

Abstract
Background
DJ‐1 is a ubiquitously expressed protein typically associated with the development of early onset Parkinson disease. Recent data suggest that it also plays a role in the cellular response to stress. Here, we sought to determine the role DJ‐1 plays in the development of heart failure.
Methods and Results
Initial studies found that DJ‐1 deficient mice (DJ‐1 knockout; male; 8–10 weeks of age) exhibited more severe left ventricular cavity dilatation, cardiac dysfunction, hypertrophy, and fibrosis in the setting of ischemia‐reperfusion–induced heart failure when compared with wild‐type littermates. In contrast, the overexpression of the active form of DJ‐1 using a viral vector approach resulted in significant improvements in the severity of heart failure when compared with mice treated with a control virus. Subsequent studies aimed at evaluating the underlying protective mechanisms found that cardiac DJ‐1 reduces the accumulation of advanced glycation end products and activation of the receptor for advanced glycation end products—thus, reducing glycative stress.
Conclusions
These results indicate that DJ‐1 is an endogenous cytoprotective protein that protects against the development of ischemia‐reperfusion–induced heart failure by reducing glycative stress. Our findings also demonstrate the feasibility of using a gene therapy approach to deliver the active form of DJ‐1 to the heart as a therapeutic strategy to protect against the consequences of ischemic injury, which is a major cause of death in western populations.
Keywords: glycative stress, heart failure, ischemic heart disease
Subject Categories: Ischemia, Myocardial Infarction, Heart Failure
Clinical Perspective
What Is New?
Our study demonstrates that the cleaved form of DJ‐1 is sufficient to provide protection against myocardial ischemia‐reperfusion injury by modulating glycative stress.
Our findings have uncovered the physiological role for the protease activity of DJ‐1.
Delayed treatment of the cleaved form of DJ‐1 abrogated the progression of heart failure.
What Are the Clinical Implications?
Therapeutic designed to induce the cleaved form DJ‐1 may serve as a viable treatment option for heart failure.
Introduction
Despite a great deal of research into the mechanisms of left ventricular (LV) remodeling following the onset of ischemia‐reperfusion (I/R) injury, we are still without an effective therapeutic strategy to coincide with timely reperfusion.1 While viable treatment options remain an unmet need, research efforts have revealed a great deal about the complexity of LV remodeling. For instance, it is known that the temporal sequence of events occurring after the onset of I/R injury must be finely tuned to promote healing while at the same time minimizing adverse remodeling.2 As such, the capacity of cardiac myocytes to maintain homeostasis following ischemic injury resides in their ability to activate or induce protective proteins that can harness the beneficial aspects of remodeling.3 DJ‐1, aka Park7 (Parkinson Disease autosomal recessive, early onset 7) is a cytoprotective protein that is activated in response to various pathological stimuli.4, 5 Because of the association of DJ‐1 with Parkinson Disease, most studies aimed at investigating its cellular actions have been confined to the brain or neuronal cells. Typically, these studies have used various model systems to characterize its actions and relate the loss of these actions to the symptoms of Parkinson Disease. However, DJ‐1 is expressed in many other tissues, including the heart,6 where it possesses cytoprotective actions.6, 7, 8
The cellular mechanisms underlying the reported actions of DJ‐1 remain largely unknown. Studies indicate that DJ‐1 plays an important role in multiple cellular processes, including oxidative stress response, anti‐apoptotic signaling, and transcriptional regulation.5, 9 Given that DJ‐1 is a small single‐domain protein, it either possesses multiple functions to account for these actions or there is a single biochemical activity to explain them all.10 DJ‐1 shares sequence homology with the PfpI family of bacterial proteases and with heat‐shock protein 31, an E coli chaperone that possesses protease activity.9 Proteases perform essential functions in regulating diverse cellular processes by catalyzing the cleavage of peptide bonds. To avoid potentially hazardous consequences of unregulated protease activity, proteases generally reside in cells as latent precursors called zymogens. In response to stimuli, a zymogen is converted into an active protease. Evidence shows that DJ‐1 possesses proteolytic activity.6 More specifically, the cleavage of DJ‐1 in response to oxidative stress converts it into an active protease that has a 26‐fold greater catalytic efficiency than the zymogen (full‐length form) and is more effective in providing cytoprotection.9 However, neither endogenous targets nor a physiological role for the protease activity of DJ‐1 have been elucidated.
α‐oxaldehydes, including glyoxal and methylglyoxal, are reactive dicarbonyls produced by glucose oxidation and lipid peroxidation.11 They react non‐enzymatically with amino groups of proteins by a process termed glycation or non‐enzymatic glycosylation to form advanced glycation end products (AGEs).11, 12 AGEs interact with the receptor for AGEs (RAGE) resulting in the propagation of stress signals and activation of mitogen‐activated protein kinases, nuclear factor‐kB, and several proapoptotic pathways.13, 14 The upregulation of RAGE also augments reactive oxygen species production, which further contributes to AGE generation and enhanced RAGE expression.15 This AGE‐RAGE signaling perpetuates a vicious cycle resulting in glycative stress and ultimately cellular and tissue injury.16 Experiments in animal models show that RAGE and its ligands are upregulated in key injuries to the heart.13, 17, 18 Pharmacological antagonism of RAGE or genetic deletion of the receptor in mice is protective.13, 14, 18 Data from human studies suggest that the AGE‐RAGE axis is implicated in heart failure19, 20 and it is predicted that therapeutic antagonism of this axis might be a unique target for therapeutic intervention.13 Recent in vitro work indicates that through its proteolytic activity DJ‐1 acts to oppose glycative stress.11 However, it is currently unknown if DJ‐1 acts in this manner in the heart, much less in the setting of heart failure. Therefore, the purpose of this study was to determine if DJ‐1 affords protection in the setting of ischemia‐reperfusion (I/R)‐induced heart failure by modulating glycative stress.
Methods
Data will be available from the authors on reasonable request.
Animals
C57BL/6J mice and DJ‐1 deficient (DJ‐1 knockout) mice21 (male; 8–12 weeks of age) were used in all experiments. Sex influences the development of cardiovascular disease.22 As such, we only used male mice in our studies. This allowed for the evaluation of DJ‐1 in a well‐controlled experimental system. All experimental protocols were approved by the Institute for Animal Care and Use Committee at Emory University and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (Publication No. 86‐23, revised 1996), and with federal and state regulations. Approximately 275 mice were included in the present study after accounting for animal deaths. All mice were randomly assigned to the treatment groups. No animals were excluded from the study.
Patient Samples
Left ventricular samples were procured from patients with advanced ischemic heart failure undergoing a heart transplant at Emory University in accordance with Institution Review Board protocols. Additional non‐failing heart failure samples were obtained from LifeLink. All patient identifiers were removed to strictly maintain donor confidentiality and anonymity. Both sample sets included male and female patients. Patient characteristics for these samples have previously been reported.23
Materials
Aminoguanidine was purchased from Sigma Aldrich (St. Louis, MO). Aminoguanidine was administered to mice in the drinking water at a concentration of 1 g/L24 starting 24 hours after reperfusion. Treatment continued for up to 7 days of reperfusion. Control mice received standard drinking water.
Heart Failure
Heart failure was induced by subjecting mice to 60 minutes of left coronary artery occlusion followed by reperfusion for up to 4 weeks. Surgical ligation of the left coronary artery was performed under anesthesia (ketamine, 100 mg/kg; sodium pentobarbital, 20 mg/kg) as previously described.25 All animals received prophylactic antibiotic therapy with cefazolin (20 mg/kg) and buprenorphine (0.05 mg/kg) for pain.
Echocardiographic Analysis
Transthoracic echocardiography was performed at baseline and 4 weeks after reperfusion using the Vevo 2100 with a 38‐MHz linear array scanhead.25
Production of Adeno‐Associated Viruses
Plasmid containing a truncated form of human DJ‐1 lacking the C‐terminal 15 amino acids (DJ1ΔC) has been previously described.9 The DJ1ΔC cDNAs was used to generate the recombinant adeno‐associated viral expression vector (AAV9) for expression of cleaved human DJ‐1 (AAV9‐DJ1ΔC) under the control of the cytomegaolovirus promoter. pAAV2/9 containing AAV2 rep and AAV9 capsid genes was kindly provided by the Penn Vector Core (University of Pennsylvania School of Medicine). Recombinant AAV‐DJ1ΔC viruses were produced by Emory Viral Vector Core, using the triple transfection method with HEK 293T cells as previously described.26 The extracted recombinant AAV9 viruses were purified by an iodixanol gradient and was dialyzed using an Amicon 15 100000 MWCO concentration unit. The titer was determined by quantitative polymerase chain reaction. AAV9‐green fluorescent protein (GFP) also was packaged and used as a control.
AAV9 Infection
For assessment of efficiency, specificity, and dose‐dependence of AAV9‐mediated transduction, 1×1011, 2×1011, and 5×1011 genome copy/mL of AAV9‐GFP were administered from the femoral vein. For experiments performed in the heart failure model, 2×1011 genome copy/mL of AAV9‐GFP or AAV9‐DJ1Δc in 50 μL of PBS were administered 2 weeks before myocardial ischemia via femoral vein injections.
Cell Culture
H9c2 cardiomyocytes were purchased from ATCC (Rockville, MD). Cells (passages 2–9) were grown in ATCC‐formulated DMEM catalog# 30‐2002 with 10% fetal bovine serum. Cells were maintained in this media until 80% confluent. Cells were then maintained in DMEM with 0.5% fetal bovine serum for 12 hours. Cells were then transfected with siRNA (10 μmol/L) against: DJ‐1 (ID:4390815; ThermoFisher Scientific) or scrambled sequence (negative control) (ID:4390843; ThermoFisher Scientific). Transfections were carried out with Lipofectamine RNAi Max (ThermoFisher Scientific) by incubating the H9c2 cells with the respective siRNA for 24 hours. Afterwards, media containing the transfecting siRNA agent was removed. The cells were washed twice with cold PBS and replenished with fresh DMEM with 0.5% fetal bovine serum. After 48 hours, the cells were washed 3 times with PBS and incubated in glucose free media (A14430‐01; Gibco) to induce energy stress.27 Cells were incubated in the glucose free media for up to 18 hours. A cohort of cells was administered aminoguanidine (100 μmol/L).28 Cell viability was evaluated at 18 hours using the MMT Assay Kit (ab211091; Abcam) according to the manufacturer's instructions.
