

Abstract
The purpose of this study was to assess the safety, tolerability, pharmacokinetics, pharmacodynamics, and immunogenicity of BOS161721, a humanized immunoglobulin G1 triple mutation (M252Y/S254T/T256E) monoclonal antibody that inhibits interleukin‐21 (IL‐21) bioactivity. This randomized, single‐center, double‐blind, placebo‐controlled study randomized healthy volunteers 3:1 to single ascending intravenous and subcutaneous doses of BOS161721 (range 1–240 mg) or placebo. BOS161721 and placebo groups had similar rates of adverse events, mostly mild; none led to study discontinuation. There were no clinically significant findings in physical examination, vital signs, or laboratory assessment. In the pooled BOS161721 population, four subjects (8.5%) tested antidrug antibody‐positive predose, and seven (14.9%) postdose. Absolute CD4+ lymphocyte count remained normal throughout follow‐up. BOS161721 administered subcutaneously was absorbed slowly, with a median time to maximum concentration (Tmax) of 144 hours across doses (range 1–15 days) and a mean apparent terminal elimination half‐life of 80–87 days for doses ≥ 30 mg. Area under the concentration‐time curve from time zero to infinity (AUC0‐inf) and maximum observed concentration (Cmax) were linear across doses > 10 mg. Subcutaneous bioavailability was 64%. Phosphorylated signal transducer and activator of transcription 3 (pSTAT3) decreased dose‐dependently with threshold characteristics at doses of ≥ 10 mg. Downregulation in BATF, IL6, LAG3, and SOCS3 genes caused by IL‐21 stimulation was reversed dose‐dependently. BOS161721 was well‐tolerated across doses, suppressed IL‐21‐induced pSTAT3 dose‐dependently, and reversed downregulation of genes critical to tolerance induction and T‐cell exhaustion induced by IL‐21. Further clinical studies are ongoing in patients with systemic lupus erythematosus, in which IL‐21 has a pathogenetic role.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THIS TOPIC?
☑ Interleukin‐21 (IL‐21) plays a critical role in promoting humoral and other immune responses, making it an important focus of potential therapeutic interventions in autoimmune conditions like systemic lupus erythematosus (SLE) that are characterized by overproduction of pathogenic autoantibodies.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Does pharmacological intervention into the IL‐21 signaling pathway have the potential for therapeutic effect in autoimmune diseases?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ BOS161721 is a humanized immunoglobulin G1 triple mutation (M252Y/S254T/T256E) monoclonal antibody that inhibits IL‐21 bioactivity. This first‐in‐human, single‐ascending‐dose trial was designed to provide initial human clinical safety, pharmacokinetic (PK), and pharmacodynamic data for BOS161721, administered either subcutaneously or intravenously to healthy subjects. BOS161721 was well‐tolerated across a wide dose range (1–240 mg), suppressed IL‐21‐induced phosphorylated signal transducer and activator of transcription 3 expression in lymphocytes in a dose‐dependent manner, and reversed the downregulation of genes (BATF, IL6, LAG3, and SOCS3) critical to tolerance induction and T‐cell exhaustion induced by IL‐21.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ An international, multicenter phase II clinical trial of BOS161721 administered subcutaneously every 4 weeks to evaluate its efficacy, safety, and PKs in patients with SLE is ongoing.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with substantial clinical heterogeneity, whose complex pathogenesis is not fully understood. B and T cells are critical in its development, and T cell‐derived cytokines, such as interleukin‐21 (IL‐21) participate in the associated inflammatory response.1 Autoantibodies are characteristic of SLE and include reactivity to Ro/SSA (anti‐Sjögren's syndrome‐related antigen) and La/SSB (anti‐Sjögren's syndrome type B antigen, also known as Lupus La protein) and double‐stranded deoxyribonucleic acid, as well as rheumatoid factor and antinuclear antibodies; some are believed to play pathogenetic roles. Importantly, T‐follicular helper cells are more abundant in patients with autoimmune diseases, such as SLE and have been correlated with IL‐21 levels, autoantibodies, and disease severity.2, 3 Furthermore, regulatory T‐cell (Treg) depletion and dysfunction thought to be modulated through IL‐21 have been associated with autoimmune diseases, such as SLE and giant cell arteritis and may be involved in their pathogenesis.7, 8
BOS161721 is a humanized immunoglobulin G1 (IgG1) monoclonal antibody (mAb) that binds to IL‐21 with sub‐picomolar affinity, preventing binding to the IL‐21 receptor (IL‐21R) and inhibiting bioactivity. The fragment crystallizable domain of BOS161721 contains a triple mutation in the constant domain of the IgG1 heavy chain that confers increased avidity and affinity for the neonatal fragment crystallizable receptor and was introduced into the parental molecule, 19E3, to prolong in vivo mean apparent terminal elimination half‐life (t1/2).9
In vitro, BOS161721 inhibited the downstream signaling, plasma cell differentiation, and natural killer cell activation induced by IL‐21 and suppressed antibody responses to keyhole limpet hemocyanin (KLH) antigen. Because of the pleiotropic nature of IL‐21, its neutralization is expected to affect several immune cell populations implicated in SLE pathogenesis.
This report presents the results of the first clinical assessment of BOS161721 in healthy subjects.
