

SUMMARY
Evolution of antibody repertoire against the Ebola virus (EBOV) proteome was characterized in an acutely infected patient receiving supportive care alone to elucidate virus-host interactions over time. Differential kinetics are observed for IgM/IgG/IgA epitope diversity, antibody binding, and affinity maturation to EBOV proteins. During acute illness, antibodies predominate to VP40 and Glycoprotein (GP). At day 13 of clinical illness a marked increase in antibody titers to most EBOV proteins and affinity maturation to GP is associated with rapid decline in viral replication and illness severity. At one-year, despite undetectable virus, a diverse IgM repertoire against VP40 and GP epitopes is observed suggesting occult viral persistence. Rabbit immunization experiments identify key immunodominant sites of GP, while challenge studies in mice found these epitopes induce EBOV-neutralizing antibodies and protect against lethal EBOV challenge. This study reveals markers of viral persistence and provides promising approaches for development and evaluation of vaccines and therapeutics.
Keywords: Ebola, Correlates of Protection, Infection, Vaccine, Neutralization, Immune Response, Antibody, GP, Epitope mapping, GFPDL, Affinity, Evolution, Virus
eTOC Blurb
Comprehensive characterization of antibody responses across the complete EBOV proteome in an EVD patient who survived with supportive care alone highlights the importance of antibody affinity in disease resolution. Khurana et al. describe the immune markers of protection to guide more effective vaccine design and targeted countermeasures against EVD.
One Sentence Summary:
Ebola immune markers of protection
INTRODUCTION
Ebola virus (EBOV) causes severe and often fatal disease in humans and remains a global public health challenge. The ongoing EBOV epidemic in the Democratic Republic of the Congo (DRC) has resulted in 3206 cases and 2143 deaths (case fatality ratio = 67%) as of October 7th, 2019 (https://www.who.int/emergencies/diseases/ebola/drc-2019), and the 2013–16 EBOV epidemic in West Africa resulted in an estimated 28,652 cases and 11,325 deaths. Given that EBOV may persist in semen of male survivors risking sexual transmission (Deen et al., 2017), and that natural spillover events continue to occur, the likelihood of recurrent severe outbreaks remains high.
Development of effective vaccines for prevention and medical countermeasures for treatment of EBOV infection remains a high global priority. Multiple vaccine candidates based on EBOV-GP are being evaluated in humans (Henao-Restrepo et al., 2015; Kennedy et al., 2017; Krause, 2015; Marzi et al., 2015; Milligan et al., 2016; Rampling et al., 2015; Regules et al., 2015), although duration of protective immunity is undetermined (Dye et al., 2012; Holtsberg et al., 2015; Marzi et al., 2013; Wilson et al., 2000). However, little is known about which sites within GP are responsible for vaccine induced antibody mediated protection against EBOV. Most studies on EBOV infection/vaccination in humans used MAb as surrogate readouts, and no vaccination studies have been performed to evaluate the contribution of each individual antigenic site within EBOV-GP as an immunogen/vaccine to generate EBOV neutralizing antibodies or protection against Ebola virus disease (EVD). It is postulated that immune responses generated during EBOV infection likely provide life-long protection among survivors, and so deeply characterizing immune responses in survivors, and correlating those responses to viral clearance and recovery might assist in rational vaccine and therapeutic design (Crook et al., 2017). An excellent example of this is the identification and isolation of monoclonal antibody (MAb) 114, from the blood of a single survivor of the 1995 EVD outbreak in Kikwit, DRC. In preclinical studies, MAb114 protected 100% nonhuman primates from lethal EBOV infection (Corti et al., 2016). MAb114 is presently being evaluated in a multi-arm Phase III randomized clinical trial in the DRC and preliminary data suggest administration early in the disease course is associated with significantly reduced mortality (Kupferschmidt, 2019).
Prior studies evaluated immune responses among EVD survivors, although all patients had received antibody treatment (ZMapp or convalescent plasma), which may confound study conclusions, and analyses primarily focused on anti-GP antibodies performed on samples collected after disease resolution, using MAbs as surrogate readouts. (Davis et al., 2019; McElroy et al., 2016; Saphire et al., 2018). Recent study showed that individual MAbs developed following rVSV-EBOV vaccination, 30–76% reacted with Sudan virus (SUDV) (Ehrhardt et al., 2019). In contrast, the polyclonal sera elicited by rVSV-EBOV vaccination appears to be specific to just EBOV, with little detectable reactivity to SUDV or other Ebolaviruses at the polyclonal level. These studies did not determine epitope specificity against the complete EBOV proteome, nor did they closely characterize antibody kinetics during disease progression and resolution. Enzyme linked immunosorbent assays (ELISA) and EBOV-neutralization tests targeting EBOV surface glycoprotein (GP) have primarily been used to characterize antibody responses in humans following EBOV infection, but provide limited insight into the diversity and quality of polyclonal antibody responses across the complete EBOV proteome and its evolution over time (Cohen and Enserink, 2015; Krause et al., 2015; Matassov et al., 2015; Wong et al., 2012). Moreover, the contribution of different antigenic sites in EBOV-GP as an immunogen or vaccine in neutralization/protection against EVD is unknown.
Previously, genome-fragment phage display library (GFPDL) spanning the entire genome of highly pathogenic avian influenza virus, respiratory syncytial virus and zika virus were used to map the antibody repertoires of convalescent sera from infected individuals and in individuals after pandemic influenza vaccinations. These studies have revealed several diagnostic and protective targets(Fuentes et al., 2016; Khurana et al., 2010; Khurana et al., 2011a; Khurana et al., 2009; Khurana et al., 2011b; Ravichandran et al., 2019).
The goal of our investigation was to perform a comprehensive longitudinal analysis of the humoral immune response across the complete EBOV proteome in a critically ill patient with EVD who survived with supportive care alone (i.e., without the use of experimental therapies) and to correlate antibody responses with viral clearance and disease resolution. After selecting immunodominant antigens in our single patient we sought to determine if synthetic peptides, reflective of these antigenic sites within surface GP, would induce EBOV-neutralizing responses in a rabbit model and provide protection from lethal EOBV infection in a mouse model. Our strategy was to use complete EBOV genome fragment-phage display libraries (GFPDL) to elucidate the epitope repertoire recognized by IgM, IgG & IgA polyclonal antibodies in sera and surface plasmon resonance (SPR) technology to measure real-time antibody binding kinetics, immunoglobulin isotypes, and affinity maturation of serum antibodies against the complete EBOV proteome daily during acute illness and intermittently during convalescence. This was done to identify the immune markers that correlate with protection and disease resolution naturally in this EVD survivor. To reveal the importance of each antigenic site within EBOV-GP as a vaccine antigen, rabbit immunization and mice challenge studies were performed with each GP antigenic site identified in our study to establish proof of concept that this approach might be used to guide rational vaccine development against EVD. Given recurrent EVD outbreaks in the DRC and ongoing risk for regional and international spread, it remains unclear if existing vector-based vaccines will provide lifelong immunity and concern exists that immunity to the vector might limit effectiveness of a re-vaccination strategy. Consequently, alternative vaccine platforms, including peptide-based vaccines, or combinatorial vaccines using these protective antigenic sites may provide an approach for better more effective vaccines that induce long-lasting immunity against EBOV.
RESULTS:
Longitudinal analysis of antibody binding kinetics of post-EBOV infection serum to EBOV proteins
Human sera collected daily from a critically ill patient with EVD during acute illness and intermittently during convalescence were evaluated, with all days being relative to the day (D) of symptom onset (Barnes et al., 2017). The patient had been medically-evacuated from Sierra Leone to the United States and treated at the NIH Clinical Center on a randomized controlled clinical trial comparing an investigational immunotherapy plus standard of care to standard of care alone; in this case the patient had randomized to receive standard of care treatment alone. The viral RNA levels, clinical symptom scores, EBOV-IgG/IgM antibodies, GP binding antibodies and neutralization titers during acute illness are shown in supplementary table 1. We performed quantitative and qualitative SPR analyses for several dilutions of polyclonal serum (Fig. S1a; shown for GP) using recombinant full-length EBOV/Makona proteins except for partial L polymerase. Uninfected control serum did not display antibody binding. Antibody binding titers to most EBOV proteins gradually increased from D7 to D12, followed by an inflection point on D13, defined by a pronounced increase in titers against all EBOV proteins was observed (Fig. 1A–H). This increase coincided with an end to viral replication in blood, as determined by strand-specific quantitative reverse transcription polymerase chain reaction testing(Kash et al., 2017), and a rapid decline in the clinical sequential organ failure assessment (SOFA) score, a validated predictor of death in critically ill patients with severe infection (Fig. S2 and Table S1). Overall, VP40 and GP induced the highest antibody binding titers, peaking around days 66/361 and 19, respectively. Serum antibodies against other EBOV proteins peaked between day 19–28 and then declined after day 66, but remained consistent against NP, VP35 and VP40 (Fig. 1 A–C).
Figure 1: SPR based analysis of human serum following EBOV-infection with purified EBOV proteins.

