

Abstract
Sodium channel blockers are used for the treatment of pain, but this is limited by the lack of selectivity for different sodium channel subtypes, which can result in central nervous system and cardiovascular side effects. As such, there is special interest in the Nav1.7 subtype, which is expressed predominantly in nociceptive and sympathetic neurons. The aim was to demonstrate analgesic properties of a potent selective Nav1.7 sodium channel blocker, PF‐05089771, alone and concomitantly with pregabalin in healthy subjects using a battery of human evoked pain models. This was a double‐blind, double‐dummy, randomized, placebo‐controlled, five‐period cross‐over study with PF‐05089771 alone and PF‐05089771 concomitantly with pregabalin as treatment arms with pregabalin, ibuprofen, and placebo as control arms (NCT02349607). A battery of human evoked pain models was used to investigate analgesic properties of PF‐05089771. Twenty‐five subjects were enrolled in the study of which 23 subjects completed all five periods. PF‐05089771 alone did not differ from placebo on the primary pain end points. The same holds when comparing PF‐05089771 concomitantly with pregabalin and pregabalin alone. Pregabalin showed significant effects relative to placebo on thermal pain on the normal skin and UVB skin (least squares means with 90% confidence interval: 0.63 (0.32–0.93) and 0.53 (0.11–0.96)), pressure stimulation (1.10 (1.04–1.18)), and cold pressor (1.22 (1.14–1.32)). Ibuprofen demonstrated significant effects on thermal pain UVB skin (1.26 (0.82–1.70)) and pressure stimulation assessment (1.08 (1.01–1.15)), consistent with historical results. This study did not demonstrate analgesic properties of PF‐05089771 alone or concomitantly with pregabalin in a battery of pain models.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Results from genetic studies suggest a link between Nav1.7 and pain signaling in humans. This battery of pain models have previously been shown to detect unique profiles of analgesic compounds.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Is a battery of human evoked pain model able to detect the analgesic profile of PF‐05089771, alone and concomitantly with pregabalin?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ PF‐05089771 did not lead to significant analgesic effects in this study in healthy volunteers. Only modest analgesic effects of PF‐05089771 have been observed in patient studies and as such no further development is currently planned—this outcome may be regarded as in favor of the predictive value of the multimodal pain test battery that was used.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The lack of a significant analgesic effect in the current study, as well as the modest effect seen in the diabetic painful neuropathy patient study, warrants a re‐evaluation of PF‐05089771 as an analgesic compound or pharmacological inhibition of Nav1.7 as an analgesic mechanism.
A significant body of evidence implicates sodium channels in mediating the pathophysiological components of both neuropathic and nociceptive pain.1, 2 This is supported by clinical evidence suggesting that drugs with sodium channel blocking properties, such as local anesthetics, certain anticonvulsants, and tricyclic antidepressants that block voltage‐gated sodium channels (VGSCs), have therapeutic utility in managing and treating pain. For example, systemic lidocaine and mexiletine have been used to treat neuropathic pain in humans.3 The use of these sodium channel blockers has, however, been limited by the lack of selectivity for different sodium channel subtypes, which can result in central nervous system (CNS) and cardiovascular side effects.2 Therefore, a key to improving on the limitations of existing sodium channel blockers is to selectively target those that are involved in pain mechanisms while sparing those channels involved in cardiovascular function and in CNS function.4
Nav1.7 channels are expressed predominantly in nociceptive and sympathetic neurons. The role of this channel in nociceptive neurons has been characterized by human genetics, which indicates an essential and nonredundant role in pain transduction and conduction following noxious stimuli.5 The importance of Nav1.7 channels has been shown by genetic linkage studies of patients suffering from congenital indifference to pain, which is the result of a loss‐of‐function mutation in the SCN9A gene encoding the Nav1.7 channel,6, 7 and by gain‐of‐function mutations in the SCN9A gene, which have been implicated in extreme pain disorders, such as inherited erythromelalgia,9 paroxysmal extreme pain disorder,10 and SCN9A‐associated idiopathic small fiber neuropathy.11
The physiological role of the Nav1.7 channel is related to the excitability of the sensory afferent terminal.1 The Nav1.7 channel amplifies small generator potentials and depolarizes the sensory terminal membrane. This causes excitability, facilitating other sodium channels (e.g., Nav1.8) to generate and conduct action potentials. In the disease states genetically linked to a gain‐of‐function of the Nav1.7 channel, the channel is mutated to increase the sodium influx resulting in a hyperexcitable sensory neuron, and a resultant sensation of pain.
