

Abstract
The oral bioavailability of ibrutinib is low and variable, mainly due to extensive first‐pass metabolism by cytochrome P450 (CYP) 3A4. The unpredictable exposure can compromise its safe and effective dosing. We examined the impact of itraconazole on ibrutinib pharmacokinetics. In a randomized crossover study, 11 healthy subjects were administered itraconazole 200 mg or placebo twice on day 1, and once on days 2–4. On day 3, 1 hour after itraconazole (placebo) and breakfast, ibrutinib (140 mg during placebo; 15 mg during itraconazole) was administered. Itraconazole increased the dose‐adjusted geometric mean area under the concentration‐time curve from zero to infinity (AUC0–∞) of ibrutinib 10.0‐fold (90% confidence interval (CI) 7.2–13.9; P < 0.001) and peak plasma concentration (Cmax) 8.8‐fold (90% CI 6.3–12.1; P < 0.001). During itraconazole, the intersubject variation for the AUC0–∞ (55%) and Cmax (53%) was around half of that during placebo (104%; 99%). In conclusion, itraconazole markedly increases ibrutinib bioavailability and decreases its interindividual variability, offering a possibility to improved dosing accuracy and cost savings.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Ibrutinib is an expensive drug used against B‐cell malignancies. Its oral bioavailability is low, variable, and dependent on cytochrome P450 (CYP) 3A4 activity. The use of strong CYP3A4 inhibitors with ibrutinib is recommended to be avoided. Effect of itraconazole on ibrutinib exposure has not been studied in humans.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Our hypothesis was that itraconazole used with a small ibrutinib dose increases ibrutinib plasma concentrations to the same level as the nearly 10‐fold higher ibrutinib dose alone and decreases intersubject variation.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Itraconazole increased ibrutinib exposure, on average, 10‐fold and diminished the intersubject variation in ibrutinib exposure by about 50%.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Concomitant use of itraconazole allows reduction of ibrutinib dose roughly by 90% and reduces intersubject variation in ibrutinib concentrations. Significant cost‐savings (US > $10,000 dollars/patient/year) could be achieved by using itraconazole as a booster with ibrutinib. In addition, the risk of fungal infections could be diminished by using itraconazole with ibrutinib.
Ibrutinib is a potent Bruton's tyrosine kinase (BTK) inhibitor used in the treatment of chronic lymphocytic leukemia (CLL), Waldenström's macroglobulinemia, certain lymphomas, and chronic graft‐vs.‐host disease.1, 2, 3 Despite substantial efficacy in treating patients with CLL, the use of ibrutinib is limited by its high cost. Owing to extensive first‐pass metabolism, its mean oral bioavailability in the fasted state is only about 3%.3, 4 The majority of ibrutinib is metabolized by cytochrome P450 (CYP) 3A4 in the intestine and liver.5 Its main metabolite, PCI‐45227, has an inhibitory activity toward BTK about 15 times lower than that of parent ibrutinib.3 The pharmacokinetics of ibrutinib displays substantial intersubject variability, which may explain some of its adverse effects.4, 5, 6
The exposure to ibrutinib is greatly increased by CYP3A4 inhibitors.3 Moreover, food enhances the bioavailability of ibrutinib about twofold, probably by increasing hepatic blood flow and thereby decreasing hepatic extraction.4, 7 Under fed conditions, the intestinal CYP3A4‐inhibitor, grapefruit juice, increased the area under the plasma concentration‐time curve (AUC) of ibrutinib 2.2‐fold in healthy adults.4 In a fasted state, the CYP3A4‐inhibitor ketoconazole raised the AUC of ibrutinib 24‐fold in healthy volunteers.6 Furthermore, in patients with B‐cell malignancy, erythromycin and voriconazole increased the exposure to ibrutinib 3.0‐fold and 5.7‐fold in a nonfasted state, respectively.8
Currently, it is recommended to avoid the concomitant use of strong CYP3A4 inhibitors with ibrutinib, unless the benefit outweighs the risk.3 However, at least theoretically, the interaction between a strong CYP3A4 inhibitor and ibrutinib could be useful in reducing the required dose of ibrutinib, and, therefore, the financial burden of the treatment, and in decreasing the high interindividual variability seen in the pharmacokinetics of ibrutinib. The antifungal agent itraconazole is a strong inhibitor of CYP3A4 with favorable tolerability and absorption properties compared with ketoconazole.9, 10 However, there is only a physiologically‐based pharmacokinetic modeling‐based estimate of the influence of itraconazole on ibrutinib pharmacokinetics, and clinical data are lacking.11
This randomized placebo‐controlled crossover study with healthy volunteers aimed to investigate the effect of clinically used doses of itraconazole on the pharmacokinetics of ibrutinib, and the potential of itraconazole to be used as a pharmacokinetic booster, allowing the use of smaller and more predictable doses of ibrutinib in the clinical practice. For safety reasons, only a 15‐mg ibrutinib dose was used in the itraconazole phase, whereas 140 mg was given with the placebo.
Methods
Study participants
Eleven healthy nonsmoking male volunteers (age range 19–38 years; body mass index range, 20.7–28.6 kg/m2) participated in the study after giving written informed consent. Their health was ascertained by medical history, clinical examination, laboratory tests, and electrocardiogram before entering the study. None of the participants used any continuous medication, and all had normal hemoglobin values and prothrombin time.
