

Abstract
Background and Purpose
The gut microbial metabolite butyrate is linked to the modulation of metabolic disease. The mechanism by which butyrate effects in atherosclerosis is unknown. Hence, the present investigation into effects of butyrate on high‐fat diet‐fed ApoE−/− mice after 16 weeks' administration.
Experimental Approach
Gut microbiota composition was analysed via 16S rRNA gene sequencing of caecal contents. The effects of butyrate on atherosclerosis were evaluated in vivo using the ApoE−/− mice model. Serum lipids and glucose were analysed for physiological changes and differentially expressed genes in liver samples were identified by hepatic transcriptome profiling. The proteins involved in reverse cholesterol transport were quantified by Western blot and immunohistochemical staining. Finally, the up‐regulatory effects of butyrate on ATP‐binding cassette sub‐family A member 1 (ABCA1) were further evaluated in RAW 264.7 cells along with role of specificity protein 1 by inhibition and silencing.
Key Results
Oral gavage of butyrate altered microbiota composition and enhanced gut microbial diversity that was decreased by high fat diet (HFD). Butyrate treatment significantly inhibited the HFD‐induced atherosclerosis as well as hepatic steatosis without changing body weight gain in ApoE−/− mice. Butyrate had metabolic effects on the liver by regulation of gene expression involved in lipid/glucose metabolism. Furthermore, ABCA1 was significantly induced by butyrate in vivo, ex vivo and in vitro and Sp1 pathway was identified as a potential mechanism.
Conclusion and Implications
Butyrate ameliorates HFD‐induced atherosclerosis in ApoE−/− mice via ABCA1‐mediated cholesterol efflux in macrophages, which suggesting a promising therapeutic strategy for protecting against atherosclerosis.
Abbreviations
- ABCA1
ATP‐binding cassette sub‐family A member 1
- ABCG1
ATP‐binding cassette sub‐family G member 1
- Acot
acyl‐CoA thioesterase
- ApoA‐I
apolipoprotein A‐I
- ApoE−/−
apolipoprotein E‐deficiency
- CVD
cardiovascular disease
- Cyp7a1
cholesterol 7‐α‐monooxygenase or cytochrome P450 7A1
- FMOs
flavin‐containing monooxygenases
- Gck
glucokinase
- HDL‐C
HDL cholesterol
- HFD
high‐fat diet
- LDL‐C
LDL cholesterol
- LDLR
LDL receptor
- LXR
liver X receptor
- MEM
Minimum Essential Medium
- NaBu
sodium butyrate
- Nsdhl
NAD(P)H steroid dehydrogenase‐lik
- Pklr
pyruvate kinase L/R
- Plin
perilipin
- RCT
reverse cholesterol transport
- SCFAs
short‐chain fatty acids
- Slc2a4
solute carrier family 2 member 4
- Sp1
specificity protein 1
- SR‐BI
scavenger receptor class B type 1
- Srebf1
sterol regulatory element binding factor 1
- TBA
total bile acids
- TC
total cholesterol
- TG
triglycerides
- TMA
trimethylamine
- TMAO
trimethylamine N‐oxide
- Vnn1
vanin‐1
What is already known
Butyrate can exert beneficial effects in metabolic diseases.
ABCA1 plays a critical role in promoting cellular cholesterol efflux in preventing atherosclerosis.
What this study adds
Butyrate ameliorates atherosclerosis at least in part by increasing ABCA1 expression and cholesterol efflux in ApoE−/− mice.
Butyrate up‐regulates ABCA1 expression and activity in macrophage cells through Sp1 pathway.
What is the clinical significance
Butyrate could be a novel target for the prevention and treatment of atherosclerosis.
1. INTRODUCTION
Atherosclerotic cardiovascular disease (CVD) contributes to the major cause of death in Western countries, and with a dramatic increase in mortality and modality in China. High levels of LDL cholesterol (LDL‐C) as well as low levels of HDL cholesterol (HDL‐C) are associated with increased CVD risk. Excessive LDL‐C in the blood are accumulated in the arterial wall, initiating the early phase of atherosclerosis and triggering inflammatory response. Despite widely used of LDL‐C lowering drugs, the substantial residual risk of atherothrombotic events remains (Kühnast et al., 2015). HDL has attracted intense interest in the development of novel therapies for prevention of atherosclerosis based on epidemiological and preclinical research. The most proposed athero‐protective functions of HDL are promoting cholesterol efflux from peripheral macrophages and subsequent reverse transport of cholesterol to the liver for catabolism (Rohatgi et al., 2014).
Atherosclerosis has long been recognized as a complex pathological process involving both dysregulated lipid metabolism and chronic inflammation. Recently, a growing amount of evidence from both animal and human studies has highlighted the essential role of intestinal microbiota in modulating various metabolic diseases, such as obesity, type 2 diabetes and atherosclerotic CVD, by affecting energy balance and inhibiting inflammation (Koren et al., 2011; Meijnikman, Gerdes, Nieuwdorp, & Herrema, 2018). The gut microbe‐derived metabolite https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5521 N‐oxide (TMAO) has been shown to promote atherosclerosis and thrombotic vascular disease in mouse models (Tang & Hazen, 2014) and is clinically relevant in human CVD (Brown & Hazen, 2017). Short‐chain fatty acids (SCFAs), mainly https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1058, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1062 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=1059, are the main end‐products from the bacterial fermentation of nondigestible dietary fibres in the large intestine (Wichmann et al., 2013). These SCFAs have been suggested to exert various metabolic effects on energy homeostasis, involving hepatic gluconeogenesis and de novo lipogenesis (Witjes, van Raalte, & Nieuwdorp, 2015). Butyrate consumption, for example, was reported to reduce body weight in obese mice by both reducing energy intake partially via regulation of appetite and increasing energy expenditure by fat oxidation (den Besten et al., 2015; Li et al., 2018). Butyrate supplementation also showed beneficial metabolic effects on improving insulin sensitivity and hepatic steatosis (Canfora, Jocken, & Blaak, 2015; Mattace Raso et al., 2013). Although the multiple effects of SCFAs on metabolic syndrome have been demonstrated, there is a paucity of studies that investigate the role of butyrate in atherosclerosis. Previous studies found that 1% butyrate‐supplemented chow diet reduced aortic lesions in mice and the anti‐inflammatory and anti‐oxidative properties of butyrate were considered responsible for such an action, however no change in serum lipid levels were observed (Aguilar et al., 2014, 2016).
Numerous studies have indicated that high‐fibre foods lower serum cholesterol levels, particularly LDL‐C, and protect against atherosclerosis (Lattimer & Haub, 2010; Adam, Thomson, Williams, & Ross, 2015; Ho et al., 2016). Some studies have reported that butyrate treatment ameliorated dyslipidaemia by decreasing cholesterol and triglycerides (TG) in high‐fat diet (HFD)‐fed rodents; however, whether the lipids‐lowering effect of butyrate observed in these conditions could contribute to atherosclerosis attenuation remains largely un‐investigated (Gao et al., 2009; Mattace Raso et al., 2013; Khan & Jena, 2016). Therefore, the underlying mechanism by which butyrate prevents atherosclerosis via regulation of hepatic cholesterol metabolism needs to be investigated in detail. Reverse cholesterol transport (RCT) is a process that involves the net movement of cholesterol from the peripheral tissues to the liver for reuse or for the final elimination via excreting into the bile. Regulating the expression of key genes that involved in the RCT pathway might enhance transport of cholesterol and retard plaque progression. Butyrate was reported to down‐regulate the expression of key genes involving in the intestinal cholesterol biosynthesis pathway (Alvaro et al., 2008). Marcil et al. (2002) demonstrated that addition of butyrate to Caco‐2 cells resulted in a consistent reduction in chylomicrons and VLDL, which modulated the cellular mechabnisms involved in lipid transport. Our previous studies have shown that sodium butyrate might induce transcriptional activity of the HDL receptor scavenger receptor class B type 1 (SR‐BI) in HepG2 cells (Bao et al., 2009). However, whether butyrate could regulate expression of RCT‐related genes and, moreover, control lipid metabolism and atherosclerosis are still not well‐understood.
In the current study, we examined the effects of orally administered butyrate on the composition of gut microbiota, hepatic steatosis and atherosclerosis progression in HFD‐induced Apolipoprotein E‐deficiency (ApoE−/−) mice. The data obtained indicates the involvement of specificity protein 1 (Sp1) in butyrate ability to stimulate ATP‐binding cassette sub‐family A member 1 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=756) expression and activity in macrophages. Our results provide new evidence that butyrate, a gut microbial metabolite, could ameliorate atherosclerosis through modulation of lipid metabolism in mice, especially via promoting reverse cholesterol transport by activating ABCA1.
2. METHODS
2.1. Animals and experimental design
All animal protocols used in this study were approved by and conducted in accordance with the guidelines of the Animal Experimentation Ethics Committee of Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China (Approval Number: IMB‐201708D3). The research protocols were consistent with the regulations of Good Laboratory Practice for non‐clinical laboratory studies of drugs issued by the National Scientific and Technological Committee of People's Republic of China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and with the recommendations made by the British Journal of Pharmacology. ApoE−/− mice on a C57BL/6 background were selected because deletion of the ApoE gene in mice results in a marked increase in the plasma cholesterol levels and the development of atherosclerotic lesions, which resembles that in human. Eight‐week‐old male ApoE−/− mice (HFK Bio, Beijing, China) were weighted (26.5 ± 0.4 g) and housed under specific pathogen free conditions (five mice per cage), in a control environment at 20 ± 2°C and humidity 50–60%. Mice were exposed to a daily 12 hr light‐12 hr dark cycle with free access to a standard chow diet (10% kcal derived from fat, 3.85 kcal·g−1, H10010, HFK Bio‐technology, Beijing) or a Western‐type HFD (41% kcal derived from fat and 0.15% cholesterol [w/w], 4.7 kcal·g−1, H10141, HFK Bio‐technology, Beijing) and drinking water. There were no significant differences in the average body weight at baseline. All the animals were randomized into five groups of equal size: chow group (n = 10); HFD‐fed group (n = 10); HFD + NaBu (200), HFD‐fed mice treated by gavage with sodium butyrate (200 mg·kg−1·day−1, n = 10); HFD + NaBu (400), (400 mg·kg−1·day−1, n = 10) and HFD + atorvastatin, HFD‐fed mice treated by gavage with atorvastatin (10 mg·kg−1·day−1, n = 10). The group sizes were selected according to previous studies (Kasahara et al., 2018). Chow and HFD mice were given a single intragastric gavage of PBS as vehicle and food intake plus body weight were measured weekly. The energy intake has been calculated by multiplying the consumption of each food by its energy density. Fasting blood was collected from the retro‐orbital plexus at 12 weeks and serum was obtained for the evaluation lipids and glucose levels with commercial assay kits on Olympus AU480 autoanalyzer. Non‐HDL‐C, providing a measure of the cholesterol content of all atherogenic lipoproteins, is calculated by subtracting HDL‐C from total cholesterol (TC). Although the study has been designed with equal group sizes, because of technical factors such as fight‐related injury resulting in animal loss, the group sizes become increasingly unequal (n = 7 for chow group, n = 8 for HFD group, n = 9 for HFD + NaBu [200 mg·kg−1·day−1] group, n = 10 for HFD + NaBu [400 mg·kg−1·day−1] group, and n = 10 for HFD + atorvastatin [10 mg·kg−1·day−1] group). At the end of treatment (16 weeks), mice were anaesthetized with sodium pentobarbital (i.p., 50 mg·kg−1) after 8‐hr fasting, the heart along with the aorta was perfused with 20 ml of PBS for posterior organ collection. Tissue samples were immediately stored at −80°C for subsequent measurements.