PF‐05089771 is a potent and selective, peripherally restricted Nav1.7 channel blocker with a half‐maximal inhibitory concentration value of 0.011 μM. It is 11‐fold, 16‐fold, and 59‐fold selective over the Nav1.2, Nav1.6, and Nav1.1 channels, respectively. The compound shows ≥ 909‐fold selectivity over Nav1.3, Nav1.4, Nav1.5, and Nav1.8.12 It has been investigated in nonclinical studies, in healthy subjects, and in clinical studies in patients with dental pain due to third molar extraction, diabetic painful neuropathy (DPN), and inherited erythromelalgia.13, 14 A phase II study to investigate the efficacy of PF‐05089771 alone, or as an add‐on therapy to pregabalin treatment for pain due to DPN has been conducted (NCT02215252). The rationale for the add‐on therapy was that the mechanism of action for PF‐05089771 is very different from that of pregabalin, the effects of which are mediated via the alpha2‐delta subunit of the voltage‐gated calcium channels and, therefore, co‐administration of both treatments could be expected to have an additive potential.
It is well known that translation of pain biomarkers to the clinic remains challenging. Insufficient understanding of the pathophysiology of pain in certain diseases and poor predictive values of current human evoked pain models are the major issues to this gap.15 It is estimated that in 43% of studies it is not possible to conclude whether or not the mechanism of action was investigated.16 Advances in the field of pain biomarkers could lead to more predictable outcomes and to a more accurate conclusion when it comes to go/no‐go decision when a compound fails to show analgesic effects in healthy subjects. From the body of literature on effects of analgesic compounds on evoked pain tests, it is clear that certain drugs may show significant results in one pain model, but not show any analgesic efficacy in another pain model.17, 18 For this reason, our study utilized a battery of multimodal pain tests.
The aim of this study was to evaluate the analgesic effects of PF‐05089771 alone and concomitantly with pregabalin in a battery of human evoked pain models. These models have demonstrated the ability to detect analgesic properties of compounds/pharmacological effects of analgesic drugs, including pregabalin and ibuprofen, in a robust manner.19, 20
Methods
Study design
The study was a double‐blind, double‐dummy, randomized, placebo‐controlled, five‐period crossover study. Subjects were to attend the clinic on seven separate occasions (screening, five study periods, and follow‐up). The five study periods were spaced apart by at least 7 days allowing sufficient time for washout of the previous treatment (pharmacokinetic (PK) and pharmacodynamic (PD) effects) based on the half‐life of each treatment. PF‐05089771 alone and given concomitantly with pregabalin was under investigation. A battery of human evoked pain models was used to demonstrate analgesic properties. The study was conducted at the clinical research unit of the Centre for Human Drug Research (CHDR) in Leiden, The Netherlands. The study was approved by the Medical Ethics Committee Stichting Beoordeling Ethiek Biomedisch Onderzoek (Assen, The Netherlands). The study was conducted according to the Dutch Act on Medical Research Involving Human Subjects (WMO) and in compliance with all International Conference on Harmonisation Good Clinical Practice (ICH‐GCP) guidelines and the Declaration of Helsinki.
Subject selections
Approximately 25 male subjects, between 18 and 55 years of age, were invited to volunteer for the study. All subjects provided written informed consent prior to undertaking any screening/study‐related activities or procedures. Subjects with a Fitzpatrick skin type I–IV, without widespread acne, tattoos, or scarring on the back, and who were willing and able to comply with all scheduled visits, treatment plan, and laboratory tests were included. Subjects were not eligible to participate if they had any existing clinically significant medical or psychiatric condition, or any condition that would affect sensitivity to pain or cold, or had a known hypersensitivity to pregabalin, ibuprofen, or any of the excipients. Also excluded were subjects who did not tolerate nociceptive assessment at screening or those who did not achieve tolerance at > 80% of maximum input intensity for any nociceptive assessment. Subjects who did not consent to abstain from excessive exposure to sunlight or sunbathing for the duration of the study were excluded.