Serial dilutions of serum samples collected at different time points from the EBOV survivor were analyzed for antibody binding to purified proteins from EBOV/Makona strain by SPR. (A-H) Total antibody binding is represented in SPR resonance units (RU) in black for binding to NP (A), VP35 (B). VP40 (B), GP (B). sGP (E), VP30 (F), VP24 (G) and L polymerase (H). Total antibody binding show is calculated RU for an undiluted serum sample. (A-H) Polyclonal antibody affinity maturation to EBOV proteins following EBOV infection in survivor was determined by SPR. Binding affinity of serially diluted post-infection serum to EBOV proteins was measured and is plotted in blue for each of the proteins as mentioned above for total antibody binding. Antibody off-rate constants that describe the fraction of antibody-antigen complexes decaying per second were determined directly from the serum sample interaction with EBOV proteins using SPR in the dissociation phase as described in Materials and Methods. All SPR experiments were performed twice and the researchers performing the assay were blinded to sample identity. The variation for each sample in duplicate SPR runs was <5%. The data shown is average value of two experimental runs. The maximum resonance units (Max RU) data shown was the calculated RU signal for the undiluted serum sample. (I - P) Antibody isotype of EBOV/Makona protein binding antibodies following EBOV infection. The isotype composition of serum antibodies bound to different proteins of EBOV/Makona isolate as measured in SPR. The resonance units for each anti-Makona protein antibody isotype (IgM in black, IgG in green, and IgA in red) was divided by the total resonance units for all antibody isotypes combined to calculate the percentage of each antibody isotype. for individual serum sample.
Technically, since antibodies are bivalent, the proper term for their binding to multivalent antigens like viruses is avidity, but here we use the term affinity throughout since we measured primarily monovalent interactions(Khurana et al., 2019). To determine the antibody affinity maturation over time against different EBOV proteins following virus infection, the dissociation kinetics (off-rate constants) of antigen-antibody complexes that are independent of antibody concentration were used as a surrogate for overall average affinity of polyclonal antibody against EBOV proteins using SPR(Khurana et al., 2016; Khurana et al., 2019; Khurana et al., 2011b). Furthermore, to ascertain that the antibody kinetics measured under optimized SPR conditions represent primarily the monovalent interactions between the antibody-antigen complex, IgG was purified from the serum and used to prepare Fab molecules and evaluated for binding to GP in the SPR. The antigen-antibody binding off-rates of the IgG and Fab interaction with Makona GP were very similar when adjusted for molecular weight of the bound IgG and Fab molecules. (Fig. S1b).
At D7, off-rates of polyclonal antibodies bound to EBOV proteins were fast (between 0.1 to 1 per sec), indicating weak antibody affinity early during illness, that matured thereafter although differentially for various EBOV proteins (Fig. 1 A–H). Anti-GP antibodies demonstrated weak affinity maturation through D12 (0.136 per sec), however, an inflection point on D13 was observed defined by a 4-fold increase in antibody affinity in a single day (0.037 per sec). A gradual increase in anti-GP antibody affinity through D31 (0.00713 per sec) was then followed by a remarkable 50-fold increase by D361 (0.00037 per sec) (Fig. 1D). We observed no GP sequence change during acute illness (serum samples) or convalescences (semen samples) in this EVD survivor (Barnes et al., 2017). The antibody binding and affinity to GP receptor binding domain (GP-RBD) follow the similar antibody kinetics to native GP (Fig. S1c). Anti-soluble (s)GP antibodies showed moderate affinity maturation without an inflection point and an off-rate of 0.0052 per sec by D361 (Fig. 1E). Anti-VP40 antibody affinity maturation demonstrated a 10-fold increase from D31 to D361 (0.0161 on D31 to 0.00109 on D361 per sec) (Fig. 1C). Binding antibodies against NP, VP35 and VP24 affinity matured slowly over time to reach off-rates of 0.01 per sec by D361 (Fig.1A, B, and G). Minimal affinity maturation was observed for anti-L and anti-VP30 antibodies with off-rate around 0.1/sec by D361 (Fig. 1F–H). The inflection point observed on day 13 for the predominant increase in anti-GP antibody affinity was followed by a rapid decline in clinical SOFA score from day 14 onwards (Fig. S2 I–P). The clinical scores and viral load decreased before an increase in neutralizing titers was measured in the PRNT80 assay, indicating that neutralizing antibodies may not be of key importance to control EBOV infection or that sensitivity of the PRNT80 assay is low in determining the functional immune response that curtails viral infection (Table S1).
Class-switching of binding antibodies to EBOV proteins after EBOV infection
Isotype analysis of binding antibodies was performed by SPR across EBOV proteins (Fig. 1 I–P) and demonstrated the presence of IgM, IgA, and IgG in post-infection sera. The absence of GP antibody binding for uninfected control human sample excluded the possibility that IgM binding may be due to polyreactive natural antibodies (i.e. sticky antibodies not induced by EBOV) in SPR (Fig. S1)(Khurana et al., 2016). On D7, the majority of the EBOV protein binding antibodies were of IgM isotype, which class switched gradually with increasing contribution from IgA and IgG isotypes over time apart from VP24 and L (Fig. 1 I–P). After D31, IgA binding antibodies declined against most EBOV proteins while IgG antibodies increased ~60% against GP and VP40, ~33% against NP, and <15% against VP35, VP30, VP24 and L proteins by D361. Further IgG subclass analysis of antibodies revealed predominance of IgG2 and IgG3 at early time points against NP, VP35, VP40, and VP24 with increased contribution by IgG4 by D66 and a predominance of IgG1 by D361 (Fig. S3).
Evolution of whole genome antigenic fingerprint generated following EBOV infection
The polyclonal antibody epitope repertoire of post-EBOV infection IgM, IgG and IgA antibodies in longitudinal sera on days 7, 13, 19, 31, 110 and 361 post-symptom onsets was analyzed by GFPDL containing sequences ranging from 50–1000 bp long from the complete EBOV/Makona genome with >107.7 unique phage clones (Fig. S4a). The EBOV-GFPDL displayed linear and conformational epitopes with random distribution of size and sequence of inserts that spanned the entire EBOV genome (Fig. S4a)(Khurana et al., 2016). The EBOV-GFPDL adsorbed >90% of EBOV-GP specific antibodies in the post-infection polyclonal human sera (Fig. S4b) supporting the use of the EBOV GFPDL for repertoire analyses of human sera (Fuentes et al., 2018; Khurana et al., 2016). Serum specimens were evaluated to delineate the IgM, IgG, and IgA epitope specificities across EBOV proteins (Table 1 and Fig. 2). An uninfected (EBOV-negative) control human sera bound very few phages. The number of bound phages was highest for IgM antibodies at early time points (D7 to D31) and remained consistent through D361 (Table 1). IgG specific phage titers increased over time reaching IgM antibody levels by D110. However, IgA specific phage titers peaked at D31 and were ~20 to 100-fold lower than phage titers for IgM antibodies.
Table 1:
Distribution of phage clones after affinity selection on post-EBOV infection sera
| D7 | D13 | D19 | D31 | D110 | D361 | |
|---|---|---|---|---|---|---|
| IgM | 9.90E+03 | 2.18E+04 | 1.77E+04 | 2.51E+04 | 2.36E+04 | 2.05E+04 |
| IgG | 1.00E+02 | 4.20E+03 | 2.40E+03 | 3.93E+03 | 1.20E+04 | 2.28E+04 |
| IgA | 1.10E+02 | 7.40E+02 | 1.60E+02 | 1.35E+03 | 2.00E+02 | 4.20E+02 |
Number of IgM, IgG and IgA bound phage clones selected using whole genome EBOV GFPDL on polyclonal sera from various days 7 (D7), 13 (D13), 19 (D19), 31 (D31), 110 (D110) and 361 (D361) following symptom onset in severely ill EVD survivor.
Figure 2: IgM, IgG and IgA antibody repertoires elicited in severely ill survivor after EBOV infection.
