

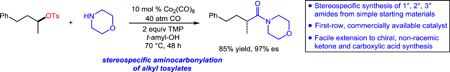
Abstract
Metal-catalyzed aminocarbonylation is a standard approach for installing amide functionality in chemical synthesis. Despite broad application of this transformation using aryl or vinyl electrophiles, few examples involve unactivated aliphatic substrates. Furthermore, there are no stereocontrolled aminocarbonylations of alkyl electrophiles known. Herein, we report a stereospecific aminocarbonylation of unactivated alkyl tosylates for the synthesis of enantioenriched amides. This cobalt-catalyzed transformation uses a remarkably broad range of amines in a convergent manner, and proceeds with excellent stereospecificity and chemoselectivity.
Keywords: alkyl tosylate, aminocarbonylation, cobalt, stereospecific, synthetic methods
Graphical Abstract
The amide functional group is ubiquitous in organic compounds, and is typically synthesized via the coupling of carboxylic acids and amines.[1] An alternative concise, catalytic approach to amide synthesis is the aminocarbonylation of activated halides or pseudohalides using amines and CO, first reported by Heck.[2] While this transformation has seen widespread use in applications up to industrial scale,[3] aminocarbonylation of widely available, unactivated aliphatic electrophiles remains a major challenge. The few reported processes require the use of alkyl iodides and either high energy irradiation or organotin initiators in radical-mediated processes (Figure 1).[4] In addition, these reactions often provide byproducts involving multiple CO insertions.[4d,5] The lack of a practical, chemoselective aminocarbonylation of unactivated alkyl electrophiles is a major limitation of this attractive approach to amide synthesis.
Figure 1.

Aminocarbonylations of unactivated alkyl electrophiles.
The control of absolute stereochemistry in the aminocarbonylation of alkyl electrophiles is another critical goal which is unlikely to be attained via radical-mediated, stereoablative transformations. Recently, we reported a new strategy for stereospecific C–C bond construction of unactivated alkyl electrophiles using nucleophilic cobaltate catalysis.[6,7] We hypothesize that this transformation involved a diene insertion of an enantioenriched acylcobalt intermediate. We proposed that an alternative catalytic mechanism involving nucleophilic substitution of this intermediate could unlock a range of new, stereospecific transformations. Herein, we report our initial studies towards this goal via a catalytic, stereospecific aminocarbonylation of unactivated alkyl electrophiles using a broad range of amines and a commercially available cobalt catalyst. This highly chemoselective, catalytic method facilitates the stereospecific synthesis of diverse amides from easily accessed chiral, non-racemic alkyl tosylate building blocks.
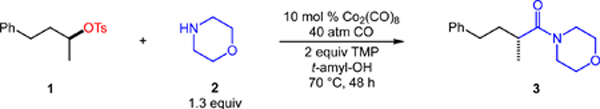
Our studies commenced with the aminocarbonylation of unactivated secondary alkyl tosylate (S)-1 with morpholine 2 (Table 1). A catalytic system comprised of 10 mol % of commercially available Co2(CO)8 and 2 equivalents of TMP (2,2,6,6-tetramethylpiperidine) provided amide 3 in good yield with high stereocontrol (86% 1H NMR yield, 97% es, entry 1) under 40 atm CO in t-amyl-OH.[8] Substituting 20 mol % of the cobaltate K[Co(CO)4] led to a slightly lower yield but similar stereospecificity (entry 2). Decreasing the loading of the catalyst to 5 mol % also decreased the yield, which we attribute to increased direct alkylation of morpholine (entry 3). Other metal carbonyls (e.g., Mn2(CO)10) were inefficient in the aminocarbonylation (entry 4). Substituting 4-(trimethylsilyl)morpholine as the nitrogen nucleophile–previously used to intercept acylcobalt intermediates[9]–provided a lower yield of the desired amide (62%, entry 5). Alternative organic bases (entry 6) or inorganic bases (entry 7) were inferior to TMP. Removing TMP from the reaction also significantly lowered the reaction efficiency (entry 8). The use of t-amyl-OH as solvent was critical, as a polar aprotic solvent (e.g., THF) gave significantly reduced yields of amide (entry 9). Lower pressures of CO (10 or 20 atm) could be used, however, the yield and enantiospecificity of the reaction decreased (entry 10—11).[10] No reaction occurred in the absence of the cobalt catalyst (entry 12).