Cardiomyocyte Isolation
Adult cardiomyocytes and non‐myocytes were isolated as previously described.29 Briefly, hearts were perfused with perfusion buffer for 4 minutes and then with perfusion buffer containing 2 mg/mL collagenase (Worthington) for ≈15 minutes at 37°C. The heart was placed in stop buffer comprising perfusion buffer plus 10% vol/vol bovine calf serum. The perfused heart was gently disaggregated by teasing apart the tissue with scissors followed by passage through pipettes of progressively smaller diameters. The cardiomyocytes and non‐myocytes were separated through density centrifugation by spinning at a low speed (18g) for 5 minutes. Cardiomyocytes sediment at this speed, but the majority of non‐myocytes (eg, fibroblasts, endothelial cells) remain in the supernatant. The supernatant was transferred into a new tube and centrifuged at 340g for 5 minutes to collect the non‐myocyte pellet. Both cardiomyocyte and non‐myocyte pellets were washed once with PBS and then snap frozen in liquid nitrogen until use.
Western Blot Analysis
Whole cell fractions were obtained as previously described.23 Protein concentrations were measured with the DC protein assay (Bio‐Rad Laboratories, Hercules, CA). Equal amounts of protein were loaded into lanes of CriterionTGX (Tris‐Glycine eXtended) Stain‐Free PAGE gels (BioRad). The gels were electrophoresed and activated using a ChemiDoc MP Visualization System (BioRad). The protein was then transferred to a polyvinylidene difluoride membrane. The membranes were then imaged using a ChemiDoc MP Visualization System to obtain an assessment of proper transfer and to obtain total protein loads. The membranes were then blocked and probed with primary antibodies overnight at 4°C. Immunoblots were next processed with secondary antibodies (Cell Signaling) for 1 hour at room temperature. Immunoblots were then probed with a Super Signal West Dura kit (Thermo Fisher Scientific) to visualize signal, followed by visualization using a ChemiDoc MP Visualization System (BioRad). Data were analyzed using Image Lab (BioRad). The total protein images were used as loading controls. For each protein of interest, the portion of the protein load image corresponding to the molecular weight of the protein of interest was used as the loading control.23
Histological Analysis
Hearts were harvested and fixed in 10% formalin and embedded in paraffin. Slices were cut at 7 μm and stained with Masson trichrome (Millipore Sigma, Burlington, MA). Fibrosis area was quantitatively analyzed with National Institutes of Health Image software.
Wheat Germ Agglutinin Staining
Cross‐sectional area (μm2) was analyzed by staining cardiac cryosections with wheat germ agglutinin‐Texas Red‐X conjugate (Life Technologies) to show myocyte membranes in histological sections. Cryosections were first washed in 1XPBS and then incubated in 10 μg wheat germ agglutinin‐Texas Red‐X conjugate for 1 hour at room temperature followed by additional washes in 1XPBS. Slides were mounted with VectaShield mounting medium (Vector Labs) and sealed. Digital images were captured, and cell surface area was assessed with Nikon NIS‐Elements Imaging Software (version 3.22.11) in at least 5 animals per group with at least 3 randomly taken sections per heart and at least 100 myocytes were counted per animal.
Oxidative Stress
Concentrations of malondialdehyde in the heart were measured using the OxiSelect MDA Adduct Competitive ELISA kit (STA‐832; Cell Biolabs, Inc, San Diego, CA) according to the manufacturer's instructions.
Glycative Stress
Concentrations of methylglyoxal, advanced glycation end products, and carboxymethyllysine in the heart tissue or cell homogenates was measured using the OxiSelect Methylglyoxal Competitive ELISA kit (STA‐811), OxiSelect Advanced Glycation End Product Competitive ELISA kit (STA‐817), and OxiSelect N‐epsilon‐(Carboxymethyl) Lysine Competitive ELISA kit (STA‐816), respectively, according to the manufacturer's (Cell Biolabs, Inc, San Diego, CA) instructions. Concentrations of RAGE in the heart tissue or cell homogenates were measured using the Mouse RAGE/AGER Elisa Kit (RAB0008) or Rat RAGE/AGER Elisa Kit (RAB0009), respectively, according to the manufacturer's (Sigma Aldrich, St. Louis, MO) instructions.
Glyoxylase Activity
The activity of cardiac Glo1 was evaluated using the Glyoxylase I Activity Assay (MAK114; Sigma Aldrich, St. Louis, MO) according to the manufacturer's instructions.
Caspase‐3/7 Activity
The activity of caspase‐3/7 in the heart tissue or cell homogenates was evaluated using the Caspase‐3 Assay Kit (ab39401; Abcam) or Apo‐One Homogeneous Caspase‐3/7 Assay kit (G7790; Promega) according to the manufacturer's instructions.
Inducible Nitric Oxide Synthase
Concentrations of inducible nitric oxide synthase in heart tissue or cell homogenates was evaluated using the Mouse iNOS ELISA kit (ab253219; Abcam) or NOS2/iNOS (Rat) Elisa Kit (E4649; BioVision) according to the manufacturer's instructions.
Tissue Processing for Histology
All tissues for histology were fixed with 4% paraformaldehyde (v/v in PBS) for 3 hours at room temperature washed with PBS and incubated in a 30% sucrose solution (w/v in PBS) overnight at 4°C. After tissues sunk down to the bottom of specimen tubes, they were trimmed, embedded in optimal cooling temperature compound and frozen with a slurry of dry ice and Isopentane. Blocks were cut into 7‐μm‐thick sections. Sections were mounted on Fisherbrand Superfrost Plus Microscope slides (Fischer Scientific). Blocks and slides were stored at −80°C until further use.
Immunohistochemistry
To quantify GFP‐labeling of cardiomyocytes on histological sections, Cryosections were stained with an antibody to GFP (183734; Abcam). The sections were then counterstained with an antibody to wheat germ agglutinin conjugated to Texas Red‐X (ThermoFisher) and DAPI. Images were acquired using a Leica DMZ6000 microscope.
TUNEL Staining
Apoptosis assay was performed by using the TUNEL Assay Kit—BrdU‐Red (Abcam; ab66110) as per the manufacturer's protocol that is based on the principle of terminal deoxynucleotidyl transferase (TdT)‐mediated dUTP nick end‐labeling (TUNEL). Sections were counterstained with an anti‐Cardiac Troponin T‐FITC antibody (Miltenyi Biotec, 130‐119‐674) to label cardiomyocytes and DAPI to label nuclei. Images were acquired with a Leica DMZ6000 microscope using a 40× objective. Images were taken of fields containing both the scar area and infarct border zone. The number of TUNEL positive cells per field of view was counted with Image J. Quantification of TUNEL positive cells was calculated as follows: (1) percentage of TUNEL positive cells per nuclei; (2) percentage of TUNEL positive cells per nuclei in non‐cardiomyocytes (troponin negative), and (3) percentage of TUNEL positive cells per nuclei in cardiomyocytes (troponin positive).
Statistical Analysis
All data are expressed as mean ± SEM. The data were first evaluated for normal distribution using the D'Agostina and Pearson omnibus normality test. Subsequent, statistical significance was evaluated as follows: (1) unpaired Student t test for comparison between 2 means; (2) a 1‐way ANOVA with a Tukey test or Dunnett test as the post hoc analysis for comparison among ≥3 groups; and (3) a 2‐way ANOVA with a Tukey test as the post hoc analysis for comparison among the means from groups of wild‐type (WT) and DJ‐1 knockout mice. For the echocardiography data, a 2‐way repeated measures ANOVA with a Bonferroni test as the post hoc analysis was used. The following comparisons were made separately: (1) baseline versus post‐baseline measurements for each group, (2) differences between each groups baseline measurements, and (3) differences between each groups post‐baseline measurements. The P value for these evaluations was adjusted by applying the Bonferroni correction for multiple comparisons. A value of P<0.05 denoted statistical significance and P values were 2‐sided. All statistical analyses were performed using Prism 7 (GraphPad Software Inc).
Results
DJ‐1 Plays a Protective Role in the Setting of I/R‐Induced Heart Failure
We recently provided the first evidence that the cleaved form of DJ‐1 is induced acutely following the onset of myocardial I/R.21 Here we found that the cleaved form of DJ‐1 persisted for the first 7 days of reperfusion (Figure 1A through 1C). To determine the role of DJ‐1 in heart failure, DJ‐1 knockout, and WT mice were subjected to 60 minutes of ischemia followed by 4 weeks of reperfusion. Echocardiography revealed that DJ‐1 knockout mice exhibited enhanced LV cavity dilatation (Figure 2A and 2B) and exacerbated cardiac dysfunction (Figure 2C) following I/R when compared with WT mice. DJ‐1 knockout also exhibited significantly greater cardiac enlargement (Figure 2D and 2E), myocyte hypertrophy (Figure 2F and 2G), and fibrosis (Figure 2H and 2I). Together these data indicate that a deficiency in DJ‐1 exacerbates I/R‐induced heart failure.
Figure 1.
