

Abstract
Background
Intravenous ferric carboxymaltose (FCM) improves symptoms, functional capacity, and quality of life in heart failure and iron deficiency. The mechanisms underlying these effects are not fully understood. The aim of this study was to examine changes in myocardial iron content after FCM administration in patients with heart failure and iron deficiency using cardiac magnetic resonance.
Methods and Results
Fifty‐three stable heart failure and iron deficiency patients were randomly assigned 1:1 to receive intravenous FCM or placebo in a multicenter, double‐blind study. T2* and T1 mapping cardiac magnetic resonance sequences, noninvasive surrogates of intramyocardial iron, were evaluated before and 7 and 30 days after randomization using linear mixed regression analysis. Results are presented as least‐square means with 95% CI. The primary end point was the change in T2* and T1 mapping at 7 and 30 days. Median age was 73 (65–78) years, with N‐terminal pro‐B‐type natriuretic peptide, ferritin, and transferrin saturation medians of 1690 pg/mL (1010–2828), 63 ng/mL (22–114), and 15.7% (11.0–19.2), respectively. Baseline T2* and T1 mapping values did not significantly differ across treatment arms. On day 7, both T2* and T1 mapping (ms) were significantly lower in the FCM arm (36.6 [34.6–38.7] versus 40 [38–42.1], P=0.025; 1061 [1051–1072] versus 1085 [1074–1095], P=0.001, respectively). A similar reduction was found at 30 days for T2* (36.3 [34.1–38.5] versus 41.1 [38.9–43.4], P=0.003), but not for T1 mapping (1075 [1065–1085] versus 1079 [1069–1089], P=0.577).
Conclusions
In patients with heart failure and iron deficiency, FCM administration was associated with changes in the T2* and T1 mapping cardiac magnetic resonance sequences, indicative of myocardial iron repletion.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT03398681.
Keywords: cardiac magnetic resonance, ferric carboxymaltose, heart failure, iron deficiency, myocardial iron
Subject Categories: Cardiomyopathy, Magnetic Resonance Imaging (MRI), Pharmacology, Treatment
Clinical Perspective
What Is New?
The exact mechanism explaining the benefits of treatment with ferric carboxymaltose in patients with heart failure remains not fully explained.
This study shows that treatment with ferric carboxymaltose resulted in significant short‐term changes in cardiac magnetic resonance sequences that are consistent with myocardial iron repletion.
What Are the Clinical Implications?
This study opens a new line of research about the utility of cardiac magnetic resonance for noninvasive quantification of myocardial iron status, including not only iron overload, but also iron deficiency and response to treatment (myocardial iron repletion).
Introduction
Iron deficiency (ID) is common in patients with heart failure (HF).1 It is associated with reduced functional capacity, quality of life, and increased risk of clinical events.1, 2, 3, 4 In patients with HF and ID (HF‐ID), with or without anemia, iron treatment with intravenous ferric carboxymaltose (FCM) has consistently been shown to improve symptoms, functional capacity, and quality of life with an acceptable safety profile.5, 6, 7, 8 The mechanisms whereby exogenous iron repletion leads to clinical improvement in HF‐ID are not completely understood.
Experimental studies have shown that iron depletion impairs mitochondrial function and adenosine triphosphate (ATP) synthesis affecting systolic and diastolic properties of cardiomyocytes, and that all these deleterious effects are reversed with iron treatment.9, 10, 11, 12 In humans, some studies have also shown that myocardial iron content is decreased in patients with advanced HF13 and this is associated with abnormal myocardial mitochondrial function.14 However, in the clinical setting, the myocardial effects of intravenous iron repletion in patients with HF‐ID are largely unknown.
A cardiac magnetic resonance (CMR) sequence, T2*, has been shown to provide a noninvasive estimation of myocardial iron overload15, 16 and myocardial iron depletion.17 Native T1 mapping has also emerged as a potential alternative tool for the assessment of myocardial iron content.18 In a recently published proof‐of‐concept pilot study of 8 patients with HF‐ID, our group reported an association between intravenous FCM administration and myocardial iron repletion estimated by T2* CMR.19 To further confirm these findings, the present study is a prospective multicenter, double‐blind, randomized clinical trial of noninvasive estimation of changes in myocardial iron after administration of FCM or placebo in patients with HF‐ID using CMR T2* and T1 mapping sequences.
Methods
Overall Study Design
The data that support the findings of this study are available from the corresponding author upon reasonable request. This investigator‐initiated, multicenter, double‐blind, randomized clinical trial was designed to evaluate the effect of intravenous FCM versus placebo on myocardial iron repletion estimated by T2* and T1 mapping CMR sequences in patients with HF‐ID. The trial was conducted in 5 academic centers in Spain.20 The patients provided signed informed consent before being randomized 1:1 to receive either FCM or placebo. Intramyocardial iron was evaluated at 3 time points: before treatment, and 7 and 30 days after treatment. At 30 days, patients assigned to placebo received intravenous FCM if ID persisted. The study design was already published.20
The investigation conforms to the principles outlined in the Declaration of Helsinki and Good Clinical Practice of the International Conference on Harmonization. The study protocol was approved by Agencia Española del Medicamento y Productos sanitarios (AEMPS) and by Comité Ético de Investigación Clínica (CEIC) del Hospital Clínico Universitario de Valencia. CMR studies were performed and analyzed by a core lab at ERESA (Valencia). Laboratory parameters were analyzed in local laboratories. This study is registered at http://clinicaltrials.gov (NCT03398681).
Study Population
The study population included patients with stable chronic HF (New York Heart Association [NYHA] II–III), left ventricular ejection fraction (LVEF) <50%, and ID (serum ferritin <100 μg/L [absolute ID] or 100–299 μg/L with transferrin saturation [TSAT] <20% [functional ID]), and hemoglobin <15 g/dL.21 The inclusion and exclusion criteria were published previously20 and are reported in detail in Table S1.
Study Procedures
Screening and eligibility assessment (visit 0)
In this visit, after reviewing the inclusion/exclusion criteria, the patients signed and dated the informed consent form. The following information was recorded: (1) clinical and demographic variables, including a complete medical history, vital signs, and a complete physical examination; (2) ECG; (3) functional capacity parameters, including NYHA functional class and 6‐minute walking test (6MWT); (4) quality of life parameters, including Kansas City Cardiomyopathy Questionnaire (KCCQ); (5) laboratory data, including hematology parameters, serum electrolytes, parameters evaluating iron status (ferritin and TSAT), creatinine, urea, estimated glomerular filtration rate, and NT‐proBNP (N‐terminal pro‐B‐type natriuretic peptide); (6) 2‐dimensional echocardiography; and (7) CMR parameters.