Study design
The study protocol was approved by the Local Ethics Committee of the Helsinki and Uusimaa Hospital District (record number HUS/93/2018), and the Finnish Medicines Agency, Fimea (EudraCT number 2016‐000150‐36). In a randomized, placebo‐controlled, 2‐phase crossover study with a washout period of 4 weeks, the participants ingested either 200 mg itraconazole (Sporanox 100 mg capsule; Janssen‐Cilag, Espoo, Finland) or placebo capsules containing microcrystallized cellulose at 8 am and 8 pm on day 1, and at 8 am on days 2–4. On day 3, following an overnight fast, itraconazole or placebo was administered at 8 am. At 9 am, participants took a single oral dose of ibrutinib with 150 mL of water. The doses of ibrutinib (Imbruvica 140 mg capsule; Janssen‐Cilag International NV, Beerse, Belgium) in the itraconazole and placebo phases were 15 mg and 140 mg, respectively. The 15‐mg ibrutinib capsules were prepared by emptying the contents of Imbruvica capsules and mixing the powder with a sufficient amount of microcrystallized cellulose by HUS Pharmacy (Helsinki University Hospital, Helsinki, Finland). The ibrutinib content of the capsules was measured using the liquid chromatography‐tandem mass spectrometry system described below. On the days of ibrutinib administration, a standard breakfast was served immediately after the administration of pretreatment, and a warm meal 3 hours and snacks 7 and 10 hours after ibrutinib ingestion. The use of other drugs was prohibited from 1 week before to 1 week after the study, along with the use of grapefruit products and alcohol for 3 days before and during the study.
Timed blood samples for drug concentration measurements were drawn prior to the administration of pretreatment, and 5 minutes before and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 11, 23, and 47 hours after ibrutinib ingestion into tubes containing EDTA. The sample tubes were placed on ice immediately thereafter, and plasma was separated within 30 minutes and stored at −70°C until analysis.
Determination of drug concentrations
The plasma samples were prepared by use of Phree phospholipid removal plate in 96‐well format (Phenomenex, Torrance, CA) according to manufacturer recommendations. In short, plasma was mixed with acetonitrile containing 1% formic acid and a deuterium‐labeled internal standard, ibrutinib‐d5 or itraconazole‐d5 and hydroxyitraconazole‐d5. The mixture was then drawn through the cartridge, evaporated to dryness in a centrifugal evaporator (GeneVac; Thermo Fisher Scientific, Waltham, MA), and reconstituted in 20% acetonitrile.
Ibrutinib and PCI‐45227
The plasma ibrutinib and its active metabolite, PCI‐45227, were quantified using a Qtrap5500 liquid chromatography‐tandem mass spectrometry system (AB Sciex, Toronto, Canada). The mobile phase consisted of 0.1% formic acid and acetonitrile, and the chromatography was performed on a Luna C18 analytical column (2.1 × 100 mm inner diameter, 1.6 µm particle size; Phenomenex) operating at 30°C. The separation was achieved in 5 minutes using a linear gradient from 20 to 77% of acetonitrile at constant 300 µL/minute flow rate. The mass spectrometer was operated in positive multiple reaction‐monitoring mode with electrospray ionization. Ibrutinib and PCI‐45227 were monitored at mass‐to‐charge ratio (m/z) 441 to 304 and m/z 475 to 304, and the lower limits of quantification were 0.05 ng/mL and 0.1 ng/mL, respectively. The inter‐day precisions (coefficient of variation (CV)) were below 9% at relevant plasma drug concentrations for both analytes.
Itraconazole and hydroxyitraconazole
Drug concentrations were determined by using a Shimadzu Nexera liquid chromatography system (Shimadzu, Kyoto, Japan) coupled to an API 3000 tandem mass spectrometer (AB Sciex). The chromatographic separation of the analytes was accomplished on a Kinetex C18 column (2.1 × 50 mm inner diameter, 2.6 µm particle size; Phenomenex) equipped with a Kinetex C18 pre‐column (2.1 × 10 mm inner diameter, 2.6 µm particle size; Phenomenex).
The mobile phase was composed of 0.1% formic acid in water and 0.1% formic acid in acetonitrile, and gradient program was set from 35 to 95% of organic phase over 5 minutes, giving a total run time of 8.1 minutes. The mobile phase flow was set at 200 µL/minute and the injection volume was 10 µL. The mass spectrometry detection was performed using electro‐spray ionization in positive mode, based on the ion transitions m/z 705 to 392 for itraconazole and m/z 721 to 408 for hydroxyitraconazole. The lower limits of quantification for itraconazole and hydroxyitraconazole were 1 ng/mL and 5 ng/mL, and the inter‐day CVs were below 10% for both analytes at relevant concentrations.
Pharmacokinetics
The pharmacokinetic calculations were performed for both the unadjusted ibrutinib and PCI‐45227 concentrations and those adjusted to a 140‐mg dose. The peak plasma concentration (Cmax), time to Cmax (Tmax), elimination half‐life (t1/2), 0–23‐hour area under the concentration‐time curve (AUC0–23 hours), and AUC from zero to infinity (AUC0–∞) were calculated with standard noncompartmental methods using Phoenix WinNonlin, version 6.4 (Certara, Princeton, NJ). For itraconazole and hydroxyitraconazole, the Cmax, AUC0–5 hours, and AUC0–24 hours were calculated.
Genotyping
Genomic DNA was extracted from buffy coats prepared from EDTA samples collected for pharmacokinetic analyses, using the Maxwell 16 LEV Blood DNA Kit on a Maxwell 16 Research automated nucleic acid extraction system (Promega, Madison, WI). All participants were genotyped for the CYP3A4*22 (rs35599367) and CYP3A5*3 (rs776746) single nucleotide variations with TaqMan genotyping assays on a QuantStudio 12K Flex Real‐Time PCR system (ThermoFisher Scientific).