Methods
Study design and treatment
This phase I first‐in‐human trial was a randomized, single‐center, double‐blind, placebo‐controlled, single‐ascending dose (SAD) study to assess the safety, pharmacokinetics (PK), pharmacodynamics (PD), and immunogenicity of i.v. and s.c. doses of BOS161721 in healthy subjects. Sixty‐one (61) male and female subjects were enrolled in eight SAD cohorts and randomized 3:1 to either BOS161721 or placebo (except one i.v. cohort without placebo). A schematic of dose escalation is presented in Figure 1.
Figure 1.
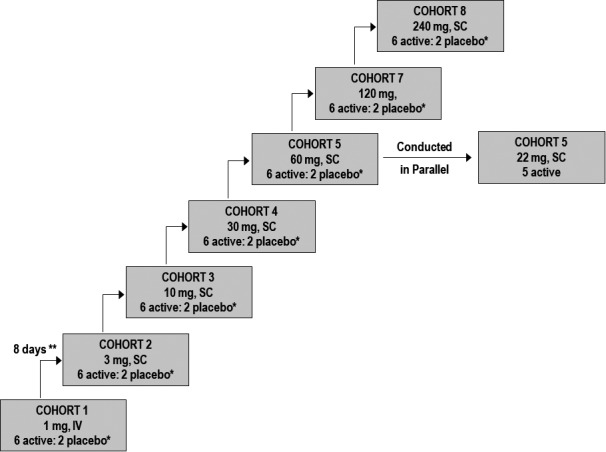
Dose escalation schematic. *One subject dosed with active and one subject dosed with placebo in a sentinel subcohort first. **For each cohort, the decision to escalate to the next higher dose level was made after all subjects were administered either BOS161721 or placebo and were followed for at least 7 days.
Following a 28‐day screening period, qualifying subjects were admitted on day –1 and received either a single s.c. dose to an abdominal site or an i.v. infusion over 30 minutes of BOS161721 or placebo on day 1. Subjects remained in‐clinic for 7 days, where they were monitored for safety, PK, and PD. After discharge, subjects returned for follow‐up evaluation of safety (including immunogenicity), PK, and PD for a total of 52 weeks.
The cohorts were administered ascending doses of either BOS161721 or placebo. Intravenous administration was examined in cohorts 1 and 6, whereas s.c. was examined in cohorts 2–5 and 7–8. Cohort 6 (i.v. 22 mg) was examined in parallel to cohort 5 (s.c. 60 mg); these 2 doses were projected to yield similar exposures, allowing estimation of bioavailability. Decisions to escalate to higher doses were made by the Safety Data Monitoring Team after the preceding cohort completed ≥ 7 days of follow‐up. This team consisted of the principal investigator from PAREXEL, a safety physician, the lead clinician, a clinical pharmacologist from Boston Pharmaceuticals, and an independent safety monitor. The investigator and the lead clinician were blinded to treatment assignment, whereas the safety physician and clinical pharmacologist were unblinded.
Study objectives
Key objectives were to assess the safety and tolerability of SADs of BOS161721 and to characterize its PK, PD, and immunogenicity. Exploratory objectives were to examine the effects of BOS161721 on the T cell‐dependent antibody response to the protein antigen KLH, plasma levels of IL‐21, and gene expression (via gene signature) in blood. In addition, levels of phosphorylated signal transducer and activator of transcription 3 (pSTAT3) in blood were assessed; pSTAT3 is a specific PD biomarker of IL‐21R signaling (see Pharmacodynamics in Results).
Subject eligibility
Subjects were male and female volunteers (N = 61) aged 18–55 years, in good health, willing and able to comply with all study procedures, with body mass index (BMI) 17.5–32 kg/m2 and weight 50–120 kg, and provided Institutional Review Board‐approved written informed consent prior to screening. Good health was determined by a responsible physician based on medical evaluation. Subjects were required to be of non‐childbearing potential status or to use contraception.
Study assessments
The study consisted of a screening visit (day –28 to day –2), clinic admission on day –1, and administration of a single dose of BOS161721 on dDay 1, and 7 days of in‐clinic observation for safety, PK, and PD. Follow‐up visits occurred on days 15, 30, 44, 60, 90, 120, 150, 180, 210, 240, 270, 300, 330, and 360. Maximal study duration for a subject was 388 days.
Safety assessments
Safety assessments included monitoring of adverse events (AEs), as well as electrocardiograms (ECGs), vital signs, clinical hematology and chemistry laboratory assessments, urinalysis, and physical examinations. Total IgG and IgM, and CD4+ counts were also measured. AEs were reported from consent through final follow‐up, with severity determined according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) criteria, version 4.03.10 AEs during or after dosing are reported herein.
Injection site reactions were assessed for redness/erythema, itching/pruritus, raised, and warmth, each scored for severity; other symptoms; size of reaction (mm); and location. Injection sites were assessed predose and 2 hours postdose on day 1 and followed until resolution.
