

Abstract
Proteolysis‐targeting chimeras are a new drug modality that exploits the endogenous ubiquitin proteasome system to degrade a protein of interest for therapeutic benefit. As the first‐generation of proteolysis‐targeting chimeras have now entered clinical trials for oncology indications, it is timely to consider the theoretical safety risks inherent with this modality which include off‐target degradation, intracellular accumulation of natural substrates for the E3 ligases used in the ubiquitin proteasome system, proteasome saturation by ubiquitinated proteins, and liabilities associated with the “hook effect” of proteolysis‐targeting chimeras This review describes in vitro and non‐clinical in vivo data that provide mechanistic insight of these safety risks and approaches being used to mitigate these risks in the next generation of proteolysis‐targeting chimera molecules to extend therapeutic applications beyond life‐threatening diseases.
Abbreviations
- Cmax
maximum concentration
- CRBN
https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=3086
- ER
oestrogen receptor
- https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=1030
immunomodulatory drugs
- PD
pharmacodynamic
- PK
pharmacokinetic
- POI
protein of interest
- PROTAC
proteolysis‐targeting chimeras
- UPS
ubiquitin proteasome system
1. INTRODUCTION
Proteolysis‐targeting chimeras (PROTACs) are drug‐like molecules that induce the degradation of proteins (Bondeson et al., 2018; Churcher, 2018; Deshaies, 2015; Mayor‐Ruiz & Winter, 2019; Paiva & Crews, 2019; Pettersson & Crews, 2019). They work by bringing into close proximity an E3 ligase and a protein of interest (POI), inducing ubiquitination of the POI and its subsequent degradation via the proteasome. Based on their mechanism of action, PROTACs can potentially target any type of protein including the previously “undruggable” class of protein therapeutic targets. As a therapeutic modality, interest in PROTACs has increased following Arvinas' announcements in 2019 of Phase 1 clinical trials with two PROTAC candidate drugs. Indeed, it has been suggested that PROTAC could be the next blockbuster therapy (Scudellari, 2019) and many pharmaceutical companies have invested in developing PROTACs as therapeutic agents. However, as for every promising new drug modality, there can be new challenges, for example, the ability to identify and investigate clinical safety risks using suitable preclinical models. Here, we assess the emerging PROTAC drug modality and identify key safety risks and questions that should be considered during discovery, optimization, and clinical development of PROTAC.
2. PROTAC MECHANISM OF ACTION AND APPLICATION IN DRUG DEVELOPMENT
PROTACs are hetero‐bifunctional molecules that consist of a ligand for a POI and a ligand for an E3 ligase connected by a linker (Figure 1). For efficient degradation of the POI, the PROTAC must first enter the cells and interact with both the POI and the E3 ligase in a ternary complex to trigger the ubiquitination of the target. Once ubiquitinated, the POI is degraded by the proteasome, which is one of the major cellular protein degradation pathways (the other being the autophagy‐lysosome system) used by eukaryotic cells. Importantly, PROTAC molecules are recycled after the ubiquitination of the POI and can engage in a new reaction. This has an important consequence from a pharmacodynamic (PD) perspective as PROTACs can therefore work in a catalytic fashion where one PROTAC molecule can degrade several targets. Furthermore, the PROTAC mode of action potentially offers a long duration of action that will depend on the stability of PROTAC inside the cell and the rate of POI resynthesis. Another PROTAC property includes the potential to target any type of protein as long as a small molecule, low MW ligand with appropriate affinity can be generated for the POI. This has been discussed in detail in several recent reviews (Churcher, 2018; Collins, Wang, Caldwell, & Chopra, 2017; Hughes & Ciulli, 2017; Mayor‐Ruiz & Winter, 2019; Ohoka, Shibata, Hattori, & Naito, 2016; Pettersson & Crews, 2019; Schapira, Calabrese, Bullock, & Crews, 2019).
Figure 1.
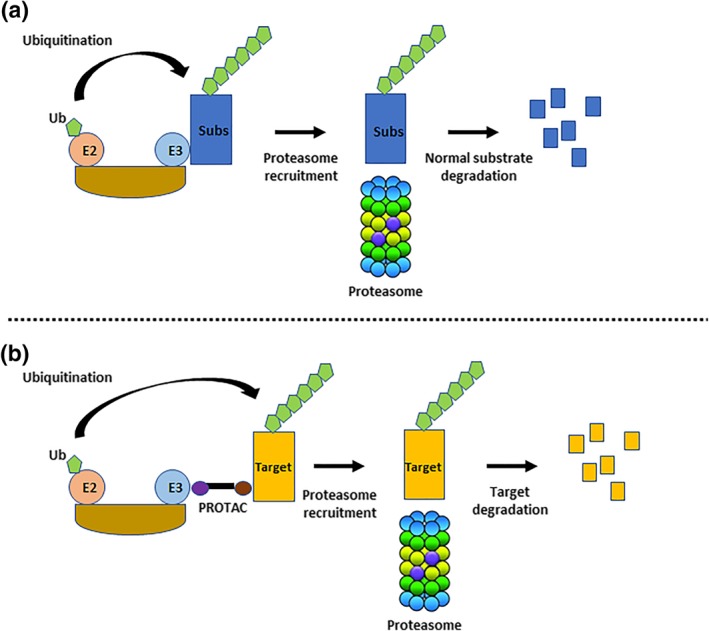
Ubiquitin‐Proteasome System and PROTAC mode of action. (a) E3 ligase substrates (Subs) are ubiquitinated leading to their targeting to the proteasome for degradation. (b) PROTAC bridges a target protein and an E3 to trigger the ubiquitination of the target and its degradation using the proteasome
At present, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=3086 and VHL are the most common E3 ligases utilized in the development of PROTACs although other E3 ligases have been utilized to a lesser extent including MDM2, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2791, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2757&familyId=876&familyType=OTHER, and RNF114 (Pettersson & Crews, 2019; Toure & Crews, 2016; Zou, Ma, & Wang, 2019). Both pharmaceutical companies and academic labs are seeking to identify new E3 ligases to serve as a platform for PROTAC lead generation. This has important consequences from a safety perspective as each E3 ligase will present its own specific safety risks that will depend on its tissue distribution, substrate specificity, biological function, and selectivity of the warhead used to bind the E3 ligase (see Section 2).