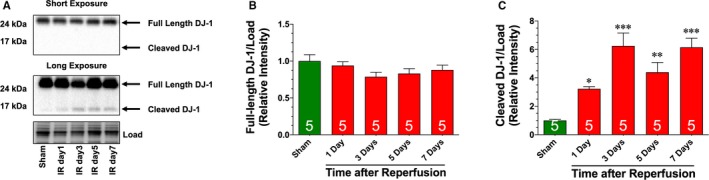
Ischemia‐reperfusion induced cleavage of DJ‐1. A, Immunoblots and analysis of the cardiac expression of the (B) full‐length and (C) cleaved forms of DJ‐1 following 60 minutes of ischemia and different periods of reperfusion. Values are means±SEM. One‐way ANOVA with Dunnett. *P<0.05, **P<0.01, and ***P<0.001 vs sham.
Figure 2.
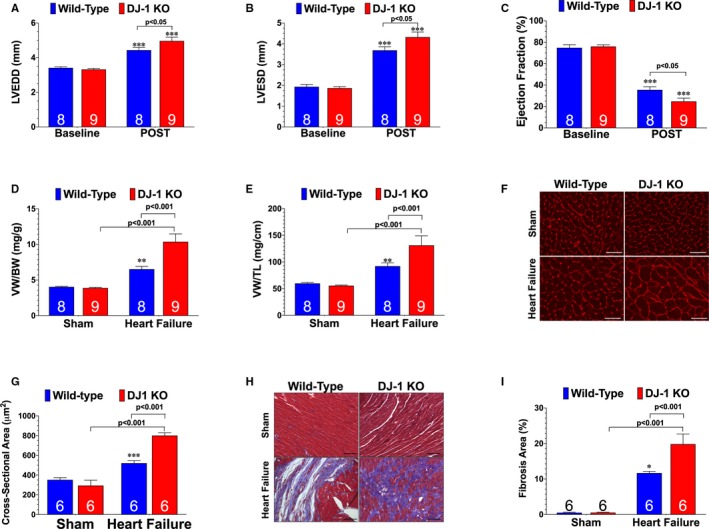
DJ‐1 Deficiency exacerbates ischemia‐reperfusion–induced heart failure. A, Left ventricular end‐diastolic diameter, (B) left ventricular end‐systolic diameter, and (C) left ventricular ejection fraction from wild‐type and DJ‐1 deficient mice (DJ‐1 knockout). Values are means±SEM. Two‐way repeated measures ANOVA with Bonferroni. ***P<0.001 vs baseline. D, Ratios of ventricular weight to body weight and (E) ventricular weight to tibia length were used as a measure of cardiac hypertrophy. F, Wheat germ agglutinin stained hearts. G, Summary of myocyte cross‐sectional area. Scale bar equals 100 μm. H, Masson Trichrome stained hearts. I, Summary of fibrosis area. Scale bar equals 100 μm. All measurements were performed in heart samples collected at 4 weeks of reperfusion. Number in bars represents sample size. Values are means±SEM. Two‐way ANOVA with Tukey. LV indicates left ventricle; LVEDD, left ventricular end‐diastolic diameter; LVESD, left ventricular end‐systolic diameter; VW:BW, ventricular weight to body weight; VW: TL, ventricular weight to tibia length, WGA, wheat germ agglutinin. *P<0.05, **P<0.01, and ***P<0.001 vs wild‐type sham.
Next, we asked whether overexpressing the cleaved form of DJ‐1 would attenuate the development of I/R‐induced heart failure. For these experiments, we created a viral vector to express human cleaved DJ‐1 (AAV9‐DJ1Δc) under the control of the cytomegaolovirus promoter (pAAV2/9 containing AAV2 rep and AAV9 capsid genes). To test the efficiency, specificity, and dose‐dependence of AAV9‐mediated transduction, we injected various doses of AAV9‐GFP into the femoral vein of C57BL/6J mice. GFP levels were measured in heart, liver, brain, and lung tissue 2 weeks after injection using an ELISA kit. A dose‐dependent increase in GFP levels was observed in the heart with 2×1011 vg/mL achieving maximal delivery to the heart with minimal delivery to other tissue (Figure S1A). Further analysis of isolated cardiomyocytes and non‐cardiomyocytes revealed that the transduced gene was restricted to cardiomyocytes (Figure S1B). Finally, GFP‐stained sections of heart tissue indicated that a substantial proportion of cardiomyocytes expressed the transduced gene (Figure S1C). Next, we found that delivery of AAV9‐DJ1Δc to uninjured hearts increased the cleaved form of cardiac DJ‐1 by 2‐fold without altering the expression of the full‐length form (Figure S2A and S2B). Further analysis revealed that these changes in DJ‐1 expression were restricted to cardiomyocytes, as no changes were evident in non‐cardiomyocytes or liver tissue (Figure S2C through S2F).
Next, we evaluated the effects of AAV9‐DJ1Δc on the development of I/R‐induced heart failure. Mice treated with AAV9‐GFP or AAV9‐DJ1Δc were followed for 2 weeks and then subjected to 60 minutes of ischemia and 4 weeks of reperfusion. Analysis revealed that AAV9‐DJ1Δc significantly improved LV function, reduced hypertrophy, and reduced fibrosis when compared with AAV9‐GFP in the setting of I/R‐induced heart failure (Figure 3). Similar to the observations in the WT mice, AAV9‐DJ1Δc improved cardiac dilatation and LV function in DJ‐1 knockout mice (Figure S3). Together these data indicate a protective role for DJ‐1 in the setting of I/R‐induced heart failure.
Figure 3.
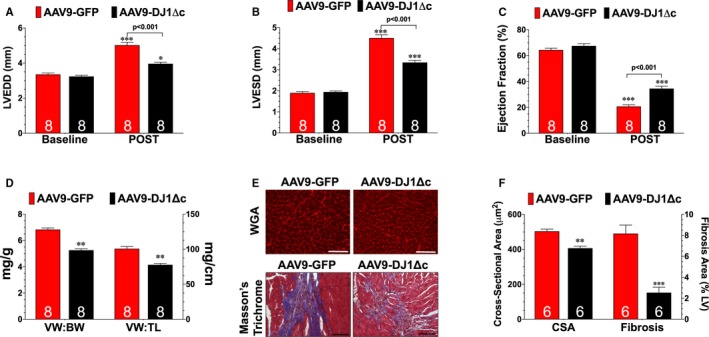
Overexpression of cleaved form of DJ‐1 attenuates ischemia‐reperfusion–induced heart failure. A, Left ventricular end‐diastolic diameter, (B) left ventricular end‐systolic diameter, and (C) left ventricular ejection fraction. Values are means±SEM. Two‐way repeated measures ANOVA with Bonferroni. *P<0.05 and ***P<0.001 vs baseline. D, Ratios of ventricular weight to body weight and ventricular weight to tibia length. E, Wheat germ agglutinin stained and Masson Trichrome stained hearts. Scale bar equals 100 μm. F, Summary of myocyte cross‐sectional area (CSA) and summary of fibrosis area. All measurements were performed in samples collected at 4 weeks of reperfusion from mice treated with either a recombinant adeno‐associated virus expressing AAV9‐GFP (green fluorescent protein) or a recombinant adeno‐associated virus expressing cleaved DJ‐1. Values are means±SEM. t test. AAV9‐DJ1ΔC indicates recombinant adeno‐associated virus expressing cleaved DJ‐1; LVEDD, left ventricular end‐diastolic diameter; LVESD, left ventricular end‐systolic diameter; VW:BW, ventricular weight to body weight; VW: TL, ventricular weight to tibia length. **P<0.01, and ***P<0.001 vs AAV9‐GFP.
DJ‐1 Alters I/R‐Induced Glycative Stress
Glycative stress (ie, accumulation of AGE and activation of RAGE) contributes to I/R‐induced cardiac injury.13, 17, 18 Given the observations that DJ‐1 possesses anti‐glycative actions in cell culture,11, 30, 31 we sought to determine if the protective actions of cardiac DJ‐1 were associated with alterations in glycative stress. Initial analysis at 3 days of reperfusion revealed significant accumulations of methylglyoxal, AGE, carboxymethyllysine (a specific AGE), and RAGE in the hearts of WT mice (Figure 4A through 4D). The levels of all 4 were further increased in DJ‐1 knockout hearts indicating enhanced glycative stress. Glycative stress is associated with apoptosis and inflammation. Consistent with the enhanced levels of glycative stress, hearts from DJ‐1 knockout hearts displayed increased levels of caspase‐3/7 activity (Figure 4E), as well as an increased number of apoptotic cells (TUNEL staining)—non‐myocytes in the scar area and infarct border zone, as well as cardiomyocytes in the infarct border zone (Figure S4). Furthermore, markers of inflammation and levels of oxidative stress were also enhanced in DJ‐1 knockout hearts (Figure S5 and Figure 4F). Together these data indicate that hearts from DJ‐1 knockout mice experience more chronic injury compared with hearts from WT mice.
Figure 4.