Pharmacokinetic assessments
For cohorts 2, 3, 4, 5, 7, and 8 (s.c. administration), plasma concentrations of total (free and bound) BOS161721 were assessed using a validated electrochemical luminescence immunoassay method and an anti‐BOS161721 detection antibody (Green Mountain Antibodies, Burlington, VT) by PRA Health Sciences (Lenexa, KS). The validation range for this assay was 160–10,000 ng/mL. Whole blood was collected in K2 EDTA tubes at predose on day 1 and following the s.c. injection at 1, 2, 4, 8, and 12 hours. Additional measurements occurred on days 2, 4, 7, 15, 30, 44, 90, 180, 210, 270, and 360. Plasma concentrations for cohorts 1 and 6 (i.v. administration) included the same time points as s.c. cohorts, with an additional measurement at 30 minutes following infusion.
The following PK parameters were determined for BOS161721: maximum observed concentration (Cmax); time to maximum concentration (Tmax); area under the concentration‐time curve (AUC), with AUC0‐last from predose (time 0) to last quantifiable concentration (Tlast), and AUC0‐inf from predose (time 0) extrapolated to infinite time; t1/2; total body clearance from plasma/serum for i.v. dosing; and volume of distribution.
Primary pharmacodynamic variables
The pSTAT3 levels in lymphocytes were evaluated using a validated flow cytometric method with a phycoerythrin‐conjugated pY705 antibody (BD Biosciences, San Jose, CA) against pSTAT3 for detection by PRA Health Sciences (Lenexa, KS). Whole blood was collected after both s.c. and i.v. administration of BOS161721 or placebo at baseline and on the following study visit days: 1, 7, 44, 90, and 360. Following IL‐21 stimulation in vitro, cells were lysed, fixed, and permeabilized before undergoing flow cytometry. The minimum and maximum fluorescence intensity of samples from each subject were determined and the area under the curve (AUC0‐360 days) for the collection period was calculated.
Exploratory variables
Blood samples for assessment of T cell‐dependent antibody response (measured by anti‐KLH IgG and IgM titers) to KLH were collected on days 15, 30, 44, 90, 180, 210, 270, and 360.
Blood samples for IL‐21 gene signature evaluations were collected at baseline and on day 60 and analyzed in duplicate by AROS Applied Biotechnology (Aarhus, Denmark). Samples were then stimulated with: (a) IL‐21 (0.5 ng); or (b) IL‐21 (0.5 ng) + BOS161721 (0.5 ng); or (c) saline (negative control) and incubated for 2 hours before being transferred to PAXgene tubes for storage at −80°C for batch analysis. Total RNA was extracted using a QIAsymphony SP Biorobot. Gene expression was quantified by real‐time polymerase chain reaction using a Fluidigm Biomark system and a panel of 46 TaqMan assays. In addition, DNA was used to genotype for three single nucleotide polymorphisms located on the IL‐21 gene.
Immunogenicity assessments
Plasma samples to determine the presence of antidrug antibody (ADA) were collected on days −1, 7, 15, 30, 60, 90, 180, 270, and 360 and assessed via a validated electrochemical luminescence immunoassay method with a lower limit of detection of 3.91 ng/mL. This ADA assay was validated for drug interference at concentrations of BOS161721 up to 15.6 μg/mL. Blood samples for ADA analysis were collected at baseline through day 60 from all subjects; subsequent samples were collected from all but assayed only if the subject was ADA‐positive on day 60. Serum samples for CD4+ count and immunoglobulin (IgG and IgM) assessments were collected at screening and thereafter and evaluated using flow cytometry, enzyme‐linked immunosorbent assay, and streptavidin coated plates from MSD (Rockville, MD).
Statistical methods
The sample size was based on empirical considerations; no formal calculation was performed.
The safety population included all randomized subjects who received any amount of BOS161721 or placebo; the PK population included all subjects in the safety population treated with BOS161721 for whom at least one PK measurement was taken or noncompartmental analysis parameter was calculated. The PD population included all subjects in the safety population (both BOS161721 and placebo subjects) with at least one PD assessment.
Results
Subject disposition and characteristics
All 61 screened subjects were randomized and completed study treatment. Three subjects (4.9%) were discontinued before completing follow‐up: one subject in the BOS161721 240 mg s.c. treatment group had a fatal pulmonary embolism (assessed as unrelated to BOS161721) 127 days after receiving a single dose; one subject in the placebo group withdrew due to a family emergency; and one subject in the BOS161721 10 mg s.c. treatment group discontinued to relocate to another country.
The study population was predominantly men (44/61; 72.1%), and the majority were black/African American (46/61; 75.4%). Median age was 36 years (range 18–55 years), mean weight was 79.4 kg (range 50–116 kg), and mean BMI was 26.1 kg/m2 (range 19–32 kg/m2). Overall demographics were consistent with healthy patient trials, with an even balance across dose cohorts and between treatment and placebo for sex, BMI, and race/ethnicity. Most subjects (60.7%) treated with BOS161721 were ADA‐negative prior to dosing.
Safety/tolerability
The highest dose of BOS161721 administered was 240 mg s.c., estimated to have a safety margin of 12.5‐fold based on the s.c. no‐observed‐AE level dose in animal studies; thus, no dose‐limiting toxicity was expected or observed. Dose escalation proceeded from 1 mg i.v. to 240 mg s.c., with no significant safety events as assessed by the Safety Data Monitoring Team.
