

Abstract
Itaconate is an immunometabolite with both anti-inflammatory and bactericidal effects. Its coenzyme A (CoA) derivative, itaconyl-CoA, inhibits B12-dependent methylmalonyl-CoA mutase (MCM) by an unknown mechanism. We demonstrate that itaconyl-CoA is a suicide inactivator of human and Mycobacterium tuberculosis MCM, which forms a markedly air-stable biradical adduct with the 5′-deoxyadenosyl moiety of the B12 coenzyme. Termination of the catalytic cycle in this way impairs communication between MCM and its auxiliary repair proteins. Crystallography and spectroscopy of the inhibited enzyme are consistent with a metal-centered cobalt radical ~6 angstroms away from the tertiary carbon-centered radical and suggest a means of controlling radical trajectories during MCM catalysis. Mycobacterial MCM thus joins enzymes in the glyoxylate shunt and the methylcitrate cycle as targets of itaconate in pathogen propionate metabolism.
The immunomodulatory and antimicrobial effects of itaconate are evincing newfound interest in a compound historically used as a precursor in polymer synthesis. Upon activation, immune cells stimulate itaconate synthesis ~10-fold via aconitate decarboxylase (Irg1)–catalyzed decarboxylation of cis-aconitate, a tricarboxylic acid cycle intermediate (1). Itaconate activates Nrf2, inhibits succinate dehydrogenase, and blocks the transcription factor Iκβξ, leading to a switch from a pro- to an anti-inflammatory state (2). The antimicrobial activity of itaconate is purportedly due to its inhibition of two microbe-specific targets: isocitrate lyase in the glyoxylate shunt and methylcitrate lyase in the methylcitrate cycle, two enzymes that are needed for pathogen survival on acetate or propionate, respectively, as the sole carbon source (Fig. 1A) (3). Propionyl-coenzyme A (CoA) is derived from cholesterol catabolism and is used by pathogens like Mycobacterium tuberculosis (Mtb) for biomass production in the glucose-limiting conditions found in phagosomes (4).
Fig. 1. I-CoA inhibits human and Mtb MCM by forming an air-stable biradical.
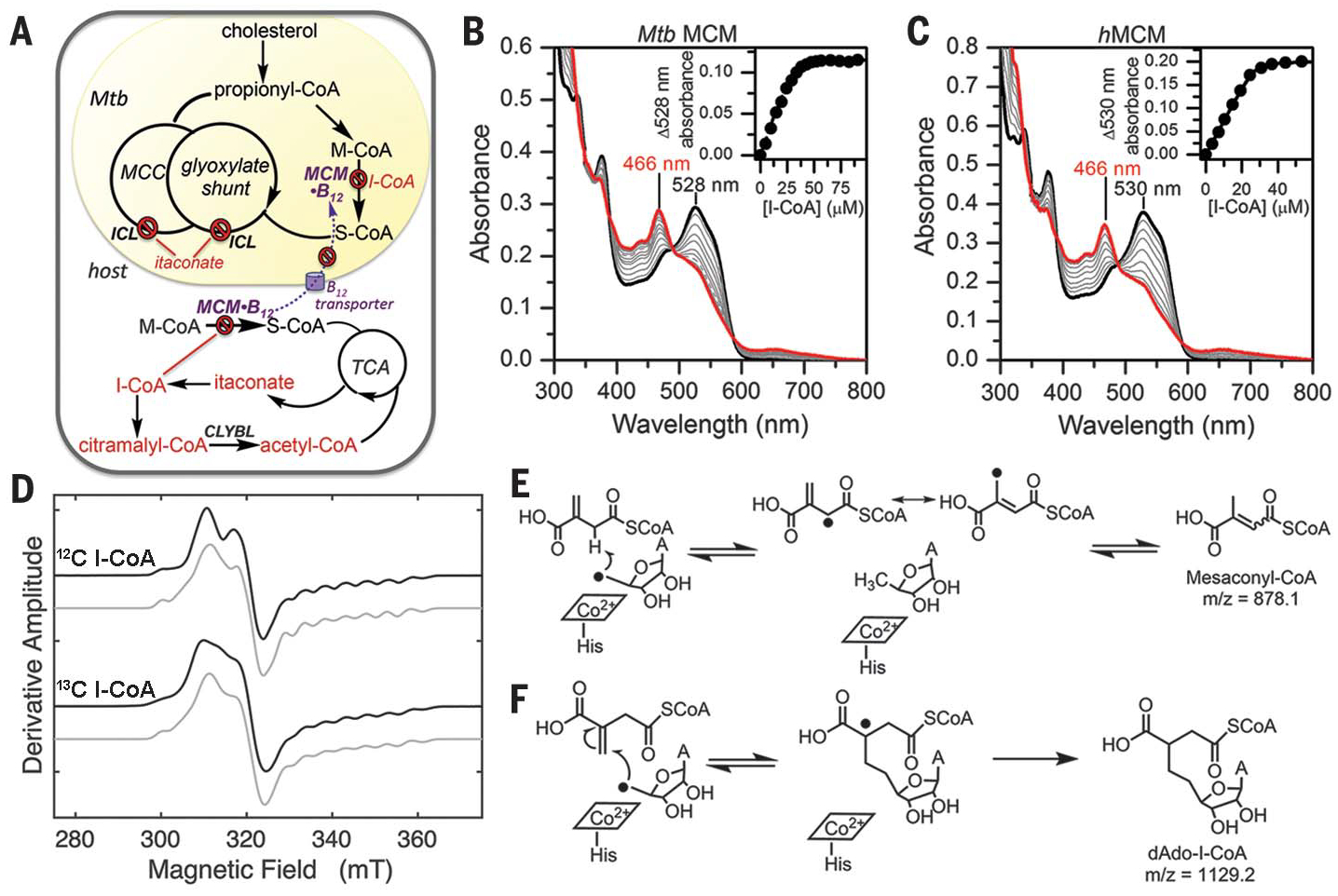
(A) Mtb pathways targetedby I-CoA. MCC, methylcitrate cycle; ICL, isocitrate lyase; S-CoA, succinyl-CoA;TCA, tricarboxylic acid cycle. (B and C) Titration of holo-Mtb MCM (B) with 30 μM bound AdoCbl or holo-hMCM (C) with 40 μM bound AdoCbl (black traces) with increasing concentrations of I-CoA. The intermediate spectra (gray) were recorded after 5 min of equilibration. (Insets) Representative plots of Δ528 or Δ530 nm versus I-CoA indicated stoichiometric binding [n = 2 (Mtb MCM); n = 3 (hMCM)]. (D) EPR spectra of the 1 mM I-CoA-induced biradical on hMCM (375 μM) in the presence of natural abundance (top) or [13C]I-CoA (bottom). The experimental and simulated spectra are in black and gray, respectively. (E and F) Possible fates of dAdo• when I-CoA behaves as substrate (E), not observed, or inhibitor (F).