The list of proteins targeted by PROTACs has increased rapidly over recent years, with most of the targets focusing on oncology indications (Table 1). This is mainly due to the repurposing of existing small‐molecule inhibitors against oncology targets and exploiting the PROTAC mode of action in an effort to achieve greater efficacy. Consequently, the first wave of PROTAC targets is not in the “undruggable” space, which was one of the distinguishing features of this modality. This illustrates a pragmatic approach used by pharmaceutical companies to develop their PROTAC programme, seeking rapid proof of concept with the first‐generation molecules to better understand the nuances of PROTAC drug discovery and development. With continued success of the first wave of PROTAC molecules, one can expect the development of PROTACs that target previously undruggable targets, which will require hit finding strategies to identify ligands for these targets (e.g., structure‐based computational methods for in silico screening and biophysical assays to quantify small‐molecule binding to target protein) and then optimizing the ligand and its coupling to an E3 ligase warhead. We anticipate that the identification of a suitable ligand for PROTAC will require a considerable effort into its validation from a potency but also safety consideration. Critically, engagement of POI by PROTAC does not require pharmacological modulation of the target in the traditional sense; that is, enzyme active site engagement is not necessary. Thus, it may be anticipated that a more diverse chemical space may be explored in discovery of POI binding leads. From a safety perspective, new ligands will need to be screened for potential safety liabilities as done for traditional small molecules, which include the important assessment of on/off target liabilities. However, the potential for newer, atypical chemistries will limit safety inference from databases compiled from more traditional leads (e.g., kinase inhibitors).
Table 1.
Examples of PROTAC targets being pursued therapeutically, their class, and the E3 ligase used in the PROTAC mode of action
| Target | Class | E3 ligase |
|---|---|---|
| BRD2/4/7/9 | Transcription factor | CRBN |
| VHL | ||
| MDM2 | ||
| RIPK2 | Kinase | VHL |
| Akt | Kinase | VHL |
| ABL | Kinase | VHL |
| BCR‐ABL | Fusion protein | CRBN |
| VHL | ||
| cIAP1 | ||
| Smad3 | Transcription factor | VHL |
| Tau | Scaffolding activity | VHL |
| Keap1 | ||
| DHODH | Enzyme | VHL |
| CDK6/9 | Kinase | CRBN |
| Sirt2 | Deacetylase | CRBN |
| BTK | Kinase | CRBN |
| MDM2 | ||
| ALK | Kinase | CRBN |
| VHL | ||
| HDAC6 | Deacetylase | CRBN |
| VHL | ||
| TRIM24 | E3 | VHL |
| EGFR | Kinase | VHL |
| CRBN | ||
| IRAK4 | Kinase | CRBN |
| VHL | ||
| STAT3 | Transcription factor | CRBN |
| MetAP‐2 | Enzyme | SCF |
| VHL | ||
| ER | Transcription factor | SCF |
| VHL | ||
| CRBN | ||
| cIAP1 | ||
| AR | Transcription factor | SCF |
| VHL | ||
| CRBN | ||
| MDM2 | ||
| cIAP1 | ||
| AHR | Transcription factor | VHL |
| CRABP | Transporter | cIAP1 |
| RAR | Transcription factor | cIAP1 |
| DAPK1 | Kinase | CMA |
| PSD‐95 | Kinase | CMA |
| α‐Synuclein | Scaffolding activity | CMA |
| TACC3 | Scaffolding activity | APC/C |
| IKZF1/3 | Transcription factors | CRBN |
| Bcl6 | Transcription factor | CRBN |
| FAK | Kinase | VHL |
| CRBN | ||
| MET | Kinase | VHL |
| Bcl‐2/Bcl‐Xl | Transcription factors | CRBN |
| VHL |
Table 1 . Examples of PROTAC targets being pursued therapeutically, their class and the E3 ligase used in the PROTAC mode of action. Met‐AP2 (methionine aminopeptidase 2); ER (estrogen receptor); AR (androgen receptor); CRABP (cellular retinoic acid‐binding protein); RAR (retinoic acid receptor); DAPK1 (death‐associated protein kinase 1); PSD‐95 (postsynaptic density protein 95); TACC3 (transforming acidic coiled‐coil‐containing protein 3); IKZF (ikaros family zinc finger protein); Bcl6 (b‐cell lymphoma 6); FAK (focal adhesion kinase); MET (proto‐Oncogene, Receptor Tyrosine Kinase); BRD (bromodomain‐containing protein); RIPK2 (receptor‐interacting serine/threonine‐protein kinase 2); Akt (protein kinase B); ABL (abelson murine leukemia); BCR‐ABL (breakpoint cluster region protein‐abelson murine leukemia); Smad3 (SMAD family member 3); DHODH (dihydroorotate dehydrogenase); CDK (cyclin dependent kinase); Sirt2 (sirtuin 2); BTK (bruton tyrosine kinase); ALK (ALK receptor tyrosine kinase); HDAC6 (histone deacetylase 6); TRIM24 (tripartite motif containing 24); EGFR (epidermal growth factor receptor); Bcl2 (B‐cell lymphoma 2); Bcl‐xL (B‐cell lymphoma‐extra large); SMARCA2/4 (SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A, Member); IRAK4 (interleukin‐1 receptor‐associated kinase 4); STAT3 (Signal transducer and activator of transcription 3).
3. SAFETY CHALLENGES FOR PROTACS
As a therapeutic modality, PROTACs achieved an important milestone in 2019 with the initiation of the first Phase I clinical trials targeting the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=628 in patients with metastatic castration‐resistant prostate cancer and the https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=96 (ER) for locally advanced or metastatic ER positive/https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2019 negative breast cancer. The authorization of these trials by the U.S. Food and Drug Administration means that the non‐clinical safety profile of these two PROTAC molecules aligns with safety guidelines to enable dosing in cancer patients. And although the non‐clinical safety characteristics of each compound has yet to be published, several safety concerns regarding this modality have been identified based on the particular mechanism of therapeutic action. These include prolonged on‐target protein degradation, off‐target protein degradation, unexpected pharmacology due to off‐target binary bindings, competition with the natural substrates of the E3 ligase used in the PROTAC mode of action, saturation of the proteasome by the ubiquitinated target, and the generation of a so‐called hook effect. Each of these topics is described in more detail in the following sections.
3.1. Prolonged target protein degradation
The PROTAC can quickly and efficiently degrade its primary target, with potential for prolonged degradation depending on the pharmacokinetic (PK) profile of the compound and the resynthesis rate of the target protein within the disease‐relevant tissue. Compared to reversible small‐molecule inhibitors, PROTACs have the potential for greater efficacy via the pronounced and extended target degradation (i.e., deeper and longer inhibition). In many ways, the non‐equilibrium PROTAC pharmacological mechanism of action resembles that of irreversible antagonists, which can produce a PD response that extends beyond that predicted from their PK profile. Irreversible inhibitors have been successfully exploited to achieve clinically effective drugs in many therapeutic areas (Strelow, 2017), so prolonged target inhibition can be tolerated, although clearly, this is target and context dependent. Optimizing dose and schedule to achieve an efficacious level of target degradation in the disease tissue can be a challenge if on‐target toxicity is a dose‐limiting concern. For oncology indications, choosing PROTAC targets that drive synthetic lethality has the potential for improved tolerability, as the tumour may be more sensitive to target degradation compared to normal tissue. In addition, choosing an E3 ligase that is preferentially expressed in the disease tissue compared to normal tissue has the potential to improve therapeutic margin for on‐target toxicity (Khan et al., 2019).