24‐hour visit
At this visit, after clinical evaluation, the patients were randomized to receive FCM or placebo. In addition, vital signs, complete physical examination, and functional class evaluation (NYHA) were noted.
7‐ and 30‐day visits
At these visits, the same explorations/variables were recorded as at visit 0, except the ECG and echocardiography, which were performed only at the 30‐day visit. During the study period, all concomitant medications and clinical adverse events (death from all causes or hospitalization for acute HF) were recorded. Optional visits were permitted at the discretion of the physician in charge, at which the same information was recorded as in the preplanned visits.
Cardiac magnetic resonance
CMR studies were performed by 2 experienced operators on a 1.5‐Tesla MR scanner (Essenza y Avanto, Siemens, Erlangen, Germany) using the spine and phased array 6‐channel surface coils. No contrast media were used. All images were obtained with electrocardiographic gating and breath‐holding. Cine images were acquired at rest in short‐axis views every 1 cm with steady‐state free precession imaging sequences (time resolution: 37 ms; voxel size: 1.7×1.7×7 mm). The LVEF was calculated by semiautomatic planimetry of endocardial and epicardial borders in short‐axis view cine images. Left ventricle diameters and volumes were also determined.
For T2* analysis, a region of interest was chosen in the mid‐left ventricular septum. The mean signal intensities of the regions of interest were measured in the series of increasing echo time images to give an exponential decay curve. The monoexponential decay model and nonlinear curve fitting algorithm were used to fit the curve to obtain the T2* measurement. T1 mapping was performed using Modified Look‐Locker Inversion Recovery sequences with motion correction (voxel size: 1.5×1.5×7 mm) in 3 short axes (basal, medial, and apical). After T1 maps were generated, a region of interest was chosen in the mid‐left ventricular septum in the 3 short axes and the average T1 values were calculated. The same protocol was repeated at 7 and 30 days. The technical specifications of the CMR sequences were described elsewhere.20 CMR operators were also blinded to treatment allocation.
Trial Intervention
Eligible patients were randomized to receive FCM or placebo. All patients received the assigned treatment with no place for crossover.
Intravenous FCM or placebo
FCM solution (Ferinject® [FCM], Vifor Pharma, Glattbrugg, Switzerland) was given as a 20‐mL perfusion (equivalent to 1000 mg of iron) diluted in a sterile saline solution (0.9% wt/vol NaCl) administered over at least 15 minutes after completion of all baseline study‐related assessments. Because FCM is a dark‐brown solution that is easily distinguishable from the saline placebo, study personnel responsible for the preparation and administration of the study drug were aware of the group assignments and, therefore, not involved in any study assessments.
In the placebo group, normal saline (0.9% weight/volume NaCl) was administered as per the instructions for active therapy. To ensure that patients were unaware of the study drug, the materials used in drug administration were covered with aluminum foil and the injection site shielded from the patient view.
Concomitant drugs
Indications for other HF‐related drugs in both treatment groups were handled according to the current recommendations for clinical practice and included the administration of angiotensin‐converting enzyme inhibitors, angiotensin receptor blockers, sacubitril/valsartan, β‐blockers, mineralocorticoid receptor antagonists, digoxin, ivabradine, diuretics, and nitrate agents. Per study protocol, changes in HF therapy during the study period were discouraged.
End Points
Primary end point
The main end point was to noninvasively estimate changes in myocardial iron content as measured by T2* and T1 mapping CMR sequences 7 and 30 days after FCM or placebo administration.
Secondary end points
The secondary end point was to correlate posttreatment changes in myocardial iron content (T2* and T1 mapping) with concomitant changes in surrogate markers of disease severity (LVEF, functional capacity [6MWT and NYHA class], quality of life [KCCQ], NT‐proBNP, and blood markers related to iron biology [ferritin and TSAT]).
Safety end points
A strict policy was implemented regarding close adverse event surveillance to ensure early detection and appropriate management. Based on previous studies,7, 8 the surveillance specifically focused on (1) general disorders and administration site conditions, (2) skin and subcutaneous tissue disorders, (3) nervous system disorders, (4) gastrointestinal disorders, (5) vascular disorders, (6) ear and labyrinth disorders, (7) injury, poisoning, and procedural complications, and (8) cardiac disorders.
Subgroup analyses
We also included as part of the analysis the effect of treatment on changes in T2* and T1 among age (≤75 years versus >75 years), ischemic heart disease (0/1), and anemia (0/1) categories.
Follow‐Up
No patients were lost to follow‐up. T2* and T1‐mapping values could not be accurately obtained in 3 and 2 patients, respectively. Reasons for unavailability included rejection for repeating the CMR test (n=1 patient) and technical issues in image acquisition (n=2 patients). In these cases, T2* and T1 mapping values were imputed.
Statistical Analysis
All statistical comparisons were made under an intention‐to‐treat principle. Continuous variables were presented as median with interquartile range. Discrete data were expressed as frequency and percentages. The χ2 test or Wilcoxon rank‐sum were used as appropriate to compare baseline characteristics among the 2 treatment groups.
The statistical comparisons for the primary efficacy end point tested the null hypothesis of no differences in changes in myocardial iron content as estimated by T2* and T1 mapping CMR in patients treated with intravenous FCM or placebo at 30 days. The alternative hypothesis stated differences in either direction. Because of the availability of 2‐time points, the 7‐day evaluation was considered a co‐primary end point.
A linear mixed regression model was used for the analysis of the primary end point. All analyses included adjustments for hospital center (as a cluster variable), the interaction term treatment×visit (7 and 30‐day), and the baseline (pretreatment) value of the regressed outcome. The inclusion of other covariates was based on clinical judgment or randomization inequalities at baseline. To limit the potential for increasing type I error rate, we focused strictly on between‐group (FMC versus placebo) comparison at 30 and 7 days, respectively.
A similar approach was used for the subgroup analyses after stratifying for the prespecified variables (age [≤75 years versus >75 years], ischemic heart disease [0/1], and anemia [0/1]). Such linear mixed regression model included a 3‐level interaction (FCM treatment×visit×subgroup variable).