One of the subjects had the CYP3A5*1/*3 genotype (CYP3A5 expressor) and the rest had the CYP3A5*3/*3 genotype. None of the subjects had the CYP3A4*22 allele. No statistical analysis of genotype data was performed due to the small sample size.
Statistical analyses
Based on the pharmacokinetic results of previous studies, 12 participants were estimated to be adequate to detect a 30% difference in the AUC of ibrutinib between the placebo and itraconazole (with dose‐adjustment for ibrutinib) phases, with a power of at least 80% (α level 5%). The results are expressed as geometric means and geometric mean ratios with geometric CV or 90% confidence intervals (CIs) unless stated otherwise. The pharmacokinetic variables, except Tmax, were logarithmically transformed before analysis. The pharmacokinetic variables (other than Tmax) were compared by repeated‐measures analysis of variance with treatment phase as a within‐subject factor. The Tmax data were compared using the Wilcoxon signed rank test. Correlations were examined using Pearson's correlation coefficients. Statistical analyses were performed using IBM SPSS Statistics for Windows version 22.0 (IBM, Armonk, NY). Differences were considered statistically significant when the P value was < 0.05.
Results
All enrolled 11 subjects completed the study, and no adverse effects were reported or observed. Itraconazole markedly increased the plasma concentrations of ibrutinib and reduced their interindividual variation (Table 1, Figures 1 and 2).
Table 1.
Pharmacokinetic variables of ibrutinib and its metabolite PCI‐45227 in 11 healthy subjects after a single 140‐mg (placebo phase) or 15‐mg (itraconazole phase; both unadjusted and dose‐adjusted values given) oral dose of ibrutinib on day 3 of a 4‐day pretreatment with 200 mg itraconazole or placebo twice daily on day 1 and once daily on days 2–4
| Variable | Placebo phase (control) | Itraconazole phase | Geometric mean ratio (90% CI) | P value | Itraconazole phase (dose‐adjusted) | Geometric mean ratio (90% CI) | P value |
|---|---|---|---|---|---|---|---|
| Ibrutinib | |||||||
| Cmax (ng/mL) | 22.0 (0.99) | 20.6 (0.53) | 0.94 (0.68, 1.30) | 0.727 | 192.2 (0.53) | 8.8 (6.34, 12.1) | < 0.001 |
| Tmax (hour) | 2 (1–4) | 3 (1–4) | — | 0.287 | — | — | — |
| t1/2 (hour) | 4.7 (0.30) | 3.8 (0.28) | 0.82 (0.72, 0.93) | 0.017 | — | — | — |
| AUC0–23 hours (ng·hour/mL) | 74.4 (1.04) | 80.9 (0.55) | 1.09 (0.79, 1.50) | 0.648 | 755.3 (0.55) | 10.2 (7.35, 14.0) | < 0.001 |
| AUC0–∞ (ng·hour/mL) | 76.5 (1.04) | 81.9 (0.55) | 1.07 (0.77, 1.49) | 0.719 | 764.0 (0.55) | 10.0 (7.18, 13.9) | < 0.001 |
| PCI‐45227 | |||||||
| Cmax (ng/mL) | 31.1 (0.33) | 2.3 (0.28) | 0.07 (0.06, 0.09) | < 0.001 | 21.2 (0.28) | 0.68 (0.59, 0.79) | < 0.001 |
| Tmax (hour) | 3 (1.5–4) | 3 (1.5–4) | — | 0.140 | — | — | — |
| t1/2 (hour) | 6.4 (0.27) | 8.1 (0.28) | 1.3 (1.10, 1.46) | 0.014 | 8.1 (0.28) | 1.3 (1.10, 1.46) | 0.014 |
| AUC0–23 hours (ng·hour/mL) | 235.2 (0.43) | 23.0 (0.36) | 0.10 (0.08, 0.11) | < 0.001 | 214.3 (0.36) | 0.91 (0.78, 1.07) | 0.307 |
| AUC0‐∞ (ng·hour/mL) | 262.8 (0.45) | 27.4 (0.42) | 0.10 (0.09, 0.12) | < 0.001 | 256.0 (0.42) | 0.97 (0.82, 1.16) | 0.792 |
|
PCI‐45227/Ibrutinib AUC0‐∞ ratio |
3.4 (0.57) | 0.34 (0.28) | 0.10 (0.08, 0.12) | < 0.001 | — | — | — |
| Ibrutinib + active metabolitea | |||||||
| AUC0‐∞ (ng·hour/mL) | 95.9 (0.89) | 83.8 (0.55) | 0.87 (0.66, 1.15) | 0.400 | 781.7 (0.55) | 8.2 (6.17, 10.8) | < 0.001 |
Data are given as geometric mean with geometric coefficient of variation, Tmax as median with range. The geometric mean ratios between the two phases are given with 90% confidence interval.
CI, confidence interval; Cmax, peak plasma concentration; Tmax, time to Cmax; t1/2, elimination half‐life; AUC0–23 hours, area under the plasma concentration‐time curve from time 0 to 23 hours; AUC0–∞, area under the plasma concentration‐time curve from time from zero to infinity.
Active metabolite is defined as of PCI‐45227 AUC0‐∞.
Figure 1.