3.2. Off‐target protein degradation
Many studies have shown that PROTACs are not entirely selective and can degrade proteins other than the primary target (Bai et al., 2017; Bondeson et al., 2018; Brand et al., 2019; Matyskiela et al., 2016; Yang et al., 2019; Zorba et al., 2018). Degradation of a protein that is not directly bound by the PROTAC can arise as a consequence of “bystander degradation” where the degradation of a protein that is not directly bound by the PROTAC becomes ubiquitinated and hence degraded as part of the same complex with the PROTAC POI (Hsu et al., 2019; Maniaci et al., 2017; Potjewyd et al., 2019). In other instances, this is an off‐target effect due to the neo‐morphic interactions with so‐called neosubstrates, which become ubiquitinated by the E3 ligase and subsequently degraded (Figure 2). Off‐target effects can also arise from the binary engagement of the target binding ligand of the PROTAC to other proteins in the same way as the POI. A well‐characterized example of off‐target activities with potential safety liabilities for PROTACs comes from the immunomodulatory drugs (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=1030) https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7327, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7348 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7331, which function as ligands for the CRBN E3 ligase. IMiDs are small molecules that have been used clinically for decades against several types of cancer without a clear understanding of their therapeutic mechanism of action. Recent studies have shown that IMiDs recruit neo‐substrates to CRBN, which induces their degradation (Figure 3). There are many neo‐substrates degraded by IMiDs: Most of them are transcription factors such as SALL4 and some Ikaros proteins (IKZF1 and IKZF3) but not all of them, as shown by the degradation of the translation regulator GSPT1 (Donovan et al., 2018; Ishoey et al., 2018; Matyskiela et al., 2016; Matyskiela et al., 2018; Sievers et al., 2018). SALL4 and p63 have recently been linked to the teratogenic effect of these IMiDs, while Ikaros proteins regulate important cell‐fate decisions during haematopoiesis, which may explain the haematotoxicity seen with the IMiDs (Asatsuma‐Okumura et al., 2019; Donovan et al., 2018; John & Ward, 2011). There are other adverse effects described with the use of IMiDs that include cardiovascular, hepatic, and neuronal toxicities, although the mechanism(s) behind these toxicities are not understood and, therefore, it is not known if they are mediated via degradation of neo‐substrates.
Figure 2.
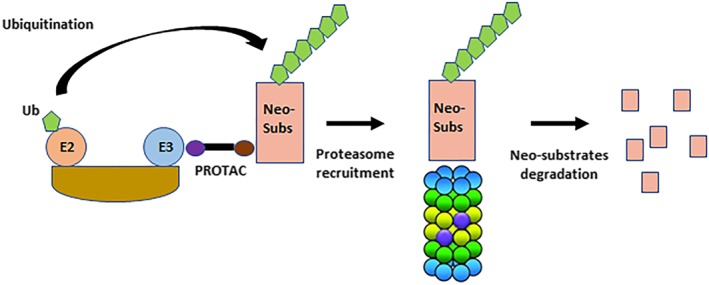
Safety challenges inherent to the PROTAC modality. PROTAC can degrade other proteins than the primary target via the recruitment of neo‐substrates (Neo‐subs) to the E3 ligase
Figure 3.
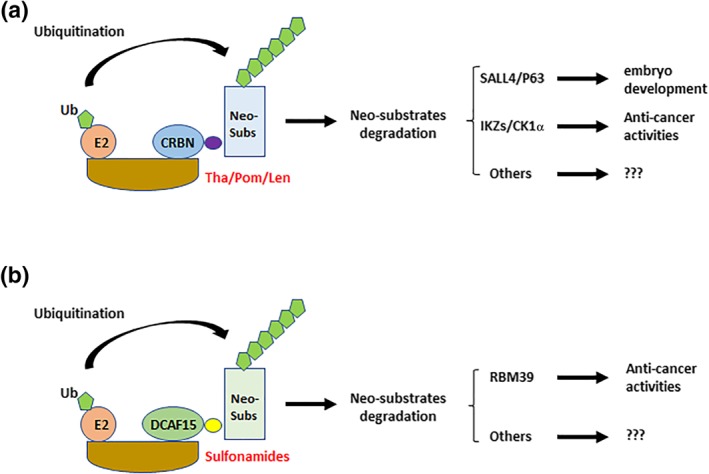
Off‐target activities of PROTAC. (a) IMiDs such as thalidomide (Tha), pomalidomide (Pom) and lenalidomide (Len) degrade neo‐substrates via their recruitment to CRBN. The IMiD substrates SALL4 and P63 are transcription factors involved in embryo development while Ikaros proteins (IKZs) and CK1α are anti‐cancer factors. (b) Anti‐cancer sulfonamides degrade neo‐substrates via their recruitment to DCAF15. The sulfonamide substrate RBM39 is a splicing regulating factor. There are many other sulfonamide substrates whose function is poorly characterised
Many PROTAC molecules have been created by incorporating an IMiD or IMiD derivative as an CRBN warhead. From a safety perspective, it is important to determine whether this imparts undesired features of SALL4 and Ikaros protein degradation and the consequent safety liabilities. Indeed, published examples indicate good reasons for concern. A BTK‐targeting PROTAC that incorporated pomalidomide as the CRBN warhead induced potent degradation of the IMiD substrates IKZF1, IKZF3, ZNF827, and ZFP91 (Zorba et al., 2018). Both IKZF1 and IKZF3 were degraded by a CDK6‐targeting PROTAC that used lenalidomide as the E3 ligase warhead as well as a HDAC6‐targeting PROTAC that incorporated a pomalidomide warhead (Brand et al., 2019; Wu et al., 2019). Although the effects of these PROTACs on haematopoiesis was not reported, it would be important to assess this safety risk using in vitro assays and/or incorporating relevant endpoints in a preclinical, investigative toxicology study.