For analysis of the correlations between changes in either T2* or T1 mapping and changes in LVEF, 6MWT, NYHA class, KCCQ, NT‐proBNP, ferritin, and TSAT, we used linear regression analysis. For this analysis, the stratification at 7 and 30 days was ignored under the assumption that these correlations may remain the same throughout follow‐up. These correlation models included the interaction between the treatment and the delta of the parameter evaluated. The normality of residuals was checked.
As a prespecified analysis, no adjustment for multiple comparisons was made in any of the analyses. Results from linear mixed regression model and linear regression analysis are presented as least‐square means with 95% CIs and P values.
Missing values in T2* and T1 mapping were imputed using a reference‐based sensitivity analysis via multiple imputation for longitudinal trials with protocol deviation.22 A 2‐sided P value of 0.05 was considered significant for all analyses. All analyses were performed using Stata 15.1 (Stata Statistical Software, College Station, TX). The multiple imputation procedure was implemented with a special module within Stata called “mimix.”
Results
Trial Population
From May 2017 to June 2018, a total of 55 patients were preselected for participation in this trial. Fifty‐three patients were finally randomized to receive FCM (n=27) or placebo (n=26) (Figure 1).
Figure 1.
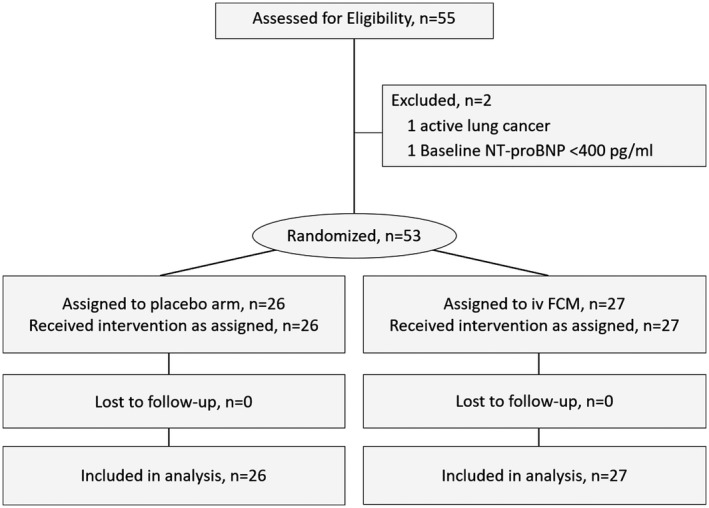
Flow chart. FCM indicates ferric carboxymaltose; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide.
The median age of the study population was 73 (interquartile range 65–78) years, 75.5% were men, 52% had type 2 diabetes mellitus, 43.4% had prior ischemic heart disease, 60.4% had a prior admission for acute heart failure in the last 12 months, and most (94.3%) were in NYHA functional class II. The median (interquartile range) CMR‐LVEF, NT‐proBNP, and estimated glomerular filtration rate were 39% (33–45), 1690 pg/mL (1010–2828), and 60 (49.3–72.9) mL/min per 1.73 m2, respectively. All patients had ID at baseline, with 37 (69.8%) and 16 (30.2%) patients exhibiting absolute and functional ID, respectively. The median (interquartile range) values of ferritin and TSAT were 63 (33–114) μg/L and 15.7% (11–19.2%), respectively. Anemia (WHO criteria) was present in 16 (30.2%) patients. Baseline ferritin and TSAT did not correlate with T2* (Spearman r=−0.08, P=0.552 and Spearman r=−0.199, P=0.153, respectively) or T1 mapping (Spearman r=−0.02, P=0.903 and Spearman r=−0.173, P=0.216, respectively). The baseline characteristics of patients in the 2 treatment groups are given in Table 1. No significant differences were found between the treatment groups. Raw data (CMR parameters [T2*, T1‐mapping, and LVEF], KCCQ, and 6MWT at different visits [baseline, 7 and 30‐day]) across treatment arms are presented in Table S2.
Table 1.
Baseline Characteristics
| Variables | Placebo (n=26) | Intravenous Iron (n=27) | P Value |
|---|---|---|---|
| Demographics and medical history | |||
| Age, y | 71 (67, 79) | 73.5 (64, 77) | 0.957 |
| Male, n (%) | 19 (73.1) | 21 (77.8) | 0.691 |
| Hypertension, n (%) | 19 (73.1) | 22 (81.5) | 0.465 |
| Dyslipidemia, n (%) | 16 (61.5) | 18 (66.7) | 0.697 |
| Diabetes mellitus, n (%) | 14 (53.8) | 15 (55.6) | 0.901 |
| Former smoker, n (%) | 16 (61.5) | 15 (55.6) | 0.659 |
| Coronary artery disease, n (%) | 10 (38.5) | 13 (48.1) | 0.477 |
| Prior admission for AHF in the last year, n (%) | 16 (61.5) | 16 (59.3) | 0.865 |
| COPD, n (%) | 6 (23.1) | 7 (25.9) | 0.810 |
| CKD, n (%) | 7 (26.9) | 8 (29.6) | 0.827 |
| Stroke, n (%) | 6 (23.1) | 2 (7.4) | 0.111 |
| Peripheral artery disease, n (%) | 4 (15.4) | 4 (14.8) | 0.954 |
| NYHA functional class, n (%) | 0.080 | ||
| II | 26 (100) | 24 (88.9) | |
| III | 0 | 3 (11.1) | |
| Vital signs | |||
| Heart rate, bpm | 68 (64, 77) | 73 (68, 82) | 0.262 |
| SBP, mm Hg | 125 (113, 146) | 117 (109, 132) | 0.142 |
| ECG | |||
| Atrial fibrillation, n (%) | 14 (53.8) | 10 (37.0) | 0.219 |
| LBBB, n (%) | 6 (23.1) | 6 (22.2) | 0.941 |
| LVEF, % | 37 (32, 43) | 40 (33.5, 45) | 0.643 |
| CMR parameters | |||
| LVEDVI, mL/m2 | 122.1 (101.5, 137.9) | 107 (80.1, 143.9) | 0.109 |
| LVESVI, mL/m2 | 72.5 (55.1, 87.6) | 63.5 (40.6, 84) | 0.096 |
| LVEDDI, mm/m2 | 30.8 (28, 33.5) | 30.9 (26.9, 31.9) | 0.493 |
| LVESDI, mm/m2 | 23.1 (21.1, 26.9) | 23.7 (23.0, 26.8) | 0.648 |
| LVEF, % | 37 (31, 45) | 43 (36, 48) | 0.128 |