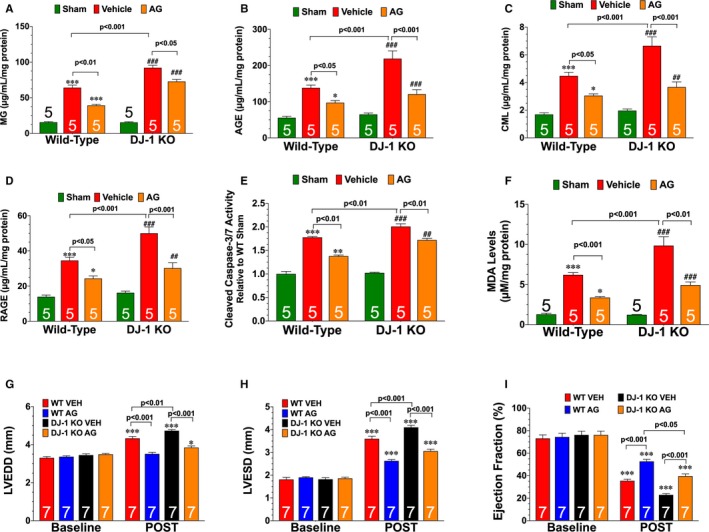
Deficiency of DJ‐1 enhances ischemia‐reperfusion–induced glycative stress. Cardiac levels of (A) methylglyoxal, (B) advanced glycation end‐product, (C) carboxymethyllysine, and (D) receptor for AGE. E, Cleaved caspase‐3/7 activity. F, Malondialdehyde levels. All measurements were performed in heart samples collected at 3 days of reperfusion from wild‐type and DJ‐1 knockout mice administered vehicle or aminoguanadine (AG; 1 g/L). Values are means±SEM. Two‐way ANOVA with Tukey. *P<0.01 and ***P<0.001 vs wild‐type sham. ## P<0.01 and ### P<0.001 vs DJ‐1 knockout sham. G, Left ventricular end‐diastolic diameter, (H) left ventricular end‐systolic diameter, and (I) left ventricular ejection fraction. All measurements were performed on hearts from wild‐type and DJ‐1 knockout mice administered vehicle or AG (1 g/L) at 4 weeks of reperfusion. Values are means±SEM. Two‐way repeated measures ANOVA with Bonferroni. AGE indicates advanced glycation end‐product; LVEDD, left ventricular end‐diastolic diameter; LVESD, left ventricular end‐systolic diameter; RAGE, receptor for AGE; WT, wild‐type. *P<0.05 and ***P<0.001 vs baseline.
AGE‐RAGE signaling induces ischemic injury in the heart via inducible nitric oxide synthase (iNOS).32 In agreement with this finding, the iNOS inhibitor, aminoguanidine, is routinely used as an antiglycation agent.24 To determine if enhanced glycative stress contributed to the more severe heart failure observed in DJ‐1 knockout mice, WT and DJ‐1 knockout mice subjected to myocardial I/R were administered aminoguanidine (1 g/L)24 in drinking water starting at 24 hours of reperfusion. Treatment continued daily through the first 7 days of reperfusion. At 3 days of reperfusion, analysis revealed that I/R‐induced iNOS levels were enhanced in DJ‐1 knockout hearts (Figure S5D). Aminoguanidine reduced iNOS levels, glycative stress, cell death (caspase‐3/7 activity and TUNEL positive cells) inflammation, and oxidative stress in both WT and DJ‐1 knockout hearts (Figure 4 and Figures S4 and S5). Importantly, aminoguanidine improved LV function in DJ‐1 knockout mice compared with vehicle treated DJ‐1 knockout mice (Figure 4G through 4I) In a similar manner, AAV9‐DJ1Δc decreased I/R‐induced glycative stress, cell death (caspase‐3/7 activity and TUNEL positive cells) inflammation, and oxidative stress in both WT and DJ‐1 knockout mice (Figure 5 and Figure S4). Together, these data indicate that aminoguanidine and AAV9‐DJ1Δc promote cardiac healing.
Figure 5.
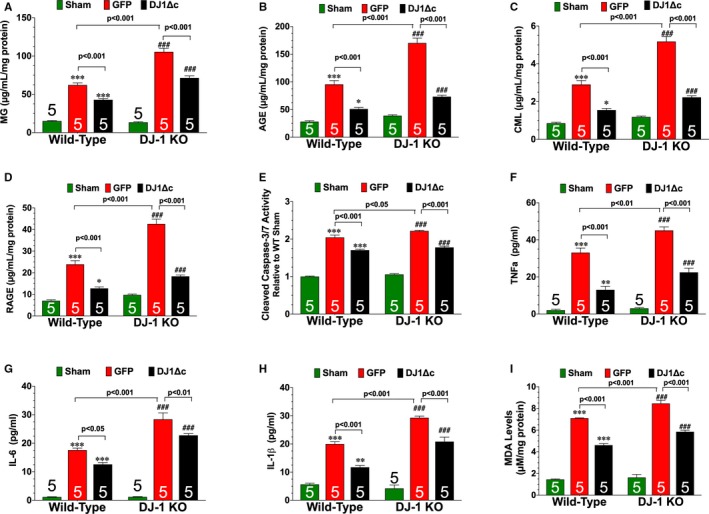
Overexpression of cleaved form of DJ‐1 reduces ischemia‐reperfusion–induced glycative stress. Cardiac levels of (A) methylglyoxal (MG), (B) advanced glycation end‐product (AGE), (C) carboxymethyllysine (CML), and (D) receptor for AGE (RAGE). E, Cleaved caspase‐3/7 activity. Cardiac levels of (F) tumor necrosis factor α (TNFα), (G) interleukin 6 (IL‐6), and (H) interleukin‐1β (IL‐1β). I, Cardiac levels of malondialdehyde, a marker of oxidative stress. All measurements were performed in samples collected at 3 days of reperfusion from wild‐type and DJ‐1 knockout (KO) mice treated with either a recombinant adeno‐associated virus expressing green fluorescent protein (GFP) or a recombinant adeno‐associated virus expressing cleaved DJ1Δc. Values are means±SEM. One‐way ANOVA with Tukey. AGE indicates advanced glycation end‐product; LVEDD, left ventricular end‐diastolic diameter; LVESD, left ventricular end‐systolic diameter; RAGE, receptor for AGE; WT, wild‐type. **P<0.01 and ***P<0.001 vs sham. ### P<0.001 vs DJ‐1 knockout sham.
To delve further into the role cardiac DJ‐1 plays in modulating glycative stress, we used an in vitro model of energy stress32 using H9c2 cells. Initial experiments validated this approach, as exposure of cells to glucose deprivation induced the cleavage of DJ‐1, as well as increased glycative stress and cell death (Figure S6). To mimic our in vivo conditions, DJ‐1 levels were reduced by transfecting cells with siRNA to DJ‐1 (siRNA‐DJ1) (Figure S6D). Remarkably, the loss of DJ‐1 was sufficient to induce glycative stress and cell death under control conditions. In response to glucose deprivation, the loss of DJ‐1 was found to further increase the levels of glycative stress and cell death when compared with cells transfected with control siRNA (siRNA‐scr). Importantly, treatment with aminoguanidine (100 μmol/L) reduced glycative stress and cell death in siRNA‐DJ1 transfected cells under both control and glucose deprivation conditions. Together these data provide further support that cardiac DJ‐1 possesses anti‐glycative actions.
DJ‐1 Does Not Alter the Expression or Activity of Glo‐1
The glyoxalase system, in which the enzyme glyoxalase‐I (Glo‐I) is the rate‐limiting step, detoxifies highly reactive carbonyls and AGE precursors to prevent the formation and accumulation of AGEs.12 The above evidence indicates that cardiac DJ‐1 possesses anti‐glycative actions in the setting of I/R‐induced heart failure. Additional experiments were performed to evaluate if DJ‐1 influenced the expression and/or activity of Glo1. In WT hearts, I/R injury did not alter the protein expression of Glo1, but it did increase Glo1 activity (Figure S7). A similar increase in Glo1 activity was evident in the hearts of DJ‐1 knockout mice and in WT hearts treated with AAV9‐DJ1Δc. Together this suggests that DJ‐1 does not influence the expression or activity of Glo1 in response to myocardial I/R injury.
Delayed Treatment With AAV9‐DJ1Δc Attenuates the Progression of I/R‐Induced Heart Failure
As noted, data from human studies suggest that the AGE‐RAGE axis is implicated in heart failure19, 20 and it is predicted that therapeutic antagonism of this axis might be a unique target for therapeutic intervention.13 This led us to evaluate the association between glycative stress and DJ‐1 expression in the setting of chronic heart failure. Using left ventricular samples obtained from end‐stage heart failure patients at the time of transplant,23 we found significantly lower levels of both the full‐length form and cleaved form of DJ‐1 compared with non‐failing control samples (Figure 6A). This was associated with elevated levels of AGE, carboxymethyllysine, and RAGE (Figure 6C and 6D). Similar results were found in hearts of mice collected at 4 weeks of reperfusion (Figure 7A through 7C). We, therefore, sought to determine if restoring the cleaved form of DJ‐1 in this chronic state would alter glycative stress and influence the development of I/R‐induced heart failure. For these experiments, mice subjected to I/R were administered AAV9‐GFP or AAV9‐DJ1Δc at 1 week of reperfusion. They were then followed for an additional 7 weeks. Our analysis revealed that AAV9‐DJ1Δc increased the expression of the cleaved form of DJ‐1 and decreased glycative stress when compared with AAV9‐GFP (Figure 7A through 7C). This was associated with improved LV function and reduced hypertrophy compared with AAV9‐GFP (Figure 7D through 7I), indicating that delayed treatment with AAV9‐DJ1Δc attenuates the progression of I/R‐induced heart failure. Interestingly, these improvements occurred in the absence of a change in fibrosis (Figure 7H through 7I).
Figure 6.
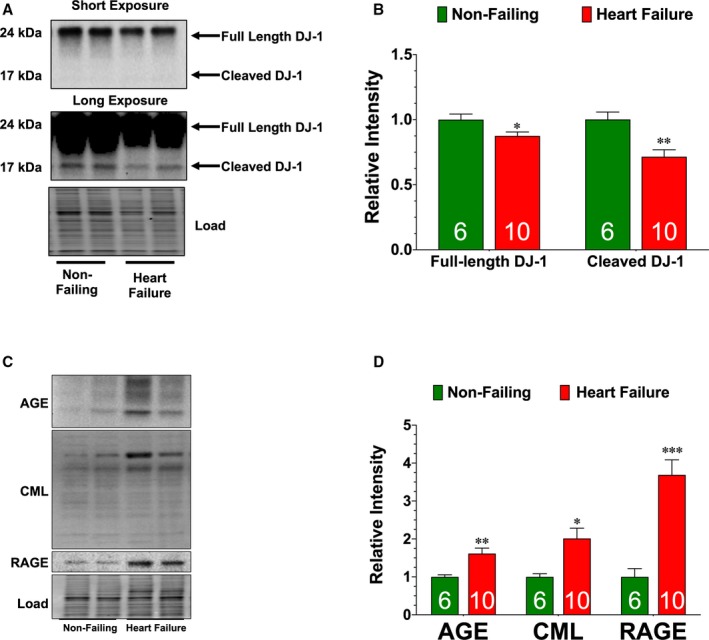
DJ‐1 expression is diminished in left‐ventricular samples collected from end‐stage heart failure patients. A, Immunoblots and (B) analysis of the expression of the full‐length and cleaved forms of DJ‐1. C, Immunoblots and (D) analysis of the expression of advanced glycation end‐product, carboxymethyllysine(, and receptor for AGE. Measurements were performed in left ventricular samples collected from non‐failing and end‐stage heart failure patients. Values are means±SEM. t test. AGE indicates advanced glycation end‐product; RAGE, receptor for AGE. *P<0.05, **P<0.01, and ***P<0.001 vs non‐failing.