Table 1.
Stereospecific, cobalt-catalyzed aminocarbonylation of an unactivated alkyl tosylate with morpholine.
 | |||
|---|---|---|---|
| entry | variation from standard conditions above | yield (%)[a] | es (%)[b] |
| 1 | none | 85 | 97 |
| 2 | 20 mol % K[Co(CO)4] instead of Co2(CO)8 | 70 | 98 |
| 3 | 5 mol % instead of 10 mol % Co2(CO)8 | 68 | 96 |
| 4 | Mn2(CO)10 instead of Co2(CO)8 | <2 | |
| 5 | 4-(trimethylsilyl)morpholine instead of morpholine | 62 | 96 |
| 6 | TEA instead of TMP | 39 | 87 |
| 7 | Cs2CO3 instead of TMP | 18 | 99 |
| 8 | No TMP | 36 | 95 |
| 9 | THF instead of t-amyl-OH | 32 | 95 |
| 10 | 20 atm CO instead of 40 atm CO | 58 | 93 |
| 11 | 10 atm CO instead of 40 atm CO | 45 | 94 |
| 12 | no Co2(CO)8 | <2 | |
Reactions were performed with [1]0 = 0.25 M.
Yields determined by 1H NMR spectroscopy of crude reaction mixture using an internal standard.
Enantiospecificity (es) = (eeproduct/eesubstrate) x 100%, determined by chiral HPLC analysis.
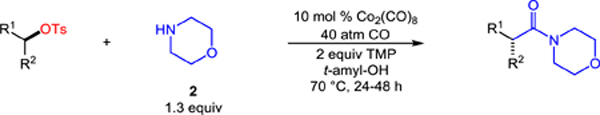
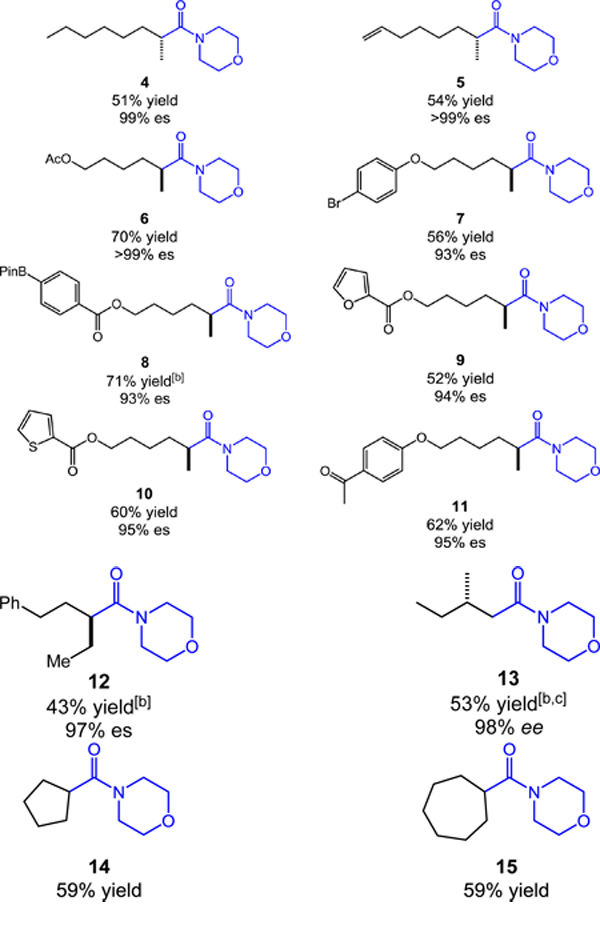
We next applied the aminocarbonylation to a range of chiral, non-racemic alkyl tosylates using morpholine as the nucleophile (Table 2). Aliphatic substrate 2-octyl tosylate provided the desired morpholine amide (4) in good yield and with near perfect stereospecificity. Pendant alkenes and acetate functionality were well tolerated, providing the respective aminocarbonylation products 5 and 6 efficiently and with excellent stereospecificity. The cobalt catalyst reacted with high chemoselectivity in the presence of common electrophilic (7) and nucleophilic (8) aryl cross-coupling partners. In addition, the reactions of heterocyclic ester substrates proceeded with good yield and high stereocontrol (9 and 10). A substrate with ketone functionality–which would be problematic in typical sequences using organometallic reagents–readily carbonylated to provide 11 in 62% yield and 95% es. Importantly, the aminocarbonylation is not limited to methyl branching, as an ethyl substituted alkyl tosylate provided amide 12 with excellent enantiospecificity (97%). Primary alkyl tosylates also participate, even in the presence of α-substitution (13). Finally, we found that the aminocarbonylation was not restricted to acyclic substrates, as cycloalkyl amides 14 and 15 were prepared in good yields.