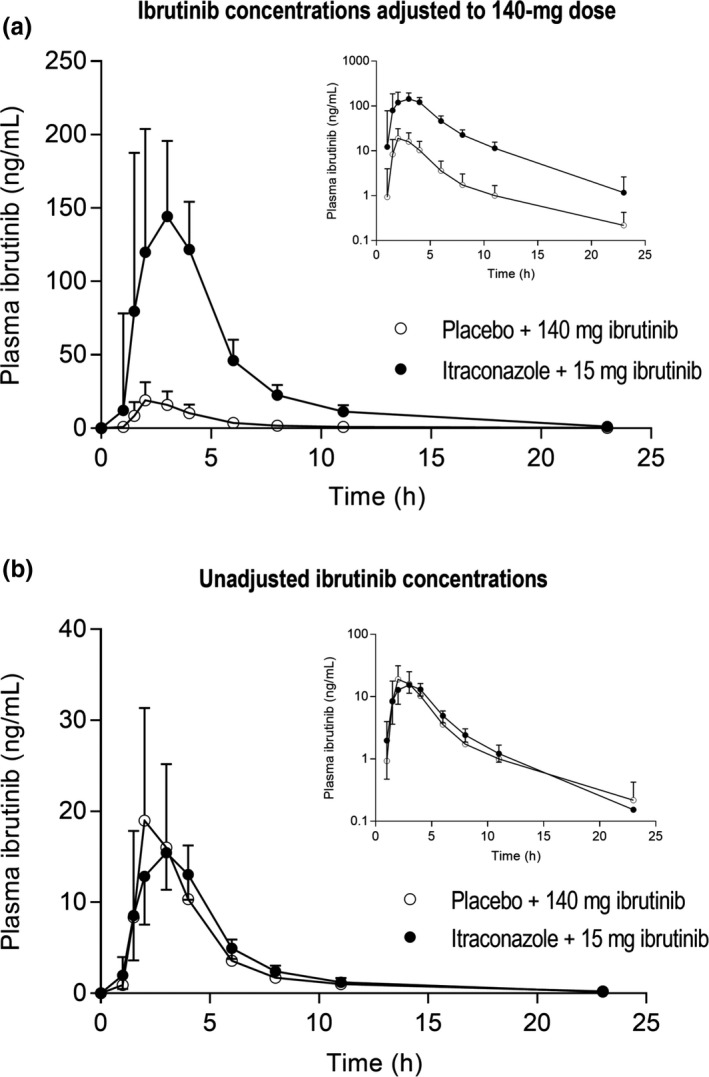
The plasma concentrations of ibrutinib in a randomized crossover study in 11 healthy subjects after a single 140‐mg (placebo phase) or 15‐mg (itraconazole phase) oral dose of ibrutinib on day 3 of a 4‐day pretreatment with 200 mg itraconazole or placebo twice daily on day 1 and once daily on days 2–4. Data are given for both the ibrutinib concentrations adjusted to a 140‐mg dose (a) and the unadjusted concentrations (b) and are presented as geometric means with 90% confidence intervals. For clarity, some error bars have been omitted. Insets depict the same data on a semilogarithmic scale.
Figure 2.
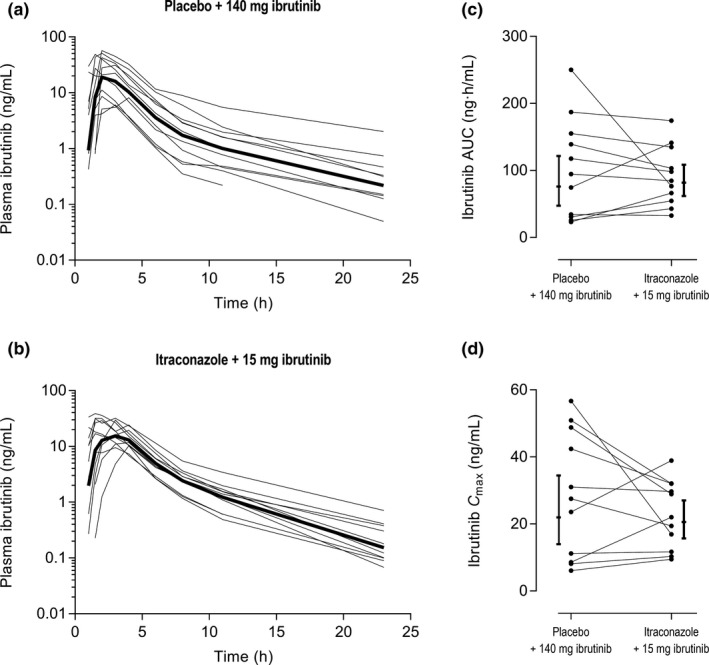
The unadjusted individual plasma concentration‐time curves of ibrutinib in placebo (a) and itraconazole (b) phases, as well as the unadjusted individual area under the plasma concentration time curves from zero to infinity (AUC0–∞) (c) and peak plasma concentrations (Cmax) (d) of ibrutinib in placebo and itraconazole phases with 90% confidence intervals. Eleven healthy subjects received in a randomized crossover study either a single 140‐mg (placebo phase) or 15‐mg (itraconazole phase) oral dose of ibrutinib on day 3 of a 4‐day pretreatment with 200 mg itraconazole or placebo twice daily on day 1 and once daily on days 2–4. The bold lines in figures (a) and (b) represent the geometric means.
Dose‐adjusted ibrutinib pharmacokinetics
Itraconazole increased the dose‐adjusted geometric mean AUC0–∞ and Cmax of ibrutinib (concentrations adjusted to a 140‐mg dose) 10.0‐fold (90% CI 7.2–13.9; P < 0.001) and 8.8‐fold (90% CI 6.3–12.1; P < 0.001), respectively (Table 1, Figure 1 a). An increase in the AUC0–∞ values was observed in all participants and the extent of interaction varied greatly from 2.9 to 26.6‐fold. The metabolite PCI‐45227/Ibrutinib AUC0–∞ ratio was decreased by itraconazole to 10% (90% CI 8–12%; P < 0.001) of control. In addition, a significant correlation (Pearson two‐tailed, r2 = 0.93; P < 0.001) was seen between the fold‐change in ibrutinib AUC0–∞ and the PCI‐45227/ibrutinib AUC0–∞ ratio in the placebo phase (Figure 3 a).