An unexpected intersection between itaconate and B12-dependent propionate metabolism was revealed recently by the demonstration that itaconyl-CoA (I-CoA) is a potent inhibitor of human 5′-deoxyadenosylcobalamin (AdoCbl)–dependent methylmalonyl-CoA mutase (hMCM) (5). Itaconate can be cleared by a C5-dicarboxylate pathway (6) via acylation to I-CoA, hydration to citramalyl-CoA, and cleavage to acetyl-CoA and pyruvate, a reaction catalyzed by citramalyl-CoA lyase, which is encoded by the recently deorphaned citrate lyase beta-like (CLYBL) gene (5) (Fig. 1A). I-CoA (or methylenesuccinyl-CoA) is an analog of succinyl-CoA, which is interconverted to methylmalonyl-CoA (M-CoA) by the isomerase MCM. A metabolic connection between CLYBL and B12 was initially revealed in a genome-wide association study (7) that showed a correlation between the biallelic loss of CLYBL, which has an ~3 to 6% prevalence in certain populations (8), and B12 deficiency. Although I-CoA inhibition of MCM provides a molecular link between mitochondrial B12 and C5-dicarboxylate metabolism, it does not explain why inhibited MCM cannot be repaired by the auxiliary protein system that is dedicated for this function (9). Given the homology between bacterial and human MCM, host-derived itaconate could also potentially target MCM in pathogenic bacteria, in which it is required for lipid breakdown.
Itaconate induces electrophilic stress and modifies small molecules and protein targets by alkylating cysteine residues (10, 11). Here, we report a radical suicide inactivation mechanism in which addition of the elusive 5′-deoxyadenosyl radical (dAdo•) to I-CoA in MCM leads to an air-stable biradical comprising a tertiary carbon radical coupled to the metal-centered cob (II)alamin radical. We visualized Mtb MCM bound to a dAdo adduct of I-CoA at 2.0-Å resolution by x-ray crystallography. In addition to identifying MCM as an antimicrobial target of itaconate and demonstrating its importance for Mtb growth on propionate, our study provides molecular insights into how MCM controls radical trajectories during catalysis with its normal substrates to promote the desired chemistry and suppress unwanted side reactions.
I-CoA inactivates Mtb MCM by forming an air-stable biradical
MCM catalyzes the reversible isomerization of M-CoA to succinyl-CoA via an AdoCbl-dependent radical mechanism. To test our hypothesis that bacterial MCM is also a target of the antimicrobial effect of itaconate, we cloned and expressed Mtb MCM and the two auxiliary proteins that load and repair AdoCbl (fig. S1A). The kinetic parameters of Mtb MCM are comparable to those of the human homolog (fig. S1, B and C). Addition of I-CoA to Mtb MCM-AdoCbl led to a rapid shift in the absorption maximum (λmax), from 528 nm to 466 nm (Fig.1B), indicating stoichiometric binding (Fig. 1B, inset), as was also seen with hMCM (Fig. 1C) (5). Homolysis of the cobalt-carbon bond in AdoCbl, the first step in the MCM-catalyzed reaction, leads to formation of the radical pair dAdo• and cob(II)alamin, albeit with a different λmax (474 nm). We therefore used electron paramagnetic resonance (EPR) spectroscopy to identify the 466-nm–absorbing products of I-CoA–inactivated MCM.
The EPR spectrum of MCM-bound cob(II) alamin displays the typical eight-line hyperfine splitting of the unpaired electron with the I =7/2 cobalt nucleus that is resolved in the highfield region of the spectrum (fig. S2, A and B). Additional superhyperfine splitting due to the coordinating I = 1 lower axial nitrogen from a histidine ligand is also observed. Notably, addition of I-CoA to human or Mtb MCM led to distinct spectra (Fig. 1D, top, and fig. S2C) that had the hallmarks of a hybrid triplet system. The spectra were reminiscent of the EPR spectrum of a transient catalytic intermediate trapped during MCM turnover, exhibiting strong electron-electron spin-coupling between the product succinyl-CoA radical and the low-spin cob(II)alamin (12). Similar biradical intermediates have also been trapped in the AdoCbl-dependent enzymes glutamate mutase (13) and 2-methyleneglutarate mutase (14). The hyperfine multiplicity and the substantial g anisotropy identified the cobalamin component in the triplet spin system.