Figure 7.
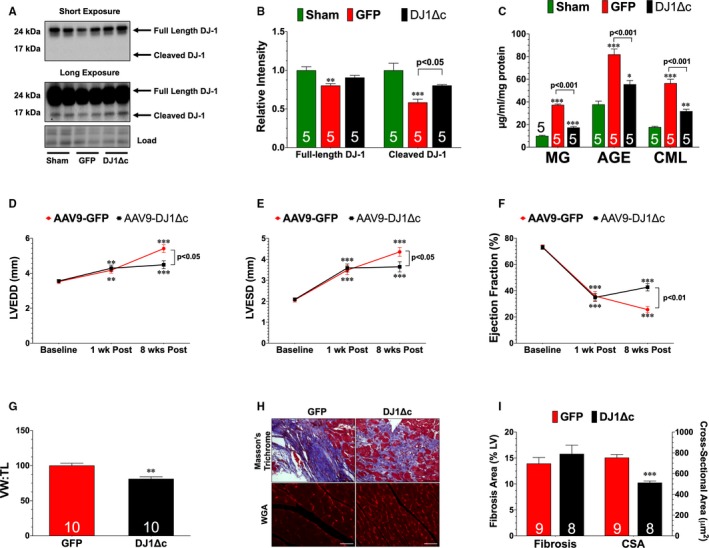
Delayed treatment with recombinant adeno‐associated virus expressing cleaved DJ‐1 reduces progression of ischemia‐reperfusion–induced heart failure. A, Immunoblots and (B) analysis of the expression of the full‐length and cleaved forms of DJ‐1. C, Cardiac levels of methylglyoxal (MG), advanced glycation end‐product (AGE), and carboxymethyllysine (CML). Values are means±SEM. One‐way ANOVA with Tukey. *P<0.05, **P<0.01, and ***P<0.001 vs Sham. D, Left ventricular end‐diastolic diameter, (E) left ventricular end‐systolic diameter, and (F) left ventricular ejection fraction. Values are means±SEM. Two‐way repeated measures ANOVA with Bonferroni. **P<0.01 and ***P<0.001 vs baseline. G, Ratio of ventricular weight to tibia length. H, Wheat germ agglutinin and Masson Trichrome stained hearts. Scale bar equals 100 μm. I, Summary of myocyte cross‐sectional area (CSA) and summary of fibrosis area. All measurements were performed in heart samples collected at 8 weeks of reperfusion from mice a treated with either a recombinant adeno‐associated virus expressing green fluorescent protein (GFP) or a recombinant adeno‐associated virus expressing cleaved DJ1Δc at 1 week of reperfusion. Values are means±SEM. t test. AAV9‐DJ1ΔC indicates recombinant adeno‐associated virus expressing cleaved DJ‐1; LV, left ventricle; LVEDD, left ventricular end‐diastolic diameter; VW:TL, ventricular weigh to tibia length; WGA, wheat germ agglutinin.**P<0.01 and ***P<0.001 vs GFP.
Discussion
Although in vitro and cell‐free systems are important for identifying the intrinsic function/properties of a protein, they do not always provide an accurate assessment of the biological/physiological role of the protein. In fact, there are numerous examples in the literature where the reported in vitro functions of a protein have not been confirmed by in vivo studies (ie, pore forming ability of α‐synuclein33). As such, it is necessary to evaluate the biological role of proteins using in vivo models to apply the acquired knowledge properly and accurately. For instance, the cytoprotective actions of DJ‐1 have been reported in various model systems ranging from in vitro oxidative stress‐induced cell death9 to ischemic injury.34 In the heart, DJ‐1 deficiency is associated with enhanced myocardial infarction following ischemia‐reperfusion injury,8, 21 as well as enhanced left ventricular dysfunction following pressure overload‐induced heart failure and permanent myocardial ischemia.7 The evidence presented here further supports a cardioprotective role for DJ‐1 with the observation that DJ‐1 deficiency leads to enhanced cardiac injury in a model of I/R‐induced heart failure, whereas the overexpression of the cleaved form of DJ‐1 lessens cardiac injury. Additionally, the current study provides important insights into the physiological role of DJ‐1 and provides a potential mechanism by which DJ‐1 attenuates the development of I/R‐induced heart failure.
In the current study, we expanded on our initial observations that DJ‐1 is cleaved in response to acute myocardial I/R injury21 with the finding that the cleaved form of DJ‐1 is present in the heart during the first 7 days of reperfusion. This is important because the first week of reperfusion is a critical time period where signaling events are important determinants of long‐term functional outcome in this model of I/R‐induced heart failure.35, 36, 37 Currently, the mechanism responsible for the cleavage of DJ‐1 is unknown. It is known that this process is redox sensitive9 and there is evidence that DJ‐1 is oxidized following myocardial ischemia.38 So, the production of reactive oxygen species during reperfusion could be a stimulus for the cleavage. Interestingly, the expression of both the full‐length and cleaved forms of DJ‐1 were found to be depressed in LV samples obtained from end‐stage heart failure patients and in hearts from mice collected at 8 weeks of reperfusion. This indicates that while DJ‐1 is active during early periods following reperfusion, it is suppressed during the late stages of heart failure. The cause of this downregulation is unclear. There is some evidence that DJ‐1 can experience irreversible methionine oxidation,39 a process that induces protein unfolding and degradation.40, 41 Furthermore, in addition to oxidation, protein S‐nitrosylation is a post‐translational modification that targets cysteine residues in proteins.42 The S‐nitrosylation of DJ‐1 is important in its dimerization43—a feature that underlies its ability to protect against oxidative stress.5 However, it is unclear if S‐nitrosylation directly affects the cleavage of DJ‐1 or only contributes to its dimerization. Future studies are, therefore, needed to fully understand how different types or levels of oxidation affect the cleavage and expression of DJ‐1, as well as how post‐translational modifications regulate its activity.
Given the cytoprotective effects of DJ‐1, therapeutic strategies designed to enhance its cellular actions are being investigated. For instance, some use phenylbuturate to target DJ‐1.44 While its use can increase the expression of DJ‐1, phenylbuturate has multiple cellular targets.45 Therefore, any beneficial effects elicited by it can't be directly attributed to DJ‐1. To overcome this deficiency, we created a viral vector that expresses the cleaved form of DJ‐1. Using both wild‐type and DJ‐1 knockout mice, we were able to demonstrate that the expression of the cleaved form of DJ‐1 is sufficient to promote healing in the setting of I/R‐induced heart failure. More importantly, we found that delayed treatment of the virus was able to reduce the progression of heart failure. As such, therapeutic overexpression of cleaved DJ‐1 may serve as a viable treatment option for heart failure.
AGE‐RAGE signaling and resultant glycative stress induces cardiac injury in various experimental models via the activation of pro‐apoptotic and proinflammatory pathways.14, 17, 18, 28, 46 While a great deal is known about the consequences of stress‐induced AGE‐RAGE signaling in the heart, little is known about how stress affects the anti‐glycation system. With that being said, the activity of Glo‐1 is decreased in the aged heart47 and diabetic heart48—conditions associated with elevated levels of glycative stress. Importantly, the overexpression of Glo‐1 is able to reduced diabetes mellitus‐induced oxidative damage, inflammation and fibrosis.48 Here, we show that the anti‐glycation system is induced by I/R, with the evidence that the activity of Glo‐1 is increased 3 days after the onset of reperfusion. More importantly, the evidence presented here indicates that DJ‐1 alters glycative stress in the setting of heart failure. Because this effect occurs independently of the I/R‐induced changes in Glo‐1 activity, it suggests that cardiac DJ‐1 directly possesses anti‐glycative actions. This notion is further supported by our in vitro data that the loss of DJ‐1 is sufficient to induce glycative stress and by our in vivo data demonstrating that the cleaved form of DJ‐1 is sufficient to oppose glycative stress following I/R injury.
There is some debate regarding the anti‐glycative actions of DJ‐1. DJ‐1 was first reported to be a glutathione‐independent glyoxalase.11 However, recent studies argue that DJ‐1 is a deglycase rather than a glyoxalase.30, 31 The distinction is important because a glyoxalase metabolizes the reactive carbonyl or AGE precursor, whereas a deglycase removes the glycation moiety from proteins. As a result, a deglycase would be considered a repair protein. It is important to note that the argument does not dispute the anti‐glycative actions of DJ‐1, just the manner in which it performs the actions. Additionally, it is important to note that these studies were performed under cell free conditions, under in vitro conditions, or in C. elegans using DJ‐1 homologs.11, 30, 31 As such it is unclear if cardiac DJ‐1 opposes glycative stress as a glyoxalase, as a deglycase, or through a combination of the 2. Future studies are, therefore, needed to understand whether the 2 proposed mechanisms by which DJ‐1 opposes glycative stress can be distinguished in the setting of I/R‐induced heart failure.