Figure 3.
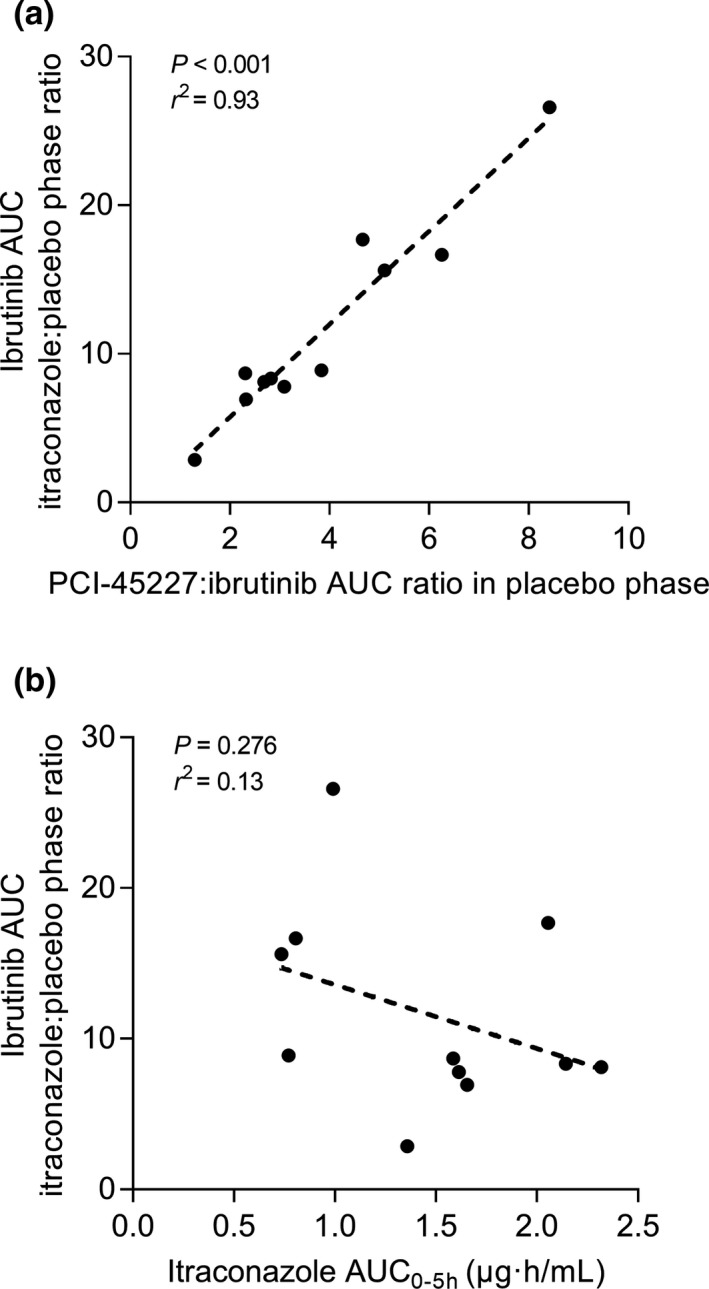
The correlation of the PCI‐45227:ibrutinib area under the plasma concentration time curve from zero to infinity (AUC0–∞) ratio in the placebo phase with the ibrutinib AUC0–∞ itraconazole to control ratio (a), and the correlation of itraconazole AUC0–5 hours with the ibrutinib AUC0–∞ itraconazole to control ratio (b).
Unadjusted ibrutinib pharmacokinetics
For the unadjusted pharmacokinetics (15 mg ibrutinib during itraconazole, 140 mg ibrutinib during placebo), similar exposures to ibrutinib were seen in both phases (Figure 1 b). The geometric mean ratios of ibrutinib mean AUC0–∞ and Cmax between the itraconazole and placebo phases were 1.07 (90% CI 0.77–1.49; P = 0.719) and 0.94 (90% CI 0.68–1.30; P = 0.727), respectively (Table 1). The geometric CVs of the ibrutinib mean AUC0–∞ and Cmax during the itraconazole phase (0.55 and 0.53, respectively) were around half of those during the placebo phase (1.04 and 0.99, respectively). Itraconazole shortened the ibrutinib mean t1/2 from 4.7 to 3.8 hours (P = 0.017). During the itraconazole phase, the AUC0‐∞ and Cmax of the ibrutinib metabolite PCI‐45227 were 10% (P < 0.001) and 7% (P < 0.001) of those in the placebo phase, respectively (Figure 4 b).
Figure 4.