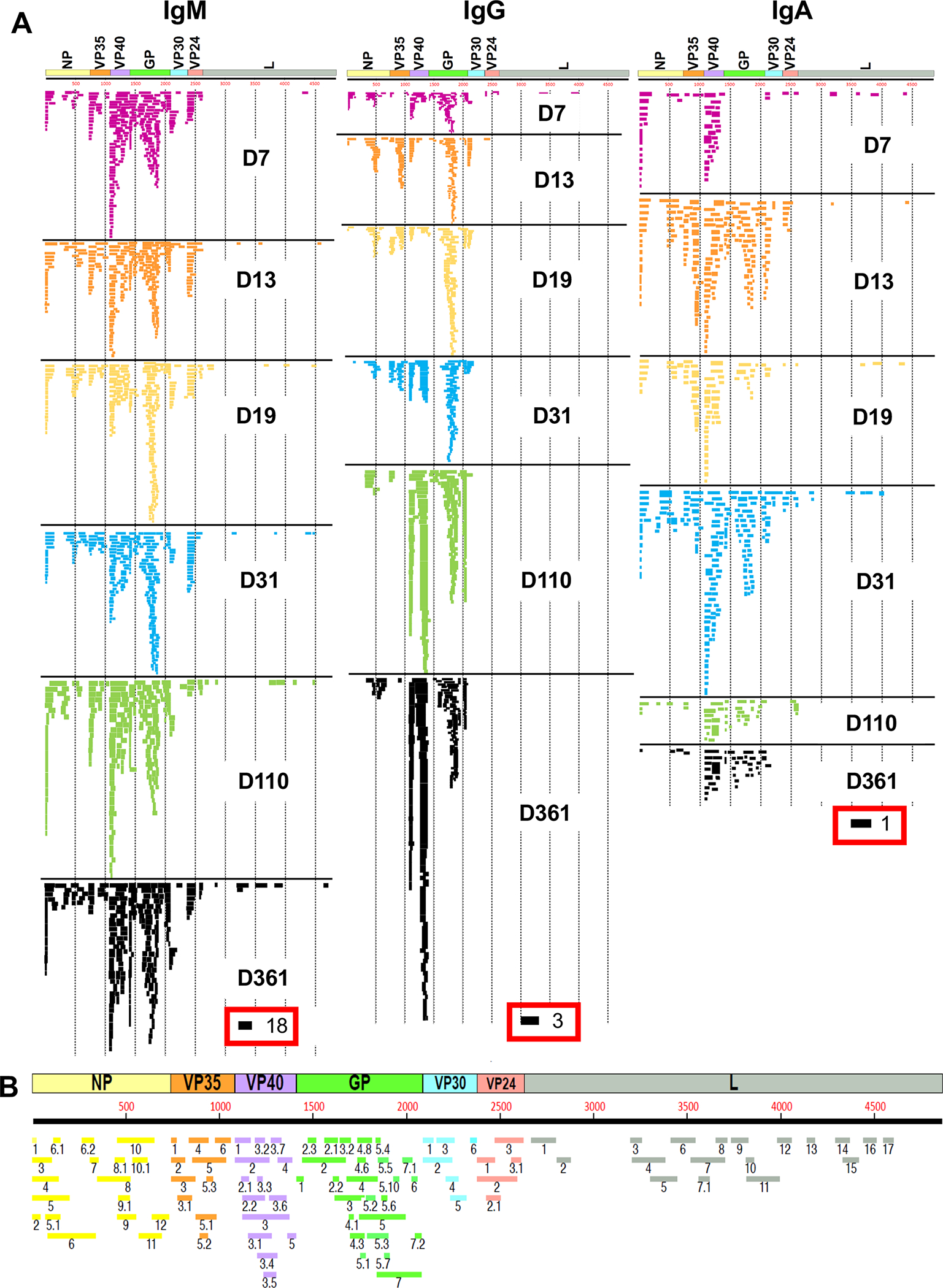
(A) IgM, IgG and IgA antibody epitope repertoire recognized in the EBOV infected sera at different days post-onset of symptoms ((D7, D13, D19, D31, D110 and D361) and their alignment to the whole proteome of EBOV showing different proteins (NP, VP35, VP40, GP, VP30, VP24 and L). Graphical distribution of representative clones with a frequency of ≥2, obtained after affinity selection, are shown. The horizontal position and the length of the bars indicate the peptide sequence displayed on the selected phage clone to its homologous sequence in the EBOV proteome on alignment. The thickness of each bar represents the frequency of repetitively isolated phage, with the scale shown below the alignment. Scale value for IgM, IgG and IgA is shown enclosed in a red box beneath the respective alignments. The GFPDL affinity selection data was performed in duplicate (two independent experiments by researcher in the lab, who was blinded to sample identity), and similar number of phage clones and epitope repertoire was observed in both phage display analysis. (B) Elucidation of antibody epitope profile against the EBOV proteome following EBOV infection. Antigenic sites within the EBOV proteins recognized by serum antibodies following EBOV infection (based on data presented in Fig. 1A). The amino acid designation is based on the EBOV protein sequence encoded by the complete EBOV/Makona genome. The antigenic regions/sites discovered in this study using the post-infection antibodies are depicted below the EBOV proteome schematic and are color coded. Epitopes of each protein are numbered in a sequential fashion indicated in black and the epitopes are color coded according to the protein color code in the proteome map.
EBOV sequences expressed by phages bound by post-infection IgM antibodies showed a diverse epitope repertoire distribution, displaying small and large sequences spanning the entire EBOV proteome, apart from L (Fig. 2A and Table S1). D7 serum IgM antibodies recognized antigenic sites in N-terminal of NP and VP35, multiple sites within VP40 and GP, and few sites within VP30 and VP24 (Figs. S5–6). By D31 IgM epitope profile evolved with additional binding to sites in C-terminus of VP35 and marginal decline in antibody binding to sites in N-terminal half of GP. By D361 the IgM repertoire evolved further with additional epitopes recognized in NP, diverse immunodominant profile in VP40 and GP, and a decline in VP24 clones (Fig. S5–6 and Table S2) in this EVD survivor.
The IgG antibody repertoire was limited at D7 with few recognized epitopes at the N- and C-terminus of NP, VP40 and GP. By D13, more IgG recognized C-terminus of NP, VP35, VP40 and GP, that evolved to predominant GP binding antibodies on days 19 and 31, with contribution from antibodies binding to additional sites focused to two regions each in VP35 and VP40 (Fig. 2A). The IgG profile further matured by days 110 and 361 with antibody binding primarily to two immunodominant regions in VP40 and several sites in GP. IgG response to VP30 was focused to the N-terminal site with few phage clones binding at early time points (D7-D19) that declined following recovery. IgG response to VP35 increased transiently at D31 but declined by D110 with minimal phage reactivity observed at D361. IgG binding phages that mapped to N- and C-terminal sites in VP40 at D7 evolved over time to a high titer anti-VP40 IgG repertoire by days 110 and 361. IgG response to GP at days 7 and 31 was focused to epitopes within the glycan cap and mucin like domain (MLD) of GP1, with limited binding to the fusion peptide and GP2. By D110 in addition to GP1 sites, the GP IgG repertoire evolved to preferentially recognize the fusion peptide and C-terminus of GP2 that predominated at D361 (Fig. 2A). Additional antibodies recognized small epitopes in the N-terminus, between the receptor binding region (RBR) and within the glycan cap domain of GP. While several antigenic sites were previously identified following recombinant vesicular stomatitis (rVSV)-EBOV GP vaccination(Khurana et al., 2016), EBOV infection induced a more diverse anti-GP antibody response across both GP1 and GP2 (Table S2; marked with asterisk).
EBOV infection generated a diverse antibody response across the EBOV proteome defined by multiple antigenic regions (Fig. 2B). Antigenic regions of 21 to 254 amino acid residues were defined based on antibody recognition by at least 4% of phage clones obtained after affinity selection on IgM/IgG/IgA antibodies with at least one serum sample at any time point. The frequency of phages expressing these antigenic sites selected by serum samples for each of the EBOV proteins are shown in Fig. S5–6 and Table S2. Most of these GFPDL identified antigenic sites were exposed on surface of the EBOV protein structures (Fig. S7–8).
Evolution of post-EBOV infection antibody binding to diverse antigenic sites in GP
To follow up on antigenic sites in GP identified using GFPDL analysis, peptides representing most of the unique antigenic sites up to 70 amino acid residues long were chemically synthesized and evaluated for antibody binding with longitudinal samples in SPR (Fig. 3). The antibody kinetics of post-infection samples to most GP peptides evolved with similar trends, however the absolute total binding antibodies against different antigenic sites varied at different time points. The measured serum sample reactivity against each peptide in SPR is possibly an aggregate sum of different antibodies recognizing overlapping epitopes in multiple antigenic sites (as defined by the GFPDL in Fig. 2) contained within that peptide sequence. The inflection point observed on D13 with intact GP with sudden increase in binding antibodies (Fig 1D) was also seen for binding antibodies against most GP peptides apart from GP 326–403 (Figs. 3H and 3K). There was a second inflection point on D25 defined by pronounced increase in antibody binding for most peptides but not for some of the antigenic sites located in the glycan cap and mucin-like domain of GP1 [GP 282–305 (Fig. 3F), GP 328–368 (Fig. 3G) and GP 424–447 (Fig. 3J)]. The increase in binding antibodies to most GP peptides corresponded with the decline in clinical SOFA scores for this patient (Fig. S9). Binding antibodies against most antigenic sites within GP peaked on D66 and D110 with highest reactivity to C-terminal of GP2 encompassing HR2-TM region (GP 617–645; 2372 RU) and C-terminus of GP1 (GP 436–491; 2348 RU) followed by peptides in the MLD and glycan cap region (GP 329–368; 689 RU, GP 372–420; 1061 RU and GP 326–403; 781 RU) and in the GP2 fusion peptide (GP 520–547; 756 RU). The antibody decay/decline after D66 was faster for antibodies targeting most sites, while the antibodies to C-terminal of GP1, fusion peptides and some GP2 antigenic sites at day 361 post-onset were at least 50% of their peak titers (Fig. 3L–Q).
Figure 3: Analysis of post-EBOV infection human serum binding to GFPDL identified GP antigenic site peptides by SPR.