| T2*, ms | 37 (31, 42) | 40 (34, 45) | 0.196 |
| T1‐mapping, ms | 1072 (1030, 1116) | 1082 (1052, 1122) | 0.173 |
| Laboratory | |||
| Hemoglobin, g/dL | 13.4 (12.7, 14.6) | 13.1 (11.9, 13.4) | 0.084 |
| Anemia (WHO), n (%) | 6 (23.1) | 10 (37.0) | 0.268 |
| TSAT, % | 15.4 (9.6, 20.0) | 15.7 (12.0, 19.2) | 0.790 |
| Ferritin, ng/mL | 47.8 (23.0, 114.0) | 73.0 (56.0, 126.0) | 0.072 |
| Absolute iron deficiency, n (%) | 19 (73.1) | 18 (66.7) | 0.611 |
| Sodium, mEq/L | 141 (140, 142) | 140 (140, 142) | 0.669 |
| Potassium, mEq/L | 4.6 (4.4, 4.8) | 4.7 (4.2, 5.0) | 0.852 |
| Urea, mEq/L | 59 (45, 84) | 59 (45, 77) | 0.669 |
| Serum creatinine, mg/dL | 1.1 (0.9, 1.5) | 1.1 (0.9, 1.4) | 0.783 |
| eGFR, mL/min per 1.73 m2 | 64.1 (48.9, 79.3) | 59.4 (50.0, 71.3) | 0.854 |
| NT‐proBNP, pg/mL | 1213 (1010, 2667) | 1990 (976, 2830) | 0.505 |
| Medical treatment | |||
| Diuretics, n (%) | 24 (92.3) | 25 (92.6) | 0.969 |
| β‐blockers, n (%) | 21 (80.8) | 25 (92.6) | 0.204 |
| ACEI, n (%) | 6 (23.1) | 7 (25.9) | 0.810 |
| ARB, n (%) | 4 (15.4) | 5 (18.5) | 0.761 |
| Sacubitril/valsartan, n (%) | 8 (30.8) | 10 (37.0) | 0.630 |
| Spironolactone, n (%) | 3 (11.5) | 2 (7.4) | 0.607 |
| Eplerenone, n (%) | 13 (50.0) | 10 (37.0) | 0.341 |
| Digoxin, n (%) | 4 (15.4) | 1 (3.7) | 0.146 |
| Ibavradine, n (%) | 1 (3.9) | 4 (14.8) | 0.172 |
| Nitrates, n (%) | 2 (7.7) | 2 (7.4) | 0.969 |
WHO criteria for anemia: adult male, hemoglobin 13 g/dL; adult, nonpregnant female, hemoglobin 12 g/dL; adult pregnant female, hemoglobin 11 g/dL. Absolute iron deficiency: ferritin <100 ng/mL. Values expressed as median (interquartile range); categorical variables are presented as percentages. ACEI indicates angiotensin‐converting enzyme inhibitors; AHF, acute heart failure; ARB, angiotensin II receptor blockers; bpm, beats per minute; CKD, chronic kidney disease; CMR, cardiac magnetic resonance; COPD, chronic pulmonary obstructive disease; eGFR, estimated glomerular filtration rate; LBBB, left bundle branch block; LVEF, left ventricular ejection fraction; LVEDDI, left ventricle end‐diastolic diameter index; LVEDVI, left ventricle end‐diastolic volume index; LVESDI, left ventricle end‐systolic diameter index; LVESVI, left ventricle end‐systolic volume index; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; NYHA, New York Heart Association; SBP, systolic blood pressure; TSAT, transferrin saturation; WHO, World Heart Organization.
Primary End Point
Compared with placebo, both 7‐day T2* and T1 mapping were significantly lower in the FCM arm (36.6 ms [34.6–38.7] versus 40 ms [38–42.1], P=0.025; and 1061 ms [1051–1072] versus 1085 ms [1074–1095], P=0.001, respectively; Figure 2A). A similar reduction was found at 30 days for T2* (36.3 ms [34.1–38.5] versus 41.1 ms [38.9–43.4], P=0.003), but this difference was no longer significant for T1 mapping (1075 ms [1065–1085] versus 1079 ms [1069–1089], P=0.577; Figure 2B).
Figure 2.
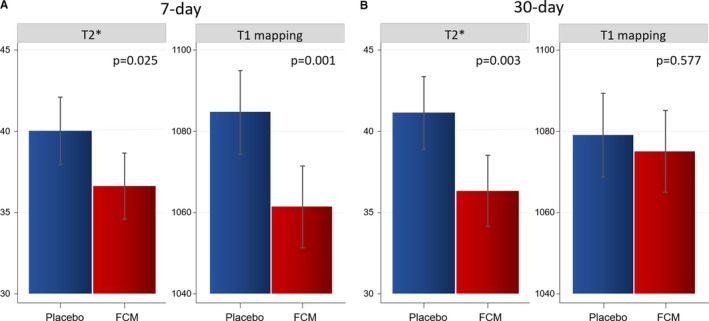
T2* and T1 mapping after administration of FCM. A, Seven‐day comparison of LSM (95% CIs) between FCM and placebo. B, Thirty‐day comparison of LSM (95% CIs) between FCM and placebo. FCM indicates ferric carboxymaltose; LSM, least‐square means from a linear mixed regression analysis.
Secondary End Points
-
1
Relationship between CMR changes in T2* and T1 mapping with concomitant changes in:
-
a
LVEF: LVEF did not significantly differ between both treatments at 7 or 30‐day (Table 2). In the FCM‐arm, a decrease in T2* was significantly associated with an increase in LVEF (Figure 4A‐left). For T1‐mapping, the direction of the association was similar and significant (Figure 4A‐right).
-
b
Quality of life: At 30 days, higher values of KCCQ were observed in the FCM‐arm (76.9 [73.6–80.1] versus 70 [66.8–73.3], P<0.001). However, there were no significant differences across treatment arms at 7‐day (Table 2). Furthermore, we found a significant association between the decrease in T1‐mapping with the increase in KCCQ in the FCM‐arm (Figure 4B‐right). The association between T2* changes was not significant (Figure 4B‐left).
-
c
Functional capacity: We failed to find significant differences in 6MWT across both treatment strategies at both time points (Table 2). However, we find a significant association between reduction in T2* and increase in 6MWT in the active arm (Figure 4C‐left). For ΔT1‐mapping, the association with Δ6MWT was not significant (Figure 4C‐right).