The identity of the organic radical component was further assessed with [13C]I-CoA (uniformly labeled in the itaconate carbons) or [13C]AdoCbl (uniformly labeled in the adenosyl moiety). Whereas 13C-labeled AdoCbl had no effect on the EPR spectrum, [13C]I-CoA led to inhomogeneous broadening throughout the line shape, which is consistent with appreciable mixing of cob(II)alamin and organic radical quantum states in the strong electron-electron coupling regime (Fig. 1D, bottom). These results indicate an absence of marked unpaired electron spin density on the dAdo moiety, which is in accord with the near-unit spin density inferred for the electron-13C interaction in the radical pair formed from [13C]I-CoA. Spectral simulations predict an interspin distance of 5 to 6 Å, while the Euler angles position the organic radical at an angle of 43° relative to the principal axis of the d 2z orbital on cobalt. In other AdoCbl-dependent isomerases, the distance between the substrate and cobalamin radicals range between 5 to 6 Å and 8 to 12 Å, respectively, necessitating small or large movements of the initially formed dAdo• to reach the substrate hydrogen atom (H-atom) destined for abstraction (15).
The I-CoA–induced biradical was air stable for over 1 hour (figs. S3 and S4), unlike the transient biradical formed during catalytic turnover with M-CoA that was trapped by freeze-quenching (12). To understand the chemical basis of its unusual stability, we further investigated the identity of the organic radical.
The adenosyl radical is stabilized by addition to itaconyl-CoA
Following generation of the dAdo•-cob(II)alamin radical pair on MCM, the isomerization reaction is initiated by H-atom abstraction from M-CoA by dAdo•, forming a substrate-centered radical that undergoes rearrangement. The dAdo• is a primary and highly reactive alkyl radical that has eluded direct detection in all AdoCbl-dependent isomerases but recently was trapped in a radical S-adenosylmethionine enzyme (16). In principle, two potential reactions between dAdo• and I-CoA can be considered (Fig. 1, E and F). To distinguish between these mechanistic possibilities, the reaction products from Mtb and hMCM inactivated by I-CoA under single-turnover and aerobic conditions were separated by high-performance liquid chromatography (fig. S5). Two major product peaks with retention times of 22.9 and 27.0 min (peaks 2 and 5) were identified; Mtb MCM showed an additional peak at 23.5 min (peak 2b).
Matrix-assisted laser desorption/ionization–mass spectrometry (MS) analysis confirmed the second reaction pathway leading to an addition product between dAdo and I-CoA (Fig. 1G), indicating that dAdo• adds to the double bond in I-CoA, yielding a tertiary carbon radical additionally stabilized by delocalization onto the π-system of the adjacent carboxylate. MS analysis revealed that peaks 2a and 2b are isomers with the same mass/charge ratio (m/z) of 1145.4 (fig. S6), which is 16 mass units higher than the expected mass of the addition product (m/z 1129), indicating incorporation of an oxygen atom. We assign peak 2 as the hydroxyl derivative of the addition product and peak 5 (m/z 1099.3), which is 30 Da lighter, indicating the formal loss of formaldehyde to the oxidative decarboxylation product of peak 2 (fig. S7A). MS analysis of the hMCM samples yielded similar results (fig. S6, D and E).
Under anaerobic conditions, additional hydrophobic products were observed (fig. S8). Peak 6 with an m/z of 1129.4 represents the expected radical addition product, while peak 5 and the minor peaks 7 and 8, with m/z values that are two mass units lower (fig. S9), were assigned to intramolecular cyclization and/or elimination products (fig. S7, B and C). Ado• cyclization products have been reported during anaerobic photolysis of AdoCbl (17) and during the reaction of hydroxyl radicals with dAdo or deoxyguanosine (18), resulting in 5′,8-cyclopurine nucleosides.
Crystallographic capture of the biradical on MCM
Given the protracted air stability of the biradical, we attempted to visualize the inhibited form by soaking AdoCbl-reconstituted Mtb MCM crystals with I-CoA. We determined structures of the unsoaked and I-CoA–soaked enzyme at1.9-Å and 2.0-Å resolution, respectively (table S1). Mtb MCM, like the Propionibacterium shermanii protein (19), is a heterodimer comprising an a subunit that binds B12 and a β subunit that is inactive (fig. S10A). In the native structure, AdoCbl is bound with its endogenous dimethylbenzimidazole tail inserted in a side pocket while His-629 serves as the lower axial ligand (Fig. 2A). On the opposite face, the 5′-carbon of the upper axial dAdo ligand is 2.5 Å away from the cobalt atom, while the adenine group is coplanar with the corrin ring and oriented above pyrrole rings A and B.
Fig. 2. Crystallographic capture of a biradical in I-CoA-inactivated Mtb MCM.