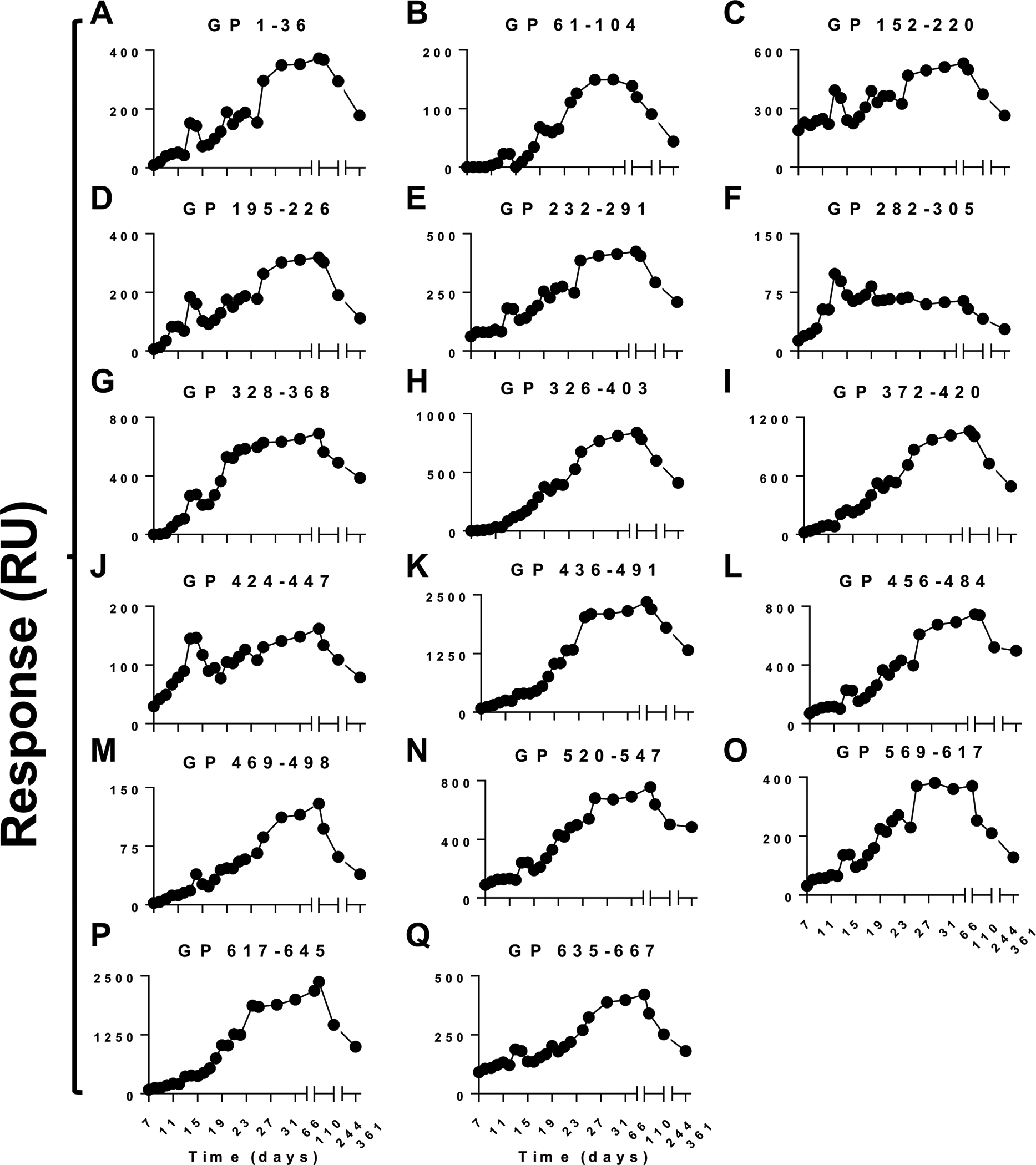
10-fold dilution of serum samples at each time point collected from EBOV survivor were analyzed for total binding to chemically synthesized peptides containing the antigenic sites identified by GFPDL (Fig 2) in SPR. (A-Q) Total antibody binding of each serum sample at different time point against the peptide is represented in SPR resonance units. The data shown is average value of two experimental SPR runs. The variation for each sample in duplicate SPR runs was <6%.
Binding, cross-reactivity & neutralization potential of EBOV-GP antigenic sites: Rabbit Immunization studies
Since EBOV-GP is target for vaccine and therapeutic development against EVD, we evaluated the contribution of these GFPDL identified GP antigenic sites as an immunogen to generate EBOV neutralizing antibodies. Rabbits were immunized with the 15-individual KLH-conjugated synthetic peptides representing most of the antigenic sites up to 70 amino acid residues long. Immunization of rabbits with selected EBOV antigenic site peptides generated strong binding antibodies against GP from the Makona and Mayinga EBOV isolates (Fig. 4A) with minimal or no binding to SUDV GP, except antibodies raised against peptide GP 617–645, likely due to high (79%) sequence conservation with SUDV GP in this region. (Table S3).
Figure 4: GP binding and EBOV neutralization by serum antibodies generated following rabbit immunization with GFPDL selected GP antigenic site peptides.

(A) 10-fold dilution of serum samples obtained from rabbits immunized thrice with KLH conjugated antigenic site peptides were analyzed for total binding to GP from different EBOV isolates (EBOV/Makona; blue, EBOV/Mayinga; black, and SUDV; red) in SPR. Total antibody binding is represented in SPR resonance units. The data shown is mean value and standard deviations of two experimental SPR runs. (B) Virus neutralization titers were measured against EBOV/Mayinga (blue bars), EBOV/Kikwit (green bars) and SUDV (black bars) by PsVN assay and authentic EBOV/Makona (red bars) in classical BSL4 based PRNT. The average end-point titers (50% neutralization titers; NT50) of rabbit sera from assay run in triplicate that demonstrated virus neutralization against either of the EBOV strains are shown. None of the post-immunization rabbit sera neutralized pseudovirus containing VSV-envelope, as specificity control. Pre-vaccination rabbit sera also did not neutralize any of the VSV particle preparations bearing different filovirus GPs tested in PSVN assay at the lowest serum dilution (10-fold). (C) The EBOV-GP structure (PDB Id −3CSY) protein used for crystallography encompasses amino acid residues 33–189, 214–278, 299–310 and 502–599 of the mature 676 a.a. GP sequence with the membrane anchor shown by the transmembrane domains (TM domain) shown at the base. (D) Structural representation of neutralizing/protective antigenic sites in EBOV-GP identified using GFPDL on the surface structures of a complete EBOV-GP model and solved EBOV-GP (PDB Id –PDC #6DZL for EBOV/Makona) structure wherever available. Sites that induced neutralizing antibodies (IV.1; GP 282–305, V.1; GP 343–368, V.7; GP 469–498, V.9; GP 520–547, and VI; GP 617–645) are depicted as a front view, and the antigenic sites in a monomer (chain A) are color coded according to Figure 2. The transmembrane domains with membrane anchor base is designated by grey bar for all structures.
To evaluate the neutralization activity of the rabbit anti-GP peptide immune sera, a pseudovirion neutralization (PsVN) assay was performed against EBOV/Mayinga. Five antigenic peptides generated neutralizing antibodies including GP 282–305, GP 343–368, GP 469–498, GP 520–547, and GP 617–645 (Fig. 4B). Three peptides (GP 469–498, GP 520–547, and GP 617–645) also neutralized EBOV/Kikwit, while only the GP2 peptide (site VI; GP 617–645) generated antibodies that neutralized SUDV in the PsVN assay. In PsVN assay without complement, neutralization titers were on average 2-fold lower than in the presence of complement (5% Guinea Pig Complement) shown in figure 4B. Two of these peptides, one from the carboxy terminus of GP1 (V.7; GP 469–498) and another in the C-terminus of GP2-HR2 domain (VI; GP 617–645), generated strong neutralizing titers against wild type EBOV/Makona in the conventional BSL4-based plaque reduction neutralization test (PRNT) with end-point titers of 640 and 320, respectively (Fig. 4B).
These rabbit studies confirmed that GFPDL identified antigenic GP peptides are immunogenic as they can elicit antibodies that bind GP from EBOV isolates. Importantly, five of these peptides including conserved antigenic sites in C-termini of GP1 and GP2 induced neutralizing antibodies. Spatial structure of these neutralizing antigenic sites on the EBOV GP crystal structure of the EBOV/Makona-GP (PDB Id #6DZL)(Murin et al., 2018); and the model of complete EBOV GP monomer are shown in Figure 4C–D. Based on the surface representation, these five targets of neutralizing antibodies discovered in this study are exposed on the native EBOV GP structures.
EBOV-GP protective antigenic sites: Mice immunization and challenge studies
To further understand the importance of these neutralizing antigenic sites in providing protection against EBOV, mouse EBOV challenge studies were performed using the 5 antigenic site peptides that generated EBOV neutralizing antibodies in rabbits. Female C57Bl/6 mice (N = 10 per group) were immunized on days 0 and 29 intra-muscularly, with 20 micrograms of the five KLH-conjugated peptides (Fig. 5A). Additionally, one group of mice was vaccinated with a combination of the KLH-peptides (at 4 microgram each). Control groups of mice were injected with 20 micrograms of KLH only (negative control), or with an EBOV GP – Venezuelan Equine Encephalitis Virus (VEEV) replicon particle-based vaccine (VRP) expressing full length EBOV GP (positive control) (Fig. 5A).
Figure 5: GP antigenic site peptide immunization and EBOV challenge study in mice.