-
d
NYHA class: we found a significant lower value at 30 days in patients allocated to FCM (1.72 [1.60–1.85] versus 1.90 [1.80–2.00], P<0.001). However, at 7‐day, the differences were not significant (Table 2). In the FCM‐arm, the greater improvement in NYHA class matched the reduction in T1‐mapping (Figure 4D‐right). However, this correlation was not significant for T2* (Figure 4D‐left).
-
e
Natriuretic peptides: NT‐proBNP did not differ across treatment strategies at 7 or 30 days (Table 2). In the active arm, we found an inverse association between CMR changes and changes in NT‐proBNP (Figure 4E).
-
a
-
2Relationship between CMR changes (T2* and T1 mapping) and changes in iron biomarkers. Least‐square means values of ferritin and TSAT values were significantly higher in the FCM‐arm at 7 and 30 days as compared with placebo (Table 2). Furthermore, in the FCM arm, there was an inverse association between ΔT2* (and ΔT1‐mapping) with Δ of surrogates of systemic iron repletion (Figure 3).
Figure 3.
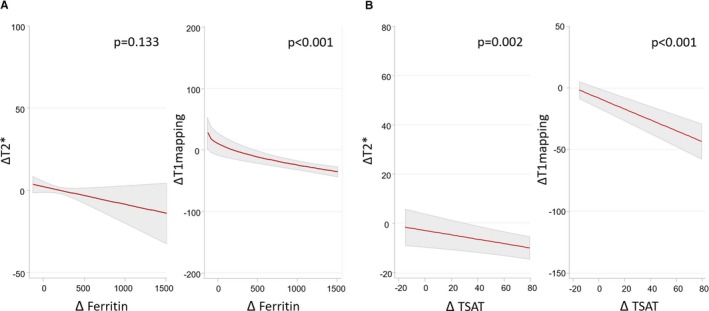 Association of posttreatment changes in myocardial iron content (T2* and T1 mapping) with concomitant changes in systemic iron status in the active arm. A, Changes in CMR sequences and changes in ferritin; B, A, Changes in CMR sequences and changes in TSAT. Values are the least‐square means (95% CIs) from each linear regression analysis (OLS). TSAT indicates transferrin saturation.
Association of posttreatment changes in myocardial iron content (T2* and T1 mapping) with concomitant changes in systemic iron status in the active arm. A, Changes in CMR sequences and changes in ferritin; B, A, Changes in CMR sequences and changes in TSAT. Values are the least‐square means (95% CIs) from each linear regression analysis (OLS). TSAT indicates transferrin saturation.
Table 2.
Effect of Intervention on Surrogates of Severity and Iron Status Markers
| Variables | Placebo (n=26) | Intravenous Iron (n=27) | P Value |
|---|---|---|---|
| 7‐d | |||
| LVEF, % | 42.6 (40.3–45) | 42.5 (40.2–44.8) | 0.916 |
| KCCQ | 72.2 (66.8–77.6) | 77.5 (72.5–82.5) | 0.203 |
| 6MWT, m | 287 (269–304) | 297 (280–313) | 0.402 |
| NYHA class | 1.91 (1.82–2.00) | 1.90 (1.84–1.96) | 0.700 |
| NT‐proBNP, pg/mL | 1870 (1609–2131) | 2305 (1859–2751) | 0.102 |
| Ferritin, ng/mL | 102 (71–132) | 993 (908–1078) | <0.001 |
| TSAT, % | 16.1 (14.1–18.1) | 41.3 (34.5–48.1) | <0.001 |
| 30‐d | |||
| LVEF, % | 40.9 (38.2–43.7) | 44.8 (42.1–47.6) | 0.056 |
| KCCQ | 70 (66.8–73.3) | 76.9 (73.6–80.1) | <0.001 |
| 6MWT, m | 299 (266–332) | 299 (279–320) | 0.992 |
| NYHA class | 1.90 (1.80–2.00) | 1.72 (1.60–1.85) | <0.001 |
| NT‐proBNP, pg/mL | 2656 (1891–3421) | 2366 (1741–2992) | 0.675 |
| Ferritin, ng/mL | 96 (68–124) | 456 (434–479) | <0.001 |
| TSAT, % | 15 (14.1–15.8) | 30.4 (26.1–34.8) | <0.001 |
Values presented are the least‐square means (95% CIs) from each mixed linear regression model. All models were adjusted by hospital center (as a cluster variable), the interaction term Tx×visit (7 and 30‐day), baseline value of hemoglobin, and the baseline (pretreatment) value of the regressed outcome. 6MWT indicates distance walked in 6 minutes; KCCQ, Kansas City Cardiomyopathy Questionnaire; LVEF, left ventricular ejection fraction; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; NYHA, New York Heart Association; TSAT, transferrin saturation.
Figure 4.
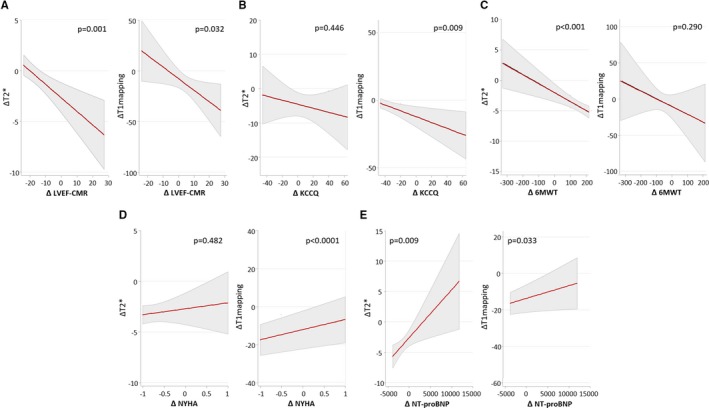
Association of posttreatment changes in myocardial iron content (T2* and T1 mapping) with concomitant changes in surrogate markers of disease severity in the active‐arm. A, Changes in CMR sequences and changes in LVEF; B, Changes in CMR sequences and changes in KCCQ; C, Changes in CMR sequences and changes in 6MWT; D, Changes in CMR sequences and changes in NYHA class; E, Changes in CMR sequences and changes in NT‐proBNP. Values are the least‐square means (95% CIs) from each linear regression analysis (OLS). 6MWT indicates distance walked in 6 minutes; CMR, cardiac magnetic resonance; KCCQ, Kansas City Cardiomyopathy Questionnaire; LVEF, left ventricular ejection fraction; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; NYHA, New York Heart Association.