Another class of small molecules that induce targeted protein degradation are the anti‐cancer sulfonamides, which include https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=7046, E7820, and CQS (Han et al., 2017; Uehara et al., 2017). These compounds function by recruiting neo‐substrates to the E3 ligase DCAF15 for degradation, in a mechanism similar to that used by the IMiDs (Figure 3). The most characterized neo‐substrate degraded by sulfonamides is the mRNA splicing factor RBM39, which has been shown to be the main target for the anti‐proliferative effect seen with anti‐cancer sulfonamide treatment (Han et al., 2017; Uehara et al., 2017). Novartis has recently solved the complex structure of DCAF15‐DDB1‐DDA1‐Indisulam‐RBM39 and from this work identified the precise mechanism by which indisulam mediates interaction between RBM39 and DCAF15 (Han et al., 2017). Indisulam forms a tripartite complex with DCAF15 and RBM39, where DCAF15 and RBM39 bind together mainly through non‐polar interactions. The observation that anti‐cancer sulfonamides make contact with both proteins simultaneously illustrates a diversity in this so‐called molecular glue mechanism and also highlights the challenge to hijack DCAF15 as a novel E3 ligase (Bussiere et al., 2020). Indisulam has been shown to be highly selective and in HCT116 cells showed degradation of RBM39 as the primary target together with RBM23 (https://doi.org/10.1101/737510). However, additional global proteomic studies have shown that other proteins are down‐regulated by anti‐cancer sulfonamides (Uehara et al., 2017).
Anti‐cancer sulfonamides have been in clinical trials although progress has been limited due to dose‐limiting myelosuppression (neutropenia and thrombocytopenia; van Kesteren et al., 2005; Zandvliet et al., 2008). Fatigue, alopecia, and injection site reactions were also observed. If anti‐cancer sulfonamides are used as a DCAF15 warhead for PROTAC applications, these toxicities would need to be carefully assessed in non‐clinical studies to ensure that the PROTAC has a safety profile to support clinical development for the particular patient population.
As described above, PROTAC molecules can induce the degradation of neo‐substrates that cannot be predicted based on the small‐molecule E3 ligase ligands alone. Therefore, ideally monitoring the degradation of the global proteome following PROTAC treatment in cell models or tissues from in vivo preclinical studies should be performed. The current method of choice to study the dynamics of protein degradation in cellular systems is a multiplexed isobaric tandem mass tag labelling approach for simultaneous identification and quantification of thousands of proteins. There are several reviews describing this technique, which has become a cornerstone “omics” technology used by most pharmaceutical companies engaged in PROTAC drug discovery (Bondeson et al., 2018; Donovan et al., 2018; Farnaby et al., 2019; Huang et al., 2018; Treumann & Thiede, 2010). A comprehensive proteomics strategy towards investigating PROTAC safety will consider two elements: selectivity and mechanism. Selectivity entails characterizing the binding and degradation selectivity of the PROTAC of interest, using affinity‐based chemical and global proteomics respectively. Chemical proteomics profiling identifies the binding partners of the PROTAC of interest, as well as the specific E3 ligase and POI ligands (along with their respective proteomics K Ds). Such information allows the assessment of safety risks derived from the degradation profile linked to specific binary binding events. Global proteomics techniques have been broadly applied in the PROTAC field (Bondeson et al., 2018; Donovan et al., 2018; Farnaby et al., 2019; Huang et al., 2018; Olson et al., 2018) as a way to measure degradation selectivity as well as identifying previously unknown degradation off targets across a proteome of 8,000–10,000 proteins, as a result of PROTAC treatment. Further, global profiling allows to monitor downstream protein dynamics and pathway effects by varying cell priming conditions and/or PROTAC treatment conditions. This parameter is particularly relevant when degrading transcription factors and epigenetic targets and delivers mechanistic insights that link degradation selectivity to in vivo observations, including adverse safety events. In addition to understanding the proteome‐wide effects of PROTAC treatment, mechanistic parameters of interest include absolute quantitation and relative stoichiometry of E3 ligase and POI levels, as well as the turnover rates of the target of interest in disease and safety relevant input material, both of which will inform mechanistic models for understanding and predicting ternary complex formation (to be discussed in a later section). A relevant consideration while using MS techniques to assess safety liabilities of protein degraders is the utilization of the most relevant biological system to answer specific questions. For example, the identification of SALL4 has remained elusive for many years until the correct biological system expressing that protein was utilized. Additionally, optimal cellular systems must be chosen for profiling against other reported off targets of CRBN warheads, such as IKZF1 and IKZF3 as mentioned earlier (Donovan et al., 2018; Matyskiela et al., 2018) as well as p63 (Asatsuma‐Okumura et al., 2019).
Following identification of off‐target proteins that are degraded by the PROTAC, it is necessary to investigate the safety risks of these undesired effects through understanding the function of the degraded proteins and the physiological consequences of reducing their levels in different tissues. This can be a formidable task, especially when the function of the degraded protein has not been characterized. For example, the BTK‐targeting PROTAC cited above degrades ZNF827, a poorly characterized protein potentially involved in telomere function and homologous recombination (Pickett & Reddel, 2015). Fortunately, toxicologists can use a collection of databases and in silico tools to learn about the tissue expression and potential biological pathways in which a particular protein functions to develop hypotheses of potential toxicities. These hypotheses could be tested experimentally by using shRNA/siRNA and genetic approaches (CRISPR knockout in cell and animal models). The resulting information can be used to influence the design of in vivo investigative studies with the PROTAC to understand safety risks in non‐clinical species by evaluating organ‐specific toxicities, to determine the exposures at which these toxicities occur relative to the predicted clinically efficacious exposure, and to monitor the reversibility of toxicological effects on PROTAC withdrawal.
3.3. Disruption of cellular proteostasis
As PROTACs utilize an E3 ligase to initiate proteasomal degradation of a POI, it is possible that PROTAC‐POI complexes compete with natural substrates for binding to the E3 ligase for ubiquitination and degradation, resulting in accumulation of those substrates and potentially perturbing specific cellular pathways (Figure 4). Furthermore, there is also a risk that PROTACs increase the cellular concentration of ubiquitinated proteins resulting in saturation of the proteasome, which could have consequences on cellular homeostasis. The proteasome controls the cellular content of proteins that regulate many aspects of cellular biology including cell cycle, cell growth, immune homeostasis, and metabolic activity (Thibaudeau & Smith, 2019) and, therefore, alterations in proteasome activity and protein accumulation could have deleterious effects. For example, the proteasome governs the presentation of MHC class I antigens which is critical for the function of the immune response (Santambrogio, Berendam, & Engelhard, 2019). Dysregulation of the proteasome is also involved in many disorders such as neurodegeneration and autoimmune diseases (Rousseau & Bertolotti, 2018).
Figure 4.