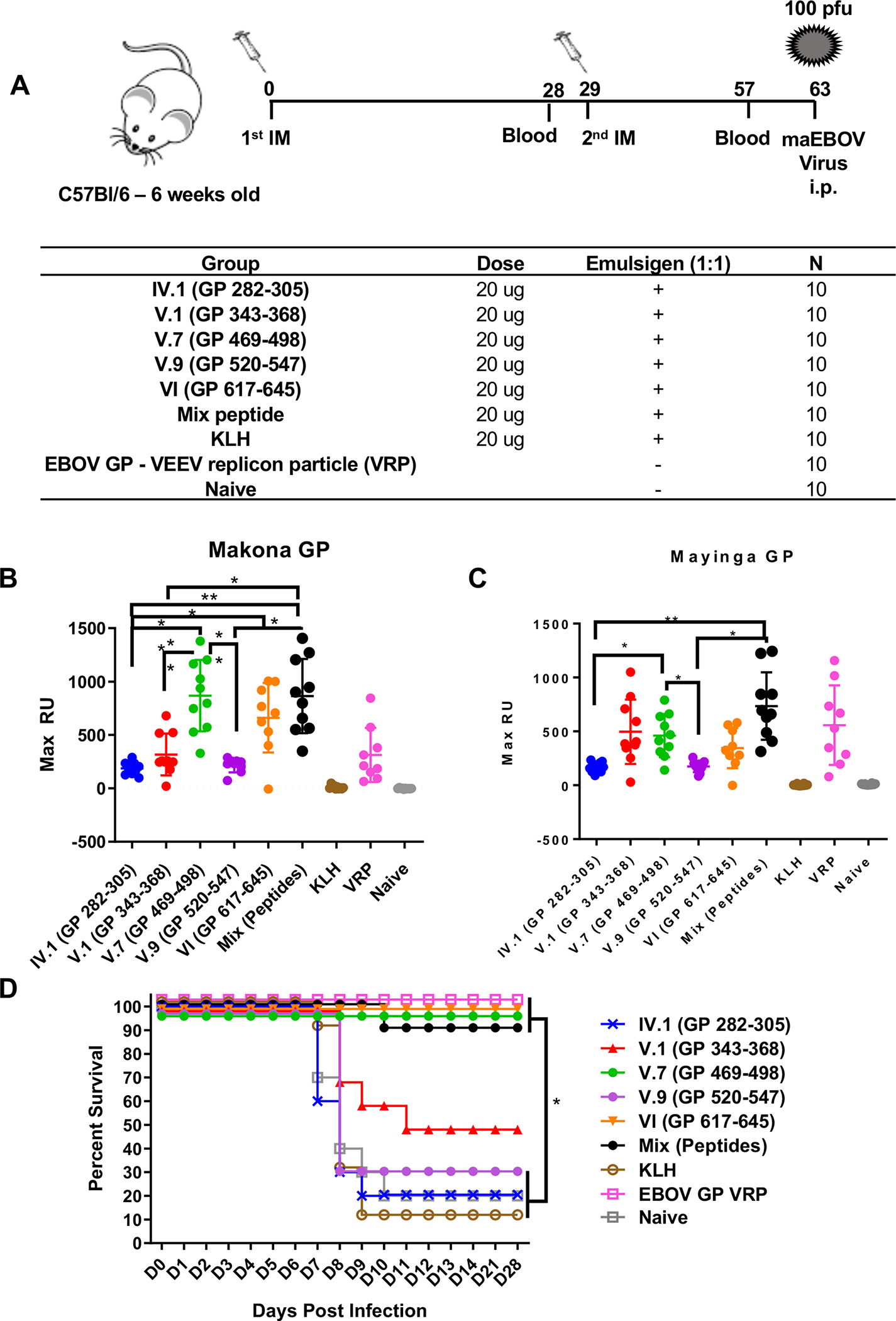
(A) Schematic representation of mouse immunization and challenge schedule. C57Bl/6 mice (N = 10 per group) were immunized intramuscularly with 20 μg of GP 282–305, GP 343–368, GP 469–498, GP 520–547, and GP 617–645 of KLH-conjugated peptides mixed with Emulsigen adjuvant, EBOV-GP VEEV replicon particles (Positive control) or with KLH alone or PBS as negative controls. After the second immunization, blood was collected on day 57 (28 days after second vaccination), and sera were analyzed for antibody binding to either GP from EBOV/Makona (B) or EBOV/Mayinga (C) in SPR. The mean values with standard deviations for each group are shown. (D) Vaccinated mice were challenged intraperitoneally with 100 PFU of mouse-adapted EBOV (maEBOV) on day 63. EBOV-challenged animals were observed daily for first 14 days and every week thereafter up to 28 days following maEBOV infection. Mice either succumbed to viral infection or were euthanized when found to be nonresponsive. Antigenic site peptides V.7 (GP 469–498), VI (GP 617–645), peptide mix and VRP provided statistically significant protection (p <0.05) compared with the control naïve group.
Following immunization but prior to the viral challenge, sera was collected on day 57 (28 days following second vaccine dose) to analyze the immune response against GP by SPR. The conserved antigenic sites in the C-terminal region of GP1 (V.7) and GP2 (VI) generated strong binding antibodies to native GP of both EBOV/Makona (Fig. 5B) and EBOV/Mayinga (Fig. 5C) comparable to that induced by the positive control VRP in SPR. The other antigenic site peptides generated moderate (V.1; GP 343–368) to weak (IV.1; GP 282–305, V.9; GP 520–547) GP binding antibodies. SPR based isotyping of the post-vaccination mouse serum revealed that >95% of GP binding antibodies were IgG (Fig. S10a). These findings were confirmed by GP-IgG ELISA that suggested that second (boost) vaccination had a minimal impact on the GP binding antibody titers (Fig. S10b). The GP-binding antibodies follow similar reactivity trends against both Makona and Mayinga strains for most post-2nd vaccination sera in SPR and ELISA. At day 63, the mice were challenged with 100 pfu wild type (mouse-adapted) EBOV-Zaire strain (maEBOV) and were followed for mortality (Fig. 5D) and weight loss (Fig. S10c). Vaccination with the conserved antigenic sites in C-terminus of GP1 (V.7; GP 469–498) and GP2 (VI; GP 617–645) provided complete protection (100% survival) against EBOV challenge, similar to the VRP control group and the group vaccinated with peptide mix (Fig. 5D) and protected against weight loss (Fig. S10c). The other antigenic site peptides only conferred moderate (V.1; GP 343–368) to minimal (IV.1; GP 282–305, V.9; GP 520–547) protection from mortality following maEBOV challenge. The peptide mixture containing 1/5th of each individual peptide provided 90% protection suggests that 4 μg of the relevant immunogen may be sufficient for protective immunity. The protection against lethality induced following maEBOV challenge mediated through neutralization and other mechanisms seems to correlate with the titer of GP binding antibodies induced following second vaccination. These protective antigenic sites identified in the current study are located either at the base (site V.7) or in the stalk domain of GP (site VI) close to the viral membrane (Fig. 4D). Analysis of sequence homology of GP showed that antigenic site VI is highly conserved between diverse EBOV species including SUDV (79%) and BDBV (92%) (Table S3). As a vaccine, these antigenic sites at the C-terminus of GP1 and in highly conserved GP2-HR2, conferred 100% protection in mice against lethal EBOV challenge.
DISCUSSION
To our knowledge this study represents the most comprehensive longitudinal characterization of antibody responses across the complete EBOV proteome in a critically ill patient with EVD who survived with supportive care alone without experimental therapies. Some of the differences between our study vs previous studies(Davis et al., 2019; McElroy et al., 2016; Saphire et al., 2018) are: i) our study describes a critically ill acutely infected EBOV patient who survived without any experimental treatment, while previous studies describe patients that received antibody treatment (ZMapp or convalescent plasma) at early time post-symptom onset (the antibody therapeutic treatment can have significant impact on the study conclusions); ii) our study performed comprehensive longitudinal analysis against complete EBOV proteome including antigenic epitope mapping of IgM, IgG and IgA every day following acute EBOV infection, while other studies primarily looked at GP responses and only IgG response to NP and VP40 without determining epitope specificity of these antibodies; iii) the daily longitudinal analysis performed in our study starts on day 7 post-symptom onset, prior to peak in symptoms and before the viral decline/disease resolution to identify the immune markers that correlate with protection naturally in this EVD survivor, while other studies described experimentally antibody treated patients primarily after their peak in symptoms; iv) to identify the immune signature providing protection during natural EBOV infection, the antigenic sites identified by GFPDL were evaluated as vaccine for their contribution to EBOV neutralization (Rabbit studies) and protective vaccine efficacy (mouse) EBOV in lethal challenge studies, while previous studies produced anti-GP MAbs once the EVD symptoms have been resolved in these EVD survivors, without any vaccination studies to evaluate the contribution of different GP antigenic sites in neutralization/protection. Our study is the primary study that shows the contribution of the different epitopes as a vaccine for EBOV neutralization and protection against EVD. We identified antibody parameters and immunodominant antibody responses to EBOV GP epitopes that coincided with disease resolution in this EVD survivor. The key antigenic sites in the C-terminus of GP1 and a highly conserved site in the base of GP2 induced neutralizing antibodies in rabbits and provided complete protection in mice from lethal EBOV challenge. Our findings indicate that close daily immunological characterization of the host-viral interaction can identify immune markers of protection that may help guide rational vaccine design and facilitate development and evaluation of more targeted immune-based countermeasures against EVD.
Over the course of our patient’s illness, we observed a differentially evolving diverse antibody response, in terms of antibody epitope repertoire, isotype class switch, and affinity maturation. An inflection point with rapid rise in antibody titers against all EBOV proteins was observed on illness D13, with GP antibodies displaying the greatest single-day rise in titer and affinity maturation that associated with a rapid decline in clinical SOFA score thereafter. VP40 and to a lesser extent VP30 also induced high titer and affinity antibodies that persisted through D361. Although the IgG response on D7 was limited, it evolved to focus on immunodominant sites within VP40 and GP. The kinetics of the IgA repertoire emerged to peak both in binding and diversity approximately one-month post-symptom onset and then declined. This differential antibody kinetics to various EBOV proteins suggests disparate expression and/or antigen exposure/recognition by the human immune system following EBOV infection in this EVD survivor that should be validated in a larger cohort. VP40 and GP antibodies accounted for >70% of IgG and IgA with high affinity during convalescence. The relevance of IgA titers is unclear but may be important at mucosal sites and in semen, and therefore should be investigated further in follow up studies. The presence of IgG2 isotype binding antibodies during acute and convalescent illness suggest a possible polysaccharide antigen-induced class switching to IgG2 following EBOV infection. IgG3 are potent mediators of effector functions, including antibody-dependent cellular cytotoxicity, complement activation, and neutralization. The observed differential antibody kinetics and class switching with early IgG3 response followed by a later IgG2 response may be related to antigen drive but equally likely to be driven by the severe cytokine storm during acute EBOV infection. Therefore, to understand the relevance of these antibody kinetics, it would be important to investigate antibody profile between EBOV-infected persons based on differential clinical outcome. High levels of IgG3 detected against GP, VP24 and VP40 at early time points post-EBOV infection potentially may have been associated with control or protection possibly by Fc-mediated functions as was observed against a range of intracellular bacteria and viruses (Lu et al., 2018).