Table 2.
Stereospecific, catalytic aminocarbonylation of alkyl tosylates with morpholine.[a]
 |
|---|
 |
Isolated yields unless otherwise noted. Enantiospecificity (es) = (eeproduct/eesubstrate) x 100%, determined by chiral HPLC or SFC analysis. [b] Reaction yield determined by 1H NMR spectroscopy of crude reaction mixtures using an internal standard. [c] The reaction time was 72 h.
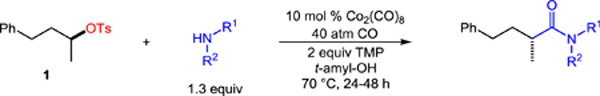
An exceptionally diverse array of amines participate in the aminocarbonylation, highlighting the broad scope of the process (Table 3). We found that a number of cyclic secondary amines readily generated the desired amides in good yield and high enantiospecificity, including piperidine, thiomorpholine, N-Boc piperazine, 4-(N-Boc-amino)piperidine, pyrrolidine, and azepane (16–21). Acyclic secondary amines are also useful nucleophiles, as products 22 and 23 were isolated in good yield. Most importantly, we found that a wealth of primary amines were viable coupling partners. Cyclohexylamine and n-hexylamine gave high yields of products 24 and 25, respectively, with good stereocontrol. Pendant functionality on the primary amines was also tolerated, including an alkene, heterocycles, and an unprotected indole (26–28 and 31). Amino acid alkyl esters were also coupled in good yield and stereospecificity (29–30). Furthermore, the aminocarbonylation was amenable to primary amide synthesis, using ammonium carbamate as an ammonia surrogate to efficiently deliver amide 32 with excellent stereocontrol.
Table 3.
Stereospecific, catalytic aminocarbonylation of (S)-1 with 1° and 2° amines and ammonium carbamate.[a]
 |
|---|
 |
Isolated yields. Enantiospecificity (es) = (eeproduct/eesubstrate) x 100%, determined by chiral HPLC analysis.
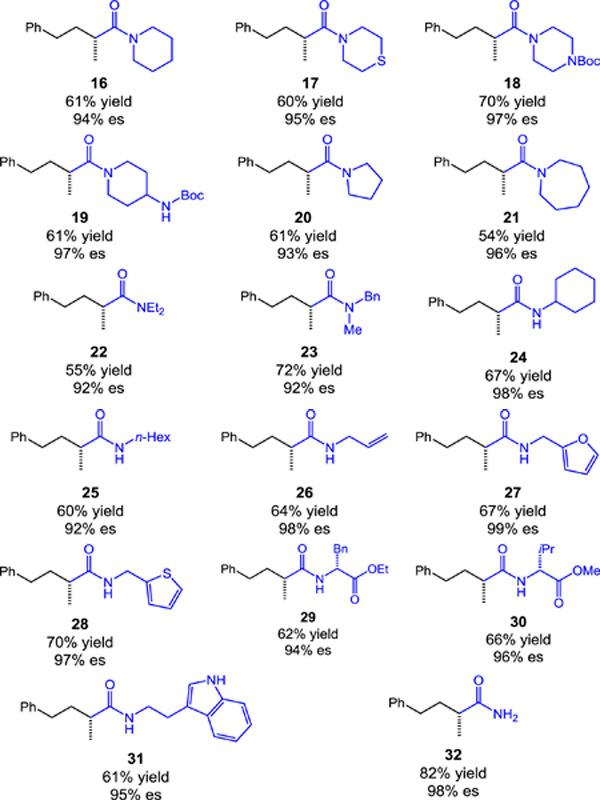
The aminocarbonylation also facilitates access to carboxylic acids or ketones in stereospecific fashion. For example, simple substitution of the amine nucleophile for 1-(trimethylsilyl)imidazole enables direct stereospecific carboxylation following acylimidazole hydrolysis during the acidic aqueous workup (eq 1). Furthermore, addition of MeMgCl to morpholine amide 3 efficiently delivers chiral, non-racemic ketone 35 with excellent stereospecificity (eq 2).[11] The asymmetric synthesis of such a non-symmetrical ketone containing an α-chiral center remains a synthetic challenge, usually requiring multistep sequences and the use of chiral auxiliaries.[12] These transformations further highlight the value of the catalytic aminocarbonylation in the asymmetric synthesis of important carbonyl compounds from easily accessed chiral building blocks.