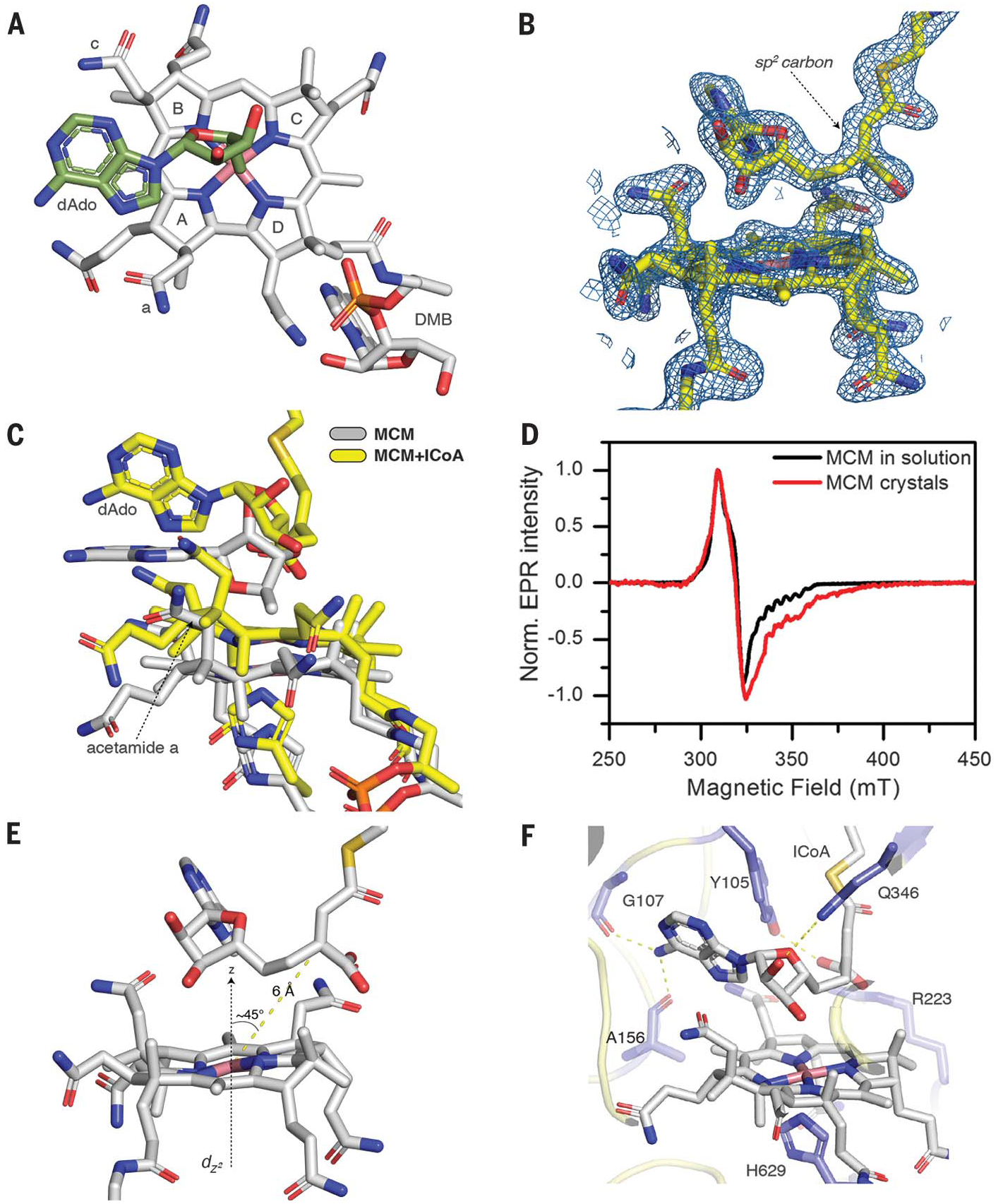
(A) Orientation of dAdo (green) in relation to the corrin ring (gray; pyrrole rings A to D and acetamides a and c are shown) in native Mtb MCM. (B) 2Fo-Fc omit maps (blue) around B12 and I-CoA contoured at 1.5s. (C) Shift in B12 and rotation of the adenine ring from the coplanar (gray) to perpendicular (yellow) position relative to the corrin ring. (D) EPR spectra of Mtb MCM + I-CoA. (E) Geometry of B12 and the I-CoA–dAdo adduct in crystal. (F) Hydrogen bonding interactions in the MCM–I-CoA structure.
I-CoA binding induces a large conformational change in the AdoCbl-binding a subunit [Cα root mean square deviation (RMSD), 1.47 Å], whereas the small subunit is almost unchanged (Cα RMSD, 0.26 Å) (fig. S10A). Soaking in I-CoA did not affect crystal stability, as there are no crystal contacts in the region affected by its binding. The α subunit collapses around the I-CoA binding pocket, with the motion being largest at the periphery and smallest where the α and β subunits are proximal. The crystal structure of hMCM, which is a homodimer with two B12 binding subunits, similarly closes in on its substrate, M-CoA (20).
We assigned the electron density in the active site to the adduct between I-CoA and the 5′-carbon of dAdo (Fig. 2B and fig. S10B). The 5′-carbon of the dAdo moiety is rotated almost 180° away from the cobalt, and the distance to the cobalt atom increases to 4.3 Å. The rotation places the 5′-carbon of dAdo 1.5 Å away from the methylene group of I-CoA, indicating the presence of a covalent bond between them. The geometry of the tertiary carbon of I-CoA is planar, as expected for an sp2 carbon (Fig. 2B and fig. S10, C and D). dAdo and corrin are shifted by 2.1 Å relative to the corrin ring in the structure without I-CoA. The acetamide group a on ring A is pushed up and in toward the adenine ring (Fig. 2C), which causes the adenine to move from a parallel to an almost perpendicular position relative to the corrin plane (fig. S10E), as predicted computationally (21). A strong biradical EPR signal associated with Mtb MCM crystals soaked with I-CoA confirmed that the spin-coupled carbon- and metal-centered radical pair can form in crystals (Fig. 2D). In the structure with the adduct, the tertiary carbon is 6 Å from the cobalt and at an ~45° angle from the principal dz2 orbital axis (Fig. 2E), in excellent agreement with EPR simulations.To our knowledge, the only other enzyme-bound, carbon-centered radical that has been crystallized is the acetyl-thiazolium cation radical in pyruvate:ferredoxin oxidoreductase, which is formed via a one-electron transfer to an iron-sulfur cluster (22).