A high titer, durable high affinity antibody response was observed against the most abundant EBOV protein VP40, which plays an essential role in virus particle assembly and budding, as well as suppression of host defenses and evasion/tolerance by EBOV of host immune response(Yamayoshi and Kawaoka, 2007). The anti-VP40 inflection point for both titers and affinity on D13 preceding decline of clinical symptoms on D14 onwards suggests a potential role of VP40 antibodies in protection from disease and, accordingly, provides support for the role of the VP40 protein as a potential vaccine or therapeutic target(Madara et al., 2015; Wilson et al., 2001). Recently it was observed that combination of GP with VP24 and VP40 induced stronger antibody responses and promoted protection in mice. VP24 was also shown to induce protection via cell-mediated immunity demonstrating potential for this as an additional vaccine antigen(Lehrer et al., 2019).
The GP specific antibody repertoire induced by EBOV infection in our patient showed greater diversity, antibody class switching, and affinity maturation compared to rVSV-EBOV GP prime-boost vaccination induced antibody responses(Khurana et al., 2016). EBOV infection-induced GP binding antibodies in our patient provided a durable response up to 12 months post-infection, in contrast to the rVSV-EBOV GP prime-boost vaccination that induced a short-lived response, with a decline to low antibody levels by six months post-vaccination(Khurana et al., 2016). Importantly, our patient demonstrated strong antibody binding to C-terminal sequences of GP1 and GP2 compared with rVSV-EBOV GP vaccination where the humoral response was primarily focused to the glycan cap and MLD sites perhaps due to the limited vector replication compared with productive EBOV infection that can stimulate the immune system over a prolonged period. One of the possible limitations of GFPDL-based assessments is that while the phage display is likely to detect both conformational and linear epitopes on EBOV proteins, they are unlikely to detect paratopic interactions that require post-translational modifications and rare quaternary epitopes that cross-protomers. However, in the in the current study (Fig. S4b) and a prior study with post-rVSV-Ebola GP vaccination serum polyclonal antibodies, 86–91% of anti-GP antibodies were removed by adsorption with the EBOV-GFPDL, supporting the use of the EBOV-GP GFPDL for analyses of human sera(Khurana et al., 2016).
During convalescence, viral RNA was detected in our patient’s semen on D32 (Ct=18.47), D66 (Ct=29.71) and D110 (Ct=30.07), but was below limit of detection by reverse transcription quantitative polymerase chain reaction (RT-qPCR) assay on days 244 and 361(Barnes et al., 2017). By D361 however, binding antibodies against 5 of 7 EBOV proteins consisted of >50% IgM antibodies, suggesting persistent immune system exposure to EBOV antigens despite inability to detect virus by RT-qPCR(Barnes et al., 2017). Moreover, the presence of EBOV specific IgG4 at different time points suggests persistent infection or long-term exposure to antigen following EBOV infection or may even reflect persistent class-switched IgM memory contributing to the serum. Finally, IgM antibody epitope repertoire generated following EBOV infection during acute illness was very diverse across the entire EBOV proteome and remained consistently diverse during convalescence, further suggesting persistent EBOV antigen exposure as late as D361. These findings are consistent with those of PREVAIL III, a longitudinal prospective cohort study of EVD survivors and controls, where 30% of 267 male survivors had viral RNA detected in semen on average 19 months following acute illness and 44% of those men had 2 negative tests followed by a positive test(Group et al., 2019). However, the potential presence of virus in immune privileged sites in this EVD survivor, suggests that antibody response induced following infection in EVD survivors is limited in providing protection/clearance of EBOV infection at these hidden sites (including eyes, testis etc.) even after virus has long been cleared in blood/plasma. Our findings of an IgM antibody signature during convalescence, despite undetectable virus by RT-qPCR assay, raise potential for GFPDL-based detection of viral persistence among survivors. Such a diagnostic assay could potentially guide use of experimental therapies to facilitate viral clearance and potentially mitigate sequela among survivors.
To identify immunodominant epitopes in GP we tested our patient’s polyclonal sera against peptides covering most antigenic sites. Antibody binding against these peptides showed differential evolution of kinetics indicative of a dynamic process of antibody-antigen interaction recognizing epitopes in overlapping antigenic sites following EBOV infection. Antibody titers generated against antigenic sites in C-terminus of GP1 (GP 436–491) and the HR2-TM region on GP2 (GP 617–645) far exceeded those of other peptides in this EVD survivor. Since most advanced EBOV vaccines in clinical trials employ GP as vaccine target, we investigated the contribution of GFPDL identified immunodominant antigenic sites in EBOV-GP to virus neutralization, peptides representing most antigenic sites up to 70 amino acid residues long were used for immunization of rabbits and screening in EBOV neutralization assays. Although all rabbit anti-peptide sera bound EBOV/Makona GP, antibodies against only 5 antigenic sites showed neutralization titers in PsVN assay, and only antibodies against sites in C-termini of GP1 (GP 469–498) and GP2 (GP 617–645) neutralized wild type EBOV/Makona virus in BSL4-based PRNT (Fig. 5B). The same two C-termini antigenic site peptides (GP 469–498 and GP 617–645) provided mice 100% protection against lethal EBOV challenge. Antibodies directed against EBOV-GP can inhibit viral entry and/or release (Kajihara et al., 2012). While the antibody impact on virus release has not been studied, the partial protection mediated by peptide V.1 in the absence of entry inhibiting antibodies could be explained, at least in part, by antibodies blocking virus release in the mice challenge studies. The focus on GP for protective vaccination studies is primarily because currently all EBOV vaccines or antibody-based therapeutics in advanced clinical development target EBOV-GP. Current EBOV vaccines generate suboptimal antibody response primarily focused to non-neutralizing sites in glycan cap and mucin-like domain, mostly IgM, and this IgM response is not durable in humans(Khurana et al., 2016). We performed animal studies to identify critical protective antigenic sites within GP identified by antibodies post-acute EBOV infection, that can be used to design immune-focused more effective Filovirus vaccines.
The rapid resolution of dissemination, intravascular coagulation and acute kidney injury(Chertow et al., 2016), causing an overall reduction in the clinical SOFA score, was immediately preceded by a surge in high-titer, high-affinity GP antibodies with immunodominance of epitopes at the C-terminus of GP1 and base of GP2 in this EVD survivor. These peptides induced neutralizing antibodies in mice and rabbits and protected mice against lethal EBOV challenge. Antibodies generated against the highly conserved site in base of GP2 (GP 617–645) induced cross-reactive binding against diverse filovirus strains, raising potential for a cross reactive vaccine across EBOV species. While others have focused on evaluation of the functional characteristics of antibody responses among EBOV survivors to guide monoclonal antibody-based therapeutic design(Saphire et al., 2018), to our knowledge this is the primary study to utilize a high-fidelity analysis of humoral responses daily during acute illness and during convalescence to identify immune markers of protection and guide peptide-based vaccine design. However, these findings are limited to the one EVD survivor and needs to be further expanded to analyses in larger acutely EBOV infected cohort. Carefully curated longitudinal samples with daily collection during acute phase of EBOV infection prior to disease resolution and later during convalescence from EVD survivors, who did not receive any experimental treatment are almost non-existent, and moreover face significant logistical challenges, preventing getting samples from Africa. Further investigation of humoral responses to our peptide target in a large cohort of EVD survivors with diverse HLA, and validation of efficacy of the peptide vaccine candidate in a nonhuman primate model of EBOV infection is warranted.
In summary, this longitudinal analysis study demonstrated a differential evolving antigenic fingerprint following EBOV infection across the EBOV proteome in terms of antibody epitope repertoire diversity, antibody isotype class switching, and antibody affinity maturation in a survivor whose natural host response was unaffected by any investigational treatments. Antibodies targeting several of these antigenic sites possessed EBOV neutralizing activity in vitro and showed an important role for the C-terminus of GP1 and highly conserved site in GP2 for providing protection in the lethal EBOV mice challenge studies. These observations provide a more in-depth understanding of quantitative and qualitative aspects of immune responses that are likely to be protective and which could aid development and evaluation of targeted more effective EBOV therapeutics and vaccines.
STAR METHODS:
Lead Contact and Materials Availability:
Surender.Khurana@fda.hhs.gov (Surender Khurana, PhD).
There are restrictions to the availability of the GFPDL technology and Ebola peptides described in this study due to US patent application.
Experimental Model and Subject Details
Proteins, Serum samples and Monoclonal Antibodies
Recombinant EBOV proteins of Makona-2014 strain were purchased from Sino Biologicals, MyBioSource, and IBT Bioservices Inc. All recombinant GP purified proteins used in the study were produced in mammalian cells. The gamma irradiated clinical serum samples were obtained from NIH (Table S1)(Wilkinson et al., 2017). In March 2015, a 34-year-old male health care worker was evacuated from Sierra Leone on day (D) 7 of documented EVD symptoms to the United States National Institutes of Health Clinical Research Center (ClinicalTrials.gov Identifier: NCT02363322; NIH IRB protocol #15-I-0083). We collected serum samples daily from D7 to 31 and then in the convalescent phase (all days are post symptom onset). Viral RNA in blood peaked at D8 (cycle threshold, Ct=23.21) and became undetectable at D24 using the EZ1 RT-qPCR assay8. Viral RNA in semen was detected by EZ1 RT-qPCR assay on D32 (Ct=18.47), 66 (Ct=29.71) and 110 (Ct=30.07) and was at or below limit of detection on D180, 244 and 361. Serum samples from these days tested negative.