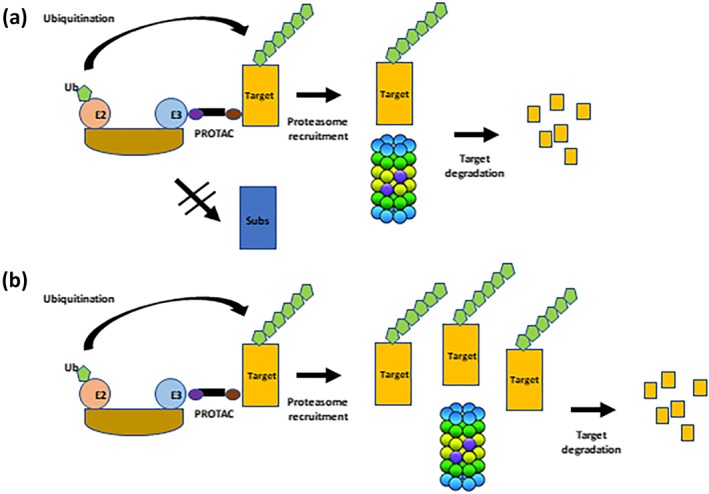
Safety challenges inherent to the PROTAC modality. (1) PROTAC may affect the degradation of the natural E3 ligase substrates. (2) PROTAC may saturate the proteasome via an increase in target occupancy
While the concern about blocking natural E3 substrate and proteasomes is theoretically relevant, experimentally, it has not been observed at low PROTAC concentrations. One reason is that as PROTAC acts catalytically and so crucially sub‐stoichiometrically. The PROTAC will only engage a sub‐population of E3 ligase molecules, particularly in the case of abundant E3 ligases (as is the case with CRBN and VHL). This means that a large proportion of E3 ligase molecules remain unbound by the PROTAC and can carry on their natural activity against their native substrate molecules, with little to no effective inhibition being observed. This has been observed at least with the E3 ligases for which natural substrates are known, such as VHL. With VHL based‐PROTACs, several studies have shown that there exists a very large concentration window between PROTAC‐mediated degradation (in the pM–nM range with the best degraders) and stabilization of HIF‐1α, which is only observed at high micromolar range, which is >1000 fold higher than that required for degradation (Bondeson et al., 2015; Frost et al., 2016; Maniaci et al., 2017; Zengerle, Chan, & Ciulli, 2015). However, the physical and PK properties of PROTACs, which, given their relatively higher MW, are distinct from traditional small‐molecule drugs and achieving appropriate PK will be challenging (Edmondson, Yang, & Fallan, 2019). Despite the catalytic mode of action, obtaining the desired exposure and exposure duration, for example, when targeting POI with high turnover, could necessitate plasma Cmax values of ten‐fold, or greater, the minimum effective concentration (see Raina et al., 2016). The relatively high Cmax therefore increases the potential for off‐target degradation and the narrowing of the therapeutic margin. This will be especially the case if the off‐target is an essential cell homeostasis or cardiovascular function gene with a slow turnover. Note that the degradation mode of action for off‐targets is likely to be more profound than traditional off‐target pharmacology where transient inhibition is a function of PK; PROTACs on the other hand have the potential for extended unintended pharmacology since recovery of the off‐target protein level is a function of protein resynthesis rather than drug PK.
Thus, further in vivo studies to better understand the PK/PD relationship between the POI and key off‐targets are therefore required. In addition, more detailed investigations of whether PROTACs affect natural E3 ligase substrate levels or induce proteasome saturation and, if so, the toxicological consequences of these events. A global proteomics approach is the method of choice to monitor the change in concentration of E3 ligase substrates following PROTAC treatment. This includes antibodies that detect and enrich for GlyGly‐modified Lys of ubiquitinated peptides, followed by MS for protein identification, enabling the mapping of tens of thousands of ubiquitination sites (Swatek et al., 2019). To monitor the proteasome activity, several commercial kits are available using a cell permeable proteasome substrate as a readout. Quantifying endoplasmic reticulum stress (e.g., by altered expression of chaperones such as Bip) can be an indirect measure of proteasome activity, as a decrease in proteasome activity would lead to the accumulation of misfolded proteins in the endoplasmic reticulum (Sun & Brodsky, 2019). Data from these experiments could then be used to infer toxicity liabilities and lead to focussed toxicological assessments.
3.4. Implications of the “hook” effect
At high concentrations, PROTACs saturate binding to the target and to the E3 ligase resulting in the formation of binary complexes instead of the productive ternary complex (E3‐PROTAC‐Target; Figure 5). This prevents target ubiquitination and degradation, a situation that has been observed with many PROTACs and has been termed the “hook” effect (Paiva & Crews, 2019; Pettersson & Crews, 2019). In theory, the formation of binary complexes could have two adverse consequences. The first concern is an increase in off‐target degradation activity via binding of the E3‐PROTAC binary complex to lower affinity off‐targets (Figure 5). The second concern is the potential for a pharmacological response driven by the PROTAC‐POI binary complex interaction that is different from target degradation. This possibility would be realized, for example, if the POI ligand (and resulting PROTAC) had agonist activity. This is a concern for PROTAC targeting androgen receptors containing mutations within the ligand binding domain, as androgen receptor antagonists function as agonists for these mutations (Prekovic et al., 2016). The hook effect is an intrinsic property of any PROTAC, and the underlying mechanisms in terms of ternary complex formation is generally understood (Douglass, Miller, Sparer, Shapiro, & Spiegel, 2013). However, while ternary complex formation is an essential step for PROTAC‐mediated protein degradation, this is not sufficient to induce degradation (Smith et al., 2019). Consequently, the onset of the hook effect observed from binding experiments may be very different from what is observed in cell‐based degradation assays. While this is clearly an issue for PROTAC efficacy, it is not clear whether the hook effect has any safety liabilities. However, on closer inspection, it may complicate toxicological assessments: In theory, the observed maximum tolerated doses in animal studies (required by regulatory authorities prior to starting clinical trials) may be a consequence of reduced POI degradation, that is, higher concentrations of PROTAC inadequately “tox the target.” Thus, careful exploration of a broad dose range may be necessary to ensure that lower doses that may result in paradoxically greater toxicity as a consequence of greater POI degradation, are properly understood. Nevertheless, it should be possible to build PK–PD models that take PROTAC‐specific properties into account and use such models to predict the onset of the hook effect. The accuracy of such predictions will depend on the model itself, but also on whether key parameters can be experimentally determined in relevant cell‐based assays. Even then the hook effect is likely to be different between tissues as it will depend on the concentration of both the target and the E3 ligase, the PROTAC exposure achieved in the tissue and possibly additional parameters as well. Until it has been demonstrated, how such PK–PD models can be useful for PROTAC dose predictions to avoid the hook effect, remains to be seen. One way to avoid this issue would be to alleviate the hook effect, for example, by increasing the protein–protein interaction or cooperativity of the ternary complex. This has been shown to be possible in vitro, but it remains to be seen whether this can be achieved in vivo.
Figure 5.