2.2. Cell culture
Murine RAW 264.7 macrophages were maintained in DMEM containing 10% FBS at 37°C in 5% CO2. ABCA1p‐Luc HepG2 cells were maintained in Minimum Essential Medium (MEM) supplemented with 10% FBS and 500 μg·ml−1 of G418. Primary peritoneal macrophages were isolated from C57BL6/J mice and cultured as previously described (Gonçalves & Mosser, 2015).
2.3. Microbiota analysis
After 16 weeks of butyrate treatment, total bacterial DNA was isolated from caecum content in mice using a Fast DNA Spin Kit (MP Biomedicals, USA) according to the manufacture's protocol. The hypervariable V3‐V4 region of the microbial 16S rRNA gene was amplified by PCR using primers 338F 5′‐ACTCCTACGGGAGGCAGCAG‐3′ and 806R 5′‐GGACTACHVGGGTWTCTAAT‐3′. In total, 1,905,221 high‐quality 16S rRNA gene sequences were obtained from 46 samples. Sequencing was performed using the Illumina MiSeq platform (Shanghai Majorbio Bio‐pharm). Closed reference operational taxonomic units were picked at 97% sequence similarity using Usearch database (version 7.1 http://drive5.com/uparse/) and aligned with SILVA128/16S bacteria database for taxonomy information. The sequencing data were analysed on the free online platform of Majorbio I‐Sanger Cloud Platform (http://www.i-sanger.com). For statistical significance, the differences between two groups were assessed using the Wilcoxon rank‐sum test, and multiple comparison was determined by the Kruskal–Wallis H test. The false discovery rate was applied to adjust P values for multiple testing.
2.4. HPLC–MS/MS detection of trimethylamine and trimethylamine N‐oxide
Serum trimethylamine (TMA) and trimethylamine N‐oxide (TMAO) levels were measured by LC–MS/MS. Fasting blood samples at 12 weeks were collected from retro‐orbital plexus and serum protein was precipitated by adding 3 volumes of 80% acetonitrile for 0.5 hr at room temperature. Samples were then centrifuged (14,000× g, 15 min, 4°C) to obtain the supernatants for filtration and analysis. LC–MS/MS analysis was performed using an Agilent 1100 series HPLC coupled to an Agilent 6410 Triple Quadrupole mass spectrometer (Agilent Technologies, USA) equipped with an electrospray ionization source. Separation of TMA and TMAO was achieved with an XBridge™ HILIC column (150 × 2.1 mm I.D., 3.5 μm; Waters, USA) with a flex capillary guard column (10 × 2.1 mm I.D., 3.5 μm; Waters). The column was then eluted with an isocratic solvent mixture of an equal volume proportion of methanol (phase A) and aqueous solution of 5‐mM ammonium formate (phase B) at a flow rate of 0.25 ml·min−1. Calibration curves consisting of calibration standards for each concentration were constructed by spiking indicated concentrations of standard into control sample. TMAO and TMA were monitored using multiple reaction monitoring mode with precursor and characteristic product ions as follow: m/z 76 → 58 for TMAO; m/z 60 → 44 for TMA. The mass spectrometric parameters were optimized to the following values: capillary voltage 4 kV and temperature 350°C.
2.5. Preparation of en face aortae and atherosclerosis quantification
Atherosclerotic lesion severity was assessed in the aortae as previously described (Lin, Bai, Chen, Zhu, et al., 2015). In brief, at euthanasia, animals were perfused with PBS and 4% paraformaldehyde. Then the entire mouse aorta was dissected from the proximal ascending aorta to the iliac bifurcation, fixed in 4% paraformaldehyde. Adventitial fat was removed, and the aorta was opened longitudinally, stained with Oil Red O (Sigma‐Aldrich, USA) and pinned out under an inverted microscope. Digital images were analysed with ImageJ software (NIH, http://rsb.info.nih.gov/ij/), and the extent of lesion area is expressed as the percentage of the total area of aorta that covered by the lesions.
2.6. Histology and immunohistochemical analyses
After perfusion with 4% paraformaldehyde, the proximal aorta and section of the heart containing the aortic root was removed and embedded in tissue freezing OCT medium. The aortic root was cross‐sectioned sequentially in 8‐μm‐thick intervals from the appearance of the aortic valve to the aortic valve cusps. The cross sections were stained with Oil Red O to detect lipid deposition and quantify foam cell formation. Macrophages and ABCA1 expression in lesions were analysed immunohistochemically with antibodies against monocyte/macrophage antibody‐3 (Mac‐3/CD107b, 550292, 1:50, BD Pharmingen, USA, RRID:AB_393587) and ABCA1 (NB400‐105, 1:100, Novus Biologicals, USA, RRID:AB_10000630). The sections were visualized using SignalStain DAB Substrate Kit (#8059, Cell Signaling Technology, USA) and further counterstained with haematoxylin to obtain well‐contrasted staining. Sections were scanned with Leica DFC 450C camera mounted on a Leica DM3000 LED microscope, and lesions were quantified with ImageJ software. The immunohistochemistry experimental procedure conforms with guidelines of British Journal of Pharmacology (Alexander et al., 2018).
2.7. Histological analysis of liver tissue
For the analysis of liver steatosis, the tissues were fixed, embedded in paraffin, and tissue sections (5 μm) were processed for haematoxylin and eosin and Oil Red O staining according to standardized protocol. Steatosis was expressed as percentage of area stained with Oil Red O, which were quantified by histomorphometry using ImageJ software.
2.8. Hepatic transcriptome analysis
Liver samples from mice were flash‐frozen in liquid nitrogen and then stored at −80°C until the time of analysis. Total RNAs were isolated with TRIzol reagent (Invitrogen, USA) according to manufacturer's protocol. mRNA was purified from total RNA using the Oligotex mRNA Midi Kit (Qiagen, Germany). Poly(A) + mRNA was obtained using magnetic poly(A)+ Dynabeads (Invitrogen, USA). Then the cDNA library was constructed with fragmented mRNA according to a TruseqTM RNA sample prep kit (Illumina, California, USA). The cDNA was then end‐repaired, adenylated and ligated to the sequencing adapters provided by Illumina. Finally, the libraries were enriched by PCR amplification and sequenced using an Illumina HiSeq 2000 platform (Majorbio Bio‐Pharm, Shanghai, China). The data were analysed on the free online platform of Majorbio I‐Sanger Cloud Platform (http://www.i-sanger.com). In brief, differentially expressed genes were defined with P adjust <.05 and a fold change of ±1.5 or more. Differentially expressed genes analysis was carried out using DESeq2, and statistical significance was assessed using a negative binomial Wald test and then corrected for multiple hypothesis testing with the Benjamini–Hochberg method. A heatmap was generated by selecting differentially expressed genes associated with lipid or glucose metabolism. A base 2 logarithm transformation was applied to the gene‐level read counts, which were then mean‐centred and hierarchically clustered for visualization.
2.9. Western blot analysis
Liver samples (20 mg) were homogenized using Fast RNA Pro Green Kit and the Fast‐Prep Instrument (MP Biochemicals, USA) in 800‐μl ice‐cold T‐PER tissue protein extraction buffer (Thermo‐Fisher, USA). Liver protein (30 μg) was separated by 8% SDS‐PAGE followed by transfer to PVDF membrane. The membranes were incubated with 5% non‐fat dry milk for 1 hr at room temperature and then incubated overnight at 4°C with primary antibodies. The following primary antibodies (catalogue number and dilutions) were used: ABCA1 (ab18180, 1:800; Abcam, USA, RRID:AB_444302), ABCG1 (ab36969, 1:2000; Abcam, USA, RRID:AB_2220181), SR‐BI (ab52629, 1:1000; Abcam, USA, RRID:AB_882458), LDL receptor (LDLR; ab30532, 1:500; Abcam, USA, RRID:AB_881272), Sp1 (sc‐59, 1:800; Santa Cruz, USA, RRID:AB_2171050), β‐tubulin (TA347064, 1:3000; Origene, USA), β‐actin (TA‐09, 1:1000; Zsbio, Beijing, China, RRID:AB_2636897), and GAPDH (TA‐08, 1:1000; Zsbio, Beijing, China, RRID:AB_2747414). Membranes were then incubated with HRP‐conjugated secondary antibodies. The signals were visualized using an enhanced chemiluminescence detection system (Millipore, MA). The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
2.10. Cholesterol efflux assay
Cholesterol efflux experiments were performed as previously described (Smith et al., 2004). Briefly, RAW 264.7 macrophages were labelled with [1,2‐3H]‐cholesterol (1.0 μCi·ml−1, PerkinElmer, USA) in media containing 0.2% BSA for 24 hr. After labelling, cells were rinsed with PBS and treated with butyrate or 9‐cis retinoic acid as indicated for further 24 hr. Cells were washed again and incubated in DMEM containing 0.2% BSA in the presence of ApoA‐I (10 μg·ml−1; Academy Bio‐Medical, USA) for 24 hr. Cells were then lysed in 0.1‐N NaOH, and radioactivities of the media and the cell lysates were quantified using MicroBeta2 scintillation counter (PerkinElmer Waltham, USA). The percentage cholesterol efflux was calculated by dividing the media‐derived radioactivity by the sum of the radioactivity in the media and the cells.
2.11. ABCA1 transcriptional activity assay
Transcriptional activity assay using ABCA1p‐Luc HepG2 cells was performed as described previously (Gao et al., 2008). This construct, containing human ABCA1 promoter region spanning the region from −819 bp to +71 bp (nucleotide +1 corresponds to the transcription start site). Briefly, ABCA1p‐Luc HepG2 cells were seeded in 96‐well plates at 50,000 cells per well and incubated for 12 hr. Then the cells were treated with indicated concentrations of butyrate or vehicle in MEM without FBS. Cells were harvested 24 hr later and the luciferase activity was detected as relative luminescence units using the Luciferase Assay System (Promega, USA).
2.12. Quantitative real‐time RT‐PCR
Then total RNA was extracted from cells using SV total RNA isolation system (Promega, USA), and 100‐ng purified RNA was reverse transcribed with SuperScript III reverse transcriptase (Invitrogen, USA) to synthesize 20‐μl cDNA template, and cDNA was diluted (5×) before use in quantitative PCR. Real‐time PCR was performed by using the FastStart Universal SYBR Green Master (Rox) reagent (Roche, Basel, Switzerland) in CFX96™ Real Time PCR Detection System (Bio‐Rad, Hercules, CA) with forward and reverse primer sets: 5′‐ATGGAGCAGGGAAGACCAC‐3′ and 5′‐GTAGGCCGTGCCAGAAGTT‐3′ for mice ABCA1 (the size of the amplicon is 71 bp), 5′‐AGCTTGTCATCAACGGGAAG‐3′ and 5′‐TTTGATGTTAGTGGGGTCTCG‐3′ for mice GAPDH (the size of the amplicon is 62 bp). mRNA expression levels were calculated by the ΔΔCt method using GAPDH as an internal control and further normalized to the mean expression level of the control group.
2.13. Knockdown of Sp1 by siRNA
Sp1 gene silencing in RAW 264.7 cells was accomplished by transfecting mouse Sp1 siRNA (MSS209281, Invitrogen, USA) using Lipofectamine™ RNAiMAX reagent (Invitrogen, USA); Scramble siRNA (12935‐300, Invitrogen, USA) was used for negative control. After 24 hr, cells were treated with butyrate or vehicle for another 24 hr. Afterwards, Western blot analysis was performed as described above.