Study drugs
Subjects received an oral dose of PF‐05089771 300 mg (2 × 150 mg tablets), alone and concomitantly with pregabalin 300 mg (1 × oral capsule) and matching placebos. The dose was justified based on margins to toxicology findings, on clinical toleration and safety data from a phase I study in healthy volunteers (NCT01259882), and a dental pain study in subjects undergoing third molar extraction where single oral doses of PF‐05089771, ranging from 1501,600 mg, were administered (NCT01529346). Additionally, a dose of 150 mg b.i.d., considered a developable dose of PF‐05089771, was used in a clinical trial for the treatment of pain due to DPN. Oral doses of ibuprofen 600 mg and pregabalin 300 mg had been used in previous studies with this battery of pain models.19, 21 These doses were well‐tolerated and were administered in accordance with European labeling and prescribing information. Pregabalin and ibuprofen were used as positive controls. Previous studies performed at our center with the same study design demonstrated consistent results, with ibuprofen significantly decreasing the pain threshold for heat pain on UVB exposed skin, and pregabalin significantly decreasing the pain thresholds for pressure stimulation and the cold pressor test.19, 20
Pharmacokinetic assessment
Blood samples (8 mL) were collected from each subject during all study periods to provide a minimum of 4 mL plasma for PK analysis. Samples were collected at predefined time points: predose, and 0.5, 1, 2, 3, 4, 5, 6, 8, and 10 hours postdose. Plasma concentration over time for PF‐05089771, pregabalin, and ibuprofen was measured and for each subject, the peak plasma concentration (Cmax), area under the concentration‐time curve from time of administration up to the time of the last quantifiable concentration (AUClast), and time of maximum plasma concentration (Tmax) were calculated for PF‐05089771. PF‐05089771, pregabalin, and ibuprofen plasma concentrations were listed and summarized descriptively (results not shown).
PD assessment
PD measurements were an integrated range of pain models for measuring different modalities of pain. Nociceptive and inflammatory pain was assessed by means of the heat and inflammatory heat pain assessments, electrical stimulation assessment, cold pressor assessment, and the pressure stimulation assessment. Detailed descriptions of the models have been described previously.20 Assessments were conducted predose and 0.5, 1, 2, 3, 4, 6, 8, and 10 hours postdose by trained personnel. Tasks were performed to measure the end points of the pain detection threshold (PDT) and pain tolerance threshold (PTT). Applicable for each pain model, except the heat pain assessments, pain intensity was measured continuously with subjects rating their pain intensity using a 100 mm electronic visual analogue scale‐slider with 1 as the PDT and 100 defined as the PTT. The intensities of the stimuli were increased until the subjects indicated their PTT, or the maximum stimulus was achieved, whichever came first, at which point the equipment was programmed to cease giving stimuli.
Sample size
Decision rules were prespecified to quantify what was required in the primary objective of the study. The criteria were based on a Bayesian interpretation of the results, assuming a noninformative prior. The prespecified decision criteria applied to each primary end point were: (i) at least 95% confident that PF‐05089771 effect is greater than placebo; and (ii) at least 95% confident that PF‐05089771 concomitantly with pregabalin effect is greater than pregabalin alone. These are equivalent to one‐sided tests for statistical significance using an alpha of 0.05. No adjustment was made for multiplicity as this was an early‐phase clinical study designed to explore the PD of PF‐05089771 and, as such, no stringent requirement to control the type 1 error rate was required for internal decision making.
The sample size was based on the mean effect over 110 hours after dosing (i.e., average of 1, 2, 3, 4, 6, 8, and 10‐hour time points) for the five primary end points: cold pressor PTT; pressure pain PTT; electrical stair PTT; normal heat PDT; and UVB heat PDT. A conservative estimate of within‐subject SD was derived from two previous methodology studies,10, 11 yielding estimates of 0.279, 0.222, 0.183, 1.86, and 1.80 for the cold pressor PTT, pressure pain PTT, electrical stair PTT, normal heat PDT, and UVB heat PDT end points, respectively. A sample size of 25 subjects was selected to ensure balance in the design, and gave at least 80% power to detect differences of 0.197, 0.157, 0.13, 1.316, and 1.276 for the five primary end points listed previously.
Statistical analysis
The primary end points of this study were the PDT for thermal pain (normal skin and UVB skin), and the PTT for electrical stair (pre‐cold pressor), pressure pain, and cold pressor.