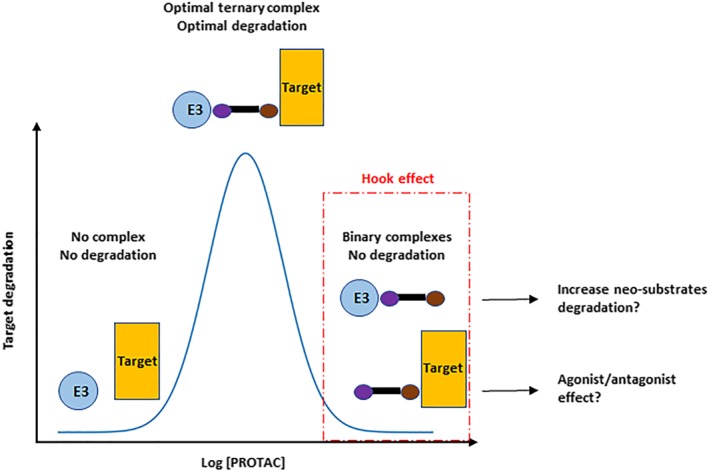
Hook effect with PROTAC. At high intracellular PROTAC concentration, binary complexes are favoured over ternary complexes, resulting in reduced target degradation. Binary complexes could cause an increase in degradation of neo‐substrates as well as the PROTAC acting as a traditional small molecule inhibitor/activator, reflecting the pharmacophore of the POI ligand
4. OPPORTUNITY TO DEVELOP “SAFER” PROTACS
Safety insight gained from the first generation of PROTACs will help direct efforts towards safer PROTAC molecules, which may be particularly important for non‐oncology indications where higher therapeutic safety margins are typically required, for example, for chronic therapy and non‐life‐threatening diseases and where other modalities provide some therapeutic benefit and therefore efficacy/safety balance is a higher hurdle for clinical use. And although no clinical safety data are yet available for PROTAC molecules, one can ask whether “safer” PROTAC can be developed. For example, it has been shown that each IMiD has a distinct degradation profile with pomalidomide inducing the degradation of a larger set of proteins, compared with those degraded with thalidomide (Donovan et al., 2018), suggesting that CRBN ligands may have different safety profiles. Therefore optimizing the E3 ligase warhead could decrease off‐target degradation activity and improve the therapeutic index of PROTAC molecules.
Another way to minimize off‐target degradation is to modify the linker and its attachment orientation to the E3 ligase warhead. This has been shown recently in several studies. For example, Brand et al. (2019) generated a potent PROTAC against CDK6 that was not adequately selective as two IMiD substrates, IKZF1 and IKZF3, were also degraded. However, modifications in the linker region resulted in a PROTAC with no observed degradation of these Ikaros proteins. Wu et al. (2019) also increased the selectivity of a PROTAC targeting HDAC6 by modifying how the linker attached to the E3 ligase warhead. Finally, the Crews lab has published examples where changes to the PROTAC linker increased PROTAC selectivity, either by modifying the interaction between the E3 ligase and the recruited protein or by modifying the orientation of the recruited ligase. Using promiscuous CRBN‐ and VHL‐PROTAC that bind to more than 50 kinases, they showed that only a few kinases were actually degraded, due to the selectivity of interactions between the E3 ubiquitin ligase and the target protein (Bondeson et al., 2018). In a second example, modifying linker attachments and lengths for a VHL PROTAC resulted in differential degradation of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1499 or https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1502&familyId=519&familyType=ENZYME MAPK (Smith et al., 2019). Finally, a recent paper from Chen's group has shown intra‐BET selectivity of PROTACs despite using pan‐selective BET targeting ligands (Jiang et al., 2020). These studies are important from a safety perspective as they provide proof of principle that PROTAC chemistry can be optimized to reduce off‐target degradation activity. However, this currently remains an empirical approach for each POI ligand and E3 ligase warhead.
Most PROTACs are designed to utilize ubiquitously expressed E3 ligases and validated ligase warheads. One way to restrict PROTAC activity to a specific tissue is to engage an E3 with a unique tissue expression. In theory, this should avoid PROTAC‐mediated degradation in tissues where the E3 ligase is not expressed. For example, Khan et al (2019) demonstrated that a https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2845‐targeting VHL PROTAC could potently degrade BCL‐XL in cancer cells and had reduced on‐target toxicity to platelets due to low VHL expression in these cells. Another strategy is to use an E3 ligase with a unique expression in a specific disease, for example, in a specific cancer (Mansour, 2018). Many pharmaceutical and biotech companies have announced plans to discover new E3 ligases for PROTAC applications with most employing proprietary strategies. However, a challenge shared by all these strategies is the difficulty and time required to identify ligands for new E3 ligases that can be used for PROTAC applications.
5. CONCLUSION
The PROTACs have progressed from an academic tool to degrade protein into a potential clinically useful therapy in about 20 years. Many pharmaceutical and biotech companies have invested in PROTACs and molecules with this mechanism of action have now entered clinical trials. Routes to assess and better understand PROTAC safety risks have been identified, which is an essential step towards delivering efficacious and safe PROTACs to patients. It will be interesting to see how the PROTAC field advances in the next 20 years and whether it develops into the blockbuster therapeutic modality with clinical benefit across a wide range of therapy areas, as envisioned by many drug developers.
5.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Cidlowski et al., 2019; Alexander, Fabbro et al., 2019a, 2019b; Alexander, Kelly et al., 2019).
CONFLICT OF INTEREST
All authors are employees of AstraZeneca.
Moreau K, Coen M, Zhang AX, et al. Proteolysis‐targeting chimeras in drug development: A safety perspective. Br J Pharmacol. 2020;177:1709–1718. 10.1111/bph.15014
Present Address Muireann Coen, Biomolecular Medicine, Systems Medicine, Department of Metabolism, Digestion and Reproduction, Imperial College, South Kensington, London, SW7 2AZ.