In the resting state, the C2′-OH in dAdo is engaged in a hydrogen-bonding interaction between Gln346 and a water-mediated hydrogen bond network to His260 and Arg223 (fig. S10F). In the I-CoA–inactivated structure, the C2′-OH maintains a hydrogen bond with only Gln346, and the adenine NH2 group forms hydrogen bonds to the backbone carbonyls of Gly107 and Ala156 (Fig. 2F). These interactions likely orient dAdo• for H-atom abstraction in the catalytic cycle when M-CoA is present; however, when I-CoA is present, it places dAdo• in close proximity to the double bond, setting up the radical addition reaction. Tyr105 forms a hydrogen bond with the terminal carboxylate of I-CoA and also flanks the dAdo moiety. In the resting enzyme, Tyr105 points in the opposite direction, i.e., away from the substrate. Arg223 also engages via electrostatic interactions with the terminal carboxylate of I-CoA and the acetamide side group c in the corrin ring. The structure has implications for how MCM controls the dAdo• radical trajectory to promote H-atom abstraction from M-CoA and concomitantly suppresses unwanted side reactions that could lead to radical extinction. As first predicted in computational studies (21), rotation and upward movement of the adenine ring (Fig. 2C) position the C5′-carbon radical for H-atom abstraction from substrate. In contrast to MCM, glutamate mutase (23) and diol dehydratase (24) use ribose pseudorotation and N-glycosidic bond rotation, respectively, to bring the C5′-carbon of dAdo• to within van der Waal’s distance of the respective substrates.
I-CoA inhibits MCM repair
While cob(II)alamin is an intermediate in the MCM-catalyzed reaction, it also represents the inactive form of the enzyme when it becomes decoupled from the dAdo moiety, and thus it fails to re-form AdoCbl at the end of the catalytic cycle (9). Under these conditions, the auxiliary proteins CblA and ATR engage with MCM to off-load cob(II)alamin onto ATR for repair (Fig. 3A). ATR catalyzes the adenosylation of cob(I)alamin to form AdoCbl and then transfers the cofactor to MCM to reconstitute the holoenzyme (25, 26). Cofactor transfer in either direction between ATR and MCM requires the heterotrimeric guanine nucleotide– binding protein chaperone CblA and is fueled by its guanosine triphosphatase (GTPase) activity (27). Because I-CoA leads to rapid inactivation of MCM and formation of cob(II)alamin, which cannot re-form AdoCbl, we assessed whether the enzyme can be repaired by the ATR-CblA system.
Fig. 3. I-CoA inactivation impairs MCM repair.
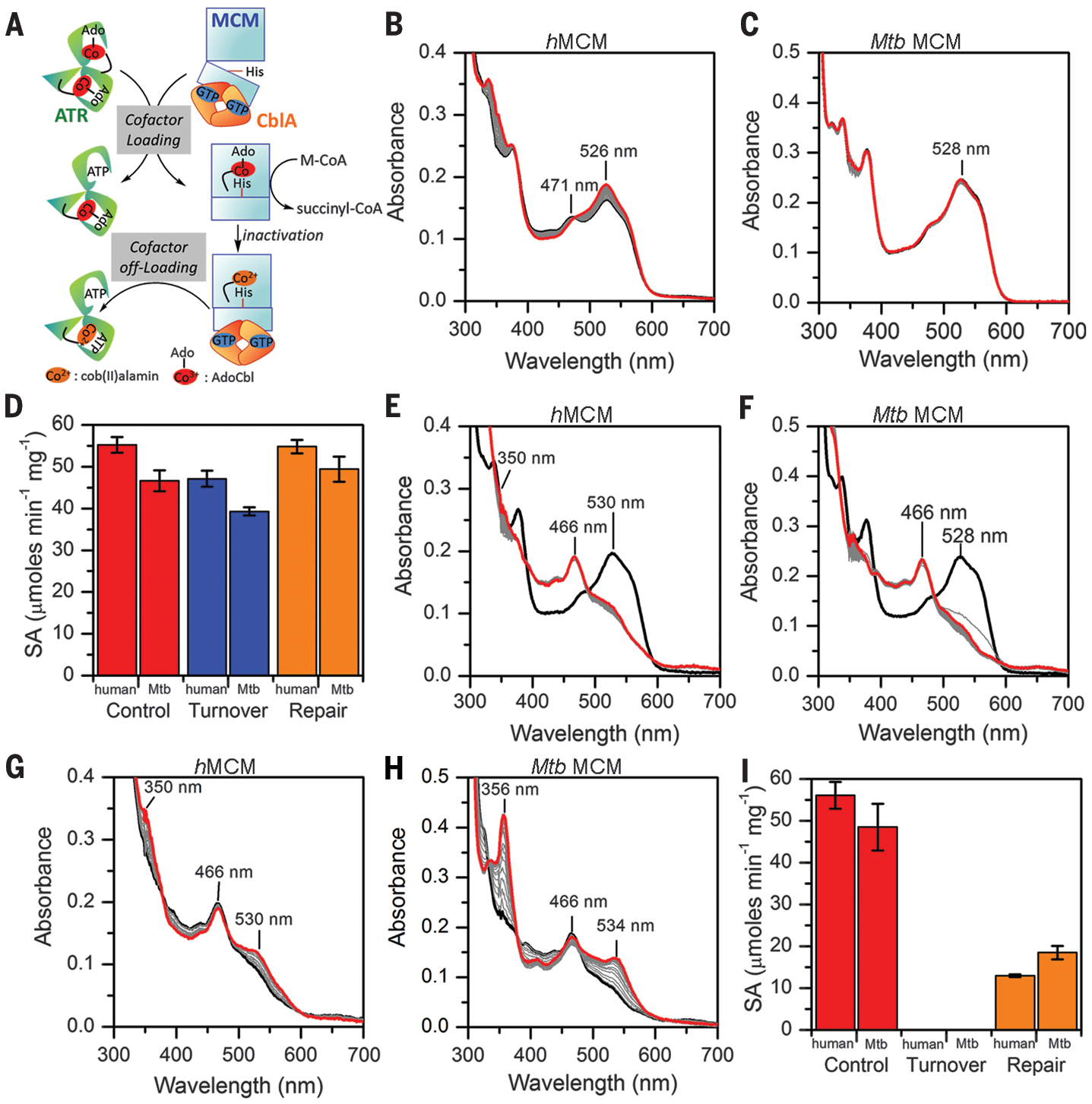
(A) Scheme showing the role of the auxiliary proteins in cofactor loading/off-loading to/from MCM. (B and C) Enzyme-monitored turnover by human (B) and Mtb (C) MCM (black spectra) in the presence of M-CoA and human or Mtb CblA-GTP. Intermediate spectra (gray) were recorded every 2 min. Final spectra (red) were recorded at 1 hour. (D) Specific activity (SA) of human and Mtb MCM after 1-hour preincubation without or with M-CoA (red versus blue) and subsequent addition of the repair system (orange). (E and F) Addition of I-CoA to hMCM-AdoCbl [black, (E)] or Mtb MCM-AdoCbl [black, (F)] results in inactive enzyme (gray). Further incubationover 1 hour causes only modest spectral changes (red). (G and H) At the end of the experiments in (E) and (F), the repair system was added for 20 min to human (G) and Mtb (H) MCM. The increase in absorbance at 350 to 356 nm is indicative of OH2Cbl formation (red). (I) Same as in (D) but with I-CoA; in both panels, data represent means ± SD (n = 3).