2.14. Statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018), and the data analysis was performed in a blinded manner. Data are presented as mean ± SEM. Statistical analysis was undertaken only when each group size has a minimum of n = 5 independent samples/individuals and performed using unpaired two‐tailed Student's t test or one‐way ANOVA with the Bonferroni's post hoc test, while post hoc tests were run only if F achieved P < .05 and there was no significant variance inhomogeneity. For data that did not pass normality testing, log transformation was applied to generate Gaussian‐distributed data set that could be subjected to parametric statistical analysis or non‐parametric statistics were used (Wilcoxon rank‐sum test or Kruskal–Wallis H test). P < .05 was considered statistically significant (*P < .05), and P value is not varied later in Section 3. Analyses were performed using GraphPad Prism 7 (GraphPad Software, USA) unless otherwise indicated.
2.15. Materials
Sodium butyrate (NaBu), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2949, and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8394 were purchased from Sigma‐Aldrich (St. Louis, USA); FBS/DMEM/MEM were obtained from Gibco; Glucose, TG, LDL‐C, HDL‐C, and TC measurement kits were from Intec PRODUCTS Inc (Xiamen, China); The TMA and TMAO standards were purchased from J&K Scientific Ltd (Beijing, China).
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019; Alexander et al., 2019).
3. RESULTS
3.1. Butyrate consumption alters the composition of gut microbiota
To evaluate the effects of butyrate treatment on atherosclerosis progression in vivo, HFD‐fed ApoE−/− mice were given a single dose of sodium butyrate daily by oral gavage for 16 weeks. Gut microbiome has been associated with plaque progression and the microbial metabolite butyrate has been proposed to exert beneficial effects, ameliorating atherosclerosis (Ohira, Tsutsui, & Fujioka, 2017). Here, to examine the further impact of butyrate administration on the composition of gut microbiota, 16S rRNA gene sequencing of caecum contents was performed using the Illumina MiSeq platform. The α diversity analyses revealed that the 400 mg·kg−1 of butyrate administration significantly increased the gut microbial diversity as observed by Shannon index and Simpson index, which might be due to the change in community evenness as indicated by Heip index (Figure 1a). At the phylum level, caecal microbial composition of all mice was dominated by Firmicutes (average: 67.3% across all groups) and Bacteroidetes (average: 22.3%). HFD intake increased the abundance of Firmicutes but reduced Bacteroidetes and Actinobacteria compared to those of the chow‐fed mice. Both 200 and 400 mg·kg−1 of butyrate‐treated mice had a relative increased abundance of Firmicutes at the expense of Bacteroidetes compared with HFD mice (Figure 1b). Principal component analysis showed a clear separation in β diversity between the chow diet, HFD and HFD plus butyrate groups (Figure 1c). Butyrate group exhibited a distinct profile of sample clusters from atorvastatin‐treated mice (Figure S1A), indicating different bacterial community structure changes between groups. The relative abundance of microbiota at the genus level was shown in Figure 1d. As compared with HFD mice, butyrate increased Lachnospiraceae UCG‐006, Lachnoclostridium and an unclassified genus under Erysipelotrichaceae etc. while decreased a genus under Bacteroidales S24.7 group and Faecalibaculum genus (Figure 1d and Figure S1B). Interestingly, comparisons of relative abundance of taxa between different groups revealed that butyrate administration effectively restored the HFD‐mediated changes in the Faecalibaculum genus and an unclassified genus under Erysipelotrichaceae in different manners (Figure S1B). In brief, the data summarized here clearly indicate that exogenous butyrate alters the composition of the caecal microbiota.
Figure 1.
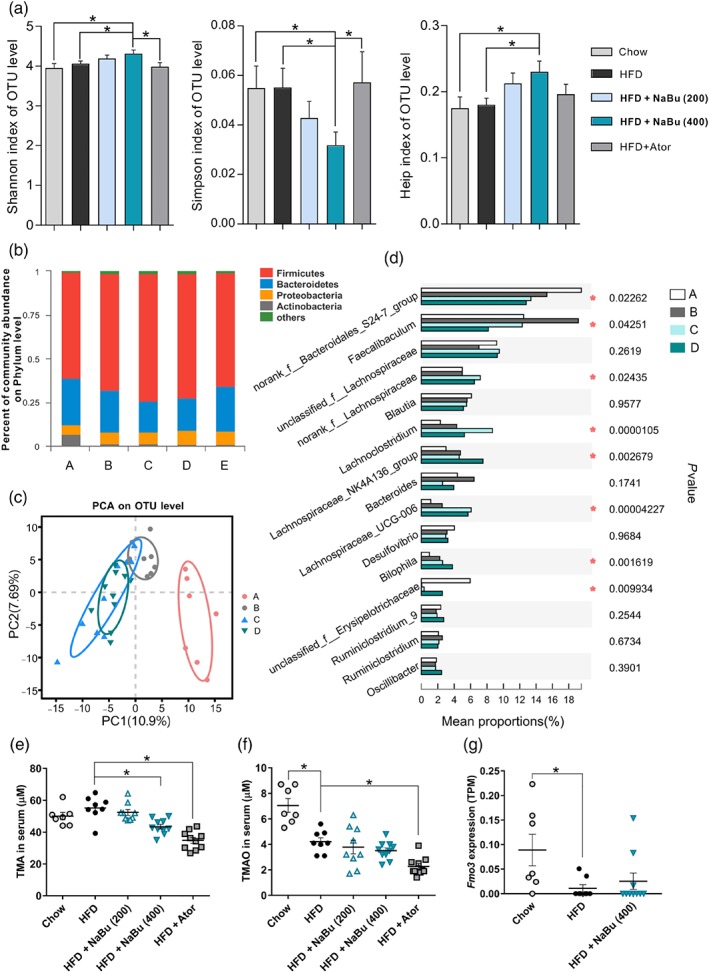
Effect of butyrate on gut microbiota composition and microbial‐derived metabolites in HFD‐fed ApoE−/− mice. After 16 weeks of treatment, total bacterial DNA was isolated from the caecum content and analysed by sequencing of 16S rRNA genes. (a) The α diversity of the gut microbiota as measured by the Shannon, Simpson, and Heip indices (Wilcoxon rank‐sum test). (b) Relative abundance of bacterial phyla. (c) Principal component analysis (PCA) plot of microbiota is shown along the first two principal coordinate (PC) axes. (d) Relative abundance of first 15 genera between different groups. Serum TMA (e) and TMAO (f) levels were determined by LC/MS; FMO3 levels (g) were assessed by liver transcriptome data. n = 7–8 for chow and HFD groups; n = 9–10 for HFD + NaBu (200 and 400 mg·kg−1) and atorvastatin groups (10 mg·kg−1). A, chow group; B, HFD group; C, HFD + 200 mg·kg−1 NaBu; D, HFD + 400 mg·kg−1 NaBu; E, HFD + atorvastatin
TMA, another well‐identified gut microbial metabolite, was converted to TMAO by host hepatic flavin‐containing monooxygenases (FMOs; Koeth et al., 2013; Wang et al., 2011). Recent studies reported that statin treatment is associated with lower TMAO levels in adults at risk of CVD via affecting gut microbiome function (Li, Wang, Li, & Hazen, 2018; Milks et al., 2018). Thus, we investigated whether the butyrate affects TMA and TMAO synthesis in HFD‐fed mice. The circulating TMA levels were significantly reduced in mice treated with 400 mg·kg−1 of butyrate as well as atorvastatin (Figure 1e), indicating that butyrate may modulate TMA‐producing bacteria in intestine. However, the serum concentrations of TMAO were not changed by butyrate in our study (Figure 1f). Notably, the serum TMAO levels were lower in all HFD diet groups as compared with chow‐fed mice, which may reflect the down‐regulation of FMO3 in liver after feeding with HFD in male ApoE−/− mice (Figure 1g).
3.2. Effects of butyrate on serum lipids
To assess whether the differences in diet intake contribute to the protective effects of butyrate treatment, the food consumption and body weight were monitored. The energy intake (expressed in kcal) of HFD group were 14.3% more than that of the chow‐fed mice, but butyrate administration did not influence energy intake in the HFD‐fed ApoE−/− mice (Figures 2a and S2A,B). The body weight of HFD group have shown a steady weight gain and induced a significant increase in body weight gain (of 24.2%) after 16 weeks, as compared to the chow group (Figure 2b). In line with the results of food intake, butyrate treatment did not inhibit HFD‐induced weight gain compared to HFD group (Figure S2C).
Figure 2.
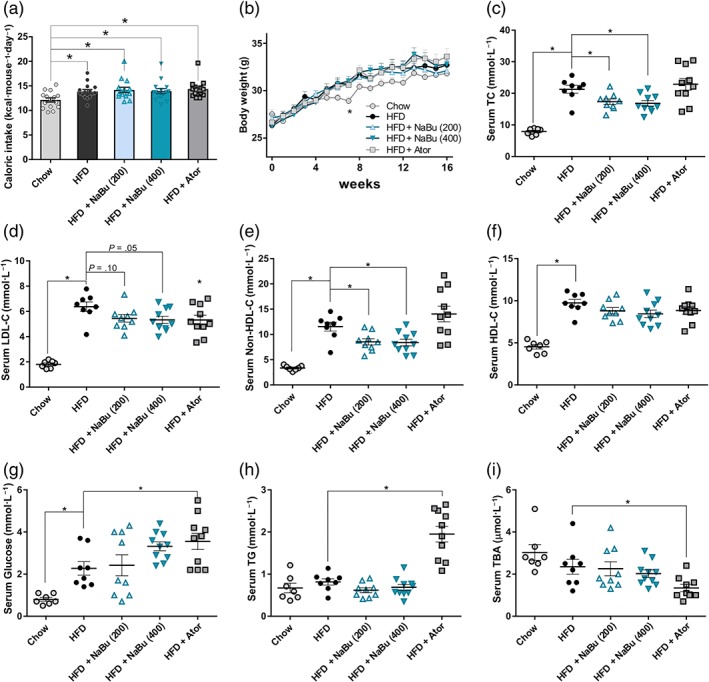
Effect of butyrate on physiological parameters and serum lipids in ApoE−/− mice. ApoE−/− mice were fed with HFD in the presence and absence of butyrate for 16 weeks. Average food intake (a) and body weight (b) per group through the entire 16‐week intervention period was measured. Serum total cholesterol (c), LDL‐cholesterol (d), non‐HDL‐cholesterol (e), HDL‐cholesterol (f), glucose (g), triglycerides (h), and total bile acid (i) were measured at 12 weeks, and average levels were calculated. Data are presented as mean ± SEM, n = 7–8 for chow and HFD groups, n = 9–10 for HFD + NaBu (200 and 400 mg·kg−1) and atorvastatin groups (10 mg·kg−1). * represent significance versus HFD
After 12 weeks of treatment, serum biochemical parameters of mice were determined. HFD diet did result in significant increases in serum TC, LDL‐C, non‐HDL‐C, HDL‐C and glucose as compared with chow group (Figure 2c–f). Both 200 and 400 mg·kg−1 of butyrate markedly decreased the HFD‐induced elevation in TC (−18.5%; −20.9%) mainly by reducing non‐HDL‐C (−26.1%; −27.3%) and more specifically LDL‐C (−14.5%; −16.4%). Butyrate treatment displayed no significant effects on serum HDL‐C, glucose, TG and total bile acids levels (Figure 2f–i). Here, atorvastatin decreased LDL‐C (−16.5%) and total bile acid (−42.8%) but significantly increased glucose and TG. Collectively, the data demonstrated that butyrate could prevent dyslipidaemia induced by HFD in ApoE−/− mice.