The incidence of AEs was similar between the pooled BOS161721 group (36.2%) and placebo group (42.9%). In both groups, the majority of AEs were mild (79.5% and 85.7%, respectively). No subjects discontinued treatment due to an AE. The AE profile was not dose‐dependent (Table 1).
Table 1.
AEs (> 1 Subject BOS161721 or PBO): All causalities by preferred term
| Adverse events | Placebo N = 14 n (%) | Pooled N = 47 n (%) | BOS161721 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 mg i.v. N = 6 n (%) | 3 mg s.c. N = 6 n (%) | 10 mg s.c. N = 6 n (%) | 30 mg s.c. N = 6 n (%) | 60 mg s.c. N = 6 n (%) | 22 mg i.v. N = 5 n (%) | 120 mg s.c. N = 6 n (%) |
240 mg s.c. N = 6 n (%) |
||||
| Total | 6 (42.9) | 17 (36.2) | 1 (16.7) | 4 (66.7) | 2 (33.3) | 2 (33.3) | 2 (33.3) | 2 (40) | 3 (50) | 1 (16.7) | |
| Diarrhea | 1 (7.1) | 6 (12.8) | 0 | 1 (16.7) | 2 (33.3) | 1 (16.7) | 0 | 1 (20) | 1 (16.7) | 0 | |
| Influenza‐like illness | 2 (14.3) | 3 (6.4) | 0 | 0 | 2 (33.3) | 0 | 0 | 0 | 1 (16.7) | 0 | |
| Feces soft | 0 | 2 (4.3) | 0 | 1 (16.7) | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | |
| URTI | 0 | 2 (4.3) | 0 | 0 | 0 | 0 | 0 | 0 | 2 (33.3) | 0 | |
| Wrist fracture | 0 | 2 (4.3) | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | |
| Headache | 2 (14.3) | 2 (4.3) | 0 | 2 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | |
| Paraesthesia | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Catheter site pain | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Catheter site swelling | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Trichomoniasis | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Viral sinusitis | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Viral URTI | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
AEs are coded using Medical Dictionary for Regulatory Activities (MedDRA), version 19.1. Safety Population, all randomized subjects who received at least one full or partial dose of BOS161721 or placebo.
AEs, adverse events; N, number of subjects in the safety population; n, number of subjects in the category; PBO, placebo; URTI, upper respiratory tract infection.
No clinically significant findings regarding physical examination, vital signs, or ECG were reported for subjects in the pooled BOS161721 population.
All absolute CD4+ lymphocyte counts were within normal levels at screening, following treatment, and throughout follow‐up. Following KLH immunization, only one subject in the 60 mg s.c. cohort (1.6% of patients) experienced a mildly raised injection site reaction that resolved ~ 8 hours later.
One subject (2.1%) in the pooled BOS161721 population had a fatal pulmonary embolism 127 days after receiving a single 240 mg s.c. dose, which the principal investigator (P.I.) assessed as unrelated to BOS161721.
Pharmacokinetics
As shown in Table 2, BOS161721 was slowly absorbed following s.c. dosing with a median Tmax of 6 days and a mean t1/2 of 77.5 to 88.3 days for doses ≥ 30 mg. AUC0‐inf and Cmax were linear across doses > 10 mg; plasma concentrations and dose proportionality are presented in Figure 2 and Figures [Link] , [Link] , [Link] , respectively. The s.c. bioavailability was estimated as 64.2% based on a comparison of 22 mg i.v. and 60 mg s.c. doses (Table S1 ).
Table 2.
Summary of plasma BOS161721 pharmacokinetic parameters following s.c. dosing
| Parameter | Statistic | BOS161721 3 mg s.c. N = 6 n (%) | BOS161721 10 mg s.c. N = 6 n (%) | BOS161721 30 mg s.c. N = 6 n (%) | BOS161721 60 mg s.c. N = 6 n (%) | BOS161721 120 mg s.c. N = 6 n (%) | BOS161721 240 mg s.c. N = 6 n (%) |
|---|---|---|---|---|---|---|---|
| AUC0-last (day•µg/mL) | n | 6 | 6 | 6 | 6 | 6 | 6 |
| Mean | 5.27 | 25.8 | 176 | 359 | 843 | 1,350 | |
| CV (%) | 169 | 44.5 | 27.4 | 41.4 | 35.6 | 45.0 | |
| Median | 1.76 | 22.7 | 188 | 294 | 872 | 1,230 | |
| AUC0-inf (day•µg/mL)a | n | 3 | 4 | 6 | 6 | 6 | 6 |
| Mean | 21.6 | 43.6 | 202 | 389 | 896 | 1,460 | |
| CV (%) | 173 | 27.2 | 24.1 | 39.1 | 34.6 | 44.3 | |
| Median | 0 | 41.2 | 214 | 333 | 965 | 1,300 | |
| Cmax (µg/mL) | n | 6 | 6 | 6 | 6 | 6 | 6 |
| Mean | 0.171 | 0.799 | 2.51 | 4.93 | 10.8 | 21.6 | |
| CV (%) | 85.1 | 31.4 | 44.8 | 47.4 | 33.9 | 38.0 | |
| Median | 0.206 | 0.742 | 2.22 | 4.4 | 10.7 | 20.4 | |
| Tmax (day) | n | 4 | 6 | 6 | 6 | 6 | 6 |
| Mean | 8 | 8.72 | 6.83 | 5.5 | 7.84 | 6.52 | |
| CV (%) | 50.1 | 48.4 | 84.4 | 22.3 | 57.4 | 68.3 | |
| Median | 6 | 6 | 4.51 | 6 | 6 | 6 | |
| t1/2 (day) | n | 1 | 4 | 6 | 6 | 6 | 6 |
| Mean | 141 | 29.7 | 86.3 | 80.2 | 87.2 | 79.5 | |
| CV (%) | ‐ | 21.1 | 9.8 | 17.6 | 32.1 | 41.6 | |
| Median | 141 | 30.3 | 85.1 | 77.5 | 87.6 | 88.3 |
Pharmacokinetic Population = All subjects in the safety population for whom at least one PK parameter was calculated.