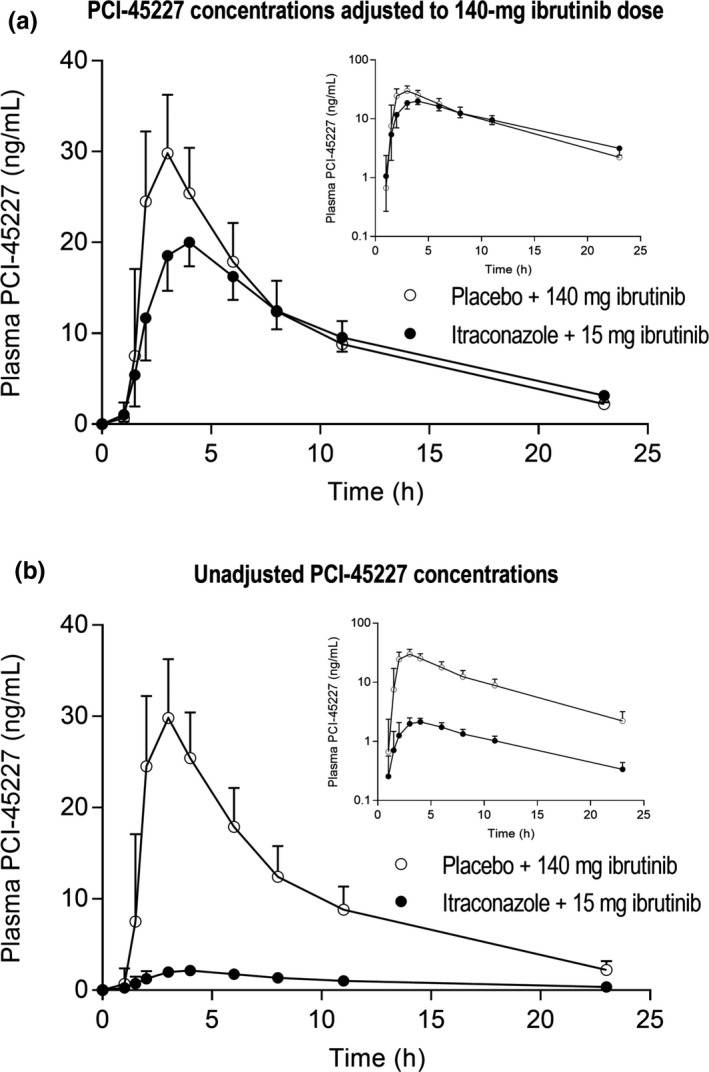
The plasma concentrations of ibrutinib's metabolite PCI‐45227 in a randomized crossover study in 11 healthy subjects after a single 140‐mg (placebo phase) or 15‐mg (itraconazole phase) oral dose of ibrutinib on day 3 of a 4‐day pretreatment with 200 mg itraconazole or placebo twice daily on day 1 and once daily on days 2–4. Data are given for both the ibrutinib concentrations adjusted to a 140‐mg dose (a) as well as the unadjusted concentrations (b) and is presented as geometric means with 90% confidence intervals. For clarity, some error bars have been omitted. Insets depict the same data on a semilogarithmic scale.
Itraconazole pharmacokinetics and the extent of interaction
The geometric means for itraconazole AUC0–24 hours and Cmax on day 3 were 4.7 (CV 0.42) µg·hour/mL and 0.39 (CV 0.41) µg/mL, respectively, and those of hydroxyitraconazole were 13.5 (CV 0.34) µg·hour/mL and 0.73 (CV 0.27) µg/mL, respectively. There was no correlation between the fold‐change in ibrutinib AUC0–∞ and itraconazole pharmacokinetics, i.e., its AUC0–5 hours, which coincides with the absorption phase of ibrutinib (Figure 3 b) nor its AUC0–24 hours or the sum AUC0–5 hours or AUC0–24 hours of itraconazole and hydroxyitraconazole (data not shown).
Discussion
Ibrutinib, an oral inhibitor of BTK, has shown considerable single‐agent efficacy in CLL.12, 13, 14 However, the high cost of long‐term ibrutinib treatment together with potential adverse effects has raised concerns.15 The use of a CYP3A4 inhibitor, such as itraconazole, as a pharmacokinetic booster could offer a tool to improve ibrutinib bioavailability and reduce the financial burden of ibrutinib treatment without the risk of compromising its therapeutic plasma concentrations.
The pharmacokinetics of ibrutinib is characterized by a low and variable oral bioavailability, due to its extensive intestinal and hepatic first‐pass metabolism by CYP3A4 and its flow‐dependent hepatic clearance.3, 4, 5 Accordingly, ibrutinib is very sensitive to drug interactions caused by CYP3A inhibitors and inducers,3, 6, 8 and even food ingestion can markedly increase its exposure.4, 7 There is evidence suggesting that concurrent use of CYP3A4 interacting medications with ibrutinib without dose‐adjustment results in treatment interruptions and shortened survival.16 Because many of the patients using ibrutinib also need other concomitant drugs, and because its therapeutic range is limited, the effect of other treatments on ibrutinib exposure should be as predictable as possible. Accordingly, dosing recommendations on the use of ibrutinib with different CYP3A4 perpetrators should be based on the most reliable evidence available.3, 17
In the present study in healthy subjects, the exposure to ibrutinib after a 15‐mg oral dose taken concomitantly with itraconazole was very close to the exposure after a 140‐mg ibrutinib dose alone. Accordingly, the effect of itraconazole on ibrutinib pharmacokinetics was strong, and there was a 10‐fold mean increase in the dose‐adjusted AUC of ibrutinib by itraconazole. The interaction was probably caused by inhibition of CYP3A4 during the absorption phase of ibrutinib, as the t1/2 of ibrutinib was not prolonged. The mechanism of the slight shortening of t1/2 is not known. Because ibrutinib is a high extraction drug, food increases its oral bioavailability likely by increasing hepatic perfusion and decreasing hepatic extraction. In this study, breakfast was served before ibrutinib ingestion to mimic the usual daily‐life situation. The effect of food can explain the smaller fold‐increase in the exposure to ibrutinib than what was expected based on previous physiologically‐based pharmacokinetic modeling (21‐fold increase in AUC), which assumed a fasted state.11
The dosages of ibrutinib in this study were smaller than those in clinical practice, particularly in the itraconazole phase, to avoid unnecessarily high exposure of healthy subjects to the potentially toxic study drug. The use of dose‐adjustment for ibrutinib was justified because its pharmacokinetics are linear.18 Moreover, the pharmacokinetic characteristics of ibrutinib have been shown to be similar in healthy subjects and in patients,7 indicating that extrapolation of our results to patients is straightforward.