Addition of M-CoA, to initiate catalytic turnover by MCM in the presence of CblA-GDP (guanosine diphosphate), led to small (hMCM) or no (Mtb MCM) changes in the absorption spectra, indicating that these enzymes are resistant to oxidative inactivation (Fig. 3, B and C). Consistent with this finding, the specific activities of both enzymes were reduced only ~15% after 1 hour of preincubation with M-CoA (Fig. 3D). By contrast, the P. shermanni andM. extorquens MCM are much more prone to inactivation and accumulate aquocobalamin (OH2Cbl) during turnover (28). Addition of the repair system (ATR, ATP, and GTP) led to com plete recovery of MCM activity (Fig. 3D), consistent with successful off-loading of inactive cob(II)alamin from MCM followed by reloading of AdoCbl from the assay mixture. In contrast to M-CoA, which supported catalytic turnover of MCM with minimal spectral changes, incubation of either human (Fig. 3E) or Mtb (Fig. 3F) MCM with I-CoA led to an immediate increase in absorption at 466 nm that did not change significantly over 1 hour and was correlated with complete loss of activity (Fig. 3I). Addition of the respective human and Mtb repair proteins led to an increase in absorbance at 350 to 356 nm and 530 to 534 nm, signaling oxidation of cob(II)alamin to OH2Cbl (Fig. 3, G and H). By contrast, addition of the repair systems to the same enzymes incubated with M-CoA did not induce cofactor oxidation (fig. S11). Following repair, only 23% (human) and 38% (Mtb) of the initial MCM activity was recovered (Fig. 3I).
To gain insights into why the repair process is impeded by I-CoA but not M-CoA, we used hMCM, which forms a stable complex with CblA when it is in need of repair but is free in the active AdoCbl-bound state (fig. S12A) (27). In size exclusion chromatography profiles, hMCM is a stand-alone dimer (173 kDa) in the presence of M-CoA and the repair mixture (fig. S12B). However, in the presence of I-CoA, the MCM peak broadens and shifts to 211 kDa (fig. S12C), indicating the presence of a 1:1 hMCM:CblA complex (mass of 237 kDa) and free MCM. The 173- and 211-kDa fractions have spectra corresponding to AdoCbl and the biradical, respectively (fig. S12, D and F). The 82-kDa ATR fractions show that with M-CoA, very little cofactor is transferred to ATR (fig. S12E), because very little inactive MCM forms. By contrast, whereas I-CoA completely inactivates MCM, very little cofactor is off-loaded to ATR (fig. S12G), indicating that the sustained presence of the dAdo-I-CoA adduct on MCM impedes cofactor repair.
Hobbling of the repair system helps explain why CLYBL deficiency is correlated with B12 deficiency. In the absence of available MCM active sites to off-load AdoCbl, ATR catalyzes an unusual sacrificial homolysis of the cobalt-carbon bond and sequesters cob(II)alamin, to which it binds more tightly than AdoCbl (25). We propose that CLYBL deficiency increases the propensity of I-CoA–dependent MCM inactivation and thereby leads to AdoCbl depletion (5). How a change in the mitochondrial B12 pool (AdoCbl) is signaled to the cytoplasm and affects B12 levels systemically is, however, not known.
Itaconate inhibits vitamin B12-stimulated Mtb growth on propionate
To corroborate the in vitro evidence that itaconate inhibits Mtb MCM, we directly tested whether exogenous itaconate can blunt B12-stimulated Mtb growth on propionate as the sole carbon source. As reported previously, vitamin B12 supplementation at concentrations as low as 1 μg/ml stimulate growth of Mtb H37Rv on propionate (Fig. 4, A and B), which has been attributed to the MCM-dependent pathway for propionate utilization (29). Growth stimulation was reduced in the presence of 1 mM itaconate and was completely inhibited at ≥5 mM itaconate (Fig. 4C). Millimolar concentrations of itaconate are endogenously produced in activated macrophages (1), and Irg1-deficient mice, unlike controls, succumb early to Mtb infection (30), suggesting that such an inhibitory mechanism could be physiologically relevant.