 |
(1) |
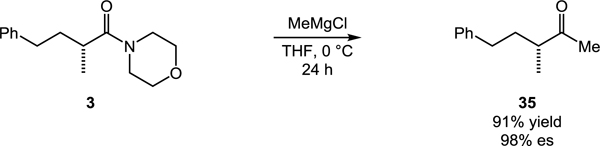 |
(2) |
 |
(3) |
 |
(4) |
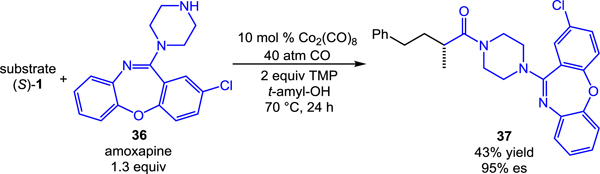
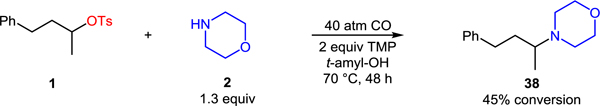
Considering the excellent chemoselectivity of the transformation, applications involving more complex substrates are easily envisioned. For example, the use of pharmaceutically relevant amine amoxapine as a nucleophile provided concise access to coupling product 37 (eq 3). It is worth noting that the aminocarbonylation outcompetes undesired alkylation of the amine. As shown in equation 4, in the absence of the cobalt catalyst, facile direct alkylation of the amine is observed.
A mechanistic proposal for the stereospecific aminocarbonylation of alkyl tosylates is shown in Scheme 1. Octacarbonyldicobalt first disproportionates to the cobaltate anion by addition of the amine or TMP.[13] Following SN2 displacement of the tosylate by the anionic cobaltate carbonyl, the alkylcobalt intermediate undergoes CO migratory insertion with retention of configuration.[8] Nucleophilic displacement of the acylcobalt species with the amine nucleophile delivers the chiral, non-racemic amide product and regenerates the catalyst.
Scheme 1.

Plausible catalytic cycle for the catalytic stereospecific aminocarbonylation.
In conclusion, we have developed a stereospecific, aminocarbonylation of alkyl tosylates with amines utilizing a commercially available, first-row transition metal catalyst. This transformation readily provides valuable chiral, non-racemic amides–as well as carboxylic acids and ketones–from readily available enantioenriched alcohols. The broad scope of the reaction is highlighted by the use of a diverse range of nitrogen nucleophiles, including the challenging use of primary amines. Ongoing efforts continue to develop unique, stereospecific transformations using first-row metal catalysis and to apply this methodology in complex synthesis.
Supplementary Material
Acknowledgements
Financial support was provided by Award R01 GM107204 from the National Institute of General Medical Sciences.