REFERENCES
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Nuclear hormone receptors. British Journal of Pharmacology, 176, S229–S246. 10.1111/bph.14750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators (2019a). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176, S247–S296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators (2019b). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Faccenda, E. , … CGTP Collaborators (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Introduction and Other Protein Targets. British Journal of Pharmacology, 176, S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asatsuma‐Okumura, T. , Ando, H. , De Simone, M. , Yamamoto, J. , Sato, T. , Shimizu, N. , et al. (2019). p63 is a cereblon substrate involved in thalidomide teratogenicity. Nature Chemical Biology, 15, 1077–1084. 10.1038/s41589-019-0366-7 [DOI] [PubMed] [Google Scholar]
- Bai, L. , Zhou, B. , Yang, C. Y. , Ji, J. , McEachern, D. , Przybranowski, S. , … Wang, S. (2017). Targeted degradation of BET proteins in triple‐negative breast cancer. Cancer Research, 77, 2476–2487. 10.1158/0008-5472.CAN-16-2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson, D. P. , Mares, A. , Smith, I. E. , Ko, E. , Campos, S. , Miah, A. H. , … Zinn, N. (2015). Catalytic in vivo protein knockdown by small‐molecule PROTACs. Nature Chemical Biology, 11, 611–617. 10.1038/nchembio.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson, D. P. , Smith, B. E. , Burslem, G. M. , Buhimschi, A. D. , Hines, J. , Jaime‐Figueroa, S. , … Crews, C. M. (2018). Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chemical Biology, 25, 78–87. e75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, M. , Jiang, B. , Bauer, S. , Donovan, K. A. , Liang, Y. , Wang, E. S. , … Müller, A. C. (2019). Homolog‐selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chemical Biology, 26, 300–306. e309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere, D. E. , Xie, L. , Srinivas, H. , Shu, W. , Burke, A. , Be, C. , … Paulk, J. (2020). Structural basis of indisulam‐mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nature Chemical Biology, 16, 15–23. 10.1038/s41589-019-0411-6 [DOI] [PubMed] [Google Scholar]
- Churcher, I. (2018). Protac‐induced protein degradation in drug discovery: Breaking the rules or just making new ones? Journal of Medicinal Chemistry, 61, 444–452. 10.1021/acs.jmedchem.7b01272 [DOI] [PubMed] [Google Scholar]
- Collins, I. , Wang, H. , Caldwell, J. J. , & Chopra, R. (2017). Chemical approaches to targeted protein degradation through modulation of the ubiquitin‐proteasome pathway. The Biochemical Journal, 474, 1127–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies, R. J. (2015). Protein degradation: prime time for PROTACs. Nature Chemical Biology, 11, 634–635. [DOI] [PubMed] [Google Scholar]
- Donovan, K. A. , An, J. , Nowak, R. P. , Yuan, J. C. , Fink, E. C. , Berry, B. C. , … Fischer, E. S. (2018). Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife, 7, e38430 10.7554/eLife.38430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass, E. F. Jr. , Miller, C. J. , Sparer, G. , Shapiro, H. , & Spiegel, D. A. (2013). A comprehensive mathematical model for three‐body binding equilibria. Journal of the American Chemical Society, 135, 6092–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson, S. D. , Yang, B. , & Fallan, C. (2019). Proteolysis targeting chimeras (PROTACs) in 'beyond rule‐of‐five' chemical space: Recent progress and future challenges. Bioorganic & Medicinal Chemistry Letters, 29, 1555–1564. [DOI] [PubMed] [Google Scholar]
- Farnaby, W. , Koegl, M. , Roy, M. J. , Whitworth, C. , Diers, E. , Trainor, N. , … Ciulli, A. (2019). BAF complex vulnerabilities in cancer demonstrated via structure‐based PROTAC design. Nature Chemical Biology, 15, 672–680. 10.1038/s41589-019-0294-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost, J. , Galdeano, C. , Soares, P. , Gadd, M. S. , Grzes, K. M. , Ellis, L. , … Ciulli, A. (2016). Potent and selective chemical probe of hypoxic signalling downstream of HIF‐α hydroxylation via VHL inhibition. Nature Communications, 7, 1–12. 10.1038/ncomms13312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, T. , Goralski, M. , Gaskill, N. , Capota, E. , Kim, J. , Ting, T. C. , … Nijhawan, D. (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science, 356, eaal3755 10.1126/science.aal3755 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, J. H. , Rasmusson, T. , Robinson, J. , Pachl, F. , Read, J. , Kawatkar, S. , … Argyrou, A. (2019). EED‐targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 complex. Cell Chemical Biology, 27(1), 41–46. [DOI] [PubMed] [Google Scholar]
- Huang, H. T. , Dobrovolsky, D. , Paulk, J. , Yang, G. , Weisberg, E. L. , Doctor, Z. M. , … Gray, N. S. (2018). A chemoproteomic approach to query the degradable kinome using a multi‐kinase degrader. Cell Chemical Biology, 25, 88–99e86. 10.1016/j.chembiol.2017.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, S. J. , & Ciulli, A. (2017). Molecular recognition of ternary complexes: A new dimension in the structure‐guided design of chemical degraders. Essays in Biochemistry, 61, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishoey, M. , Chorn, S. , Singh, N. , Jaeger, M. G. , Brand, M. , Paulk, J. , … Winter, G. E. (2018). Translation termination factor GSPT1 is a phenotypically relevant off‐target of heterobifunctional phthalimide degraders. ACS Chemical Biology, 13, 553–560. 10.1021/acschembio.7b00969 [DOI] [PubMed] [Google Scholar]
- Jiang, F. , Wei, Q. , Li, H. , Li, H. , Cui, Y. , Ma, Y. , … Chen, Y. (2020). Discovery of novel small molecule induced selective degradation of the bromodomain and extra‐terminal (BET) bromodomain protein BRD4 and BRD2 with cellular potencies. Bioorganic & Medicinal Chemistry, 28, 115181. [DOI] [PubMed] [Google Scholar]
- John, L. B. , & Ward, A. C. (2011). The Ikaros gene family: Transcriptional regulators of hematopoiesis and immunity. Molecular Immunology, 48, 1272–1278. 10.1016/j.molimm.2011.03.006 [DOI] [PubMed] [Google Scholar]
- van Kesteren, C. , Zandvliet, A. S. , Karlsson, M. O. , Mathot, R. A. , Punt, C. J. , Armand, J. P. , … Roché, H. H. (2005). Semi‐physiological model describing the hematological toxicity of the anti‐cancer agent indisulam. Investigational New Drugs, 23, 225–234. 10.1007/s10637-005-6730-3 [DOI] [PubMed] [Google Scholar]
- Khan, S. , Zhang, X. , Lv, D. , Zhang, Q. , He, Y. , Zhang, P. , … Zhou, D. (2019). A selective BCL‐XL PROTAC degrader achieves safe and potent antitumor activity. Nature Medicine, 25, 1938–1947. 10.1038/s41591-019-0668-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci, C. , Hughes, S. J. , Testa, A. , Chen, W. , Lamont, D. J. , Rocha, S. , … Ciulli, A. (2017). Homo‐PROTACs: Bivalent small‐molecule dimerizers of the VHL E3 ubiquitin ligase to induce self‐degradation. Nature Communications, 8, 1–14. 10.1038/s41467-017-00954-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour, M. A. (2018). Ubiquitination: Friend and foe in cancer. The International Journal of Biochemistry & Cell Biology, 101, 80–93. [DOI] [PubMed] [Google Scholar]
