negative allosteric modulators promote biased signaling at the calcium‐sensing receptor. Endocrinology, 153, 1232–1241. 10.1210/en.2011-1426 [DOI] [PubMed] [Google Scholar]
- Dore, A. S. , Okrasa, K. , Patel, J. C. , Serrano‐Vega, M. , Bennett, K. , Cooke, R. M. , … Marshall, F. H. (2014). Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature, 511, 557–562. 10.1038/nature13396 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, L. A. , Dabrowski, C. E. , Cicconetti, G. , Gordon, D. N. , Fuerst, T. , Engelke, K. , & Genant, H. K. (2012). Ronacaleret, a calcium‐sensing receptor antagonist, increases trabecular but not cortical bone in postmenopausal women. Journal of Bone and Mineral Research, 27, 255–262. 10.1002/jbmr.554 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, L. A. , Dabrowski, C. E. , Cicconetti, G. , Gordon, D. N. , Papapoulos, S. , Bone, H. G. 3rd , & Bilezikian, J. P. (2011). The effects of ronacaleret, a calcium‐sensing receptor antagonist, on bone mineral density and biochemical markers of bone turnover in postmenopausal women with low bone mineral density. The Journal of Clinical Endocrinology and Metabolism, 96, 2441–2449. 10.1210/jc.2010-2855 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, L. A. , Smith, P. L. , McBride, T. A. , Fries, M. A. , Hossain, M. , Dabrowski, C. E. , & Gordon, D. N. (2011). Ronacaleret, a calcium‐sensing receptor antagonist, has no significant effect on radial fracture healing time: Results of a randomized, double‐blinded, placebo‐controlled Phase II clinical trial. Bone, 49, 845–852. 10.1016/j.bone.2011.06.017 [DOI] [PubMed] [Google Scholar]
- Geng, Y. , Mosyak, L. , Kurinov, I. , Zuo, H. , Sturchler, E. , Cheng, T. C. , … Fan, Q. R. (2016). Structural mechanism of ligand activation in human calcium‐sensing receptor. eLife, 5, E13662 10.7554/eLife.13662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowen, M. , Stroup, G. B. , Dodds, R. A. , James, I. E. , Votta, B. J. , Smith, B. R. , … Fox, J. (2000). Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. Journal of Clinical Investigation, 105, 1595–1604. 10.1172/JCI9038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, K. J. , Giraldo, J. , Daio, J. , Christopoulos, A. , & Leach, K. (2019). Evaluation of operational models of agonism and allosterism at receptors with multiple orthosteric binding sites. Molecular Pharmacology, 97, 35–45. 10.1124/mol.119.118091 [DOI] [PubMed] [Google Scholar]
- Gregory, K. J. , Kufareva, I. , Keller, A. N. , Khajehali, E. , Mun, H. C. , Goolam, M. A. , … Leach, K. (2018). Dual action calcium‐sensing receptor modulator unmasks novel mode‐switching mechanism. ACS Pharmacology and Translational Science, 1, 96–109. 10.1021/acsptsci.8b00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halse, J. , Greenspan, S. , Cosman, F. , Ellis, G. , Santora, A. , Leung, A. , … Denker, A. E. (2014). A phase 2, randomized, placebo‐controlled, dose‐ranging study of the calcium‐sensing receptor antagonist MK‐5442 in the treatment of postmenopausal women with osteoporosis. The Journal of Clinical Endocrinology and Metabolism, 99, E2207–E2215. 10.1210/jc.2013-4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Jiang, J. , Costanzi, S. , Thomas, C. , Yang, W. , Feyen, J. H. , … Spiegel, A. M. (2006). A missense mutation in the seven‐transmembrane domain of the human Ca2+ receptor converts a negative allosteric modulator into a positive allosteric modulator. The Journal of Biological Chemistry, 281, 21558–21565. 10.1074/jbc.M603682200 [DOI] [PubMed] [Google Scholar]
- Jacobsen, S. E. , Gether, U. , & Bräuner‐Osborne, H. (2017). Investigating the molecular mechanism of positive and negative allosteric modulators in the calcium‐sensing receptor dimer. Scientific Reports, 7, 46355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John, M. R. , Harfst, E. , Loeffler, J. , Belleli, R. , Mason, J. , Bruin, G. J. , … Kneissel, M. (2014). AXT914 a novel, orally‐active parathyroid hormone‐releasing drug in two early studies of healthy volunteers and postmenopausal women. Bone, 64, 204–210. 10.1016/j.bone.2014.04.015 [DOI] [PubMed] [Google Scholar]