Samples were tested in different antibody assays with approval from the U.S. Food and Drug Administration’s Research Involving Human Subjects Committee (FDA-RIHSC) under exemption protocol #15–0B. Day 110 and 361 samples were analyzed before and after gamma irradiation in both GFPDL and SPR that demonstrated identical antibody reactivity.
Rabbit Husbandry
Female naïve New Zealand white rabbits (4–6 weeks old; KBL(NZW)BR) were obtained from Charles River Labs and housed in microisolator cages and given feed and water. At FDA, all work with animals was conducted in compliance with the Animal Welfare Act and other Federal statutes and regulations relating to animals and experiments involving animals. All procedures were reviewed and approved by the U.S. FDA Institutional Animal Care and Use Committee (IACUC) under Protocol #2008–10. The animal care and use protocol meets National Institutes of Health (NIH) guidelines.
Mouse Husbandry
Female naïve C57Bl/6 mice, aged 6 to 8 weeks-old (Charles River Laboratory) housed in microisolator cages and given chow and water ad libitum were used for in vivo protection assays. All animal work involving infectious live virus was performed in the BSL-4 laboratories at USAMRIID. At USAMRIID, all work with animals was conducted in compliance with the Animal Welfare Act and other Federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 1996 edition. All procedures were reviewed and approved by the Institutional Animal Care and Use Committee at USAMRIID.
Methods Details
Clinical Illness Severity Scoring
A modified sequential organ failure assessment (SOFA) score was used to quantify clinical illness severity daily. A score of 0 to +4 was applied for each of the respiratory, neurological, cardiovascular, hepatic, coagulation, and renal systems as per the validated assessment tool with higher scores indicative of greater organ dysfunction and illness severity(Ferreira et al., 2001). The following modifications to the scoring system were applied: 1) conversion tables estimating partial pressure of oxygen in blood from peripheral oxygen saturation, and estimating fraction of inspired oxygen from oxygen flow rate and delivery method were applied and 2) the Glasgow verbal score during intubation was estimated from the Glasgow eye and motor scores using a validated linear regression prediction model(Meredith et al., 1998).
Antibody binding kinetics of post-EBOV infection human sera to recombinant EBOV proteins by Surface Plasmon Resonance (SPR)
Steady-state equilibrium binding of post-EBOV infected human polyclonal serum was monitored at 25°C using a ProteOn surface plasmon resonance (BioRad). The purified recombinant Makona proteins were captured to a Ni-NTA sensor chip with 200 resonance units (RU) in the test flow channels. The protein density on the chip was optimized such as to measure monovalent interactions independent of the antibody isotype. Serial dilutions (10-, 100-, 200-, 400- and 800-fold) of freshly prepared sera in BSA-PBST buffer (PBS pH 7.4 buffer with Tween-20 and BSA) were injected at a flow rate of 50 μl/min (120 sec contact duration) for association, and disassociation was performed over a 1200-second interval. Responses from the protein surface were corrected for the response from a mock surface and for responses from a buffer-only injection. SPR was performed with serially diluted serum of each individual time point in this study. Antibody isotype analysis for the EBOV protein bound antibodies in the polyclonal serum was performed using SPR. Total antibody binding and antibody isotype analysis were calculated with BioRad ProteOn manager software (version 3.1). All SPR experiments were performed twice and the researchers performing the assay were blinded to sample identity. In these optimized SPR conditions, the variation for each sample in duplicate SPR runs was <5%. The maximum resonance units (Max RU) data shown in the figures was the calculated RU signal for the undiluted serum sample.
Antibody off-rate constants, which describe the stability of the antigen-antibody complex, i.e. the fraction of complexes that decays per second, were determined directly from the human polyclonal sera sample interaction with recombinant purified EBOV proteins using SPR. To that end, serially diluted sera at 10-, 100-, 200-, 400- and 800-fold dilutions were analyzed to determine antibody off-rate constants, which describe the fraction of antigen-antibody complexes that decay per second in the dissociation phase, only for the sensorgrams with maximum RU in range of 10–100 RU (Fig. S1a). and calculated using the BioRad ProteOn manager software for the heterogeneous sample model.
To confirm that the intact polyclonal IgG interacts with GP via monomeric interaction under the defined SPR conditions, binding kinetics of purified IgG from D361 sera and Fab fragments were compared. To that end, 10 μg/mL or 2 μg/mL each of purified IgG and purified Fab fractions of serum sample were analyzed for binding to Makona GP under optimized conditions in SPR as described above (Fig. S1b).
Purification of IgG from serum and preparation of Fab molecules
IgG was purified from serum using Protein A chromatography per manufacturer’s instructions (Pierce/Thermofisher). Purified IgG was digested with Papain and the cleaved Fc was removed using Nab Protein A Plus Spin column kit (Thermofisher) and Fab fraction was collected as the flow-through fraction.
Gene Fragment Phage Display Library (GFPDL) construction
cDNA complementary to all EBOV gene of EBOV/Makona strain was chemically synthesized and used for cloning. A gIII display-based phage vector, fSK-9–3, was used where the desired polypeptide can be displayed on the surface of the phage as a gIII-fusion protein. Purified DNA containing each EBOV genes were digested separately with DNase I to obtain gene fragments of 50–1000 bp size range, then combined at equimolar amounts and used for GFPDL construction as described previously(Khurana et al., 2016). The phage libraries were constructed from the whole EBOV genome potentially display viral protein segments ranging in size from 15 to 350 amino acids, as fusion protein on the surface of bacteriophage (Fig. S4).
Affinity selection of EBOV GFPDL phages with polyclonal human serum
Prior to panning of GFPDL with polyclonal serum antibodies, serum components that could non-specifically interact with phage proteins were removed by incubation with UV-killed M13K07 phage-coated Petri dishes. Equal volumes of each human serum were used for GFPDL panning. GFPDL affinity selection was carried out in-solution with anti-IgM, or protein A/G (IgG), or anti-IgA specific affinity resin as previously described (Khurana et al., 2016; Khurana et al., 2009; Khurana et al., 2011b). Briefly, the individual serum was incubated with the GFPDL and the specific resin, the unbound phages were removed by PBST (PBS containing 0.1 % Tween-20) wash followed by washes with PBS. Bound phages were eluted by addition of 0.1 N Gly-HCl pH 2.2 and neutralized by adding 8 μl of 2 M Tris solution per 100 μl eluate. After panning, antibody-bound phage clones were amplified, the inserts were sequenced, and the sequences were aligned to the EBOV genome, to define the fine epitope specificity in this EVD survivors. The GFPDL affinity selection data was performed in duplicate (two independent experiments by research fellow in the lab, who was blinded to sample identity), and similar number of phage clones and epitope repertoire observed in both phage display analysis.
Binding of GP antigenic site peptides to post-EBOV infection sera by SPR
Steady-state equilibrium binding of GP antigenic site peptide with each time -point post-EBOV infection from this severely ill patient was monitored at 25°C using a ProteOn surface plasmon resonance (Bio Rad). The biotinylated GP peptides were captured to an NLC sensor chip via avidin interaction with 500 resonance units (RU) in the test flow channels. Samples of 300 μl freshly prepared sera at 10-fold dilution in BSA-PBST buffer (PBS pH 7.4 buffer with Tween-20 and BSA) were injected at a flow rate of 50 μl/min (120 sec contact duration) for association, and disassociation was performed over a 600-second interval. Responses from the protein surface were corrected for the response from a mock surface and for responses from a buffer-only injection. The maximum resonance units (Max RU) data shown in the figures was the observed RU signal for each serum sample.
Adsorption of polyclonal human post-infection sera on EBOV GFPDL phages and residual reactivity to Makona-GP
Prior to panning of GFPDL, 500 μl of 10-fold diluted serum antibodies from post-infection sample was adsorbed by incubation with UV-inactivated M13K07 phage-coated petri dishes. To ascertain the residual antibodies specificity, an ELISA was performed with wells coated with 200 ng/100 μl of recombinant Makona-GP. After blocking with PBST containing 2% milk, serial dilutions of human serum (with or without adsorption) in blocking solution were added to each well, incubated for 1 hr at RT, followed by addition of 5000-fold diluted HRP-conjugated goat anti-human IgA + IgG + IgM specific antibody and developed by 100 μl of OPD substrate solution. Absorbance was measured at 490 nm.
Peptide fragment conjugation to KLH carrier protein
The peptide conjugation to Maleimide Activated KLH was performed as described in the product manual of Imject Maleimide Activated KLH (Product 77605, Thermo Scientific). Briefly, 10 mg/mL solution of peptide and KLH were mixed at pH 6.5 for 2 hours at room temperature. The maleimide groups in the maleimide-activated KLH react with free sulfhydryls in the sulfhydryl-containing peptide to form stable thioether bonds of KLH-conjugated peptide.
Rabbit immunization Studies
Female New Zealand white rabbits were immunized thrice intra-muscularly at 21-days interval with 25 μg of KLH-conjugated peptides mixed with Emulsigen Adjuvant. Sera were collected before (pre-vaccination) and after 3rd vaccination and analyzed for binding antibodies in Surface Plasmon Resonance (SPR) and neutralization assay.