A mixed effects repeated measures model was fitted for each end point, using data collected during the first 10 hours post‐treatment. The fixed effects included in the model were baseline, period, time, treatment, and treatment by time interaction, with baseline as covariate. Subject was fitted as a random effect and time repeated within each subject × period as a repeated effect. Baseline was included as two separate variables, the average baseline for the subject, and the deviation of each period baseline from the average baseline for each subject.23 The Kenward–Roger approximation was used for estimating degrees of freedom for the model parameters. The primary analysis included all subjects randomized into the study. The PDT and PTT end points for the electrical stimulation, pressure stimulation, and cold pressor tests were log transformed prior to analysis.
The least squares means (LSMeans) together with 90% confidence intervals (CIs) were obtained for each treatment averaged across time points that covered the peak exposure of each treatment. The average effect across 110 hours was obtained for PF‐05089771, PF‐05089771 + pregabalin and placebo. The average for pregabalin and placebo was obtained from both 110 hours and the first 6 hours. The average for ibuprofen and placebo was obtained from the first 4 hours. Differences between treatments and placebo or pregabalin were, therefore, made using the appropriate average (i.e., ibuprofen was compared with the placebo 4‐hour average, whereas PF‐05089771 was compared with the placebo 110‐hour average). The probabilities for the decision criteria for each of the pain model end points were calculated directly from the results of the mixed model, assuming a noninformative prior.
As a sensitivity analysis to the primary analysis, a mixed effects model was also fitted for the maximum (over 10 hours post‐treatment) change from baseline for each pain model end point. The fixed effects included in the model were baseline, period, and treatment. Baseline was included as two separate variables, the average baseline for the subject, and the deviation of each period baseline from the average baseline for each subject. Subject was fitted as a random effect. In cases where a subject did not show an increase, the minimum decrease was taken. LSMeans together with 90% CIs were obtained for each treatment, and differences in the LSMeans and 90% CIs were obtained for the comparisons mentioned above.
Results
Demographics
A total of 25 male subjects were randomized to receive study treatments. Twenty‐three subjects each received PF‐05089771 300 mg + pregabalin 300 mg, ibuprofen 600 mg, and placebo; 24 subjects received PF‐05089771 300 mg and pregabalin 300 mg. One subject was discontinued from the study due to a positive urine drug screen for abuse. A second subject discontinued as he was no longer willing to participate in the study. None of the subjects were replaced in this study (Figure S1 ).
A summary of the demographic characteristics is provided in Table S1 .
PDs
A summary of the result for the primary end points are presented in Table 1. PF‐05089771 alone did not meet the decision criterion of at least 95% CI that the effect was greater than placebo for any of the primary end points. PF‐05089771 concomitantly administered with pregabalin did not differ from pregabalin alone on the primary end points. Pregabalin showed evidence (> 95% probability) of effects relative to placebo on thermal pain (normal skin and UVB skin) PDT, pressure stimulation PTT, and cold pressor PTT. Ibuprofen showed evidence (> 95% probability) of effects relative to placebo on thermal pain (UVB skin) PDT, and pressure stimulation PTT. The overall LSMeans with 90% CIs for all pain models is plotted in Figure 1.
Table 1.
Summary of results for the primary analysis
| End point | PF‐05089771 | PF‐05089771 + pregabalin | Pregabalin | Ibuprofen | ||
|---|---|---|---|---|---|---|
| LSMeans difference (90% CI)a | Probabilities associated with decision | LSMeans difference (90% CI)a | Probabilities associated with decision | LSMeans difference (90% CI)a | LSMeans difference (90% CI)a | |
| Normal heat PDT | 0.08 (−0.25, 0.42) | 0.66 | −0.24 (−0.57, 0.08) | 0.11 | 0.63 (0.32, 0.93) | 0.18 (−0.13, 0.50) |
| UVB heat PDT | −0.01 (−0.44, 0.43) | 0.49 | 0.30 (−0.14, 0.74) | 0.87 | 0.53 (0.11, 0.96) | 1.26 (0.82, 1.70) |
| Electrical stimulation PTT | 0.97 (0.91, 1.03) | 0.22 | 0.97 (0.91, 1.04) | 0.23 | 1.03 (0.98, 1.09) | 0.98 (0.93, 1.04) |
| Pressure stimulation PTT | 1.01 (0.95, 1.07) | 0.58 | 1.03 (0.97, 1.10) | 0.81 | 1.10 (1.04, 1.18) | 1.08 (1.01, 1.15) |
| Cold pressor PTT | 1.03 (0.95, 1.11) | 0.70 | 1.01 (0.93, 1.09) | 0.54 | 1.22 (1.14, 1.32) | 1.08 (1.00, 1.17) |
Criteria 1: At least 95% confident that PF‐05089771 effect was greater than placebo.