Discussion
For the first time in a clinical setting, we found that treatment with FCM resulted in significant and short‐term changes in CMR sequences that are consistent with myocardial iron repletion. Interestingly, these surrogates of myocardial iron repletion were, in most cases, related to systemic iron repletion and short‐term improvement in LVEF and other surrogates of severity such as 6MWT and quality of life. Our results are in line with prior studies showing the clinical utility of iron supplementation in HF‐ID patients, and more importantly, provide new insights into the role that myocardial iron repletion may play in these beneficial effects.
ID and Heart Failure
Iron is a micronutrient that plays a crucial role in oxygen transport and storage, and as a component of the mitochondrial respiratory chain is involved in energy production.10, 11, 12, 13, 23 In vitro studies have shown that cellular iron depletion in cardiomyocytes is associated with mitochondrial dysfunction, impaired ATP synthesis, increased oxidative stress in these cells, and that all these factors promote a profound impairment of systolic and diastolic properties of the cardiomyocytes.12 In the clinical context, ID is highly prevalent in patients with HF and commonly associated with adverse outcomes.1, 2, 3, 4 In these patients, treatment of ID with FCM improves symptoms, exercise capacity, and quality of life, and reduces the risk of readmission.1, 5, 6, 7, 8 However, the pathophysiological link between exogenous iron repletion and clinical improvement is not totally elucidated. A subanalysis of the FAIR‐HF (Ferrinject assessment in patients with iron deficiency and chronic heart failure) study and other studies showed that clinical improvement in response to intravenous iron administration occurs in patients with or without anemia, suggesting that the beneficial effect of iron administration is, at least in part, independent of the erythropoietic response.24
Iron Supplementation and Myocardial Performance in HF Patients
In the clinical setting, the evidence regarding the myocardial effect of iron supplementation is scarce. For example, in a small randomized trial with HF, ID, and chronic kidney disease, Toblli et al25 showed that iron sucrose administration translated into a significant 6‐month improvement in LVEF estimated by echocardiography (6.6 ± 3.8%). Other studies have also shown a significant reduction in the plasma level of NT‐proBNP and attenuation of hypertrophic cardiac remodeling.26, 27 The present study goes a step further in this regard and provides evidence that the improvement is related to benefits at the cardiac level with myocardial iron repletion that translates in improvements in cardiac function and patient functioning.
Quantification of Myocardial Iron Changes by CMR
CMR is an accurate and noninvasive technique that enables the characterization of cardiac structure, function, and risk stratification.28 This technique has been used to estimate myocardial iron content.15, 16 For example, lower T2* CMR sequence values have been shown to provide a reliable assessment of myocardial iron overload.15, 16 More interestingly, changes in T2* following chelation therapy are strongly related to the response to therapy in patients with iron overload.29 Regarding ID, in a small case–control study, Nagao et al17 suggested that cardiac T2* may also play a role in the quantification of myocardial ID in nonischemic HF. In addition, these authors reported that T2* CMR was related to an increased risk of adverse outcomes.17 Furthermore, new CMR techniques, such as T1 mapping, have been found to be a potential alternative tool for myocardial iron quantification.18 To the best of our knowledge, this is the first controlled clinical trial showing that treatment with FCM is associated with significant short‐term decreases in T1 mapping and T2* CMR values, changes that are indicative of myocardial iron repletion. Indeed, we found a 7‐day decrease in both CMR sequences; however, at 30 days only T2* was decreased. Interestingly, these changes were strongly related to a concomitant improvement in LVEF and other surrogates of disease severity. Unfortunately, our study was not designed to further evaluate this discrepancy among T2* and T1 mapping at 30 days. Beyond the potential role of type II error, we speculate that changes in T1 mapping at later times may also reflect changes in other tissue characteristics. T1‐mapping values are influenced by several other conditions, such as fibrosis, inflammation/edema, amyloid, and Fabry disease.18 Despite the fact that all enrolled patients exhibited systemic ID, baseline T2* values did not correlate with systemic markers of ID. This fact may reflect a lack of agreement between traditional markers of ID and myocardial tissue iron, as shown in previous publications30 or, perhaps, a lack of specificity of the baseline values of these CMR sequences for identifying myocardial ID in clinical practice. We do not believe the present findings have a direct translation into daily clinical practice. However, given the emerging role of iron biology in the pathophysiology of HF, we believe further research should test a clinical utility of the present findings or focus on the development of more specific and accurate noninvasive imaging techniques and better biomarkers for estimating myocardial iron content. This would translate into a better selection of HF patients who would most benefit from iron supplementation.
Limitations
This trial has some limitations to be acknowledged. First, some negative results may be explained by insufficient statistical power. Second, CMR end points are well‐established imaging sequences for evaluating myocardial iron overload but not myocardial iron deficiency. In addition, their values and changes are not specific to myocardial iron content and may reflect the turnover of other tissue. Neither CMR sequence can discriminate whether tissue changes occur in an intra‐ or extracellular compartment. Third, as limited by the study design, we cannot prove any correlation between myocardial histological or myocardial energetic efficiency aspects with the administration of FCM. Performing a myocardial biopsy to conduct such studies would be considered ethically conflicting. Fourth, in the present study we did not evaluate the effect of iron treatment on skeletal muscle function, a potential mechanism that may also explain the beneficial effect of iron in HF patients. Fifth, in order to avoid interference with CMR sequences, patients with cardiac devices were excluded from this study. Sixth, the subgroup analysis was not reported given the small size of the study. Finally, despite the fact that the magnitude of changes we found in surrogates of the disease (LVEF, quality of life, functional capacity, and NYHA class) appears not to be clinically relevant, this study was not designed to quantify the effect of FCM nor magnitude of these changes.
Conclusions
In patients with stable HF and ID, administration of FCM led to a greater reduction in CMR T2* and T1‐mapping values compared with placebo in the short‐term. Both changes are suggestive of myocardial iron repletion. Further studies are warranted to confirm these findings and to evaluate the potential utility of CMR for assessing myocardial iron content.
Appendix
Myocardial Iron Study Group Investigators
Cardiology Department, Hospital Clínico Universitario de Valencia, Universidad de Valencia, INCLIVA, Valencia, Spain: Julio Núñez, MD, PhD, Gema Miñana, MD, PhD, Ingrid Cardells, MD, Martina Amiguet, MD, Jessika González, MD, Ernesto Valero, MD, Sergio García‐Blas, MD, Vicent Bodí, MD, PhD, Rafael de la Espriella‐Juan, MD, Jorge Navarro, MD, PhD, Juan Sanchis, MD, PhD, Francisco J. Chorro, MD, PhD, Meritxell Soler, MD, Amparo Villaescusa, Jose Civera, Anna Mollar, PhD.