- Matyskiela, M. E. , Couto, S. , Zheng, X. , Lu, G. , Hui, J. , Stamp, K. , … Chamberlain, P. P. (2018). SALL4 mediates teratogenicity as a thalidomide‐dependent cereblon substrate. Nature Chemical Biology, 14, 981–987. 10.1038/s41589-018-0129-x [DOI] [PubMed] [Google Scholar]
- Matyskiela, M. E. , Lu, G. , Ito, T. , Pagarigan, B. , Lu, C. C. , Miller, K. , … Chamberlain, P. P. (2016). A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature, 535, 252–257. 10.1038/nature18611 [DOI] [PubMed] [Google Scholar]
- Mayor‐Ruiz, C. , & Winter, G. E. (2019). Identification and characterization of cancer vulnerabilities via targeted protein degradation. Drug Discovery Today: Technologies, 31, 81–90. [DOI] [PubMed] [Google Scholar]
- Ohoka, N. , Shibata, N. , Hattori, T. , & Naito, M. (2016). Protein knockdown technology: Application of ubiquitin ligase to cancer therapy. Current Cancer Drug Targets, 16, 136–146. [DOI] [PubMed] [Google Scholar]
- Olson, C. M. , Jiang, B. , Erb, M. A. , Liang, Y. , Doctor, Z. M. , Zhang, Z. , … Gray, N. S. (2018). Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nature Chemical Biology, 14, 163–170. 10.1038/nchembio.2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiva, S. L. , & Crews, C. M. (2019). Targeted protein degradation: elements of PROTAC design. Current Opinion in Chemical Biology, 50, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson, M. , & Crews, C. M. (2019). PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discovery Today: Technologies, 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett, H. A. , & Reddel, R. R. (2015). Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nature Structural & Molecular Biology, 22, 875–880. [DOI] [PubMed] [Google Scholar]
- Potjewyd, F. , Turner, A. W. , Beri, J. , Rectenwald, J. M. , Norris‐Drouin, J. L. , Cholensky, S. H. , … James, L. I. (2019). Degradation of polycomb repressive complex 2 with an EED‐targeted bivalent chemical degrader. Cell Chemical Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekovic, S. , van Royen, M. E. , Voet, A. R. , Geverts, B. , Houtman, R. , Melchers, D. , … Helsen, C. (2016). The effect of F877L and T878A mutations on androgen receptor response to enzalutamide. Molecular Cancer Therapeutics, 15, 1702–1712. 10.1158/1535-7163.MCT-15-0892 [DOI] [PubMed] [Google Scholar]
- Raina, K. , Lu, J. , Qian, Y. , Altieri, M. , Gordon, D. , Rossi, A. M. , … Coleman, K. G. (2016). PROTAC‐induced BET protein degradation as a therapy for castration‐resistant prostate cancer. Proceedings of the National Academy of Sciences of the United States of America, 113, 7124–7129. 10.1073/pnas.1521738113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau, A. , & Bertolotti, A. (2018). Regulation of proteasome assembly and activity in health and disease. Nature Reviews. Molecular Cell Biology, 19, 697–712. 10.1038/s41580-018-0040-z [DOI] [PubMed] [Google Scholar]
- Santambrogio, L. , Berendam, S. J. , & Engelhard, V. H. (2019). The antigen processing and presentation machinery in lymphatic endothelial cells. Frontiers in Immunology, 10, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira, M. , Calabrese, M. F. , Bullock, A. N. , & Crews, C. M. (2019). Targeted protein degradation: Expanding the toolbox. Nature Reviews. Drug Discovery, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- Scudellari, M. (2019). Protein‐slaying drugs could be the next blockbuster therapies. Nature, 567, 298–300. 10.1038/d41586-019-00879-3 [DOI] [PubMed] [Google Scholar]
- Sievers, Q. L. , Petzold, G. , Bunker, R. D. , Renneville, A. , Slabicki, M. , Liddicoat, B. J. , … Thomä, N. H. (2018). Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science, 362, eaat0572 10.1126/science.aat0572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, B. E. , Wang, S. L. , Jaime‐Figueroa, S. , Harbin, A. , Wang, J. , Hamman, B. D. , & Crews, C. M. (2019). Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nature Communications, 10, 1–13. 10.1038/s41467-018-08027-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelow, J. M. (2017). A perspective on the kinetics of covalent and irreversible inhibition. SLAS Discov, 22, 3–20. 10.1177/1087057116671509 [DOI] [PubMed] [Google Scholar]
- Sun, Z. , & Brodsky, J. L. (2019). Protein quality control in the secretory pathway. The Journal of Cell Biology, 218, 3171–3187. 10.1083/jcb.201906047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek, K. N. , Usher, J. L. , Kueck, A. F. , Gladkova, C. , Mevissen, T. E. T. , Pruneda, J. N. , … Komander, D. (2019). Insights into ubiquitin chain architecture using Ub‐clipping. Nature, 572, 533–537. 10.1038/s41586-019-1482-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibaudeau, T. A. , & Smith, D. M. (2019). A practical review of proteasome pharmacology. Pharmacological Reviews, 71, 170–197. 10.1124/pr.117.015370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure, M. , & Crews, C. M. (2016). Small‐molecule PROTACs: New approaches to protein degradation. Angewandte Chemie (International Ed. in English), 55, 1966–1973. 10.1002/anie.201507978 [DOI] [PubMed] [Google Scholar]
- Treumann, A. , & Thiede, B. (2010). Isobaric protein and peptide quantification: Perspectives and issues. Expert Review of Proteomics, 7, 647–653. 10.1586/epr.10.29 [DOI] [PubMed] [Google Scholar]
- Uehara, T. , Minoshima, Y. , Sagane, K. , Sugi, N. H. , Mitsuhashi, K. O. , Yamamoto, N. , … Owa, T. (2017). Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nature Chemical Biology, 13, 675–680. 10.1038/nchembio.2363 [DOI] [PubMed] [Google Scholar]
- Wu, H. , Yang, K. , Zhang, Z. , Leisten, E. D. , Li, Z. , Xie, H. , … Tang, W. (2019). Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. Journal of Medicinal Chemistry, 62, 7042–7057. 10.1021/acs.jmedchem.9b00516 [DOI] [PubMed] [Google Scholar]
- Yang, J. , Li, Y. , Aguilar, A. , Liu, Z. , Yang, C. Y. , & Wang, S. (2019). Simple structural modifications converting a bona fide MDM2 PROTAC degrader into a molecular glue molecule: A cautionary tale in the design of PROTAC degraders. Journal of Medicinal Chemistry, 62, 9471–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandvliet, A. S. , Schellens, J. H. , Dittrich, C. , Wanders, J. , Beijnen, J. H. , & Huitema, A. D. (2008). Population pharmacokinetic and pharmacodynamic analysis to support treatment optimization of combination chemotherapy with indisulam and carboplatin. British Journal of Clinical Pharmacology, 66, 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle, M. , Chan, K. H. , & Ciulli, A. (2015). Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chemical Biology, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba, A. , Nguyen, C. , Xu, Y. , Starr, J. , Borzilleri, K. , Smith, J. , … Calabrese, M. F. (2018). Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proceedings of the National Academy of Sciences of the United States of America, 115, E7285–E7292. 10.1073/pnas.1803662115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, Y. , Ma, D. , & Wang, Y. (2019). The PROTAC technology in drug development. Cell Biochemistry and Function, 37, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]