3.3. Butyrate consumption reduces atherosclerosis
To further elucidate the involvement of butyrate in protecting against atherosclerosis, both en face analyses of the aorta and the cross‐sectional analyses of the aortic sinus area were evaluated. As represented in Figure 3a, the atherosclerotic lesion area in the HFD group was significantly increased compared with chow group (4.36 ± 0.42% vs. 19.53 ± 2.83%) as indicated by lipid‐positive area. Both dose of butyrate significantly reduced atherosclerotic lesion formation (−37.4%, −43.5%). The total lesion area of the atorvastatin group was decreased to a level similar to that of 400 mg·kg−1 of butyrate group (−47.5%). Although aortic sinus plaque volume was frequently used to assess mild or early stage of atherosclerosis, the cross‐sectional lesion areas were still moderately reduced after 400 mg·kg−1 of butyrate treatment, indicating a decreasing tendency of lipid deposition and foam cell formation in the aortic root area (Figure 3b). Furthermore, butyrate reduced macrophage infiltration into the atherosclerotic plaques as judged by MAC‐3‐positive macrophages (Figure 3c), indicating butyrate inhibits inflammatory content in atherosclerotic lesion of ApoE−/− mice. Then we assessed the relationship between atherosclerosis development and changes of lipid levels. Linear regression analyses showed that the percentage aortic lesion area was positively correlated with the non‐HDL‐C, TC and LDL‐C (Figure S2D–F). These data further support that alteration of serum lipids by butyrate favourably contributes to the inhibition of atherosclerotic lesion development.
Figure 3.
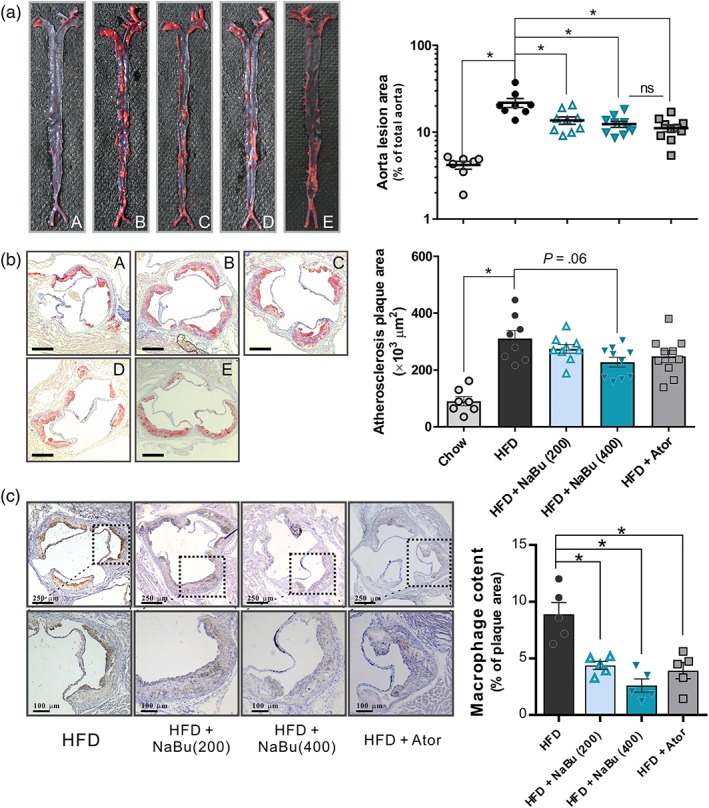
Effect of butyrate on development of atherosclerotic lesion in ApoE−/− mice. Representative photographs (left) and quantifications (right) of en face atherosclerotic lesions in the entire aorta, the data were log transformed to generate a Gaussian‐distributed data set (a) and those of aortic root cross sections (b) stained with Oil Red O to determine foam cell formation for the chow group (A), HFD group (B), HFD + 200 mg·kg−1 NaBu (C), HFD + 400 mg·kg−1 NaBu (D), and HFD + atorvastatin group (E) after 16 weeks of treatment. Scale bar, 250 μm. n = 7–8 for chow and HFD groups, n = 9–10 for HFD + NaBu and atorvastatin groups. (c) Representative photographs immunohistochemical staining MAC‐3 in aortic root sections and quantifications. (n = 5). * represent significance versus HFD
3.4. Butyrate inhibits HFD‐induced liver steatosis
Next, to investigate the impacts of butyrate on hepatic lipid metabolism, we evaluated the degree of hepatic steatosis in all experimental groups. As illustrated in Figure 4a, exposure to HFD developed a remarkable lipid accumulation in the liver, characterized by the presence of massive micro‐ and macro‐vesicular steatosis in liver sections. However, the size and content of lipid droplets were small and reduced in the butyrate treated group. Consistent with focal fatty change observed in liver, Oil Red O analyses confirmed that butyrate significantly prevent HFD‐induced hepatic steatosis (−63.7% and −70.5%, for 200 and 400 mg·kg−1, respectively, Figure 4b). The above results indicated that butyrate administration could exert beneficial metabolic effects on alleviation of fatty liver. It has been reported that hepatic steatosis is independently associated with carotid atherosclerotic plaques and non‐alcoholic fatty liver disease is an independent risk factor for CVD (Sookoian & Pirola, 2008; Mahfood Haddad, Hamdeh, Kanmanthareddy, & Alla, 2017). Therefore, the beneficial effects on hepatic steatosis could potentially attenuate the progression of atherosclerosis.
Figure 4.
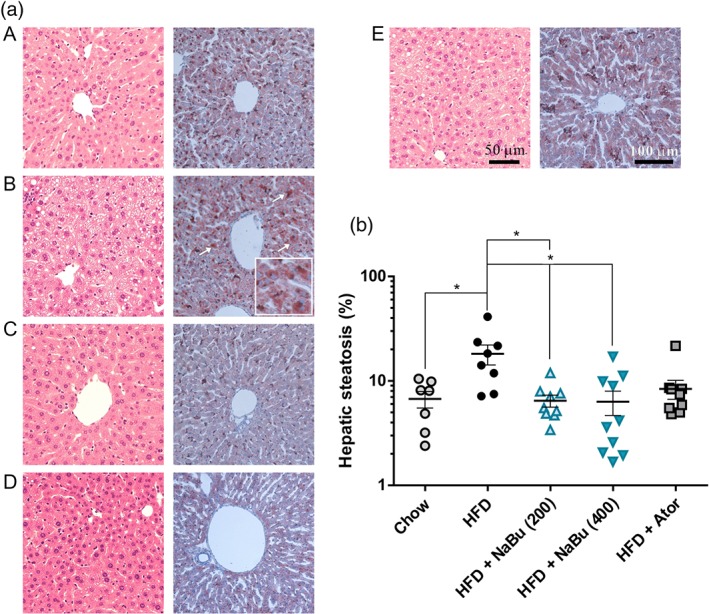
Effect of butyrate on HFD‐induced hepatic steatosis in ApoE−/− mice. Representative photomicrographs of the liver sections from chow group (A), HFD group (B), HFD + 200 mg·kg−1 NaBu (C), HFD + 400 mg·kg−1 NaBu (D), and HFD + atorvastatin group (E) stained with haematoxylin and eosin (H&E) (right) and Oil Red O (left, staining area indicated by the arrow) (a) and quantitative analysis of lipid accumulation in mice liver (b). n = 7–8 for chow and HFD groups, n = 9–10 for HFD + NaBu and atorvastatin groups. * represent significance versus HFD. The data were log transformed to fit a Gaussian distribution
3.5. Butyrate alters the liver transcriptome
To better understand the effect of butyrate on hepatic steatosis, liver transcriptome analysis was performed with the mice of HFD and HFD plus 400 mg·kg−1 of butyrate. Under the butyrate treatment, 88 genes were identified to express differentially with significance, of which 62.5% were down‐regulated. Gene ontology analysis revealed that these differentially expressed genes are significantly enriched in monocarboxylic acid metabolism, small‐molecule metabolism, lipid metabolic process and regulation of lipid biosynthetic process (Figures 5a and S3A,B). Accordingly, 27 differentially expressed genes (31% of the total) were implicated in lipid and/or glucose metabolism as illustrated by the heatmap (Figure 5b). Fifteen of the 22 butyrate‐regulated genes mainly involved in lipid metabolism were down‐regulated compared to the HFD control. There was a significant down‐regulation of lipogenic genes, such as Acot1 (acyl‐CoA thioesterase 1), Acot2, Plin2 (Perilipin 2) and Plin5. Fatty acid degradation‐associated genes were also down‐regulated in butyrate treated mice, including Cyp4a10, Cyp4a14 and Cyp4a31. Meanwhile, the expression of the Slc2a4 (solute carrier family 2 member 4, also known as glucose transporter type 4, Glut4) and Gck (glucokinase) genes, responsible for glucose homeostasis, were increased. In brief, changes in hepatic gene expression suggest that butyrate not only alters lipid metabolism but also affects glucose homeostasis. Thus, the correlation analysis between differentially expressed genes and phenotypes of atherosclerosis (including serum lipid levels) was performed (Figure 5c). We observed that liver steatosis was most negatively correlated with the expression of Srebf1 (sterol regulatory element binding factor 1) and Pklr (pyruvate kinase L/R) genes while most positively correlated with the Vnn1 (vanin‐1). The gene expression of Nsdhl (NAD(P)H steroid dehydrogenase‐like that encodes a 3β‐hydroxysterol dehydrogenase) and Acot2 showed a significant correlation with both hepatic steatosis and the level of TC and non‐HDL‐C, possibly reflecting the interacting regulatory mechanisms.
Figure 5.