Fig. 4. Itaconate inhibits B12-dependent Mtb and macrophage metabolism.
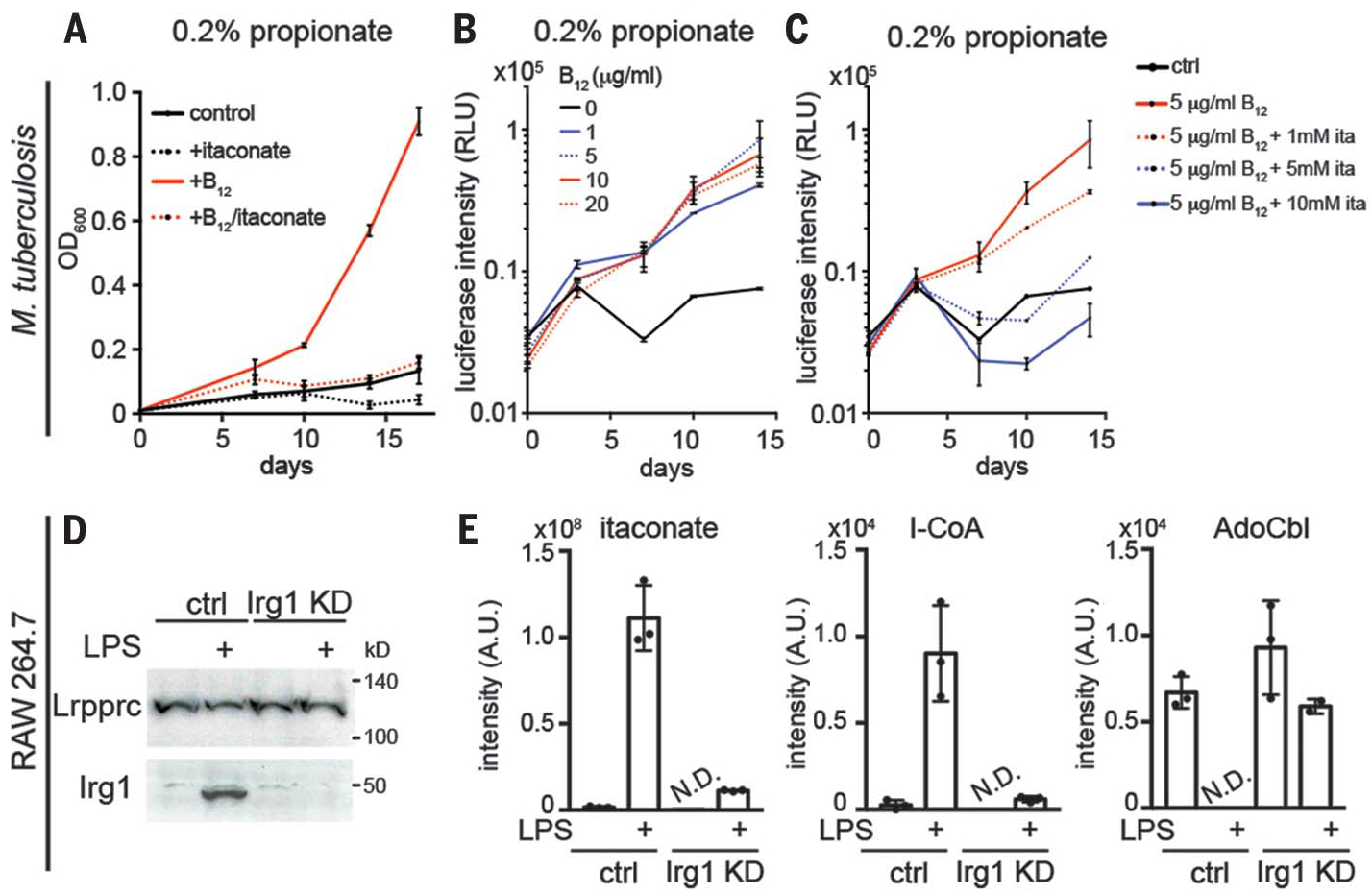
(A) Vitamin B12 (10 μg/ml) stimulates growth of wild-type Mtb strain H37Rv on 0.2% propionate as the carbon source. OD, optical density. (B and C) B12 concentration dependence of Mtb growth and its inhibition by itaconate. (D) Western blot of Irg1 in Irg1 CRISPR knockdown (KD) RAW264.7 cells with or without LPS (10 ng/ml) stimulation for 6 hours. Lrpprc, a mitochondrial protein, was used as the loading control. (E) Liquid chromatography–MS of itaconate, I-CoA, and AdoCbl in control and Irg1 KD RAW264.7 cells with or without LPS stimulation for 6 hours. Data represent means ± SD of three independent experiments. N.D., not detected.
MCM inhibition and AdoCbl depletion in macrophages require endogenous I-CoA synthesis
Mtb infection in mice elevates itaconate production in lungs (31), presumably through Irg1 induction. To recapitulate the metabolic consequence of endogenous itaconate production, we stimulated RAW264.7 cells with lipopolysaccharide (LPS), a potent activator of Irg1 transcription and itaconate production (1). We previously found that LPS stimulation depletes AdoCbl in macrophages (5). Using a CRISPR knockdown of Irg1 in RAW264.7 cells (Fig. 4D), we observed that AdoCbl depletion is dependent on Irg1, which is transcriptionally upregulated in LPS-stimulated macrophages (1, 32). Treatment with LPS induced an increase in itaconate and I-CoA levels, which was significantly attenuated in Irg1 knockdown cells (Fig. 4E). Although LPS stimulation reduced AdoCbl to undetectable levels in control cells, it was not significantly changed in the Irg1 knockdown cells (Fig. 4E). Together, these data suggest that AdoCbl depletion and MCM inhibition in macrophages is caused by endogenous itaconate produced by Irg1 and induced during LPS stimulation.