According to prescribing information, the use of ibrutinib concomitantly with strong inhibitors of CYP3A4 should be avoided unless the benefit outweighs the risk.3 However, dose recommendations for the concomitant use of ibrutinib with the CYP3A4 inhibitors voriconazole and posaconazole as well as moderate CYP3A inhibitors have been given.3 According to the European Medicines Agency summary of product characteristics, if a strong CYP3A4 inhibitor must be used, the treatment with ibrutinib should be either withheld temporarily or its dose should be reduced to 140 mg.17 Our results suggest that the reduced 140‐mg dose of ibrutinib is too high when used concomitantly with itraconazole, and that about one‐tenth of the regular treatment dose of ibrutinib (420 mg or 560 mg) would result in optimal therapeutic plasma concentrations when used with itraconazole. The activity of the main metabolite of ibrutinib, PCI‐45227, is marginal. Thus, the reduction in PCI‐45227 AUC0‐∞ by itraconazole should not influence the dose recommendation as indicated by the AUC0‐∞ of active compounds (ibrutinib + of PCI‐45227) (Table 1). Of note, the fold‐increase in ibrutinib AUC showed no correlation with itraconazole pharmacokinetics. This indicates that the inhibition of presystemic CYP3A4‐mediated metabolism by itraconazole was nearly maximal and is not susceptible to moderate changes in itraconazole doses or interindividual variability in its pharmacokinetics.
The exposure to ibrutinib varies greatly with standard dosing. In this study, the intersubject CV values for both the Cmax and AUC of ibrutinib were reduced from about 100% in the placebo phase to 55% in the itraconazole phase. Hence, extremely high or low ibrutinib concentrations could be avoided by using itraconazole with appropriately reduced ibrutinib doses. Of interest, the extent of the interaction correlated with the metabolite‐to‐parent AUC ratio in the placebo phase (Figure 3 a; i.e., the interaction was greatest in individuals with a high rate of ibrutinib metabolism). Similar findings have been observed in the ketoconazole‐ibrutinib interaction study.6 Accordingly, the high interindividual variability seems to be related to differences in CYP3A activity in the intestine and liver as well as to variation in hepatic blood flow, and it is possible to considerably reduce this variability by CYP3A4 inhibitors.
Patients with hematologic disorders are prone to infections due to impaired immunity related either to the disease itself or to its treatments. Therefore, antifungal prophylaxis is widely used in this patient population in high‐risk situations. A high number of invasive fungal infections, in particular invasive aspergillosis with frequent cerebral involvement, has been reported in patients treated with ibrutinib.19, 20, 21 However, antifungal prophylaxis in patients using ibrutinib is considered problematic because many antifungal agents interact strongly with ibrutinib.21, 22 Voriconazole is effective in the treatment of invasive fungal infections but it is generally not recommended for primary prophylaxis.23, 24 Itraconazole and fluconazole can be used to reduce the incidence of invasive fungal infections, although in high‐risk hematologic patients their efficacy is weaker than that of posaconazole.23, 24, 25 Accordingly, itraconazole could be an alternative to fluconazole in the prevention of fungal infections in low‐risk patients on ibrutinib treatment when used together with greatly reduced ibrutinib doses. It should be noted that itraconazole may also increase plasma concentrations of other drugs, which are substrates of CYP3A4 or P‐glycoprotein. However, its drug interactions are well‐documented, and can often be managed with dose adjustments.
Compared with conventional chemotherapy, ibrutinib is generally well‐tolerated, but some serious adverse effects, such as cytopenias, major bleeding, atrial fibrillation, and infectious complications, have been described in relation to its use.3, 26 In a retrospective study of 616 patients with CLL, 41% of the patients discontinued ibrutinib, with intolerance rather than CLL progression being the major cause of discontinuation.26 In another recent retrospective analysis, major bleeding occurred in 19% of patients receiving ibrutinib.27 Inhibition of other kinases than BTK has been suggested to be involved in some of these adverse effects, such as inhibitory effect on platelet function.28 Based on our present results, the concomitant use of currently available 70‐mg and 140‐mg capsules of ibrutinib with itraconazole or other strong CYP3A4 inhibitors increases ibrutinib exposure to a higher level than what is observed when the usual 420‐mg or 560‐mg daily doses are taken without CYP3A4 inhibitors. Such a high exposure could increase the risk of concentration‐dependent adverse effects and lead to discontinuation of the treatment and potentially affect clinical outcomes.
In conclusion, co‐administering itraconazole as a pharmacokinetic booster with a markedly lowered ibrutinib dose may offer advantages. First, by knowing the extent of the interaction, a smaller dose of ibrutinib can be used together with itraconazole. This could serve as a cost‐effective means to provide efficient treatment to a larger number of patients as the financial burden is lessened. Second, itraconazole reduces the interindividual variation in the exposure to ibrutinib. This provides more predictable pharmacokinetics, which potentially results in more reliable efficacy and less adverse effects. Third, because patients treated with ibrutinib are more prone to fungal infections, itraconazole could give patients some prophylaxis against fungal infections.
Funding
This study was supported by grants from the Academy of Finland (Grant decision 278123, 2014), Sigrid Jusélius Foundation (Helsinki, Finland), and by State funding for university‐level health research (Hospital District of Helsinki and Uusimaa, Finland).
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
T.T., A.M.O., A.T., M.N., E.E., P.J.N., M.N., and J.T.B. wrote the manuscript. T.T., A.M.O., A.T., M.N., E.E., P.J.N., M.N., and J.T.B. designed the research. T.T., A.M.O., A.T., M.N., M.N., and J.T.B. performed the research. T.T., A.M.O., A.T., M.N., P.J.N., M.N., and J.T.B. analyzed the data.