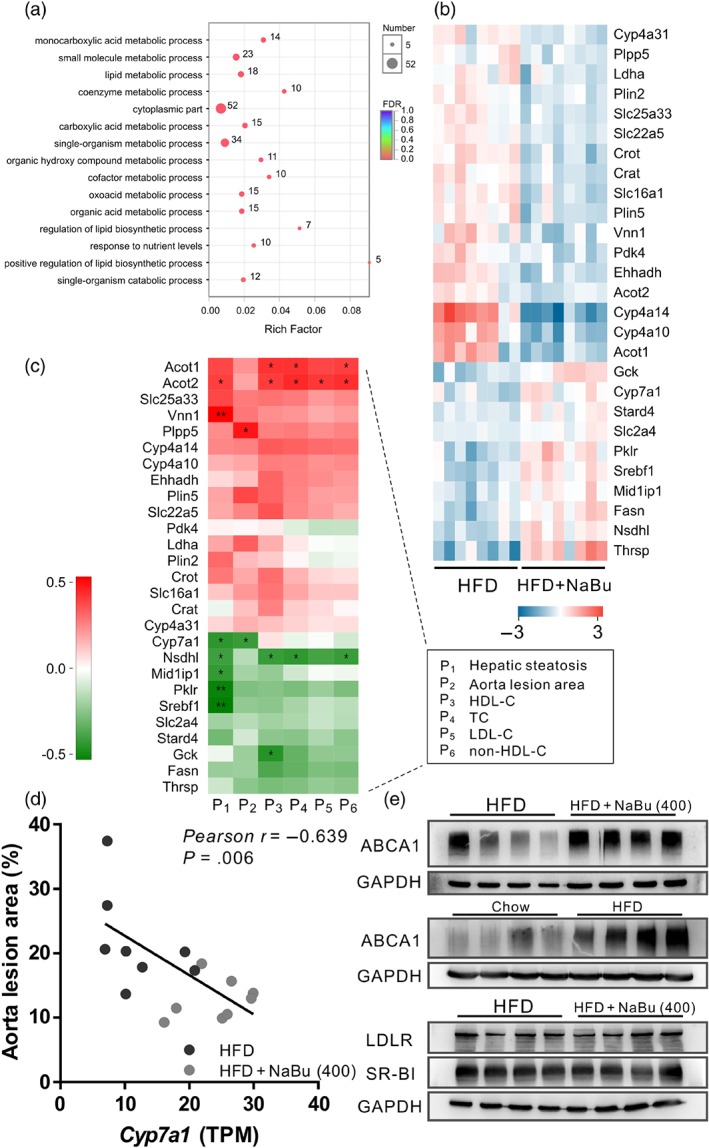
Effect of butyrate on liver glucose and lipid metabolism in vivo. Liver transcriptome was compared between HFD and HFD + 400 mg·kg−1 NaBu groups. (a) Gene ontology term enrichments with Bonferroni correction for differentially expressed genes. (b) Heatmap of differentially expressed gene levels (log2) involved in glucose and lipid metabolism as assessed by RNA‐sequencing analysis. (c) Pearson correlation analysis between differentially expressed genes and phenotypes of atherosclerosis in mice treated with HFD or HFD + 400 mg·kg−1 NaBu. (d) Pearson correlation between aorta lesion area and Cyp7a1 levels in liver of mice treated with HFD or HFD + 400 mg·kg−1 NaBu. (e) Representative Western blot analysis of the indicated hepatic protein levels that involved in RCT. (n = 7 for chow group, n = 8 for other groups)
https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=265#1354 (also known as cytochrome P450 7A1, CYP7A1) is a key rate‐limiting enzyme for bile acid biosynthesis that is responsible for the final elimination of cholesterol in the liver. Butyrate treatment resulted in up‐regulation of Cyp7a1 as revealed by liver transcriptome data. We further found a significant negative correlation between the aorta lesion area and Cyp7a1 (r = −0.639, Figure 5d) as well as between hepatic steatosis and Cyp7a1 (r = −0.554), suggesting a possible link between the therapeutic effect of butyrate and the modulation of cholesterol catabolism and thus RCT pathway. Therefore, we focused on whether butyrate enhanced the RCT process and analysed the expression of several RCT‐related hepatic genes. ABCA1 is a plasma membrane transporter that mediates the efflux of free cholesterol and phospholipids to lipid‐poor apolipoprotein A‐I (ApoA‐I) and it is critical for HDL particle biogenesis, which initializes the early steps in RCT. LDLR and HDL receptor SR‐BI play a critical role in the clearance of lipoproteins from blood. The results showed that butyrate treatment significantly increased the ABCA1 level in the mice liver without affecting the LDLR or SR‐BI levels (Figure 5e).
3.6. Butyrate induces ABCA1 activation in peripheral macrophages via a Sp1‐mediated pathway
Cholesterol efflux from peripheral macrophages accumulating excess cholesterol is the initial and important step of RCT. We then assessed the expression of ABCA1 in aortic atherosclerotic lesions from mice. As compared with HFD‐fed mice, the percentages of ABCA1‐staining area to the plaque area in aortic roots were significantly increased in butyrate‐treated groups (Figure 6a,b). Furthermore, peritoneal macrophages incubated with butyrate showed remarked increases in ABCA1 protein level in comparison with vehicle control (Figure 6c). Next, we performed lipid efflux experiments to determine the impact of butyrate on macrophage cholesterol efflux mediated by ABCA1. Butyrate increased the cholesterol efflux in RAW 264.7 macrophages in a dose‐dependent manner (Figure 6d). Additionally, Western blot analysis showed that the protein level of ABCA1, but not of SR‐BI or ABCG1, was significantly elevated under butyrate treatment (Figure 6e). SR‐BI and/or ABCG1 is required for the efflux of cholesterol from macrophages to HDL and the present results suggest that butyrate‐increased macrophage cholesterol efflux is ABCA1‐dependent.
Figure 6.
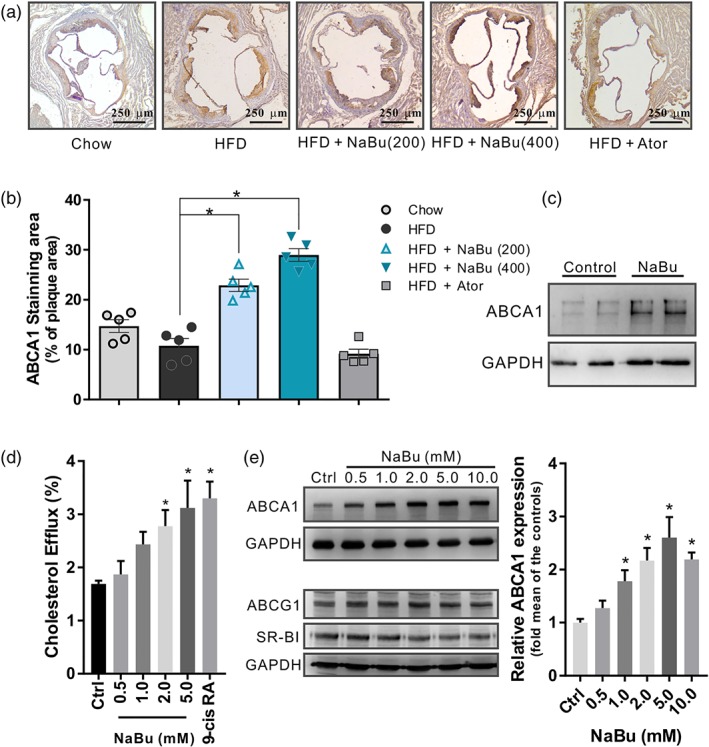
Effect of butyrate on ABCA1 induction in vivo and in vitro. (a) Representative immunohistochemistry sections and (b) quantitative analysis of ABCA1‐positive area (% of plaque area) in the lesions of aortic sinus, data are expressed as mean ± SEM (n = 5). (c) Western blot analysis of the ABCA1 levels in butyrate‐treated peritoneal macrophages from C57BL6/J mice (n = 4 per group). (d) [3H]‐Cholesterol efflux induced by butyrate was evaluated by incubating with ApoA‐I (10 μg·ml−1) in RAW 264.7 macrophages and calculated as a percentage of the radioactivity in the supernatant to the total radioactivity in the supernatant and cell lysates, 9‐cis RA: 9‐cis retinoic acid 10 μmol·L−1 (n = 5). (e) Western blots of ABCA1, ABCG1, and SR‐BI with corresponding quantifications in RAW 264.7 macrophages incubated with indicated butyrate for 24 hr (n = 5). Data are presented as the mean ± SEM, * represent significance versus vehicle control (Ctrl)
Then ABCA1 mRNA transcript level and promoter activity was investigated to delineate the molecular mechanism of butyrate on ABCA1 expression (Figure 7a,b). The results showed that butyrate cause a concentration‐dependent upregulation in both mRNA levels and luciferase activity of ABCA1 promoter. Sp1 and liver https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=89 (LXR) α are known to play an essential role in the transcriptional induction of the ABCA1 (Chawla et al., 2001; Thymiakou, Zannis, & Kardassis, 2007). Here, we observed butyrate decreased the level of Sp1 in a dose‐dependent manner in RAW 264.7 cells, but not LXRα (Figure 7c). We then investigated whether Sp1 is involved in the butyrate‐induced ABCA1 expression. To confirm if reduction of Sp1 will increase ABCA1 expression, siRNA transfection assays were performed. We found that siSp1 transfected for 24 hr enhanced the induction of butyrate on ABCA1 expression while the basal level of ABCA1 was not affected (Figure 7d). Thereafter, Mithramycin A, a GC specific DNA binding antibiotic that blocks binding of Sp1, was utilized. The results clearly showed that Mithramycin A alone did not inhibit the level of Sp1 but completely abolished the butyrate‐mediated induction of ABCA1 in RAW 264.7 cells (Figure 7e). These findings indicate that butyrate might increase ABCA1 expression by decreasing the level of Sp1, but the interaction of Sp1 with its target DNA is required.
Figure 7.
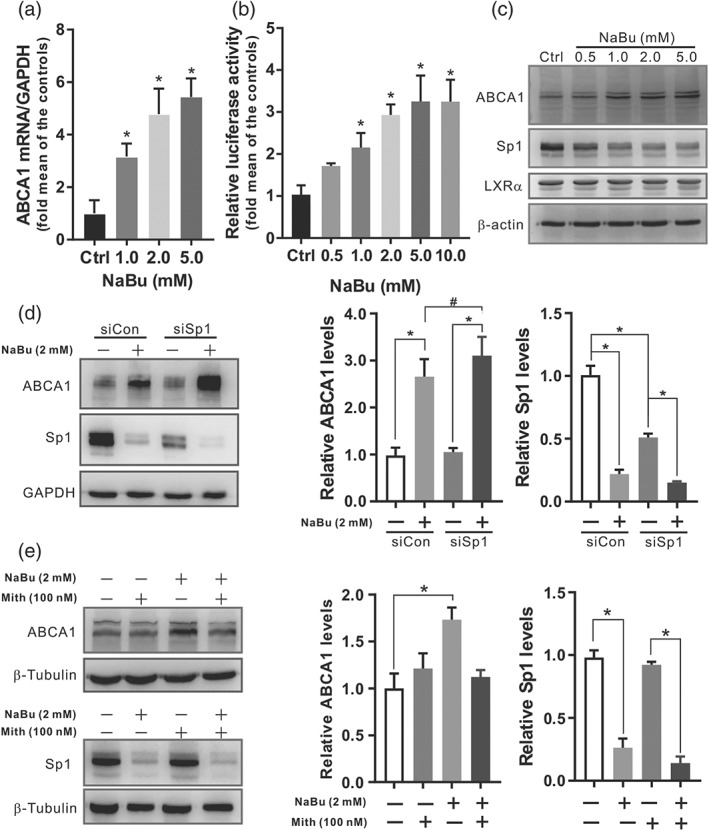
Butyrate regulates ABCA1 expression at transcriptional level in which Sp1 is involved. Cultured cells were incubated with indicated concentrations of butyrate for 24 hr. (a) The mRNA levels of the ABCA1 in RAW 264.7 cells were detected by real‐time PCR. (b) The relative luciferase activity of ABCA1 promoter was determined in ABCA1p‐Luc HepG2 cells. (c) Representative Western blot of ABCA1, Sp1, and LXRα proteins from RAW 264.7 cells. (d) Western blots of ABCA1 and Sp1 with corresponding quantifications of RAW 264.7 cell lysates following transfection with 10 nmol·L−1 of Sp1 siRNA (siSp1) or negative control (siCon) for 24 hr and incubated with or without 2 mmol·L−1 of butyrate for additional 24 hr. (E) Western blots of ABCA1 and Sp1 with corresponding quantifications of RAW 264.7 cell lysates following pretreatment with 100 nmol·L−1 of Mithramycin A (Mith), and further incubated with or without 2 mmol·L−1 of butyrate for 24 hr. Data were presented as means ± SEM, (n = 5). * represent significance versus vehicle control
4. DISCUSSION
Short‐chain fatty acids (SCFAs), especially butyrate, have been suggested to have crucial roles in host immunity and metabolic disease (Canfora, Jocken, & Blaak, 2015; Koh, De Vadder, Kovatcheva‐Datchary, & Bäckhed, 2016). Previous studies revealed that butyrate supplementation suppresses atherosclerotic lesions in chow diet‐fed ApoE−/− mice mainly through its anti‐inflammatory properties (Aguilar et al., 2014, 2016). In one recent study, gnotobiotic ApoE−/− mice with synthetic microbial communities colonized with butyrate‐producing bacteria Roseburia intestinalis inhibited the progression of atherosclerotic plaque formation in a standard chow diet with high content of plant polysaccharides. Although the exact mechanism was not fully elucidated, the results suggested the athero‐protective effect of R. intestinalis was mediated at least in part by butyrate (Kasahara et al., 2018). In spite of no significant changes in serum lipid levels in the above studies, however other animal studies have shown promising results of cholesterol‐lowering by butyrate (Gao et al., 2009; Mollica et al., 2017). This discrepancy may be due to differences in diet composition, especially the contents of fat and cholesterol. In the present study, with the utilization of Western‐type of HFD, we noted that 12‐week butyrate treatment reduced the serum TC, LDL‐C and non‐HDL‐C in ApoE−/− mice. Moreover, the developments of atherosclerotic lesions both in the aorta and in the aortic root were inhibited upon 16 weeks of butyrate intervention. We infer that the role of butyrate in suppression of atherosclerosis is mediated, at least in part, through improving dyslipidaemia in the present study.