Criteria 2: At least 95% confident that PF‐05089771 + pregabalin effect was greater than pregabalin.
Statistically significant result in bold.
CI, confidence interval; LSMeans, least squares means; PDT, Pain Detection Threshold; PTT, Pain Tolerance Threshold.
LSMeans differences for PF‐05089771 alone, pregabalin and ibuprofen are relative to placebo, whereas for PF‐05089771 + pregabalin the differences are relative to pregabalin. PTT end points were analyzed on the log scale, so results are presented as back‐transformed LSMeans ratios and 90% CIs for treatment differences.
Figure 1.
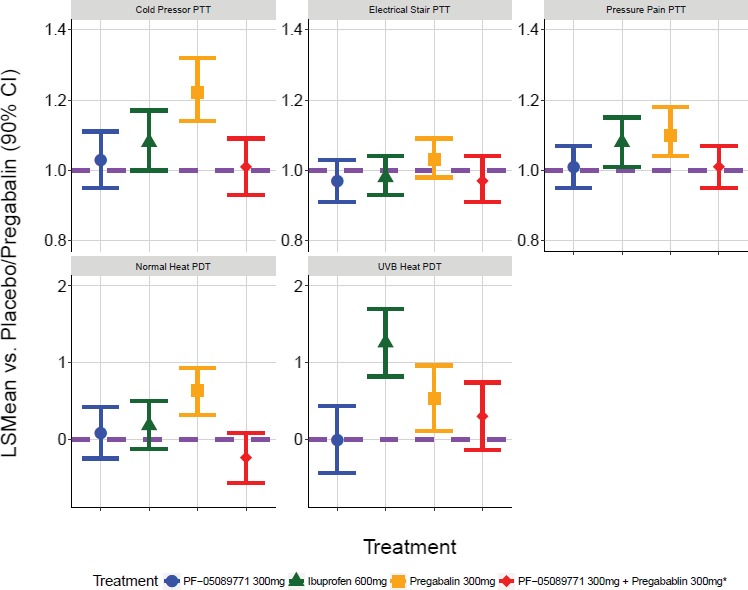
Primary analysis results. The comparisons of PF‐05089771 vs. placebo, and PF‐05089771 + pregabalin vs. pregabalin alone (*) was made with least squares means (LSMeans) averaged over 110 hours. The comparison of pregabalin vs. placebo was made with LSMeans averaged over 6 hours. The comparison of ibuprofen vs. placebo was made with LSMeans averaged over 4 hours. The purple horizontal dashed line represents no effect relative to placebo/pregabalin. Pain tolerance threshold (PTT) end points are presented on the fold‐change to placebo scale, whereas pain detection threshold (PDT) end points are presented on the absolute difference to placebo scale. CI: confidence interval.
PKs
Following oral administration of a 300 mg dose of PF‐05089771 administered alone or in conjunction with 300 mg of pregabalin, PF‐05089771 absorption was similar for both treatment arms. Cmax was achieved within a median Tmax of 2 hours postdose for PF‐05089771 administered alone (individual range 2–5 hours) and within a median Tmax of 3 hours postdose for PF‐05089771 administered in combination with pregabalin (individual range 2–5 hours). Overall, PF‐05089771 exposure based on geometric mean AUClast and Cmax values seemed to be similar for both treatments (Table S2 ).
Safety
Single doses of PF‐05089771 were considered safe and well‐tolerated in this study. None of the subjects experienced a serious adverse event (AE), dose reduction, or temporary or permanent discontinuation due to an AE. The most frequently reported all causality (treatment‐related) AEs were somnolence, dizziness, headache, fatigue, and euphoric mood (Table S3 ). AEs were mild in severity, except one subject in the pregabalin 300 mg treatment group who experienced somnolence, which was moderate in severity and considered treatment‐related. There were no clinically significant changes in safety laboratory assessments, vital signs, and echocardiograms.