AUC0‐inf, area under the concentration‐time curve from time zero to infinity; AUC0‐last, area under the concentration‐time curve from the last measurable plasma concentration; Cmax, maximum observed concentration; CV, coefficient of variation; t1/2, terminal elimination half‐life; Tmax, time to maximum concentration.
AUC0‐inf represents an approximation with a high degree of extrapolation.
Figure 2.
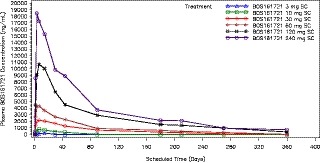
Median BOS161721 plasma concentration vs. time following s.c. dosing.
Pharmacodynamics
Upon IL‐21 binding, IL‐21R signaling initially involves phosphorylation of JAK1/JAK3, which dissociates from the receptor complex and pSTAT3, which, in turn, translocates to the nucleus and drives gene expression regulated by IL‐21. Although JAK1/JAK3 represents a more proximal biomarker for IL‐21 binding to its receptor, it is a less specific PD biomarker of IL‐21R signaling than is pSTAT3, because JAK1/JAK3 stimulation initiates intracellular signaling for the common gamma chain family of cytokine receptors, which includes receptors for IL‐2, IL‐4, IL‐7, IL‐9, IL‐15, and IL‐21. Regardless of the route of administration, when blood samples from subjects receiving BOS161721 were subjected to an ex vivo IL‐21 stimulation assay, minimum percentages of pSTAT3‐positive lymphocytes were reduced in a dose‐responsive manner, with threshold characteristics at doses ≥ 10 mg (Figure 3). The median pSTAT3 AUC0‐last decreased dose‐dependently among subjects receiving BOS161721 (Figure 4). The dose‐dependent suppression of pSTAT3 is consistent with a strong PD response, reflected by the ability of BOS161721 at doses ≥ 10 mg to efficiently block signaling through IL‐21R. There was no discernible trend in median AUC0‐last or Cmax of anti‐KLH antibodies among those receiving BOS161721 s.c. (data not shown).
Figure 3.
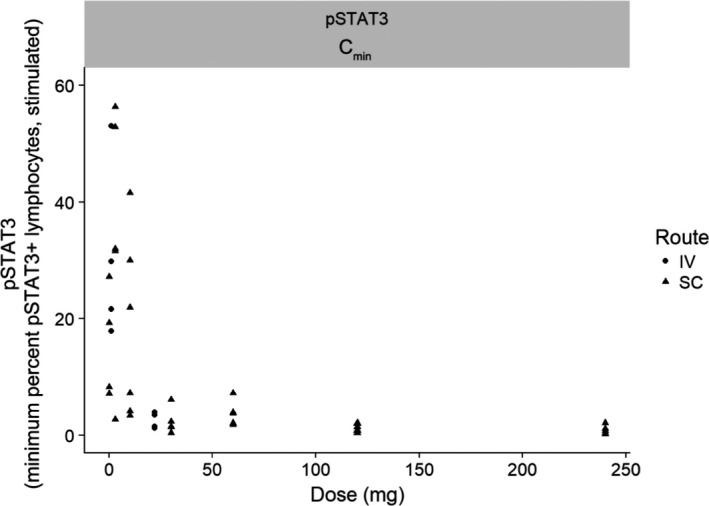
Phosphorylated signal transducer and activator of transcription 3 (pSTAT3) Cmin vs. BOS161721 dose. CI, confidence interval, Cmin, minimum percentage of pSTAT3 positive lymphocytes. Simple linear regression predicted natural log of parameter with 95% CI on the predicted mean.
Figure 4.
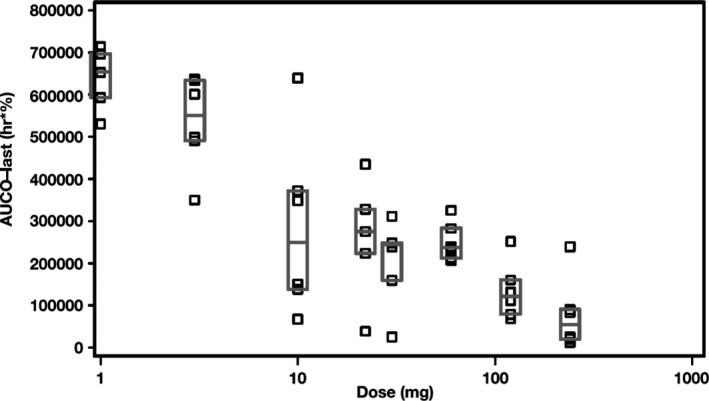
Phosphorylated signal transducer and activator of transcription 3 AUC0-last vs. BOS161721 dose. AUC0-last = area under the plasma concentration time curve from predose (time = 0) to last quantifiable concentration.