- Keller, A. N. , Kufareva, I. , Josephs, T. M. , Diao, J. , Mai, V. T. , Conigrave, A. D. , … Leach, K. (2018). Identification of global and ligand‐specific calcium sensing receptor activation mechanisms. Molecular Pharmacology, 93, 619–630. 10.1124/mol.118.112086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach, K. , & Gregory, K. J. (2017). Molecular insights into allosteric modulation of Class C G protein‐coupled receptors. Pharmacological Research, 116, 105–118. 10.1016/j.phrs.2016.12.006 [DOI] [PubMed] [Google Scholar]
- Leach, K. , Gregory, K. J. , Kufareva, I. , Khajehali, E. , Cook, A. E. , Abagyan, R. , … Christopoulos, A. (2016). Towards a structural understanding of allosteric drugs at the human calcium sensing receptor. Cell Research, 26, 574–592. 10.1038/cr.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach, K. , Wen, A. , Davey, A. E. , Sexton, P. M. , Conigrave, A. D. , & Christopoulos, A. (2012). Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium‐sensing receptor. Endocrinology, 153, 4304–4316. 10.1210/en.2012-1449 [DOI] [PubMed] [Google Scholar]
- Li, B. , Samp, L. , Sagal, J. , Hayward, C. M. , Yang, C. , & Zhang, Z. (2013). Synthesis of quinazolin‐4(3H)‐ones via amidine N‐arylation. Journal of Organic Chemistry, 78, 1273–1277. 10.1021/jo302515c [DOI] [PubMed] [Google Scholar]
- Miedlich, S. U. , Gama, L. , Seuwen, K. , Wolf, R. M. , & Breitwieser, G. E. (2004). Homology modeling of the transmembrane domain of the human calcium sensing receptor and localization of an allosteric binding site. The Journal of Biological Chemistry, 279, 7254–7263. 10.1074/jbc.M307191200 [DOI] [PubMed] [Google Scholar]
- Neves, M. A. , Totrov, M. , & Abagyan, R. (2012). Docking and scoring with ICM: The benchmarking results and strategies for improvement. Journal of Computer‐Aided Molecular Design, 26, 675–686. 10.1007/s10822-012-9547-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel, M. J. , Gregory, K. J. , Vinson, P. N. , Manka, J. T. , Stauffer, S. R. , Lindsley, C. W. , … Conn, P. J. (2013). A novel metabotropic glutamate receptor 5 positive allosteric modulator acts at a unique site and confers stimulus bias to mGlu5 signaling. Molecular Pharmacology, 83, 835–847. 10.1124/mol.112.082891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrel, C. , Kessler, A. , Dauban, P. , Dodd, R. H. , Rognan, D. , & Ruat, M. (2004). Positive and negative allosteric modulators of the Ca2+‐sensing receptor interact within overlapping but not identical binding sites in the transmembrane domain. The Journal of Biological Chemistry, 279, 18990–18997. 10.1074/jbc.M400724200 [DOI] [PubMed] [Google Scholar]
- Petrel, C. , Kessler, A. , Maslah, F. , Dauban, P. , Dodd, R. H. , Rognan, D. , & Ruat, M. (2003). Modeling and mutagenesis of the binding site of Calhex 231, a novel negative allosteric modulator of the extracellular Ca2+‐sensing receptor. The Journal of Biological Chemistry, 278, 49487–49494. 10.1074/jbc.M308010200 [DOI] [PubMed] [Google Scholar]
- Roberts, M. S. , Gafni, R. I. , Brillante, B. , Guthrie, L. C. , Streit, J. , Gash, D. , … Collins, M. T. (2019). Treatment of autosomal dominant hypocalcemia type 1 with the calcilytic NPSP795 (SHP635). Journal of Bone and Mineral ResearchE Pub, 34, 1609–1618. 10.1002/jbmr.3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira, M. , Totrov, M. , & Abagyan, R. (1999). Prediction of the binding energy for small molecules, peptides and proteins. Journal of Molecular Recognition, 12, 177–190. 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.0.CO;2-Z [DOI] [PubMed] [Google Scholar]
- Smithkline Beecham Corporation (2002). Calcilytic compounds. Patent WO/2002/038106