Discussion
In the current study, we found no significant analgesic effects of PF‐05089771 alone or of PF‐05089771 given concomitantly with pregabalin. This is an argument in favor of a strong predictive value of the PainCart with respect to efficacy in patients with clinical pain. A possible explanation for the negative findings could be that the battery of pain models used in the current study is not sufficiently sensitive to detect the analgesic effects of VGSC blockers, however, analgesic effects of another VGSC blocker, phenytoin, a potent sodium channel blocker, were demonstrated using the same battery of pain models.19, 24 In a previous study, phenytoin was able to significantly increase the pain detection and pain tolerance thresholds in an electrical stimulation paradigm.19 In addition, analgesic effects have previously been reported where phenytoin significantly affected the PTT in the cold pressor assessment.25 We cannot rule out that the pain models used in the current pain test battery may not have been sufficiently sensitive, nor have had the dynamic range required to detect analgesic effects of selective Nav1.7 channel blockers. Alternatively, an axonal excitability measurement (e.g., using threshold tracking), might have been able to detect analgesic effect of PF‐05089771, because dysfunction of the Nav1.7 may lead to changes in the conduction velocity.26, 27
It is also possible that the sample size may have been too small to detect smaller effect sizes than were previously reported, although the clinical significance of such effects may be questionable. The sample size for this study was based on conservative estimates from two previous methodology studies with the same test battery and is more commonly used in single‐dose human evoked pain model testing.
Another possible explanation for the negative findings in our study may be related to the dose of PF‐05089771 administered in the current study, which could have been too low to exert analgesic effects large enough to measure using this analgesic test battery in healthy volunteers. Analgesic effects were demonstrated in a phase II clinical trial for the treatment of patients with inherited erythromelalgia, but these patients were administered a single dose of 1,600 mg.14 The dose used in the current study was based on the selected dose for the DPN study (150 mg b.i.d., oral dose), which was conducted in parallel to the current study. The dose selected for the DPN study was based on results from a prior dental pain study (NCT01529346). Although both the dental pain study and the DPN study13 demonstrated trends toward pain relief, the magnitude of efficacy observed in either study did not meet prespecified decision criteria. The current study confirmed these results suggesting that PF‐05089771 does not have a strong analgesic effect. The current study highlights the predictive value of the battery of pain models as a tool that may contribute to go/no go decisions.
It is to be considered whether peripheral Nav1.7 blockage is sufficient or if CNS access is required to produce sufficient analgesia. Peripheral blockage was believed to be adequate to achieve analgesic efficacy because Nav1.7 plays an important role in activation of the threshold current for action potential initiation in the peripheral terminals of primary afferent nociceptors.5, 13 A more complex role for Nav1.7 in primary afferent nociceptors, with evidence for a contribution to the upstroke of the action potential and consequential role in axonal conduction as well as both central and peripheral neurotransmitter release, has been recently highlighted.28 An inability to penetrate through the blood–brain barrier may be a key factor as to why PF‐05089771 is unable to satisfactorily modulate C‐fiber nociceptive transmission, both along the sheathed peripheral nerve as well as neurotransmitter release in the dorsal horn of the spinal cord.13 Despite the limited success to date with Nav1.7 blockers, it is likely that they will remain a key analgesic target. It remains to be seen whether it is sufficient to selectively target Nav1.7 blockade or whether co‐targeting other Nav channels involved in nociceptive transmission, such as Nav1.8, Nav1.9, and even Nav1.3, is required, while avoiding subtypes Nav1.1 and Nav1.2 for their CNS side effects, and Nav1.4 and Nav1.5 for their cardiovascular effects. Alternatively, combining a Nav1.7 blocker with an opioid may be promising.29 Research with Nav1.7 null mutant mice showed that mechanical and cold allodynia can still be induced under certain conditions.30 Nav1.7‐related analgesia may be enhanced by opioid signaling.31, 32 Minett et al. were able to reverse a pain free state in mice with a deletion of SCN9A encoding for Nav1.7 with naloxone, which was replicated in one human subject with an identical deletion.32 The exact mechanism by which Nav1.7 influences the opioid system is unclear and needs further investigation, but it may hold promise for development of more effective Nav1.7 channel blockers.