Gene expression
Upon in vivo BOS161721 treatment, gene downregulation with ex vivo IL‐21 stimulation was reversed in a dose‐dependent manner in 4 of the 29 genes analyzed (BATF, IL6, LAG3, and SOCS3; Figure 5 and Table S2 ). Notably, the reversal of downregulation produced by BOS161721 is expected to induce T‐cell exhaustion and tolerance in vivo, as these genes control T‐cell exhaustion, peripheral Treg fitness, and multiple cytokine signaling.
Figure 5.
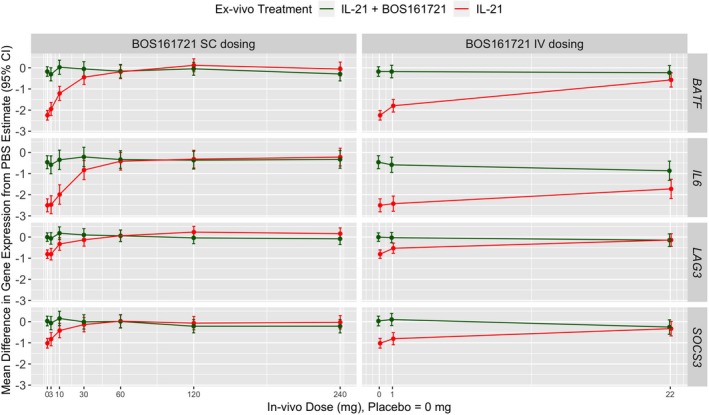
In vivo BOS161721 reverses ex vivo interleukin (IL)‐21‐induced downmodulation of BATF, IL6, LAG3, and SOCS3 expression. Blood from subjects treated with placebo or single dose of BOS161721 by s.c. or i.v. routes were collected as assessed for gene expression in a stepwise manner. First, predose samples from subjects were evaluated for differential gene expression resulting from IL‐21 stimulation in presence and absence of BOS161721. A total of 29 genes were identified for further analysis using a P < 0.025 level of significance with a Benjamini‐Hochberg adjustment for testing of multiple genes. BOS1617821 dosing‐related differential expression of these 29 genes was assessed in a second step by comparing baseline and postdose (day 60) samples from subjects. Only four genes demonstrated a statistically significant (P > 0.01) after adjusting for multiplicity as in the first step. Data are expressed as threshold cycle (Ct) values and the absolute differences in the means of gene expression are determined in comparison to unstimulated (saline treated) negative control samples. Error bars reflect 95% confidence intervals around the expression estimates.
Immunogenicity
In the pooled BOS161721 (i.v. and s.c. dosing) population, a positive ADA response was observed in four subjects (8.5%) predose and seven subjects (14.9%) postdose. Of the 14 AEs in subjects with confirmed ADA, only 5 were related to the study drug, indicating a lower percentage of response to BOS161721 than other mAbs.11 ADA was not detected at baseline in any placebo‐dosed (n = 14) or i.v.‐dosed subject (n = 11), but was found in 11.1% (4/36) of s.c.‐dosed subjects. No CD4+ count findings were clinically significant for any subjects in the pooled BOS161721 population. Detection of KLH IgG and IgM was not dose responsive.
Conclusions
Overall, BOS161721 was safe and well‐tolerated, with no dose‐related trends in the AE profile. ADA responses were positive for four subjects (8.5%) predose and seven subjects (14.9%) postdose; percentages were similar between i.v. and s.c. administration and compared with placebo. No CD4+ findings were clinically significant; detection of KLH IgG and IgM was not dose responsive. The median pSTAT3 AUC0‐last decreased in a dose‐dependent manner but showed evidence of a threshold effect at doses ≥ 10 mg. Downregulation from IL‐21 stimulation was reversed dose‐dependently in BATF, IL6, LAG3, and SOCS3 genes. Based on these findings, a multiple ascending dose study in patients with SLE has been completed and is being followed by an ongoing phase II proof‐of‐concept study in patients with SLE.
Discussion
IL‐21 promotes CD4+ T‑cell differentiation into specialized T‐follicular helper cells12, 13 and promotes the generation of T helper 17 cells.14 One principal nonredundant role of IL‐21 is the promotion of B‐cell activation, differentiation, or death during humoral immune responses.15 B‑cells are a critical component of SLE autoimmunity and clearly a major target for IL‐21. In immune diseases, elevations in IL‐21 and autoantibodies are correlated.3, 4 Patients with SLE have elevated serum IL‐21 that correlates with disease severity. Recent genome‐wide association studies provide convincing evidence that the chromosomal 4q27 region harbors the IL‐21 genes and is associated with chronic inflammatory disorders, including SLE.6
Evidence supporting the critical role of IL‐21 in promoting humoral and other immune responses makes it an important focus of potential therapeutic interventions in conditions like SLE that are characterized by overproduction of pathogenic autoantibodies. Notably, the dose‐dependent reversal of IL‐21‐induced downregulation of BATF, IL6, LAG3, and SOC3 produced by BOS161721 is expected to induce T‐cell exhaustion and tolerance in vivo, imparting potentially superior efficacy in SLE compared with current biologic therapies, such as belimumab.