Conclusions
AdoCbl is a radical initiator that generates the “working” dAdo• and “spectator” cob(II)alamin radical by homolytic cleavage of its cobalt-carbon bond. We found that I-CoA triggers homolytic cleavage of the cobalt-carbon bond in AdoCbl as in the normal MCM catalytic cycle, but proximity effects promote suicidal addition of dAdo• into its double bond. A chemically akin, albeit nonspecific, Michael addition mechanism has been invoked to explain itaconate-induced electrophilic stress (10, 11). The combined action of itaconate and I-CoA on Mtb propionate metabolism would be predicted to result in increased levels of toxic propionate/propionyl-CoA derived from cholesterol-dependent growth of this pathogen in host phagosomes (29). Although the conditions are not known under which the Mtb pathway for de novo B12 biosynthesis might be operative (33), Mtb can scavenge B12 from its host (34). Itaconate-induced B12 deficiency in host macrophages thus might be a strategy for restricting pathogen growth, in addition to targeting pathogen enzymes involved in propionate metabolism (Fig. 1A); the relative importance of each inhibitory arm is unknown, however. We speculate that the CLYBL null background could boost the efficacy of this pathogen containment strategy, explaining the prevalence of the null genotype in human populations (5). In this context, it is noteworthy that the incidence of active tuberculosis is reported to be markedly lower in patients with B12 deficiency due to pernicious anemia (35).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge H. Sharma (University of Michigan) for his assistance with crystallization and the GM/CA CAT at the Advanced Light Source for beam time. We also acknowledge the NIH Common Fund Metabolite Standards Synthesis Core (NHLBI contract no. HHSN268201300022C) for providing [13C]itaconate.
Funding: This work was supported in part by grants from the National Institutes of Health (RO1-DK45776 to R.B., 5 F32 GM113405 to G.C.C., K99-GM124296 to H.S., R35GM122455 to V.K.M., R01-DK054514 to K.W., and U19AI107774 to E.J.R.) and start-up funds from the University of Michigan (to M.K.). V.K.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: V.K.M. is a paid advisor to Janssen Pharmaceuticals and 5AM Ventures and owns equity in Raze Therapeutics.
Data and materials availability: All data are available in the manuscript or supplementary materials. The structure factors and coordinates for Mtb MCM (6OXC) and Mtb MCM + I-CoA (6OXD) have been deposited in the Protein Data Bank.
REFERENCES AND NOTES
- 1.Michelucci A et al. , Proc. Natl. Acad. Sci. U.S.A 110, 7820–7825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Neill LAJ, Artyomov MN, Nat. Rev. Immunol 19, 273–281 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Kumar R, Int. J. Integr. Biol 7, 69–72 (2009). [Google Scholar]
- 4.McKinney JD et al. , Nature 406, 735–738 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Shen H et al. , Cell 171, 771–782.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasikaran J, Ziemski M, Zadora PK, Fleig A, Berg IA, Nat. Chem. Biol 10, 371–377 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Grarup N et al. , PLOS Genet. 9, e1003530 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lek M et al. , Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Padovani D, Banerjee R, Proc. Natl. Acad. Sci. U.S.A 106, 21567–21572 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mills EL et al. , Nature 556, 113–117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bambouskova M et al. , Nature 556, 501–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mansoorabadi SO et al. , Biochemistry 44, 3153–3158 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bothe H et al. , Biochemistry 37, 4105–4113 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Buckel W et al. , Eur. J. Inorg. Chem 2006, 3622–3626 (2006). [Google Scholar]
- 15.Banerjee R, Biochemistry 40, 6191–6198 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Yang H et al. , J. Am. Chem. Soc 141, 12139–12146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hogenkamp HPC, J. Biol. Chem 238, 477–480 (1963). [Google Scholar]
- 18.Jaruga P, Dizdaroglu M, DNA Repair 7, 1413–1425 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Mancia F et al. , Structure 4, 339–350 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Froese DS et al. , J. Biol. Chem 285, 38204–38213 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwiecien RA et al. , J. Am. Chem. Soc 128, 1287–1292 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Chabrière E et al. , Science 294, 2559–2563 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Gruber K, Reitzer R, Kratky C, Angew. Chem. Int. Ed 40, 3377–3380 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Masuda J, Shibata N, Morimoto Y, Toraya T, Yasuoka N, Structure 8, 775–788 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Campanello GC et al. , J. Am. Chem. Soc 140, 13205–13208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Padovani D, Labunska T, Palfey BA, Ballou DP,Banerjee R, Nat. Chem. Biol 4, 194–196 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Ruetz M et al. , Cell Chem. Biol 26, 960–969.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Padovani D, Banerjee R, Biochemistry 45, 9300–9306 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Savvi S et al. , J. Bacteriol 190, 3886–3895 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair S et al. , J. Exp. Med 215, 1035–1045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin JH et al. , J. Proteome Res 10, 2238–2247 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Jha AK et al. , Immunity 42, 419–430 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Gopinath K, Moosa A, Mizrahi V, Warner DF, Future Microbiol. 8, 1405–1418 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Gopinath K et al. , Open Biol. 3, 120175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barron M, JAMA 100, 1590–1592 (1933). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.