Administration of PF‐05089771 concomitantly with pregabalin was included in this study because it was to be considered as a treatment for neuropathic pain, especially in patients with painful DPN. Pregabalin and other alpha2‐delta anticonvulsants, such as gabapentin, are useful as an adjuvant therapy together with opioids for neuropathic (cancer) pain.33, 34 With the current dose levels in this study, no additive PD effect was observed compared with pregabalin alone.
Conclusion
This study did not demonstrate analgesic properties of PF‐05089771, alone or when administered concomitantly with pregabalin, compared with placebo or pregabalin, respectively. The use of the battery of human evoked pain models did confirm the analgesic profile of pregabalin and ibuprofen, as established in previous studies with the PainCart test battery.
Funding
The trial was sponsored by Pfizer.
Conflict of Interest
All authors completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and P.S., G.A., J.H., A.M., D.G., R.B., and G.G. declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could seem to have influenced the submitted work. A.M., D.G., and R.B. were employees of Pfizer Ltd. during study execution and may own stock in the company; the Centre for Human Drug Research received funding from Pfizer Ltd. for execution of the study. The funder reviewed and provided feedback on the manuscript. All other authors declared no competing interests for this work.
Author Contributions
P.S., G.A., G.G., D.G., A.M., J.H., and R.B. wrote the manuscript. P.S., G.A., J.H., R.B., and G.G. designed the research. P.S., G.A., J.H., and G.G. performed the research. D.G. analyzed the data.
Trial Registration
The trial was registered in the trial register of the Committee on Research Involving Human Subjects (CCMO, https://www.toetsingonline.nl, NL51495.056.14).
Supporting information
Figure S1 . Disposition of subjects.
Table S1.
Table S2.
Table S3.
Acknowledgments
The authors wish to thank the subjects who participated in the studies.
References
- 1. Amir, R. et al The role of sodium channels in chronic inflammatory and neuropathic pain. J. Pain. 7, 1–29 (2006). [DOI] [PubMed] [Google Scholar]
- 2. Lai, J. , Hunter, J.C. & Porreca, F. The role of voltage‐gated sodium channels in neuropathic pain. Curr. Opin. Neurobiol. 13, 291–297 (2003). [DOI] [PubMed] [Google Scholar]
- 3. Kalso, E. Sodium channel blockers in neuropathic pain. Curr. Pharm. Des. 11, 3005–3011 (2005). [DOI] [PubMed] [Google Scholar]
- 4. Priest, B.T. Future potential and status of selective sodium channel blockers for the treatment of pain. Curr. Opin. Drug Discov. Devel. 12, 682–692 (2009). [PubMed] [Google Scholar]
- 5. Dib‐Hajj, S.D. , Black, J.A. & Waxman, S.G. Voltage‐gated sodium channels: therapeutic targets for pain. Pain Med. 10, 1260–1269 (2009). [DOI] [PubMed] [Google Scholar]
- 6. Cox, J.J. et al A SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldberg, Y.P. et al Loss‐of‐function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 71, 311–319 (2007). [DOI] [PubMed] [Google Scholar]
- 8. Ahmad, S. et al A stop codon mutation in SCN9A causes lack of pain sensation. Hum. Mol. Genet. 16, 2114–2121 (2007). [DOI] [PubMed] [Google Scholar]
- 9. Yang, Y. et al Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermelalgia. J. Med. Genet. 41, 171–174 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fertleman, C.R. et al SCN9A mutation in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774 (2006). [DOI] [PubMed] [Google Scholar]
- 11. Faber, C.G. et al Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 71, 26–39 (2012). [DOI] [PubMed] [Google Scholar]
- 12. Bagall, S.K. et al Ion channels as therapeutic targets: a drug discovery perspective. J. Med. Chem. 56, 593–624 (2013). [DOI] [PubMed] [Google Scholar]
- 13. McDonnell, A. et al Efficacy of the Nav1.7 blocker PF‐05089771 in a randomised, placebo‐controlled, doubleblind clinical study in subjects with painful diabetic peripheral neuropathy. Pain 159, 1465–1476 (2018). [DOI] [PubMed] [Google Scholar]
- 14. Cao, L. et al Pharmacological reversal of a pain phenotype in iPSC‐derived sensory neurons and patients with inherited erythromelalgia. Sci. Transl. Med. 8, 335ra56 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Taneja, A. , Di Iorio, V.L. , Danhof, M. & Della Pasqua, O. Translation of drug effects from experimental models of neuropathic pain and analgesia to humans. Drug Discov. Today 17, 837–849 (2012). [DOI] [PubMed] [Google Scholar]