Cardiology Department, Hospital General de Castellón. Universitat Jaume I, Castellón, Spain: Patricia Palau, MD, PhD, Alicia Serrano, MD.
Internal Medicine Department, Hospital de Manises, Manises, Spain: Pau Llàcer, MD, PhD, Maria del Carmen Moreno, MD.
Cardiology Department, Hospital General Universitario de Valencia, Valencia, Spain: Lorenzo Fácila, MD, PhD, Vicente Montagud, MD, Veronica Vidal, MD.
Cardiology Department, Hospital Universitario La Fe de Valencia, Valencia, Spain: Luis Almenar, MD, PhD, Raquel López‐Vilella, MD.
Unidad de Imagen Cardiaca (ERESA) Hospital Clínico Universitario de Valencia, Valencia, Spain: Maria P. López‐Lereu, MD, PhD, Jose V. Monmeneu, MD, PhD.
Cardiology Department and Heart Failure Unit, Hospital Universitari Germans Trias i Pujol, Badalona, Spain. Universitat Autonoma de Barcelona, Barcelona, Spain: Antoni Bayés‐Genís, MD, PhD, Josep Lupón, MD, PhD.
Nephrology Department, Hospital Clínico Universitario de Valencia, Universidad de Valencia, INCLIVA, Valencia, Spain: José Luis Górriz, MD, PhD.
Department of Cardiology, Hospital del Mar, Barcelona, Spain Heart Diseases Biomedical Research Group, IMIM (Hospital del Mar Medical Research Institute), Barcelona, Spain. Department of Medicine, Universitat Autònoma de Barcelona, Spain: Josep Comín‐Colet, MD, PhD.
Sources of Funding
This work was supported in part by an unrestricted grant from Vifor Pharma, CIBER Cardiovascular (grant numbers 16/11/00420 and 16/11/00403), and Proyectos de Investigación de la Sección de Insuficiencia Cardiaca 2017 from Sociedad Española de Cardiología.
Disclosures
J. Núñez received board speaker fees and travel expenses from Novartis, Roche Diagnostics, Abbott, Rovi, Vifor Pharma, Daiichi Sankyo, Boehringer Ingelheim, and Astra‐Zeneca (modest). L. Fácila received speaker fees and travel expenses from Novartis (modest). J. Sanchis received speaker fees from Astra‐Zeneca, Abbott, and Edwards Lifesciences (modest). J. L. Gorriz received board membership fees, speaker fees, and consulting fees from Novartis, Astra‐Zeneca, and Vifor Fresenius Medical Care Renal Pharma (modest). J. Comín‐Colet received speaker fees from Vifor Pharma and an unrestricted grant from Vifor Pharma (modest). A. Bayés‐Genís received board membership fees and travel expenses from Novartis, Roche Diagnostics, Vifor Pharma, and Critical Diagnostics (modest). The remaining authors have no disclosures to report.
Supporting information
Table S1. Inclusion and Exclusion Criteria
Table S2. Patients’ Characteristics by Visit
(J Am Heart Assoc. 2020;9:e014254 DOI: 10.1161/JAHA.119.014254.)
Contributor Information
Julio Núñez, Email: yulnunez@gmail.com.
Antoni Bayés‐Genís, Email: abayesgenis@gmail.com.
the Myocardial‐IRON Investigators*:
Meritxell Soler, Amparo Villaescusa, Jose Civera, Anna Mollar, Maria del Carmen Moreno, and Veronica Vidal
References
- 1. Rocha BML, Cunha GJL, Menezes Falcão LF. The burden of iron deficiency in heart failure: therapeutic approach. J Am Coll Cardiol. 2018;71:782–793. [DOI] [PubMed] [Google Scholar]
- 2. Comín‐Colet J, Enjuanes C, González G, Torrens A, Cladellas M, Meroño O, Ribas N, Ruiz S, Gómez M, Verdú JM, Bruguera J. Iron deficiency is a key determinant of health‐related quality of life in patients with chronic heart failure regardless of anaemia status. Eur J Heart Fail. 2013;15:1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jankowska EA, Rozentryt P, Witkowska A, Nowak J, Hartmann O, Ponikowska B, Borodulin‐Nadzieja L, Banasiak W, Polonski L, Filippatos G, McMurray JJ, Anker SD, Ponikowski P. Iron deficiency: an ominous sign in patients with systolic chronic heart failure. Eur Heart J. 2010;31:1872–1880. [DOI] [PubMed] [Google Scholar]
- 4. Núñez J, Comín‐Colet J, Miñana G, Núñez E, Santas E, Mollar A, Valero E, García‐Blas S, Cardells I, Bodí V, Chorro FJ, Sanchis J. Iron deficiency and risk of early readmission following a hospitalization for acute heart failure. Eur J Heart Fail. 2016;18:798–802. [DOI] [PubMed] [Google Scholar]
- 5. von Haehling S, Ebner N, Evertz R, Ponikowski P, Anker SD. Iron deficiency in heart failure: an overview. JACC Heart Fail. 2019;7:36–46. [DOI] [PubMed] [Google Scholar]
- 6. Anker SD, Comin Colet J, Filippatos G, Willenheimer R, Dickstein K, Drexler H, Lüscher TF, Bart B, Banasiak W, Niegowska J, Kirwan BA, Mori C, von Eisenhart Rothe B, Pocock SJ, Poole‐Wilson PA, Ponikowski P; FAIR‐HF Trial Investigators . Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med. 2009;361:2436–2448. [DOI] [PubMed] [Google Scholar]
- 7. Ponikowski P, van Veldhuisen DJ, Comin‐Colet J, Ertl G, Komajda M, Mareev V, McDonagh T, Parkhomenko A, Tavazzi L, Levesque V, Mori C, Roubert B, Filippatos G, Ruschitzka F, Anker SD; CONFIRM‐HF Investigators . Beneficial effects of long‐term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur Heart J. 2015;36:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Veldhuisen DJ, Ponikowski P, van der Meer P, Metra M, Böhm M, Doletsky A, Voors AA, Macdougall IC, Anker SD, Roubert B, Zakin L, Cohen‐Solal A; EFFECT‐HF Investigators . Effect of ferric carboxymaltose on exercise capacity in patients with chronic heart failure and iron deficiency. Circulation. 2017;136:1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong F, Zhang X, Culver B, Chew HG Jr, Kelley RO, Ren J. Dietary iron deficiency induces ventricular dilation, mitochondrial ultrastructural aberrations and cytochrome c release: involvement of nitric oxide synthase and protein tyrosine nitration. Clin Sci (Lond). 2005;109:277–286. [DOI] [PubMed] [Google Scholar]