Furthermore, we found that butyrate administration may further altered the gut microbiota composition, improved the gut microbial diversity and increased the abundance of the phylum Firmicutes compared with HFD mice, which may exhibit potential protections against atherosclerosis development. Butyrate can be generated by some well‐classified species related to Clostridial clusters IV and XIVa within the Firmicutes phylum (Louis, Young, Holtrop, & Flint, 2010). Notably, the present study indicated that butyrate treatment significantly altered the relative abundance of Faecalibaculum, Lachnoclostridium, Lachnospiraceae UCG‐006 and an unclassified genus under Erysipelotrichaceae etc. at the genus level. Furthermore, these changes of microbial composition influenced by butyrate have significant functional implications as indicated in Spearman correlation analysis (Figure S4). The above‐mentioned genera are highly correlated with expression levels of lipid and/or glucose metabolic genes, which is in agreement with earlier findings showing their involvement in lipid/glucose metabolism (Martínez et al., 2013; Wang et al., 2019; Zhu et al., 2018). Microbiota‐derived metabolite TMAO was reported to profoundly modulate atherosclerosis by impairing RCT and inducing the formation of foam cells (Koeth et al., 2013). Here, we found butyrate reduced TMA production. Although very low levels of TMAO were detected in this study due to male mice having lower FMO3 (Wang et al., 2011), these observations raise the possibility that the alteration in the composition of microbiota by butyrate may beneficially affect TMA production and hence TMAO level, ultimately leading to the inhibition of atherogenesis.
Consumption of dietary fat promotes the development of hepatic steatosis, which is closely associated with obesity and carotid atherosclerosis (Bhatia et al., 2016; Mahfood Haddad, Hamdeh, Kanmanthareddy, & Alla, 2017). Butyrate administration significantly alleviated the HFD‐induced hepatic steatosis in ApoE−/− mice, as previously reported (Matheus, Monteiro, Oliveira, Maschio, & Collares‐Buzato, 2017; Mattace Raso et al., 2013). To find the differential expressed genes that may affect fatty liver and atherosclerosis, transcriptome profiling of mice liver was investigated. The observed changes in gene expression suggest butyrate has the metabolic effects in the liver, where butyrate can directly reach via portal circulation or secondarily through improving gut microbiota environment. Acot1 and Acot2 hydrolyses long‐chain fatty acyl‐CoA thioester bond, responsible for fatty acid synthesis (Franklin, Sathyanarayan, & Mashek, 2017; Moffat et al., 2014). Cyp4a14 hydroxylate medium‐chain fatty acids and gene disruption of CYP4A14 attenuated HFD‐induced liver lipid accumulation (Zhang et al., 2017). Here, we found that Acot1, Acot2, Cyp4a14, etc. decreased significantly by butyrate, suggesting the down‐regulation of genes involved in fatty acid synthesis and oxidation. Given that the profound effect of butyrate on microbiota composition, significant correlations exist between the specific genera and the expression of differentially expressed genes involved in lipid and glucose metabolism (Figure S4). In a word, these findings strengthen the beneficial actions of butyrate on both lipid and glucose homeostasis.
RCT process is termed as the HDL transport of excessive cholesterol from peripheral macrophages to the liver for metabolism and excretion in the faeces, which has been regarded crucial for prevention and treatment of atherosclerosis (Rosenson et al., 2012). Our present data show that Cyp7a1 expression in liver was increased with butyrate treatment and was inversely correlated with aorta lesion area and hepatic steatosis, possibly by accelerating liver cholesterol elimination via bile acids route in faeces and consequently promoting RCT process in mice. To elucidate the potential role of butyrate in RCT, expression of several key RCT transporters was investigated, and ABCA1 was identified significantly regulated by butyrate both in liver and peripheral macrophages. This finding is consistent with previous studies that CYP7A1 and ABCA1 were co‐ordinately regulated via multiple mechanisms to maintain hepatic cholesterol homeostasis (Lan et al., 2016; Li, Francl, Boehme, et al., 2013). ABCA1 promotes the efflux of cellular cholesterol to lipid‐free apolipoprotein A‐I and other apolipoproteins and thus emerged as a key player in RCT. A number of studies have demonstrated that ABCA1 inhibit the foam cells formation, which involved in the pathogenesis of atherosclerotic lesions, as summarized in a review (Chistiakov, Bobryshev, & Orekhov, 2016). Hence, it is generally acknowledged that promoting cholesterol efflux from macrophages is the primary mechanism by which ABCA1 protect against atherosclerosis (Genest & Choi, 2017; Voloshyna & Reiss, 2011). Overexpression of human ABCA1 in C57BL/6 mice resulted in an athero‐protective lipoprotein profiles and decreased atherosclerosis, when fed a high‐cholesterol diet (Joyce et al., 2002). In particular, specifically upregulation of ABCA1 in macrophages inhibited the atherogenesis (Van Eck et al., 2006). Therefore, up‐regulation of ABCA1 has been proposed as a therapeutic strategy for treating and preventing atherosclerosis.
We observed that butyrate enhanced ABCA1 expression in the liver, lesions from aortic roots and peritoneal macrophages of ApoE−/− mice, and elevated ABCA1‐mediated cholesterol efflux in RAW 264.7 cells. Of note, butyrate decreased cholesterol deposition and atherosclerotic lesion sizes in the aortae of ApoE−/− mice. Although the free cholesterol and cholesterol ester delivered by HDL via RCT process might contributed to the liver pool of cholesterol, Sahoo et al. (2004) reported that ABCA1‐mediated cholesterol efflux to ApoA‐I depleted the regulatory pool of hepatocyte cholesterol and hence decreased the secretion of triacylglycerol and ApoB in primary mouse and rat hepatocytes. Thus, the improved homeostasis of cellular cholesterol in liver may be responsible for the reduction of hepatic steatosis that is consistent with a previous study (Jeon et al., 2015). These findings suggest that butyrate‐induced ABCA1 expression could contribute to the reduction of hepatic steatosis and atherosclerosis. Sp1 plays an important role in the regulation of ABCA1 gene expression (Thymiakou, Zannis, & Kardassis, 2007) by binding to the ABCA1 promoter (Chen et al., 2011). Here, we found butyrate decreased Sp1 protein level while increased ABCA1 expression in RAW 264.7 cells. Inhibition Sp1 using siRNA further enhanced ABCA1 expression with butyrate, whereas blocking Sp1 binding to ABCA1 promoter with Mithramycin A abolished butyrate‐induced transactivation of ABCA1. We speculate that Sp1 binding to ABCA1 promoter is essential for transcriptional activation by butyrate. Nevertheless, there is still possibility that other transcription factor might be involved in the butyrate's induction of ABCA1. In addition, although butyrate directly increased the ABCA1 expression as evaluated in RAW 264.7 cells, the present study does not exclude the possible involvement of the gut microbiota manipulations in activation of ABCA1, for instance, the effect of microbiota‐generated butyrate, and further investigation is warranted.
In summary, we show that oral administration of microbiota metabolite butyrate significantly attenuates HFD‐induced atherosclerosis in ApoE−/− mice, possibly by further remodelling gut microbiota, reducing serum cholesterol and hepatic steatosis. More specifically, butyrate increased ABCA1 expression both in liver and in plaque (peripheral macrophages), via a Sp1 pathway, which may accelerate RCT and thus preventing atherosclerosis development (Figure 8). In conclusion, our findings suggest that butyrate plays a multi‐faceted role in its athero‐protective effect, at least an important part of which is mediated by modulation of lipid metabolism, especially through improving RCT. Although completely understanding the impact of butyrate on atherosclerosis will require further investigation, the present study may provide novel insights into metabolic effects of butyrate on lipid homeostasis and atherosclerosis.
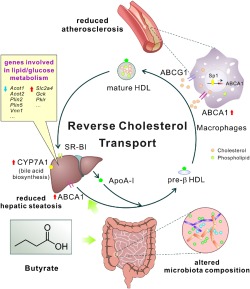
Figure 8.
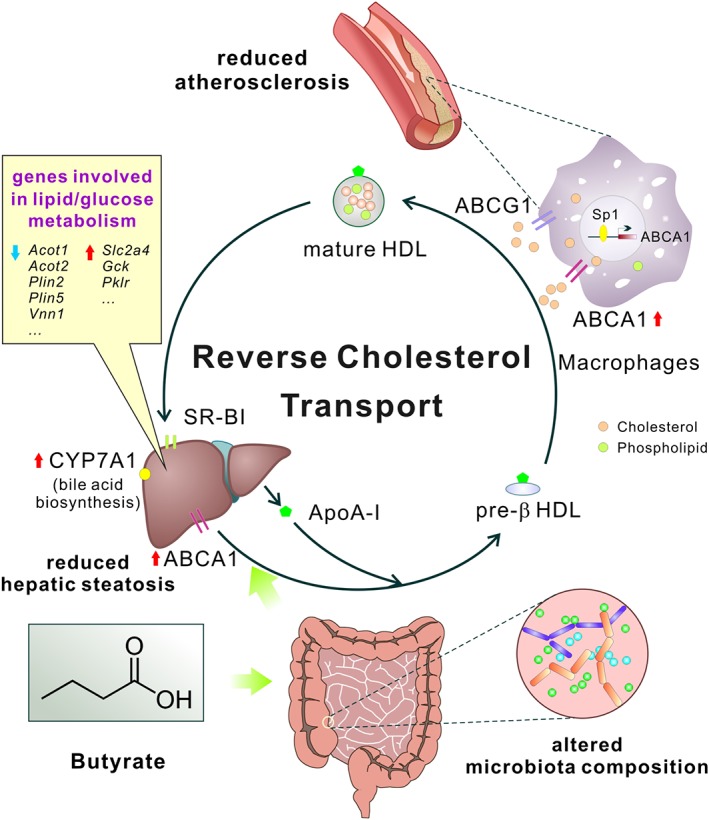
Schema showing the major roles of butyrate in inhibiting atherosclerosis of mice in this study. Butyrate treatment exerts multi‐faceted beneficial metabolic effects, such as remodelling gut microbiota, reducing hepatic steatosis, modulating lipid/glucose metabolism and, especially, improving RCT by up‐regulating ABCA1 through Sp1 pathway, all of which may eventually lead to ameliorating the development of atherosclerosis
AUTHOR CONTRIBUTIONS
B.H. and Y.D. conceived and designed the experiments. Y.D., C.‐Y.S., M.X., X.‐M.Z., X.‐X.L., Z.‐B.J., and L.W. performed the experiments. Y.D., X.‐X.L., and C.‐Y.S. analysed the data. Y.D. and B.H. wrote the paper.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Effect of butyrate on gut microbiota composition in HFD‐fed ApoE−/− mice. (A) Principal component analysis plot of microbiota is shown along the first two principal coordinate (PC) axes for HFD and HFD with butyrate or atorvastatin group. (B) Relative abundance of several genera between different groups. n = 7–8 for chow (group A) and HFD (group B) groups, n = 9–10 for HFD + butyrate (200 and 400 mg·kg‐1, group C and D). * P < 0.05
Figure S2. Effect of butyrate on food intake (A), total caloric intake (B) and body weight gain (C) in ApoE−/− mice after 16 weeks treatment. (D‐F) Correlation between aortae lesion area and serum non‐HDL‐cholesterol, total cholesterol, LDL‐cholesterol, respectively. Black circle: HFD; light gray circle: HFD + 200 mg·kg‐1 NaBu; dark grey circle: HFD + 400 mg·kg−1 NaBu.