This placebo‐controlled study assessed the safety and tolerability, PK, PD, and immunogenicity of BOS161721, a mAb designed to inhibit IL‐21 bioactivity. The vast majority of treatment‐emergent adverse events (TEAEs) in the pooled BOS161721 population were mild and caused no discontinuations. One subject experienced a serious TEAE (pulmonary embolism) and died 127 days after receiving a single 240 mg s.c. dose of BOS161721; this was assessed as unrelated to the study drug. A positive ADA response was reported for four subjects (8.5%) predose and seven subjects (14.9%) postdose. Only one subject experienced an injection site reaction. No findings in physical examination, vital signs, ECG, ADA, total IgG and IgM levels, or CD4+ count were reported as clinically significant for any subject who received BOS161721. The PK of s.c.‐administered BOS161721, the intended clinical route, was linear with doses > 10 mg, with no evidence of target‐mediated disposition, yielding a mean t1/2 of 80–87 days for doses ≥ 30 mg. The relative bioavailability of s.c.‐administered doses was 64.2%. A dose‐dependent decrease was reported for the PD biomarker pSTAT3 parameter AUC0‐last. Downregulation of four genes (BATF, IL6, LAG3, and SOCS3) caused by IL‐21 stimulation was reversed by BOS161721 in a dose‐dependent manner. Notably, the potent effect of IL‐21 in suppressing gene expression of BATF, LAG3, and SOCS3 could lead to fostering autoimmunity, as BATF regulates the action of FoxP3 Tregs in the periphery, and lack of BATF expression renders Tregs incapable of suppressive function. Downmodulating LAG3 (a checkpoint in T‐cell activation) results in T‐cell overactivity, fueling inflammation. Similarly, SOCS3 is an endogenous inhibitor of excessive cytokine signaling, and suppression of its expression by IL‐21 contributes to autoimmunity. A single dose of BOS161721 completely reverses these deleterious effects on the expression of these critical regulatory molecules in healthy subjects, suggesting that the pharmacologic action of BOS161721 treatment may be to restore immunologic tolerance, the ultimate goal in the therapy for autoimmune diseases, such as SLE. These findings will need to be confirmed in a multiple‐dose setting in patients with SLE.
Two investigational drug products, ATR‐107 (Pfizer) and NNC01140006 (Novo Nordisk), are in the same pharmacological class as BOS161721 based on inhibition of IL‐21 signaling. No potential risk generalizable across the class has been observed with these investigational products.11 ATR‐107 is an anti‐IL‐21R mAb that Pfizer discontinued after high titers of ADA were detected in the first‐in‐human SAD trial, although no safety concerns were reported. NNC01140006 is also an mAb targeting IL‐21 for which early clinical development has been conducted in both healthy subjects and specific patient populations (rheumatoid arthritis, SLE, and diabetes mellitus).16 Results from healthy subjects and patients with rheumatoid arthritis (126 subjects dosed) did not reflect significant untoward safety risks or significant changes in laboratory safety parameters.17
The multifunctional effects of IL‐21 on the immune system make it an attractive therapeutic target for SLE and other autoimmune diseases, such as rheumatoid arthritis.18 In this phase I clinical trial, the anti‐IL‐21 mAb BOS161721 seemed to be well‐tolerated in healthy subjects after single‐dose i.v. and s.c. administration, and no safety signals emerged during the trial.
The PK of anti‐IL‐21 increased dose proportionally, indicating linearity within the tested dose ranges > 10 mg. The study met its primary end point by assessing the safety and tolerability of BOS161721 in healthy subjects via physical examinations, incidence and severity of TEAEs and serious TEAEs, ECGs and vital signs, and laboratory assessments, including ADA, IgG/IgM, and CD4+ count.
BOS161721 has acceptable safety and tolerability and is suitable for subsequent clinical exploration in individuals with autoimmune conditions, including SLE. Based on the results from this study, a clinical trial (NCT03036865) to explore the efficacy of anti‐IL‐21 in patients with SLE has been initiated and is ongoing.19
Funding
Funding for this study was provided by Boston Pharmaceuticals.
Conflicts of Interest
During this clinical trial, P.T.C.H., D.M.B., and Ri.M. were paid employees of Boston Pharmaceuticals, the study sponsor, and have equity holdings in Boston Pharmaceuticals. A.H., Ra.M., and G.G. were directly compensated by Boston Pharmaceuticals for their contributions. D.M.B. is currently affiliated with The Bill & Melinda Gates Medical Research Institute.
Author Contributions
P.T.C.H. and Ri.M. wrote the manuscript. Ri.M. designed the research. A.H., Ri.M., and D.M.B. performed the research. C.G., Ri.M., Ra.M., and P.T.CH. analyzed the data.