While our current study focused on the ability of DJ‐1 to alter glycative stress, other mechanisms of action are likely involved given the complexity of post‐ischemic cardiac remodeling. For instance, DJ‐1 can alter mitochondrial morphology, inhibit mPTP opening, influence transcription factors, and bind RNA.49 All of these actions could contribute to the protective effects of DJ‐1. It is also possible that the anti‐glycative actions of DJ‐1 contribute in part to a number of the reported actions of DJ‐1. For instance, by reducing AGE‐RAGE signaling, DJ‐1 could reduce glycative stress‐induced oxidative stress and apoptosis, which in turn could result in less mPTP opening. Additionally, the glycation of nucleotides results in nucleotide AGEs,50 so the RNA binding actions of DJ‐1 could be related to its anti‐glycative effects. Finally, DJ‐1 interacts with thioredoxin‐151 and glycative stress inhibits the antioxidant and cardioprotective actions16 of thioredoxin‐1. So, the antioxidant actions of DJ‐1 could be related to its ability to maintain the activity of thioredoxin. Therefore, future studies are warranted to fully investigate the actions of DJ‐1 in the heart.
In summary, our findings provide novel insights into the physiological role for the protease activity of DJ‐1 and support the emerging concept that the cleavage of DJ‐1 into a proteolytic active form is an endogenous stress response designed to protect the heart from injury. Harnessing this activation may be of clinical importance in the treatment of heart failure.
Sources of Funding
This work was funded by grants from the American Heart Association (15POST25610016 to Shimizu and 16GRNT31190016 to Calvert) and the National Institutes of Health (R01DK115213 to Calvert, NS092343 to Li, and R01HL136915 to Calvert and Li). This work was also supported by funding from the Carlyle Fraser Heart Center of Emory University Hospital Midtown to Calvert.
Disclosures
None.
Supporting information
Figure S1. Delivery of AAV9‐GFP increases the expression of GFP in the heart. A, GFP levels in heart, liver, and lung tissue collected from mice administered different doses of AAV9‐GFP. B, Immunoblots of GFP in cardiomyocytes and non‐cardiomyocytes isolated from mice treated with either PBS or AAV9‐GFP. C, Representative images of heart sections from PBS and AAV9‐GFP treated mice stained with wheat germ agglutinin, GFP, and DAPI. Scale bar equals 100 μm. Values are means±SEM. One‐way ANOVA with Dunnett. *P<0.0, **P<0.01, and ***P<0.001 vs PBS.
Figure S2. Delivery of AAV9‐DJ1 or AAV9‐DJ1Δc increases the expression of DJ‐1 only in the heart. A, Immunoblots and (B) densitometric analysis of the expression of the fulllength and cleaved forms of DJ‐1 from hearts administered AAV9‐GFP or AAV9‐DJ1Δc. Values are means±SEM. t test. **P<0.01 vs AAV9‐GFP. C, Immunoblots and (D) densitometric analysis of the expression of the full‐length and cleaved forms of DJ‐1 in cardiomyocytes and non‐cardiomyocytes isolated from mice treated with either AAV9‐GFP or AAV9‐DJ1ΔC (DJ1ΔC). Values are means±SEM. Lane in between cardiomyocytes and non‐cardiomyocytes was intentionally left blank. t test. **P<0.01 vs AAV9‐GFP. E, Immunoblots and (F) densitometric analysis of the expression of the full‐length and cleaved forms of DJ‐1 in livers collected from mice treated with AAV9‐GFP, or AAV9‐DJ1ΔC. Values are means±SEM. One‐way ANOVA with Tukey.
Figure S3. Delivery of AAV9‐DJ1Δc attenuates ischemia‐reperfusion–induced heart failure in DJ‐1 deficient mice. A, Left ventricular end‐diastolic diameter (LVEDD), (B) LV end‐systolic diameter (LVESD), and (C) LV ejection fraction. All measurements were performed on hearts from AAV9‐GFP (GFP) or AAV9‐DJ1Δc treated DJ‐1 deficient (DJ‐1 knockout) mice at 4 weeks of reperfusion. Values are means±SEM. Two‐way Repeated Measures ANOVA with Bonferroni. *P<0.05 and ***P<0.001 vs Baseline. D, Improvements in ejection fraction (EF) observed post‐I/R in wild‐type and DJ‐1 knockout mice treated with AAV9‐DJ1Δc when compared with matched AAV9‐GFP treated mice. Values are means±SEM. t test. *P<0.05 and **P<0.01 vs AAV9‐DJ1.
Figure S4. Aminoguanadine and AAV9‐DJ1Δc Reduce Apoptotic Cells in Wild‐Type and DJ‐1 Deficient Hearts. A, Representative images of heart sections showing TUNEL positive cells (red) in the scar area and infarct border zone. Nuclei are stained with DAPI (blue) and cardiomyocytes are stained with cardiac troponin (green). Arrowheads indicate TUNEL positive nuclei in cardiomyocytes. Scale bar equals 50 μm. Control images to the left represent sections stained with either TUNEL or troponin antibody only. B, Summary of percentage of TUNEL positive cells per nuclei in all cells in the field of interest. C, Summary of percentage of TUNEL positive cells per nuclei in non‐cardiomyocyte. D, Summary of percentage of TUNEL positive cells per nuclei in cardiomyocytes. All measurements were performed in heart samples collected at 3 days of reperfusion from Wild‐Type and DJ‐1 deficient mice (DJ‐1 knockout) administered vehicle, aminoguanadine (1g/L), or AAV9‐DJ1Δc. Values are means±SEM. Two‐way ANOVA with Tukey.
Figure S5. Aminoguanadine reduces inflammation and iNOS levels in wild‐type and DJ‐1 deficient hearts. Cardiac levels of (A) tumor necrosis factor α (TNFα), (B) interleukin 6 (IL‐6), (C) IL‐1β, and (D) inducible nitric oxide synthase (iNOS) levels. All measurements were performed in heart samples collected at 3 days of reperfusion from wild‐type and DJ‐1 deficient mice (DJ‐1 knockout) administered vehicle or aminoguanadine (1g/L). Values are means±SEM. Two‐way ANOVA with Tukey. *P<0.01 and ***P<0.001 vs wild‐type sham. ### P<0.001 vs DJ‐1 knockout sham.
Figure S6. Knockdown of DJ‐1 in H9c2 cells enhances glycative stress and cell death in response to energy stress. A, Immunoblots and densitometric analysis of the expression of the (B) full‐length and (C) cleaved forms of DJ‐1 in H9c2 cells exposed to glucose free media for up to 18 hours. Values are means±SEM. One‐way ANOVA with Dunnett. *P<0.01 and ***P<0.001 vs control. D, Immunoblot and densitometric analysis of DJ‐1 from cells transfected with either control siRNA (siRNA‐scr) or siRNA to DJ‐1 (siRNA‐DJ1) for 48 hours. Levels of (E) advanced glycation end products (AGE), (F) the receptor for AGEs (RAGE), and (G) inducible nitric oxide synthase (iNOS). H, Cleaved caspase‐3/7 activity. Cells were transfected with siRNA‐scr or siRNA‐DJ1. All measurements were performed in samples exposed to glucose free media for 4 hours in the presence or absence of aminoguanadine (100 μmol/L). Values are means±SEM of 3 independent experiments of at least 3 biological replicates. Two‐way ANOVA with Tukey. I, Cell viability was determined in samples exposed to glucose free media for 18 hours. Values are means±SEM of 3 independent experiments of at least 3 biological replicates. Two‐way ANOVA with Tukey.
Figure S7. DJ‐1 Does not alter the expression of activity of glyoxylase‐1. A, Immunoblots and (B) densitometric analysis of the expression of glyoxylase‐1 (Glo1). C, Glo1 activity. Measurements were performed in heart samples collected at 3 days of reperfusion from Wild‐Type or DJ‐1 deficient mice (DJ‐1 knockout). Values are means±SEM. Two‐way ANOVA with Tukey. *P<0.01 vs Wild‐Type Sham. D, Immunoblots and (E) densitometric analysis of the expression of glyoxylase‐1 (Glo1). F, Glo1 activity. Measurements were performed in samples collected at 3 days of reperfusion from AAV9‐GFP (GFP) or AAV9‐DJ1Δc (DJ1ΔC) treated mice. Values are means±SEM. One‐way ANOVA with Tukey. *P<0.05 vs Sham.
Acknowledgments
We acknowledge the Viral Vector Core at Emory University for their assistance in generating the viral vectors.
(J Am Heart Assoc. 2020;9:e014691 DOI: 10.1161/JAHA.119.014691.)