- 16. Morgan, P. et al Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving phase II survival. Drug Discov. Today 17, 419–424 (2012). [DOI] [PubMed] [Google Scholar]
- 17. van Amerongen, G. , de Boer, M.W. , Groeneveld, G.J. & Hay, J.L. A literature review on the pharmacological sensitivity of human evoked hyperalgesia pain models. Br. J. Clin. Pharmacol. 82, 903–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arendt‐Nielsen, L. , Curatolo, M. & Drewes, A. Human experimental pain models in drug development: translational pain research. Curr. Opin. Investig. Drugs 8, 41–53 (2007). [PubMed] [Google Scholar]
- 19. Okkerse, P. et al The use of a battery of pain models to detect analgesic properties of compounds: a two‐part four‐way crossover study. Brit. J. Clin. Pharmacol. 83, 976–990 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hay, J.L. , Okkerse, P. , van Amerongen, G. & Groeneveld, G.J. Determining pain detection and tolerance thresholds using an integrated multi‐modal pain task battery. J. Vis. Exp. 14, 110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loudon, P. et al Demonstration of an anti‐hyperalgesic effect of a novel pan Trk inhibitor PF‐06273340 in a battery of human evoked pain models. Brit. J. Clin. Pharmacol. 84, 301–309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siebenga, P.S. et al Reproducibility of a battery of human evoked pain models to detect pharmacological effects of analgesic drugs. Eur. J. Pain 21, 1129–1140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kenward, M.G. & Roger, J.H. The use of baseline covariates in crossover studies. Biostatistics 11, 1–17 (2011). [DOI] [PubMed] [Google Scholar]
- 24. Mantegazza, M. , Curia, G. , Biagini, G. , Ragsdale, D.S. & Avoli, M. Voltage‐gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 9, 413–424 (2010). [DOI] [PubMed] [Google Scholar]
- 25. Webb, J. & Kamali, F. Analgesic effects of lamotrigine and phenytoin on cold‐induced pain: a crossover placebo‐controlled study in healthy volunteers. Pain 76, 357–363 (1998). [DOI] [PubMed] [Google Scholar]
- 26. Hoffmann, T. et al NaV1.7 and pain: contribution of peripheral nerves. Pain 159, 496–506 (2018). [DOI] [PubMed] [Google Scholar]
- 27. Farrar, M.A. , Lee, M.J. , Howells, J. , Andrews, P.I. & Lin, C.S. Burning pain: axonal dysfunction in erythromelalgia. Pain 158, 900–911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexandrou, A.J. et al Subtype‐selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and pre‐synaptic release. PLoS One 11, e0152405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emery, E.C. , Luiz, A.P. & Wood, J.N. Nav1.7 and other voltage‐gated sodium channels as drug targets for pain relief. Expert Opin. Ther. Targets 20, 975–983 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Minett, M.S. et al Pain without nociceptors? Nav1.7‐independent pain mechanisms. Cell. Rep. 6, 301–312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dehen, H. , Willer, J.C. , Prier, S. , Boureau, F. & Cambier, J. Congenital insensitivity to pain and the "morphine‐like" analgesic system. Pain 5, 351–358 (1978). [DOI] [PubMed] [Google Scholar]
- 32. Minett, M.S. et al Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat. Commun. 6, 8967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Finnerup, N.B. et al Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurol. 14, 162–173 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Finnerup, N.B. , Otto, M. , McQuay, H.J. , Jensen, T.S. & Sindrup, S.H. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain 118, 289–305 (2005). [DOI] [PubMed] [Google Scholar]
- 35. Garassino, M.C. et al Randomised phase II trial (NCT00637975) evaluating activity and toxicity of two different escalating strategies for pregabalin and oxycodone combination therapy for neuropathic pain in cancer patients. PLoS One 8, e59981 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen, D.L. , Li, Y.H. , Wang, Z.J. & Zhu, Y.K. The research on long‐term clinical effects and patients' satisfaction of gabapentin combined with oxycontin in treatment of severe cancer pain. Medicine (Baltimore) 95, e5144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 . Disposition of subjects.
Table S1.
Table S2.
Table S3.