Figure S3. Liver transcriptome analyses compared between HFD and HFD + 400 mg·kg‐1 butyrate groups. Gene Ontology term enrichments with Bonferroni tests for differentially expressed genes that up‐regulated (A) or down‐regulated (B) by butyrate (Fold change >1.5, FDR < 0.05).
Figure S4. Heat map of Spearman correlations between the differentially expressed genes involved in lipid and/or glucose metabolism and the abundance of microbiota at the genus level. * P < 0.05
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (81402929, 81473214 and 81621064), the National Mega‐Project for Innovative Drugs (2018ZX09711001‐007 and 2018ZX09711001‐003‐006), CAMS Innovation Fund for Medical Sciences (2017‐I2M‐1‐008 and 2016‐I2M‐2‐002) and Beijing Natural Science Foundation (7162129).
Du Y, Li X, Su C, et al. Butyrate protects against high‐fat diet‐induced atherosclerosis via up‐regulating ABCA1 expression in apolipoprotein E‐deficiency mice. Br J Pharmacol. 2020;177:1754–1772. 10.1111/bph.14933
REFERENCES
- Adam, C. L. , Thomson, L. M. , Williams, P. A. , & Ross, A. W. (2015). Soluble fermentable dietary fibre (Pectin) decreases caloric intake, adiposity and lipidaemia in high‐fat diet‐induced obese rats. PLoS ONE, 10(10), 1–14, e0140392 10.1371/journal.pone.0140392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar, E. C. , dos Santos, L. C. , Leonel, A. J. , de Oliveira, J. S. , Santos, E. A. , Navia‐Pelaez, J. M. , … Alvarez‐Leite, J. I. (2016). Oral butyrate reduces oxidative stress in atherosclerotic lesion sites by a mechanism involving NADPH oxidase down‐regulation in endothelial cells. The Journal of Nutritional Biochemistry, 34, 99–105. 10.1016/j.jnutbio.2016.05.002 [DOI] [PubMed] [Google Scholar]
- Aguilar, E. C. , Leonel, A. J. , Teixeira, L. G. , Silva, A. R. , Silva, J. F. , Pelaez, J. M. N. , … Alvarez‐Leite, J. I. (2014). Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutrition, Metabolism, and Cardiovascular Diseases, 24(6), 606–613. 10.1016/j.numecd.2014.01.002 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP collaborators (2019). The Concise Guide to pharmacology 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … GTP collaborators (2019). The Concise Guide to pharmacology 2019/20: Transporters. British Journal of Pharmacology, 176, S397–S493. 10.1111/bph.14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvaro, A. , Solà, R. , Rosales, R. , Ribalta, J. , Anguera, A. , Masana, L. , … Vallvé, J. C. (2008). Gene expression analysis of a human enterocyte cell line reveals downregulation of cholesterol biosynthesis in response to short‐chain fatty acids. IUBMB Life, 60(11), 757–764. 10.1002/iub.110 [DOI] [PubMed] [Google Scholar]
- Bao, Y. , Yang, Y. , Wang, L. , Gao, L. , Jiang, W. , Wang, L. , … Hong, B. (2009). Identification of trichostatin A as a novel transcriptional up‐regulator of scavenger receptor BI both in HepG2 and RAW 264.7 cells. Atherosclerosis, 204(1), 127–135. 10.1016/j.atherosclerosis.2008.08.041 [DOI] [PubMed] [Google Scholar]
- den Besten, G. , Bleeker, A. , Gerding, A. , van Eunen, K. , Havinga, R. , van Dijk, T. H. , … Bakker, B. M. (2015). Short‐chain fatty acids protect against high‐fat diet‐induced obesity via a PPARγ‐dependent switch from lipogenesis to fat oxidation. Diabetes, 64(7), 2398–2408. 10.2337/db14-1213 [DOI] [PubMed] [Google Scholar]
- Bhatia, L. , Scorletti, E. , Curzen, N. , Clough, G. F. , Calder, P. C. , & Byrne, C. D. (2016). Improvement in non‐alcoholic fatty liver disease severity is associated with a reduction in carotid intima‐media thickness progression. Atherosclerosis, 246, 13–20. 10.1016/j.atherosclerosis.2015.12.028 [DOI] [PubMed] [Google Scholar]
- Brown, J. M. , & Hazen, S. L. (2017). Targeting of microbe derived metabolites to improve human health: the next frontier for drug discovery. The Journal of Biological Chemistry, 292(21), 8560–8568. 10.1074/jbc.R116.765388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfora, E. E. , Jocken, J. W. , & Blaak, E. E. (2015). Short‐chain fatty acids in control of body weight and insulin sensitivity. Nature Reviews. Endocrinology, 11(10), 577–591. 10.1038/nrendo.2015.128 [DOI] [PubMed] [Google Scholar]
- Chawla, A. , Boisvert, W. A. , Lee, C. H. , Laffitte, B. A. , Barak, Y. , Joseph, S. B. , … Tontonoz, P. (2001). A PPAR γ‐LXR‐ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Molecular Cell, 7(1), 161–171. 10.1016/s1097-2765(01)00164-2 [DOI] [PubMed] [Google Scholar]
- Chen, X. , Zhao, Y. , Guo, Z. , Zhou, L. , Okoro, E. U. , & Yang, H. (2011). Transcriptional regulation of ATP‐binding cassette transporter A1 expression by a novel signaling pathway. The Journal of Biological Chemistry, 286(11), 8917–8923. 10.1074/jbc.M110.214429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov, D. A. , Bobryshev, Y. V. , & Orekhov, A. N. (2016). Macrophage‐mediated cholesterol handling in atherosclerosis. Journal of Cellular and Molecular Medicine, 20(1), 17–28. 10.1111/jcmm.12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin, M. P. , Sathyanarayan, A. , & Mashek, D. G. (2017). Acyl‐CoA thioesterase 1 (ACOT1) regulates PPARα to couple fatty acid flux with oxidative capacity during fasting. Diabetes, 66(8), 2112–2123. 10.2337/db16-1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, J. , Xu, Y. , Yang, Y. , Yang, Y. , Zheng, Z. , Jiang, W. , … Si, S. (2008). Identification of upregulators of human ATP‐binding cassette transporter A1 via high‐throughput screening of a synthetic and natural compound library. Journal of Biomolecular Screening, 13(7), 648–656. 10.1177/1087057108320545 [DOI] [PubMed] [Google Scholar]
- Gao, Z. , Yin, J. , Zhang, J. , Ward, R. E. , Martin, R. J. , Lefevre, M. , … Ye, J. (2009). Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes, 58(7), 1509–1517. 10.2337/db08-1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genest, J. , & Choi, H. Y. (2017). Novel approaches for HDL‐directed therapies. Current Atherosclerosis Reports, 19(12), 55 10.1007/s11883-017-0699-1 [DOI] [PubMed] [Google Scholar]
- Gonçalves, R. , & Mosser, D. M. (2015). The isolation and characterization of murine macrophages. Current Protocols in Immunology, 83, 14.1.1–14.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, H. V. , Sievenpiper, J. L. , Zurbau, A. , Blanco Mejia, S. , Jovanovski, E. , Au‐Yeung, F. , … Vuksan, V. (2016). The effect of oat β‐glucan on LDL‐cholesterol, non‐HDL‐cholesterol and apoB for CVD risk reduction: A systematic review and meta‐analysis of randomised‐controlled trials. The British Journal of Nutrition, 116(8), 1369–1382. 10.1017/S000711451600341X [DOI] [PubMed] [Google Scholar]
- Jeon, B. H. , Lee, Y. , Yun, M. R. , Kim, S. H. , Lee, B. W. , Kang, E. S. , … Cha, B. S. (2015). Increased expression of ATP‐binding cassette transporter A1 (ABCA1) as a possible mechanism for the protective effect of cilostazol against hepatic steatosis. Metabolism, 64(11), 1444–1453. 10.1016/j.metabol.2015.07.014 [DOI] [PubMed] [Google Scholar]
- Joyce, C. W. , Amar, M. J. , Lambert, G. , Vaisman, B. L. , Paigen, B. , Najib‐Fruchart, J. , … Santamarina‐Fojo, S. (2002). The ATP binding cassette transporter A1 (ABCA1) modulates the development of aortic atherosclerosis in C57BL/6 and apoE‐knockout mice. Proceedings of the National Academy of Sciences of the United States of America, 99(1), 407–412. 10.1073/pnas.012587699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara, K. , Krautkramer, K. A. , Org, E. , Romano, K. A. , Kerby, R. L. , Vivas, E. I. , … Rey, F. E. (2018). Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nature Microbiology, 3(12), 1461–1471. 10.1038/s41564-018-0272-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. , & Jena, G. (2016). Sodium butyrate reduces insulin‐resistance, fat accumulation and dyslipidemia in type‐2 diabetic rat: A comparative study with metformin. Chemico‐Biological Interactions, 254, 124–134. 10.1016/j.cbi.2016.06.007 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth, R. A. , Wang, Z. , Levison, B. S. , Buffa, J. A. , Org, E. , Sheehy, B. T. , … Hazen, S. L. (2013). Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nature Medicine, 19(5), 576–585. 10.1038/nm.3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh, A. , De Vadder, F. , Kovatcheva‐Datchary, P. , & Bäckhed, F. (2016). From dietary fiber to host physiology: Short‐chain fatty acids as key bacterial metabolites. Cell, 165(6), 1332–1345. 10.1016/j.cell.2016.05.041 [DOI] [PubMed] [Google Scholar]
- Koren, O. , Spor, A. , Felin, J. , Fåk, F. , Stombaugh, J. , Tremaroli, V. , … Bäckhed, F. (2011). Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proceedings of the National Academy of Sciences, 108(Supplement_1), 4592–4598. 10.1073/pnas.1011383107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühnast, S. , van der Tuin, S. J. , van der Hoorn, J. W. , van Klinken, J. B. , Simic, B. , Pieterman, E. , … Princen, H. M. (2015). Anacetrapib reduces progression of atherosclerosis, mainly by reducing non‐HDL‐cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. European Heart Journal, 36(1), 39–48. 10.1093/eurheartj/ehu319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan, X. , Yan, J. , Ren, J. , Zhong, B. , Li, J. , Li, Y. , … Lu, S. (2016). A novel long noncoding RNA Lnc‐HC binds hnRNPA2B1 to regulate expressions of Cyp7a1 and Abca1 in hepatocytic cholesterol metabolism. Hepatology, 64(1), 58–72. 10.1002/hep.28391 [DOI] [PubMed] [Google Scholar]
- Lattimer, J. M. , & Haub, M. D. (2010). Effects of dietary fiber and its components on metabolic health. Nutrients, 2(12), 1266–1289. 10.3390/nu2121266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. Y. , Wang, Z. , Li, X. S. , Hazen, S. L. , & Tang, W. H. W. (2018). Relationship between statin use and trimethylamine n‐oxide in cardiovascular risk assessment. Journal of the American College of Cardiology, 71(Supplement 11), 115 10.1016/S0735-1097(18)30656-9 [DOI] [Google Scholar]