Supporting information
Table S1. Summary of Plasma BOS161721 Pharmacokinetic Parameters following 60 mg s.c. or 22 mg i.v. Dosing.
Table S2. Gene Expression Profiles following BOS161721 Dosing.
Figure S1. BOS161721 Dose Proportionality: Cmax.
Figure S2. BOS161721 Dose Proportionality: AUC(0‐inf).
Figure S3. Median BOS161721 Plasma Concentration vs. Time following s.c. Dosing.
Acknowledgments
We acknowledge the participation of Xavier Valencia, Santiago Arroyo, and Ashley Milton in the design and conduct of this study and of Xavier Valencia in the preparation of the manuscript. Medical writing and editing support was provided by Paul Guttry, Acumen Medical Communications.
References
- 1. Sarra, M. & Monteleone, G. Interleukin‐21: a new mediator of inflammation in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 294582 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nakou, M. et al. Interleukin‐21 is increased in active systemic lupus erythematosus patients and contributes to the generation of plasma B‐cells. Clin. Exp. Rheumatol. 31, 172–179 (2013). [PubMed] [Google Scholar]
- 3. Li, Q. et al Follicular helper T‐cells (Tfh) and IL‐21 involvement in the pathogenesis of bullous pemphigoid. PLoS One 8, e68145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang, H. et al Functional interleukin‐21 polymorphism is a protective factor of diffuse large B‐cell lymphoma. DNA Cell Biol. 33, 775–780 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Liu, R. et al A regulatory effect of IL‐21 on T follicular helper‐like cell and B‐cell in rheumatoid arthritis. Arthritis Res. Ther. 14, R255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kang, K.Y. et al Impact of interleukin‐21 in the pathogenesis of primary Sjögren's syndrome: increased serum levels of interleukin‐21 and its expression in the labial salivary glands. Arthritis Res. Ther. 13, R179 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Terrier, B. et al Interleukin‐21 modulates Th1 and Th17 responses in giant‐cell arteritis. Arthritis Rheum. 64, 2001–2011 (2012). [DOI] [PubMed] [Google Scholar]
- 8. Terrier, B. et al Interleukin 21 correlates with T‐cell and B‐cell subset alterations in systemic lupus erythematosus. J. Rheumatol. 39, 1819–1828 (2012). [DOI] [PubMed] [Google Scholar]
- 9. Dall'Acqua, W.F. et al Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J. Immunol. 169, 5171–5180 (2002). [DOI] [PubMed] [Google Scholar]
- 10. NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. <https://evs.nci.nih.gov/ftp1/CTCAE/About.html>.
- 11. Hua, F. et al Anti‐IL21 receptor monoclonal antibody (ATR‐107): safety, pharmacokinetics, and pharmacodynamic evaluation in healthy volunteers: a phase I, first‐in‐human study. J. Clin. Pharmacol. 54, 14–22 (2014). [DOI] [PubMed] [Google Scholar]
- 12. Tangye, S.G. , Ma, C.S. , Brink, R. & Deenick, E.K. The good, the bad and the ugly – TFH cells in human health and disease. Nat. Rev. Immunol. 13, 412–426 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Spolski, R. & Leonard, W.J. IL‐21 and T follicular helper cells. Int. Immunol. 22, 7–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang, Y. , Xu, J. , Niu, Y. , Bromberg, J.S. & Ding, Y. T‐bet and eomesodermin play critical roles in directing T cell differentiation to Th1 versus Th17. J. Immunol. 181, 8700–8710 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ettinger, R. , Kuchen, S. & Lipsky, P.E. The role of IL‐21 in regulating B‐cell function in health and disease. Immunol. Rev. 223, 60–86 (2008). [DOI] [PubMed] [Google Scholar]
- 16. A randomised, double‐blind, double‐dummy, placebo‐controlled, parallel‐group multi‐centre clinical proof‐of‐principle trial in adult subjects with newly diagnosed type 1 diabetes mellitus investigating the effect of NNC0114‐0006 and liraglutide on preservation of beta‐cell function. ClinicalTrials.gov: NCT02443155 <http://clinicaltrials.gov/ct2/show/NCT02443155>.
- 17. Ignatenko, S. , Skrumsager, B.K. & Mouritzen, U. Safety, PK, and PD of recombinant anti‐interleukin‐21 monoclonal antibody in a first‐in‐human trial. Int. J. Clin. Pharmacol. Ther. 54, 243–252 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Yuan, F.L. et al Targeting interleukin‐21 in rheumatoid arthritis. Mol. Biol. Rep. 38, 1717–1721 (2011). [DOI] [PubMed] [Google Scholar]
- 19. Study of BOS161721 in systemic lupus erythematosus (SLE) patients on a background of limited standard of care. ClinicalTrials.gov: NCT03371251 <http://clinicaltrials.gov/ct2/show/NCT03371251>.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of Plasma BOS161721 Pharmacokinetic Parameters following 60 mg s.c. or 22 mg i.v. Dosing.
Table S2. Gene Expression Profiles following BOS161721 Dosing.
Figure S1. BOS161721 Dose Proportionality: Cmax.
Figure S2. BOS161721 Dose Proportionality: AUC(0‐inf).
Figure S3. Median BOS161721 Plasma Concentration vs. Time following s.c. Dosing.