- 10. Kobak KA, Radwańska M, Dzięgała M, Kasztura M, Josiak K, Banasiak W, Ponikowski P, Jankowska EA. Structural and functional abnormalities in iron‐depleted heart. Heart Fail Rev. 2019;24:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haddad S, Wang Y, Galy B, Korf‐Klingebiel M, Hirsch V, Baru AM, Rostami F, Reboll MR, Heineke J, Flögel U, Groos S, Renner A, Toischer K, Zimmermann F, Engeli S, Jordan J, Bauersachs J, Hentze MW, Wollert KC, Kempf T. Iron‐regulatory proteins secure iron availability in cardiomyocytes to prevent heart failure. Eur Heart J. 2017;38:362–372. [DOI] [PubMed] [Google Scholar]
- 12. Hoes MF, Grote Beverborg N, Kijlstra JD, Kuipers J, Swinkels DW, Giepmans BNG, Rodenburg RJ, van Veldhuisen DJ, de Boer RA, van der Meer P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail. 2018;20:910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maeder MT, Khammy O, dos Remedios C, Kaye DM. Myocardial and systemic iron depletion in heart failure implications for anemia accompanying heart failure. J Am Coll Cardiol. 2011;58:474–480. [DOI] [PubMed] [Google Scholar]
- 14. Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H, Pluhacek T, Spatenka J, Kovalcikova J, Drahota Z, Kautzner J, Pirk J, Houstek J. Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. Eur J Heart Fail. 2017;19:522–530. [DOI] [PubMed] [Google Scholar]
- 15. Anderson LJ, Holden S, Davis B, Prescott E, Charrier CC, Bunce NH, Firmin DN, Wonke B, Porter J, Walker JM, Pennell DJ. Cardiovascular T2‐star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171–2179. [DOI] [PubMed] [Google Scholar]
- 16. He T. Cardiovascular magnetic resonance T2* for tissue iron assessment in the heart. Quant Imaging Med Surg. 2014;4:407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagao M, Matsuo Y, Kamitani T, Yonezawa M, Yamasaki Y, Kawanami S, Abe K, Mukai Y, Higo T, Yabuuchi H, Takemura A, Yoshiura T, Sunagawa K, Honda H. Quantification of myocardial iron deficiency in nonischemic heart failure by cardiac T2* magnetic resonance imaging. Am J Cardiol. 2014;113:1024–1030. [DOI] [PubMed] [Google Scholar]
- 18. Taylor AJ, Salerno M, Dharmakumar R, Jerosch‐Herold M. T1 mapping: basic techniques and clinical applications. JACC Cardiovasc Imaging. 2016;9:67–81. [DOI] [PubMed] [Google Scholar]
- 19. Núñez J, Monmeneu JV, Mollar A, Núñez E, Bodí V, Miñana G, García‐Blas S, Santas E, Agüero J, Chorro FJ, Sanchis J, López‐Lereu MP. Left ventricular ejection fraction recovery in patients with heart failure treated with intravenous iron: a pilot study. ESC Heart Fail. 2016;3:293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miñana G, Cardells I, Palau P, Llàcer P, Fácila L, Almenar L, López‐Lereu MP, Monmeneu JV, Amiguet M, González J, Serrano A, Montagud V, López‐Vilella R, Valero E, García‐Blas S, Bodí V, de la Espriella‐Juan R, Sanchis J, Chorro FJ, Bayés‐Genís A, Núñez J; Myocardial‐IRON Investigators . Changes in myocardial iron content following administration of intravenous iron (Myocardial‐IRON): study design. Clin Cardiol. 2018;41:729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P; Authors/Task Force Members; Document Reviewers . 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 22. Carpenter JR, Kenward MG. Multiple Imputation and Its Application. Chichester, UK: Wiley; 2013. ISBN: 978‐0‐470‐74052‐1. [Google Scholar]
- 23. Cohen‐Solal A, Leclercq C, Deray G, Lasocki S, Zambrowski JJ, Mebazaa A, de Groote P, Damy T, Galinier M. Iron deficiency: an emerging therapeutic target in heart failure. Heart. 2014;100:1414–1420. [DOI] [PubMed] [Google Scholar]
- 24. Filippatos G, Farmakis D, Colet JC, Dickstein K, Lüscher TF, Willenheimer R, Parissis J, Gaudesius G, Mori C, von Eisenhart Rothe B, Greenlaw N, Ford I, Ponikowski P, Anker SD. Intravenous ferric carboxymaltose in iron‐deficient chronic heart failure patients with and without anaemia: a subanalysis of the FAIR‐HF trial. Eur J Heart Fail. 2013;15:1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Toblli JE, Di Gennaro F, Rivas C. Changes in echocardiographic parameters in iron deficiency patients with heart failure and chronic kidney disease treated with intravenous iron. Heart Lung Circ. 2015;24:686–695. [DOI] [PubMed] [Google Scholar]
- 26. Toblli JE, Lombraña A, Duarte P, Di Gennaro F. Intravenous iron reduces NT‐pro‐brain natriuretic peptide in anemic patients with chronic heart failure and renal insufficiency. J Am Coll Cardiol. 2007;50:1657–1665. [DOI] [PubMed] [Google Scholar]
- 27. Usmanov RI, Zueva EB, Silverberg DS, Shaked M. Intravenous iron without erythropoietin for the treatment of iron deficiency anemia in patients with moderate to severe congestive heart failure and chronic kidney insufficiency. J Nephrol. 2008;21:236–242. [PubMed] [Google Scholar]
- 28. Pennell DJ. Cardiovascular magnetic resonance. Circulation. 2010;121:692–705. [DOI] [PubMed] [Google Scholar]
- 29. Carpenter JP, Pennell DJ. Role of T2* magnetic resonance in monitoring iron chelation therapy. Acta Haematol. 2009;122:146–154. [DOI] [PubMed] [Google Scholar]
- 30. Leszek P, Sochanowicz B, Szperl M, Kolsut P, Brzóska K, Piotrowski W, Rywik TM, Danko B, Polkowska‐Motrenko H, Różański JM, Kruszewski M. Myocardial iron homeostasis in advanced chronic heart failure patients. Int J Cardiol. 2012;159:47–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Inclusion and Exclusion Criteria
Table S2. Patients’ Characteristics by Visit