- Totrov, M. , & Abagyan, R. (1999). Derivation of sensitive discrimination potential for virtual ligand screening RECOMB99 3rd International Conference on Computational Molecular Biology (p. 312). Lyon, France. [Google Scholar]
- Widler, L. , Altmann, E. , Beerli, R. , Breitenstein, W. , Bouhelal, R. , Buhl, T. , … Seuwen, K. (2010). 1‐Alkyl‐4‐phenyl‐6‐alkoxy‐1H‐quinazolin‐2‐ones: A novel series of potent calcium‐sensing receptor antagonists. Journal of Medicinal Chemistry, 53, 2250–2263. 10.1021/jm901811v [DOI] [PubMed] [Google Scholar]
- Widler, L. G. R. , Seuwen, K. , Buhl, T. , Beerli, R. , Breitenstein, W. , Gyaw, S. , … John, M. R. (2008). A novel calcium‐sensing receptor antagonist leads to dose‐dependent transient release of parathyroid hormone after oral administration to healthy volunteers—An initial proof‐of‐concept for a potential new class of anabolic osteoporosis therapeutics. Journal of Bone and Mineral Research, 23, S49. ASBMR 30th Annual Meeting, abstract number 1173. [Google Scholar]
- Wu, H. , Wang, C. , Gregory, K. J. , Han, G. W. , Cho, H. P. , Xia, Y. , … Stevens, R. C. (2014). Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science, 344, 58–64. 10.1126/science.1249489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W. , Ruan, Z. , Wang, Y. , Van Kirk, K. , Ma, Z. , Arey, B. J. , … Dickson, J. K. Jr. (2009). Discovery and structure‐activity relationships of trisubstituted pyrimidines/pyridines as novel calcium‐sensing receptor antagonists. Journal of Medicinal Chemistry, 52, 1204–1208. 10.1021/jm801178c [DOI] [PubMed] [Google Scholar]
- Yarova, P. L. , Stewart, A. L. , Sathish, V. , Britt, R. D. Jr. , Thompson, M. A. , P Lowe, A. P. , … Riccardi, D. (2015). Calcium‐sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Science Translational Medicine, 7, 284ra260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Zhang, T. , Zou, J. , Miller, C. L. , Gorkhali, R. , Yang, J. Y. , … Yang, J. J. (2016). Structural basis for regulation of human calcium‐sensing receptor by magnesium ions and an unexpected tryptophan derivative co‐agonist. Science Advances, 2, E1600241 10.1126/sciadv.1600241 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 BMS compound 1 retains activity at an N terminally‐truncated CaS receptor . Ca2 + i mobilisation measured in response to the PAM‐agonist, AC265347, in the absence and presence of BMS compound 1. Curves are the best fit of a 3 parameter Hill equation to the data. Vehicle (buffer) has been plotted as the smallest [AC265347] in accordance with a logarithmic scale. Data are mean + SEM pooled from an n of 3 independent experiments performed in duplicate.
Figure S2 Alternative NAM docking poses. Alternative poses and associated docking scores for ronacaleret (A), ATF936 (B), and Pfizer compound 1 (C) in comparison to our proposed poses (ghosted) shown in Figures 4 and 5. Alternative poses are representative of those that are compatible with the mutagenesis and SAR data while being the most distinct alternative to our proposed pose. Also shown are two poses for BMS compound 1 (D) that were not supported by mutagenesis or SAR data. Scores for each pose were calculated as described in Totrov & Abagyan (1999), where lower scores represent lower ligand binding energies and therefore a more favourable interaction. Residues are those that alter NAM affinity as shown in Figure 4.
Table S1 WT and mutant Ca2 + o potency (pEC50), maximal response (Emax) and Hillslope determined in Ca2 + i mobilisation assays, and previously published mutant cell surface expression levels determined in FACS experiments. Data are mean ± s.e.m from the indicated number of experiments (n).
Table S2 Functional affinity (pKB) and cooperativity (Logαβ) parameters for BMS compound 1 at WT and ECL3 mutants determined in Ca2 + i mobilisation assays as described in the methods section. Data are mean ± s.e.m from the indicated number of independent experiments (n).