Acknowledgments
The authors thank Ms. Eija Mäkinen‐Pulli and Ms. Lisbet Partanen for skillful technical assistance and HUS Pharmacy for preparation of the capsules for this work.
References
- 1. Honigberg, L.A. et al. The Bruton tyrosine kinase inhibitor PCI‐32765 blocks B‐cell activation and is efficacious in models of autoimmune disease and B‐cell malignancy. Proc. Natl. Acad. Sci. USA 107, 13075–13080 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Byrd, J.C. et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 369, 32–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Janssen Biotech, Inc. Imbruvica prescribing information. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210563s000lbl.pdf> (2018). Accessed June 5, 2019.
- 4. de Vries, R. et al. Stable isotope‐labelled intravenous microdose for absolute bioavailability and effect of grapefruit juice on ibrutinib in healthy adults. Br. J. Clin. Pharmacol. 81, 235–245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scheers, E. et al. Absorption, metabolism, and excretion of oral 14C radiolabeled ibrutinib: an open‐label, phase I single‐dose study in healthy men. Drug Metab. Dispos. 43, 289–297 (2015). [DOI] [PubMed] [Google Scholar]
- 6. de Jong, J. et al. Effect of CYP3A perpetrators on ibrutinib exposure in healthy participants. Pharmacol. Res. Perspect. 3, e00156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Jong, J. et al. The effect of food on the pharmacokinetics of oral ibrutinib in healthy participants and patients with chronic lymphocytic leukemia. Cancer Chemother. Pharmacol. 75, 907–916 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Jong, J. et al. A drug‐drug interaction study of ibrutinib with moderate/strong CYP3A inhibitors in patients with B‐cell malignancies. Leuk. Lymphoma 59, 2888–2895 (2018). [DOI] [PubMed] [Google Scholar]
- 9. Olkkola, K.T. , Backman, J.T. & Neuvonen, P.J. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin. Pharmacol. Ther. 55, 481–485 (1994). [DOI] [PubMed] [Google Scholar]
- 10. Liu, L. et al. Best practices for the use of itraconazole as a replacement for ketoconazole in drug‐drug interaction studies. J. Clin. Pharmacol. 56, 143–151 (2016). [DOI] [PubMed] [Google Scholar]
- 11. de Zwart, L. , Snoeys, J. , De Jong, J. , Sukbuntherng, J. , Mannaert, E. & Monshouwer, M. Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin. Pharmacol. Ther. 100, 548–557 (2016). [DOI] [PubMed] [Google Scholar]
- 12. Byrd, J.C. et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 371, 213–223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burger, J.A. et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N. Engl. J. Med. 373, 2425–2437 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Brien, S. et al. Single‐agent ibrutinib in treatment‐naïve and relapsed/refractory chronic lymphocytic leukemia: a 5‐year experience. Blood 131, 1910–1919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen, L.S. et al. A pilot study of lower doses of ibrutinib in patients with chronic lymphocytic leukemia. Blood 132, 2249–2259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kittai, A. et al. Effect of concurrent CYP3A4 interacting medications on ibrutinib outcomes in patients with CLL [abstract no. e19514]. J. Clin. Oncol. 36 (15 suppl.) (2018). [Google Scholar]
- 17. European public assessment report for Imbruvica. <https://www.ema.europa.eu/en/medicines/human/EPAR/imbruvica> (2019). Accessed June 5, 2019.
- 18. Marostica, E. et al. Population pharmacokinetic model of ibrutinib, a Bruton tyrosine kinase inhibitor, in patients with B cell malignancies. Cancer Chemother. Pharmacol. 75, 111–121 (2015). [DOI] [PubMed] [Google Scholar]
- 19. Ghez, D. et al. Early‐onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood 131, 1955–1959 (2018). [DOI] [PubMed] [Google Scholar]
- 20. Varughese, T. et al. Serious infections in patients receiving ibrutinib for treatment of lymphoid cancer. Clin. Infect. Dis. 67, 687–692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maschmeyer, G. et al. Infections associated with immunotherapeutic and molecular targeted agents in hematology and oncology. A position paper by the European Conference on Infections in Leukemia (ECIL). Leukemia 33, 844–862 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown, J.R. How I treat CLL patients with ibrutinib. Blood 131, 379–386 (2018). [DOI] [PubMed] [Google Scholar]
- 23. Mellinghoff, S.C. et al. Primary prophylaxis of invasive fungal infections in patients with haematological malignancies: 2017 update of the recommendations of the Infectious Diseases Working Party (AGIHO) of the German Society for Haematology and Medical Oncology (DGHO). Ann. Hematol. 97, 197–207 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maertens, J.A. et al. European guidelines for primary antifungal prophylaxis in adult haematology patients: summary of the updated recommendations from the European Conference on Infections in Leukaemia. J. Antimicrob. Chemother. 73, 3221–3230 (2018). [DOI] [PubMed] [Google Scholar]
- 25. Cornely, O.A. et al. Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N. Engl. J. Med. 356, 348–359 (2007). [DOI] [PubMed] [Google Scholar]
- 26. Mato, A.R. et al. Toxicities and outcomes of 616 ibrutinib‐treated patients in the United States: a real‐world analysis. Haematologica 103, 874–879 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mock, J. et al. Risk of major bleeding with ibrutinib. Clin. Lymphoma Myeloma Leuk. 18, 755–761 (2018). [DOI] [PubMed] [Google Scholar]
- 28. Bye, A.P. et al. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 1, 2610–2623 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]