References
- 1. Yamaguchi O, Higuchi Y, Hirotani S, Kashiwase K, Nakayama H, Hikoso S, Takeda T, Watanabe T, Asahi M, Taniike M, Matsumura Y, Tsujimoto I, Hongo K, Kusakari Y, Kurihara S, Nishida K, Ichijo H, Hori M, Otsu K. Targeted deletion of apoptosis signal‐regulating kinase 1 attenuates left ventricular remodeling. Proc Natl Acad Sci USA. 2003;100:15883–15888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, Kass DA. Cardiomyocyte‐specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res. 2014;114:1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takagi H, Hsu CP, Kajimoto K, Shao D, Yang Y, Maejima Y, Zhai P, Yehia G, Yamada C, Zablocki D, Sadoshima J. Activation of PKN mediates survival of cardiac myocytes in the heart during ischemia/reperfusion. Circ Res. 2010;107:642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yanagisawa D, Kitamura Y, Inden M, Takata K, Taniguchi T, Morikawa S, Morita M, Inubushi T, Tooyama I, Taira T, Iguchi‐Ariga SM, Akaike A, Ariga H. DJ‐1 protects against neurodegeneration caused by focal cerebral ischemia and reperfusion in rats. J Cereb Blood Flow Metab. 2008;28:563–578. [DOI] [PubMed] [Google Scholar]
- 5. Taira T, Saito Y, Niki T, Iguchi‐Ariga SM, Takahashi K, Ariga H. DJ‐1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olzmann JA, Brown K, Wilkinson KD, Rees HD, Huai Q, Ke H, Levey AI, Li L, Chin LS. Familial Parkinson's disease‐associated l166p mutation disrupts DJ‐1 protein folding and function. J Biol Chem. 2004;279:8506–8515. [DOI] [PubMed] [Google Scholar]
- 7. Billia F, Hauck L, Grothe D, Konecny F, Rao V, Kim RH, Mak TW. Parkinson‐susceptibility gene DJ‐1/PARK7 protects the murine heart from oxidative damage in vivo. Proc Natl Acad Sci USA. 2013;110:6085–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dongworth RK, Mukherjee UA, Hall AR, Astin R, Ong SB, Yao Z, Dyson A, Szabadkai G, Davidson SM, Yellon DM, Hausenloy DJ. DJ‐1 protects against cell death following acute cardiac ischemia‐reperfusion injury. Cell Death Dis. 2014;5:e1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen J, Li L, Chin LS. Parkinson disease protein DJ‐1 converts from a zymogen to a protease by carboxyl‐terminal cleavage. Hum Mol Genet. 2010;19:2395–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Brug MP, Blackinton J, Chandran J, Hao LY, Lal A, Mazan‐Mamczarz K, Martindale J, Xie C, Ahmad R, Thomas KJ, Beilina A, Gibbs JR, Ding J, Myers AJ, Zhan M, Cai H, Bonini NM, Gorospe M, Cookson MR. Rna binding activity of the recessive parkinsonism protein DJ‐1 supports involvement in multiple cellular pathways. Proc Natl Acad Sci USA. 2008;105:10244–10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee JY, Song J, Kwon K, Jang S, Kim C, Baek K, Kim J, Park C. Human DJ‐1 and its homologs are novel glyoxalases. Hum Mol Genet. 2012;21:3215–3225. [DOI] [PubMed] [Google Scholar]
- 12. Sousa Silva M, Gomes RA, Ferreira AE, Ponces Freire A, Cordeiro C. The glyoxalase pathway: the first hundred years…. and beyond. Biochem J. 2013;453:1–15. [DOI] [PubMed] [Google Scholar]
- 13. Ramasamy R, Schmidt AM. Receptor for advanced glycation end products (rage) and implications for the pathophysiology of heart failure. Curr Heart Fail Rep. 2012;9:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aleshin A, Ananthakrishnan R, Li Q, Rosario R, Lu Y, Qu W, Song F, Bakr S, Szabolcs M, D'Agati V, Liu R, Homma S, Schmidt AM, Yan SF, Ramasamy R. Rage modulates myocardial injury consequent to lad infarction via impact on JNK and stat signaling in a murine model. Am J Physiol Heart Circ Physiol. 2008;294:H1823–H1832. [DOI] [PubMed] [Google Scholar]
- 15. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. [DOI] [PubMed] [Google Scholar]
- 16. Liu Y, Qu Y, Wang R, Ma Y, Xia C, Gao C, Liu J, Lian K, Xu A, Lu X, Sun L, Yang L, Lau WB, Gao E, Koch W, Wang H, Tao L. The alternative crosstalk between rage and nitrative thioredoxin inactivation during diabetic myocardial ischemia‐reperfusion injury. Am J Physiol Endocrinol Metab. 2012;303:E841–E852. [DOI] [PubMed] [Google Scholar]
- 17. Shang L, Ananthakrishnan R, Li Q, Quadri N, Abdillahi M, Zhu Z, Qu W, Rosario R, Toure F, Yan SF, Schmidt AM, Ramasamy R. Rage modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK‐3beta signaling pathways. PLoS One. 2010;5:e10092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bucciarelli LG, Kaneko M, Ananthakrishnan R, Harja E, Lee LK, Hwang YC, Lerner S, Bakr S, Li Q, Lu Y, Song F, Qu W, Gomez T, Zou YS, Yan SF, Schmidt AM, Ramasamy R. Receptor for advanced‐glycation end products: key modulator of myocardial ischemic injury. Circulation. 2006;113:1226–1234. [DOI] [PubMed] [Google Scholar]
- 19. Koyama Y, Takeishi Y, Arimoto T, Niizeki T, Shishido T, Takahashi H, Nozaki N, Hirono O, Tsunoda Y, Nitobe J, Watanabe T, Kubota I. High serum level of pentosidine, an advanced glycation end product (age), is a risk factor of patients with heart failure. J Card Fail. 2007;13:199–206. [DOI] [PubMed] [Google Scholar]
- 20. Wang LJ, Lu L, Zhang FR, Chen QJ, De Caterina R, Shen WF. Increased serum high‐mobility group box‐1 and cleaved receptor for advanced glycation endproducts levels and decreased endogenous secretory receptor for advanced glycation endproducts levels in diabetic and non‐diabetic patients with heart failure. Eur J Heart Fail. 2011;13:440–449. [DOI] [PubMed] [Google Scholar]
- 21. Shimizu Y, Lambert JP, Nicholson CK, Kim JJ, Wolfson DW, Cho HC, Husain A, Naqvi N, Chin LS, Li L, Calvert JW. DJ‐1 protects the heart against ischemia‐reperfusion injury by regulating mitochondrial fission. J Mol Cell Cardiol. 2016;97:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murphy E, Lagranha C, Deschamps A, Kohr M, Nguyen T, Wong R, Sun J, Steenbergen C. Mechanism of cardioprotection: what can we learn from females? Pediatr Cardiol. 2011;32:354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shimizu Y, Polavarapu R, Eskla KL, Nicholson CK, Koczor CA, Wang R, Lewis W, Shiva S, Lefer DJ, Calvert JW. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J Mol Cell Cardiol. 2018;116:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein‐7 and advanced glycation end product inhibitors. Diabetes. 2007;56:1825–1833. [DOI] [PubMed] [Google Scholar]
- 25. Shimizu Y, Polavarapu R, Eskla KL, Pantner Y, Nicholson CK, Ishii M, Brunnhoelzl D, Mauria R, Husain A, Naqvi N, Murohara T, Calvert JW. Impact of lymphangiogenesis on cardiac remodeling after ischemia and reperfusion injury. J Am Heart Assoc. 2018;7:e009565 DOI: 10.1161/JAHA.118.009565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang X, Hartley AV, Yin Y, Herskowitz JH, Lah JJ, Ressler KJ. AAV2 production with optimized N/P ratio and pei‐mediated transfection results in low toxicity and high titer for in vitro and in vivo applications. J Virol Methods. 2013;193:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous DRP1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. [DOI] [PubMed] [Google Scholar]
- 28. Wang XL, Lau WB, Yuan YX, Wang YJ, Yi W, Christopher TA, Lopez BL, Liu HR, Ma XL. Methylglyoxal increases cardiomyocyte ischemia‐reperfusion injury via glycative inhibition of thioredoxin activity. Am J Physiol Endocrinol Metab. 2010;299:E207–E214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tejada T, Tan L, Torres RA, Calvert JW, Lambert JP, Zaidi M, Husain M, Berce MD, Naib H, Pejler G, Abrink M, Graham RM, Lefer DJ, Naqvi N, Husain A. IGF‐1 degradation by mouse mast cell protease 4 promotes cell death and adverse cardiac remodeling days after a myocardial infarction. Proc Natl Acad Sci USA. 2016;113:6949–6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Richarme G, Mihoub M, Dairou J, Bui LC, Leger T, Lamouri A. Parkinsonism‐associated protein DJ‐1/PARK7 is a major protein deglycase that repairs methylglyoxal‐ and glyoxal‐glycated cysteine, arginine, and lysine residues. J Biol Chem. 2015;290:1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Advedissian T, Deshayes F, Poirier F, Viguier M, Richarme G. The parkinsonism‐associated protein DJ‐1/PARK7 prevents glycation damage in human keratinocyte. Biochem Biophys Res Commun. 2016;473:87–91. [DOI] [PubMed] [Google Scholar]
- 32. Bucciarelli LG, Ananthakrishnan R, Hwang YC, Kaneko M, Song F, Sell DR, Strauch C, Monnier VM, Yan SF, Schmidt AM, Ramasamy R. Rage and modulation of ischemic injury in the diabetic myocardium. Diabetes. 2008;57:1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Furukawa K, Matsuzaki‐Kobayashi M, Hasegawa T, Kikuchi A, Sugeno N, Itoyama Y, Wang Y, Yao PJ, Bushlin I, Takeda A. Plasma membrane ion permeability induced by mutant alpha‐synuclein contributes to the degeneration of neural cells. J Neurochem. 2006;97:1071–1077. [DOI] [PubMed] [Google Scholar]