References
- [1].Pattabiraman VR, Bode JW, Nature 2011, 480, 471. [DOI] [PubMed] [Google Scholar]
- [2].a) Schoenberg A, Heck RF, J. Org. Chem. 1974, 39, 3327; [Google Scholar]; b) Brennführer A, Neumann H, Beller M, Angew. Chem. Int. Ed. 2009, 48, 4114; Angew. Chem. 2009, 121, 4176. For an alternative approach to aminocarbonylation involving alkenes, see: [DOI] [PubMed] [Google Scholar]; c) Cheng J, Qi X, Li M, Chen P, Liu G, J. Am. Chem. Soc. 2015, 137, 2480. [DOI] [PubMed] [Google Scholar]
- [3].a) Atkins RJ, Banks A, Bellingham RK, Breen GF, Carey JS, Etridge SK, Hayes JF, Hussain N, Morgan DO, Oxley P, Passey SC, Walsgrove TC, Wells AS, Org. Process Res. Dev. 2003, 7, 663; [Google Scholar]; b) Barnard CFJ, Organometallics 2008, 27, 5402. [Google Scholar]
- [4].a) Ryu I, Nagahara K, Kambe N, Sonoda N, Kreimerman S, Komatsu M, Chem. Commun. 1998, 1953; [Google Scholar]; b) Itsenko O, Kihlberg T, Långström B, J. Org. Chem. 2004, 69, 4356; [DOI] [PubMed] [Google Scholar]; c) Fukuyama T, Nishitani S, Inouye T, Morimoto K, Ryu I, Org. Lett. 2006, 8, 1383; [DOI] [PubMed] [Google Scholar]; d) Fukuyama T, Inouye T, Ryu I, J. Organomet. Chem. 2007, 692, 685; [Google Scholar]; e) Chow SY, Stevens MY, Åkerbladh L, Bergman S, Odell LR, Chem. – Eur. J. 2016, 9155; [DOI] [PubMed] [Google Scholar]; f) Chow SY, Odell LR, Eriksson J, Chem. – Eur. J. 2016, 5980; [Google Scholar]; g) Rahman O, Långström B, Halldin C, ChemistrySelect 2016, 1, 2498 For aminocarbonylation of alkyl iodides to generate imides, see: [Google Scholar]; h) Li Y, Zhu F, Wang Z, Rabeah J, Brückner A, Wu X-F, ChemCatChem 2017, 9, 915 For an alternative approach to racemic amides using alkyl bromides and isocyanate addition, see: [Google Scholar]; i) Serrano E, Martin R, Angew. Chem. Int. Ed. 2016, 55, 11207; Angew. Chem. 2016, 128, 11373. For amidation of an alkylthium with stereocontrol, see: [DOI] [PubMed] [Google Scholar]; j) Dagousset G, Moriya K, Mose R, Berionni G, Karaghiosoff K, Knochel P, Angew. Chem. Int. Ed. 2014, 53, 1425; Angew. Chem. 2014, 126, 1449. [DOI] [PubMed] [Google Scholar]
- [5].Urata H, Ishii Y, Fuchikami T, Tetrahedron Lett. 1989, 30, 4407. [Google Scholar]
- [6].Sargent BT, Alexanian EJ, J. Am. Chem. Soc. 2017, 139, 12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For an alternative platform for catalytic, stereospecific C–C construction, see: Tollefson EJ, Hanna LE, Jarvo ER, Acc. Chem. Res. 2015, 48, 2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]. See the Supporting Information for determination of the absolute stereochemistry.
- [9].a) Goodman SN, Jacobsen EN, Angew. Chem. Int. Ed. 2002, 41, 4703; Angew. Chem. 2002, 114, 4897; [DOI] [PubMed] [Google Scholar]; b) Watanabe Y, Nishiyama K, Zhang K, Okuda F, Kondo T, Tsuji Y, Bull. Chem. Soc. Jpn. 1994, 67, 879. [Google Scholar]
- [10]. Under lower pressures of CO, isomerization to the primary amide was observed. See the Supporting Information for reaction details.
- [11].Martín R, Romea P, Tey C, Urpí F, Vilarrasa J, Synlett 1997, 12, 1414. [Google Scholar]
- [12].a) Kohler MC, Wengryniuk SE, Coltart DM, In Stereoselective Synthesis of Drugs and Natural Products; American Cancer Society, 2013; 1–31; [Google Scholar]; b) Cano R, Zakarian A, McGlacken GP, Angew. Chem. Int. Ed. 2017, 56, 9278; Angew. Chem. 2017, 129, 9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pauson PL, Stambuli JP, Chou T-C, Hong B-C, Octacarbonyldicobalt. In Encyclopedia of Reagents for Organic Synthesis; American Cancer Society, 2014; 1–26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.