- Li, T. , Francl, J. M. , Boehme, S. , & Chiang, J. Y. (2013). Regulation of cholesterol and bile acid homeostasis by the cholesterol 7α‐hydroxylase/steroid response element‐binding protein 2/microRNA‐33a axis in mice. Hepatology, 58(3), 1111–1121. 10.1002/hep.26427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Yi, C. X. , Katiraei, S. , Kooijman, S. , Zhou, E. , Chung, C. K. , … Wang, Y. (2018). Butyrate reduces appetite and activates brown adipose tissue via the gut‐brain neural circuit. Gut, 67(7), 1269–1279. 10.1136/gutjnl-2017-314050 [DOI] [PubMed] [Google Scholar]
- Lin, Y. , Bai, L. , Chen, Y. , Zhu, N. , Bai, Y. , Li, Q. , … Liu, E. (2015). Practical assessment of the quantification of atherosclerotic lesions in apoE−/− mice. Molecular Medicine Reports, 12(4), 5298–5306. 10.3892/mmr.2015.4084 [DOI] [PubMed] [Google Scholar]
- Louis, P. , Young, P. , Holtrop, G. , & Flint, H. J. (2010). Diversity of human colonic butyrate‐producing bacteria revealed by analysis of the butyryl‐CoA: Acetate CoA‐transferase gene. Environmental Microbiology, 12(2), 304–314. 10.1111/j.1462-2920.2009.02066.x [DOI] [PubMed] [Google Scholar]
- Mahfood Haddad, T. , Hamdeh, S. , Kanmanthareddy, A. , & Alla, V. M. (2017). Nonalcoholic fatty liver disease and the risk of clinical cardiovascular events: A systematic review and meta‐analysis. Diabetes and Metabolic Syndrome: Clinical Research and Reviews, 11(Suppl 1), S209–S216. 10.1016/j.dsx.2016.12.033 [DOI] [PubMed] [Google Scholar]
- Marcil, V. , Delvin, E. , Seidman, E. , Poitras, L. , Zoltowska, M. , Garofalo, C. , … Levy, E. (2002). Modulation of lipid synthesis, apolipoprotein biogenesis, and lipoprotein assembly by butyrate. American Journal of Physiology. Gastrointestinal and Liver Physiology, 283(2), G340–G346. 10.1152/ajpgi.00440.2001 [DOI] [PubMed] [Google Scholar]
- Martínez, I. , Perdicaro, D. J. , Brown, A. W. , Hammons, S. , Carden, T. J. , Carr, T. P. , … Walter, J. (2013). Diet‐induced alterations of host cholesterol metabolism are likely to affect the gut microbiota composition in hamsters. Applied and Environmental Microbiology, 79(2), 516–524. 10.1128/AEM.03046-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheus, V. A. , Monteiro, L. , Oliveira, R. B. , Maschio, D. A. , & Collares‐Buzato, C. B. (2017). Butyrate reduces high‐fat diet‐induced metabolic alterations, hepatic steatosis and pancreatic beta cell and intestinal barrier dysfunctions in prediabetic mice. Experimental Biology and Medicine (Maywood, N.J.), 242(12), 1214–1226. 10.1177/1535370217708188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattace Raso, G. , Simeoli, R. , Russo, R. , Iacono, A. , Santoro, A. , Paciello, O. , … Meli, R. (2013). Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet. PLoS ONE, 8(7), 1–13, e68626 10.1371/journal.pone.0068626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijnikman, A. S. , Gerdes, V. E. , Nieuwdorp, M. , & Herrema, H. (2018). Evaluating causality of gut microbiota in obesity and diabetes in humans. Endocrine Reviews, 39(2), 133–153. 10.1210/er.2017-00192 [DOI] [PubMed] [Google Scholar]
- Milks, M. , He, X. , Sharkey‐Toppen, T. , Smart, S. , McCarthy, B. , Somogyi, A. , … Raman, S. (2018). Statin use is associated with lower trimethylamine‐n‐oxide (TMAO) level in adults at risk of atherosclerotic cardiovascular disease, independent of serum cholesterol and renal function. Journal of Clinical Lipidology, 12(2), 567–568. 10.1016/j.jacl.2018.03.074 [DOI] [Google Scholar]
- Moffat, C. , Bhatia, L. , Nguyen, T. , Lynch, P. , Wang, M. , Wang, D. , … Seifert, E. L. (2014). Acyl‐CoA thioesterase‐2 facilitates mitochondrial fatty acid oxidation in the liver. Journal of Lipid Research, 55(12), 2458–2470. 10.1194/jlr.M046961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica, M. P. , Mattace Raso, G. , Cavaliere, G. , Trinchese, G. , De Filippo, C. , Aceto, S. , … Meli, R. (2017). Butyrate regulates liver mitochondrial function, efficiency, and dynamics in insulin‐resistant obese mice. Diabetes, 66(5), 1405–1418. 10.2337/db16-0924 [DOI] [PubMed] [Google Scholar]
- Ohira, H. , Tsutsui, W. , & Fujioka, Y. (2017). Are short chain fatty acids in gut microbiota defensive players for inflammation and atherosclerosis? Journal of Atherosclerosis and Thrombosis, 24(7), 660–672. 10.5551/jat.RV17006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi, A. , Khera, A. , Berry, J. D. , Givens, E. G. , Ayers, C. R. , Wedin, K. E. , … Shaul, P. W. (2014). HDL cholesterol efflux capacity and incident cardiovascular events. The New England Journal of Medicine, 371(25), 2383–2393. 10.1056/NEJMoa1409065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenson, R. S. , Brewer, H. B. Jr. , Davidson, W. S. , Fayad, Z. A. , Fuster, V. , Goldstein, J. , … Yvan‐Charvet, L. (2012). Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation, 125(15), 1905–1919. 10.1161/CIRCULATIONAHA.111.066589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo, D. , Trischuk, T. C. , Chan, T. , Drover, V. A. , Ho, S. , Chimini, G. , … Lehner, R. (2004). ABCA1‐dependent lipid efflux to apolipoprotein AI mediates HDL particle formation and decreases VLDL secretion from murine hepatocytes. Journal of Lipid Research, 45(6), 1122–1131. 10.1194/jlr.M300529-JLR200 [DOI] [PubMed] [Google Scholar]
- Smith, J. D. , Le Goff, W. , Settle, M. , Brubaker, G. , Waelde, C. , Horwitz, A. , & Oda (2004). ABCA1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein A‐I. Journal of Lipid Research, 45(4), 635–644. 10.1194/jlr.M300336-JLR200 [DOI] [PubMed] [Google Scholar]
- Sookoian, S. , & Pirola, C. J. (2008). Non‐alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: A systematic review. Journal of Hepatology, 49(4), 600–607. 10.1016/j.jhep.2008.06.012 [DOI] [PubMed] [Google Scholar]
- Tang, W. H. , & Hazen, S. L. (2014). The contributory role of gut microbiota in cardiovascular disease. The Journal of Clinical Investigation, 124(10), 4204–4211. 10.1172/JCI72331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thymiakou, E. , Zannis, V. I. , & Kardassis, D. (2007). Physical and functional interactions between liver X receptor/retinoid X receptor and Sp1 modulate the transcriptional induction of the human ATP binding cassette transporter A1 gene by oxysterols and retinoids. Biochemistry, 46(41), 11473–11483. 10.1021/bi700994m [DOI] [PubMed] [Google Scholar]
- Van Eck, M. , Singaraja, R. R. , Ye, D. , Hildebrand, R. B. , James, E. R. , Hayden, M. R. , & Van Berkel, T. J. (2006). Macrophage ATP‐binding cassette transporter A1 overexpression inhibits atherosclerotic lesion progression in low‐density lipoprotein receptor knockout mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 26(4), 929–934. [DOI] [PubMed] [Google Scholar]
- Voloshyna, I. , & Reiss, A. B. (2011). The ABC transporters in lipid flux and atherosclerosis. Progress in Lipid Research, 50(3), 213–224. 10.1016/j.plipres.2011.02.001 [DOI] [PubMed] [Google Scholar]
- Wang, P. , Li, D. , Ke, W. , Liang, D. , Hu, X. , & Chen, F. (2019). Resveratrol‐induced gut microbiota reduces obesity in high‐fat diet‐fed mice. International Journal of Obesity, 1–13. 10.1038/s41366-019-0332-1 [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Klipfell, E. , Bennett, B. J. , Koeth, R. , Levison, B. S. , Dugar, B. , … Hazen, S. L. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature, 472(7341), 57–63. 10.1038/nature09922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann, A. , Allahyar, A. , Greiner, T. U. , Plovier, H. , Lundén, G. Ö. , Larsson, T. , … Bäckhed, F. (2013). Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host & Microbe, 14(5), 582–590. 10.1016/j.chom.2013.09.012 [DOI] [PubMed] [Google Scholar]
- Witjes, J. J. , van Raalte, D. H. , & Nieuwdorp, M. (2015). About the gut microbiome as a pharmacological target in atherosclerosis. European Journal of Pharmacology, 763(Pt A), 75–78. 10.1016/j.ejphar.2015.06.023 [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Li, S. , Zhou, Y. , Su, W. , Ruan, X. , Wang, B. , … Guan, Y. (2017). Ablation of cytochrome P450 omega‐hydroxylase 4A14 gene attenuates hepatic steatosis and fibrosis. Proceedings of the National Academy of Sciences of the United States of America, 114(12), 3181–3185. 10.1073/pnas.1700172114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Z. , Zhu, B. , Sun, Y. , Ai, C. , Wang, L. , Wen, C. , … Liu, X. (2018). Sulfated polysaccharide from sea cucumber and its depolymerized derivative prevent obesity in association with modification of gut microbiota in high‐fat diet‐fed mice. Molecular Nutrition & Food Research, 62(23), 1–12, e1800446 10.1002/mnfr.201800446 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of butyrate on gut microbiota composition in HFD‐fed ApoE−/− mice. (A) Principal component analysis plot of microbiota is shown along the first two principal coordinate (PC) axes for HFD and HFD with butyrate or atorvastatin group. (B) Relative abundance of several genera between different groups. n = 7–8 for chow (group A) and HFD (group B) groups, n = 9–10 for HFD + butyrate (200 and 400 mg·kg‐1, group C and D). * P < 0.05
Figure S2. Effect of butyrate on food intake (A), total caloric intake (B) and body weight gain (C) in ApoE−/− mice after 16 weeks treatment. (D‐F) Correlation between aortae lesion area and serum non‐HDL‐cholesterol, total cholesterol, LDL‐cholesterol, respectively. Black circle: HFD; light gray circle: HFD + 200 mg·kg‐1 NaBu; dark grey circle: HFD + 400 mg·kg−1 NaBu.
Figure S3. Liver transcriptome analyses compared between HFD and HFD + 400 mg·kg‐1 butyrate groups. Gene Ontology term enrichments with Bonferroni tests for differentially expressed genes that up‐regulated (A) or down‐regulated (B) by butyrate (Fold change >1.5, FDR < 0.05).
Figure S4. Heat map of Spearman correlations between the differentially expressed genes involved in lipid and/or glucose metabolism and the abundance of microbiota at the genus level. * P < 0.05