- 34. Aleyasin H, Rousseaux MW, Phillips M, Kim RH, Bland RJ, Callaghan S, Slack RS, During MJ, Mak TW, Park DS. The parkinson's disease gene DJ‐1 is also a key regulator of stroke‐induced damage. Proc Natl Acad Sci USA. 2007;104:18748–18753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia‐induced heart failure in mice. Circulation. 2010;122:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nicholson CK, Lambert JP, Molkentin JD, Sadoshima J, Calvert JW. Thioredoxin 1 is essential for sodium sulfide‐mediated cardioprotection in the setting of heart failure. Arterioscler Thromb Vasc Biol. 2013;33:744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shimizu Y, Nicholson CK, Lambert JP, Barr LA, Kuek N, Herszenhaut D, Tan L, Murohara T, Hansen JM, Husain A, Naqvi N, Calvert JW. Sodium sulfide attenuates ischemic‐induced heart failure by enhancing proteasomal function in an NRF2‐dependent manner. Circ Heart Fail. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C. Simultaneous measurement of protein oxidation and S‐nitrosylation during preconditioning and ischemia/reperfusion injury with resin‐assisted capture. Circ Res. 2011;108:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, Gearing M, Levey AI, Chin LS, Li L. Oxidative damage of DJ‐1 is linked to sporadic parkinson and Alzheimer diseases. J Biol Chem. 2006;281:10816–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tarrago L, Kaya A, Weerapana E, Marino SM, Gladyshev VN. Methionine sulfoxide reductases preferentially reduce unfolded oxidized proteins and protect cells from oxidative protein unfolding. J Biol Chem. 2012;287:24448–24459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petropoulos I, Friguet B. Maintenance of proteins and aging: the role of oxidized protein repair. Free Radic Res. 2006;40:1269–1276. [DOI] [PubMed] [Google Scholar]
- 42. Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S‐nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA. 1992;89:444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ito G, Ariga H, Nakagawa Y, Iwatsubo T. Roles of distinct cysteine residues in s‐nitrosylation and dimerization of DJ‐1. Biochem Biophys Res Commun. 2006;339:667–672. [DOI] [PubMed] [Google Scholar]
- 44. Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, Freed CR. Phenylbutyrate up‐regulates the DJ‐1 protein and protects neurons in cell culture and in animal models of parkinson disease. J Biol Chem. 2011;286:14941–14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kusaczuk M, Bartoszewicz M, Cechowska‐Pasko M. Phenylbutyric acid: simple structure—multiple effects. Curr Pharm Des. 2015;21:2147–2166. [DOI] [PubMed] [Google Scholar]
- 46. Gao W, Zhou Z, Liang B, Huang Y, Yang Z, Chen Y, Zhang L, Yan C, Wang J, Lu L, Wen Z, Xian S, Wang L. Inhibiting receptor of advanced glycation end products attenuates pressure overload‐induced cardiac dysfunction by preventing excessive autophagy. Front Physiol. 2018;9:1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ruiz‐Meana M, Minguet M, Bou‐Teen D, Miro‐Casas E, Castans C, Castellano J, Bonzon‐Kulichenko E, Igual A, Rodriguez‐Lecoq R, Vazquez J, Garcia‐Dorado D. Ryanodine receptor glycation favors mitochondrial damage in the senescent heart. Circulation. 2019;139:949–964. [DOI] [PubMed] [Google Scholar]
- 48. Brouwers O, de Vos‐Houben JM, Niessen PM, Miyata T, van Nieuwenhoven F, Janssen BJ, Hageman G, Stehouwer CD, Schalkwijk CG. Mild oxidative damage in the diabetic rat heart is attenuated by glyoxalase‐1 overexpression. Int J Mol Sci. 2013;14:15724–15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ariga H, Takahashi‐Niki K, Kato I, Maita H, Niki T, Iguchi‐Ariga SM. Neuroprotective function of DJ‐1 in parkinson's disease. Oxid Med Cell Longev. 2013;2013:683920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thornalley PJ. Protecting the genome: defence against nucleotide glycation and emerging role of glyoxalase I overexpression in multidrug resistance in cancer chemotherapy. Biochem Soc Trans. 2003;31:1372–1377. [DOI] [PubMed] [Google Scholar]
- 51. Fu C, Wu C, Liu T, Ago T, Zhai P, Sadoshima J, Li H. Elucidation of thioredoxin target protein networks in mouse. Mol Cell Proteomics. 2009;8:1674–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Delivery of AAV9‐GFP increases the expression of GFP in the heart. A, GFP levels in heart, liver, and lung tissue collected from mice administered different doses of AAV9‐GFP. B, Immunoblots of GFP in cardiomyocytes and non‐cardiomyocytes isolated from mice treated with either PBS or AAV9‐GFP. C, Representative images of heart sections from PBS and AAV9‐GFP treated mice stained with wheat germ agglutinin, GFP, and DAPI. Scale bar equals 100 μm. Values are means±SEM. One‐way ANOVA with Dunnett. *P<0.0, **P<0.01, and ***P<0.001 vs PBS.
Figure S2. Delivery of AAV9‐DJ1 or AAV9‐DJ1Δc increases the expression of DJ‐1 only in the heart. A, Immunoblots and (B) densitometric analysis of the expression of the fulllength and cleaved forms of DJ‐1 from hearts administered AAV9‐GFP or AAV9‐DJ1Δc. Values are means±SEM. t test. **P<0.01 vs AAV9‐GFP. C, Immunoblots and (D) densitometric analysis of the expression of the full‐length and cleaved forms of DJ‐1 in cardiomyocytes and non‐cardiomyocytes isolated from mice treated with either AAV9‐GFP or AAV9‐DJ1ΔC (DJ1ΔC). Values are means±SEM. Lane in between cardiomyocytes and non‐cardiomyocytes was intentionally left blank. t test. **P<0.01 vs AAV9‐GFP. E, Immunoblots and (F) densitometric analysis of the expression of the full‐length and cleaved forms of DJ‐1 in livers collected from mice treated with AAV9‐GFP, or AAV9‐DJ1ΔC. Values are means±SEM. One‐way ANOVA with Tukey.
Figure S3. Delivery of AAV9‐DJ1Δc attenuates ischemia‐reperfusion–induced heart failure in DJ‐1 deficient mice. A, Left ventricular end‐diastolic diameter (LVEDD), (B) LV end‐systolic diameter (LVESD), and (C) LV ejection fraction. All measurements were performed on hearts from AAV9‐GFP (GFP) or AAV9‐DJ1Δc treated DJ‐1 deficient (DJ‐1 knockout) mice at 4 weeks of reperfusion. Values are means±SEM. Two‐way Repeated Measures ANOVA with Bonferroni. *P<0.05 and ***P<0.001 vs Baseline. D, Improvements in ejection fraction (EF) observed post‐I/R in wild‐type and DJ‐1 knockout mice treated with AAV9‐DJ1Δc when compared with matched AAV9‐GFP treated mice. Values are means±SEM. t test. *P<0.05 and **P<0.01 vs AAV9‐DJ1.
Figure S4. Aminoguanadine and AAV9‐DJ1Δc Reduce Apoptotic Cells in Wild‐Type and DJ‐1 Deficient Hearts. A, Representative images of heart sections showing TUNEL positive cells (red) in the scar area and infarct border zone. Nuclei are stained with DAPI (blue) and cardiomyocytes are stained with cardiac troponin (green). Arrowheads indicate TUNEL positive nuclei in cardiomyocytes. Scale bar equals 50 μm. Control images to the left represent sections stained with either TUNEL or troponin antibody only. B, Summary of percentage of TUNEL positive cells per nuclei in all cells in the field of interest. C, Summary of percentage of TUNEL positive cells per nuclei in non‐cardiomyocyte. D, Summary of percentage of TUNEL positive cells per nuclei in cardiomyocytes. All measurements were performed in heart samples collected at 3 days of reperfusion from Wild‐Type and DJ‐1 deficient mice (DJ‐1 knockout) administered vehicle, aminoguanadine (1g/L), or AAV9‐DJ1Δc. Values are means±SEM. Two‐way ANOVA with Tukey.
Figure S5. Aminoguanadine reduces inflammation and iNOS levels in wild‐type and DJ‐1 deficient hearts. Cardiac levels of (A) tumor necrosis factor α (TNFα), (B) interleukin 6 (IL‐6), (C) IL‐1β, and (D) inducible nitric oxide synthase (iNOS) levels. All measurements were performed in heart samples collected at 3 days of reperfusion from wild‐type and DJ‐1 deficient mice (DJ‐1 knockout) administered vehicle or aminoguanadine (1g/L). Values are means±SEM. Two‐way ANOVA with Tukey. *P<0.01 and ***P<0.001 vs wild‐type sham. ### P<0.001 vs DJ‐1 knockout sham.
Figure S6. Knockdown of DJ‐1 in H9c2 cells enhances glycative stress and cell death in response to energy stress. A, Immunoblots and densitometric analysis of the expression of the (B) full‐length and (C) cleaved forms of DJ‐1 in H9c2 cells exposed to glucose free media for up to 18 hours. Values are means±SEM. One‐way ANOVA with Dunnett. *P<0.01 and ***P<0.001 vs control. D, Immunoblot and densitometric analysis of DJ‐1 from cells transfected with either control siRNA (siRNA‐scr) or siRNA to DJ‐1 (siRNA‐DJ1) for 48 hours. Levels of (E) advanced glycation end products (AGE), (F) the receptor for AGEs (RAGE), and (G) inducible nitric oxide synthase (iNOS). H, Cleaved caspase‐3/7 activity. Cells were transfected with siRNA‐scr or siRNA‐DJ1. All measurements were performed in samples exposed to glucose free media for 4 hours in the presence or absence of aminoguanadine (100 μmol/L). Values are means±SEM of 3 independent experiments of at least 3 biological replicates. Two‐way ANOVA with Tukey. I, Cell viability was determined in samples exposed to glucose free media for 18 hours. Values are means±SEM of 3 independent experiments of at least 3 biological replicates. Two‐way ANOVA with Tukey.
Figure S7. DJ‐1 Does not alter the expression of activity of glyoxylase‐1. A, Immunoblots and (B) densitometric analysis of the expression of glyoxylase‐1 (Glo1). C, Glo1 activity. Measurements were performed in heart samples collected at 3 days of reperfusion from Wild‐Type or DJ‐1 deficient mice (DJ‐1 knockout). Values are means±SEM. Two‐way ANOVA with Tukey. *P<0.01 vs Wild‐Type Sham. D, Immunoblots and (E) densitometric analysis of the expression of glyoxylase‐1 (Glo1). F, Glo1 activity. Measurements were performed in samples collected at 3 days of reperfusion from AAV9‐GFP (GFP) or AAV9‐DJ1Δc (DJ1ΔC) treated mice. Values are means±SEM. One‐way ANOVA with Tukey. *P<0.05 vs Sham.