PsVN Assay
Pseudovirion neutralization (PsVN) assay was performed using different VSV hybrid viruses expressing various filovirus glycoproteins (either EBOV/Mayinga, EBOV/Kikwit or SUDV) in absence or presence of complement (5% guinea pig complement). Briefly 2.5× 104 293T cells/100 μL /well were plated in 96-well plates. The next day, 5-fold serial dilutions of serum (starting with 10-fold dilution) were made in 293T media (for 50 μL per well) and mixed with an equal volume of virus. After incubation at 37 °C for 1 h, the virus-serum mixture was added to 293T cells. Cells were incubated for 48 h, the medium aspirated, and the cells washed twice with PBS before lysis with Renilla lysis buffer while shaking for 30 min, and quantified for luciferase reporter activity by luminometer. The neutralization endpoint titer was determined by linear regression analysis and is defined as the inhibitory concentration (IC) required to effect a 50% reduction (IC50) of the luciferase signal compared with virus-control samples (infected cells in the absence of antibody/serum).
EBOV microneutralization Assay
A 2-fold series of dilutions of the rabbit sera were prepared (starting dilution of 1:5). An equal volume of authentic EBOV/Makona was added to the diluted serum samples and incubated for 1hr at 37°C. The virus was diluted to provide a MOI of 0.2 when added to the cells in triplicate. The dilutions of serum after mixing with virus were 1:10 and 2-fold thereafter. After the pre-incubation of virus and serum, this mixture was added to Vero cells and incubated for 1hr at 37°C. Following this inoculation period, the virus/serum mixture was discarded, and fresh medium added to the cells. Following a 48hr incubation at 37°C, cells were fixed in formalin and transferred to BSL2. Plates were blocked overnight at 4°C in a PBS/FBS buffer. EBOV-infected cells were detected by MAb KZ52 followed by an anti-human IgG labelled with Alexa fluor 488. Hoechst dye was added to stain nuclei. Serum dilutions that inhibit EBOV infection by 50% were determined and represent the IC50 values.
Mouse Immunization and Challenge study
6–8 weeks old C57Bl/6 mice (n = 10 per group) were immunized twice intramuscularly with 20 μg of GP 282–305, GP 343–368, GP 469–498, GP 520–547, and GP 617–645 KLH-conjugated peptides mixed with Emulsigen adjuvant, EBOV-GP VEEV replicon particle (1×106 focus-forming units, sub-cutaneous inoculation), or with KLH or PBS as a negative control. An additional group received a mixture of GP peptides 282–305, 343–368, 469–498, 520–547, and 617–645 (20 μg total). Vaccinations were performed 29 days apart. 34 days following the second vaccination, mice were infected with 100pfu of mouse-adapted EBOV-Zaire/1976 strain (maEBOV; Bray et al, 1998) by the intraperitoneal route. Mice were monitored daily for clinical symptoms and daily cage weights were recorded.
ELISA
96 well polystyrene plates were coated with 50uL of recombinant EBOV GP (amino acids 1–649, produced in HEK293 cells) at 10 μg/mL in PBS overnight at 4°C. Starting at a 1:100 dilution, serum samples were serially diluted 1:3 and applied to the EBOV GP-coated plate in 50 μL for 2 hr at ambient temperature. Serum samples were assayed in duplicate. Naïve serum samples were assayed the same as experimental samples. After three washes with PBS/0.02% Tween 20, EBOV GP-specific antibodies were detected with an anti-mouse IgG (H+L) HRP-conjugated antibody. After 1hr, plates were washed as before and ABTS was added for 30min. Absorbance was measured at 405nm. End titer was determined by averaging the absorbance values of the naïve serum samples and adding three standard deviations. The end titer is reported as the last serum dilution that was above this cutoff.
Quantification and Statistical Analyses
The statistical significances of group differences were determined using an Ordinary oneway ANOVA and Tukey’s multiple comparisons method. p-values less than 0.05 were considered significant with a 95% confidence interval. Correlations were calculated with a Pearson method and P value for correlation was calculated by two-tailed test. Specific details of each experiment, including experimental repeats, are noted in the associated figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human anti-EBOV GP mAb (KZ52) | IBT Bioservices | 0260–001 |
| HRP-conjugated goat anti-human IgA + IgG + IgM | Jackson ImmunoResearch | 109-035-064 |
| Bacterial and Virus Strains | ||
| E coli TG1 | Khurana et al, 2011b | In house |
| maEBOV | Bray et al, 1998 | In house |
| EBOV/Makona | USAMRIID | In house |
| EBOV/Makona GFPDL | This manuscript | N/A |
| M13K07 | NEB | N0315S |
| Biological Samples | ||
| EBOV post-infection samples | Chertow et al, 2016 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Zaire EBOV VP24 Protein | Sino Biologicals | 40454-V07E |
| Recombinant Zaire EBOV Minor nucleotide VP30 | My BioSource | MBS1288327 |
| Recombinant Zaire EBOV polymerase cofactor VP35 | My BioSource | MBS1308847 |
| Zaire EBOV VP40 Matrix protein | Sino Biologicals | 40446-V07E |
| Zaire EBOV RNA directed RNA polymerase L | My BioSource | MBS1414035 |
| Recombinant Zaire EBOV NP | Alpha Diagnostics | EVNP15-R-10 |
| EBOV H.sapiens-wt/GIN/2014/ Kissidougou-C15 GP | Sino Biologicals | 40442-V08H1 |
| Recombinant EBOV Soluble GP | IBT | 0565–001 |
| Recombinant Mayinga 1976 EBOV GP | Sino Biologicals | 40304-V08B1 |
| Recombinant EBOV Kikwit-95 GP | Acros Bio | ZE5-V5221 |
| Recombinant SUDV GP | Immune Tech | IT-014–007p |
| Peptides & KLH-Peptides | This manuscript | N/A |
| Protein A UltraLink™ Resin | Thermo Scientific | 53139 |
| Fab Preparation Kit | Thermo Scientific | 44985 |
| Emulsigen | MVP Adjuvants | N/A |
| Experimental Models: Cell Lines | ||
| Vero E6 cells | ATCC | CCL-81 |
| 293T cells | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Female New Zealand white rabbits | Charles River Labs | KBL(NZW)BR |
| Female C57Bl/6 mice | Charles River Labs | N/A |
HIGHLIGHTS.
Longitudinal analysis of Ebola survivor reveals long-lasting IgM/IgG/IgA epitope diversity
Despite undetectable virus, an EBOV-specific IgM suggests occult viral persistence Antibody affinity and immunodominant sites associated with EVD resolution are revealed
Sites in C-terminus of GP1 and GP2 are promising therapeutic and vaccine targets
ACKNOWLEDGEMENTS:
We thank K. Peden and H. Golding for their insightful review of the manuscript.
Funding: The antibody characterization work described in this manuscript was supported by FDA Office of Counterterrorism and Emerging Threats (OCET) - Medical Countermeasures initiative (MCMi) grant- OCET 2019–1018 and Defense Threat Reduction Agency (HDTRA1930447) funds to S. K. This work was in part supported by the intramural research programs of the National Institutes of Health, Bethesda, MD. We would like to thank Defense Threat Reduction Agency (CB4088) for supporting the containment experiments. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of Interests: The authors declare no competing interests. The GFPDL technology and Ebola peptide sequences described in this study are covered under US patent application.
Ethics Statement: The study at CBER, FDA was conducted with de-identified samples (ClinicalTrials.gov Identifier: NCT02363322; NIH IRB protocol #15-I-0083) under Research Involving Human Subjects (RIHSC) exemption #15–0B; and all assays performed fell within the permissible usages in the original consent.
REFERENCES
- Barnes KG, Kindrachuk J, Lin AE, Wohl S, Qu J, Tostenson SD, Dorman WR, Busby M, Siddle KJ, Luo CY, et al. (2017). Evidence of Ebola Virus Replication and High Concentration in Semen of a Patient During Recovery. Clin Infect Dis. 65(8), 1400–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. (1998). A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis. 178(3):651–61. [DOI] [PubMed] [Google Scholar]
- Chertow DS, Nath A, Suffredini AF, Danner RL, Reich DS, Bishop RJ, Childs RW, Arai AE, Palmore TN, Lane HC, et al. (2016). Severe Meningoencephalitis in a Case of Ebola Virus Disease: A Case Report. Ann Intern Med. 165(4), 301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J, and Enserink M (2015). CLINICAL TRIALS. Ebola vaccines face daunting path to approval. Science. 349(6254), 1272–1273. [DOI] [PubMed] [Google Scholar]
- Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M, et al. (2016). Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science. 351(6279), 1339–1342. [DOI] [PubMed] [Google Scholar]
- Crook P, Smith-Palmer A, Maguire H, McCarthy N, Kirkbride H, Court B, Kanagarajah S, Turbitt D, Ahmed S, Cosford P, et al. (2017). Lack of Secondary Transmission of Ebola Virus from Healthcare Worker to 238 Contacts, United Kingdom, December 2014. Emerg Infect Dis. 23(12), 2081–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CW, Jackson KJL, McElroy AK, Halfmann P, Huang J, Chennareddy C, Piper AE, Leung Y, Albarino CG, Crozier I, et al. (2019). Longitudinal Analysis of the Human B Cell Response to Ebola Virus Infection. Cell. 177(6), 1566–1582 e1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deen GF, Broutet N, Xu W, Knust B, Sesay FR, McDonald SLR, Ervin E, Marrinan JE, Gaillard P, Habib N, et al. (2017). Ebola RNA Persistence in Semen of Ebola Virus Disease Survivors - Final Report. N Engl J Med. 377(15), 1428–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye JM, Herbert AS, Kuehne AI, Barth JF, Muhammad MA, Zak SE, Ortiz RA, Prugar LI, and Pratt WD (2012). Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc Natl Acad Sci U S A. 109(13), 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt SA, Zehner M, Krahling V, Cohen-Dvashi H, Kreer C, Elad N, Gruell H, Ercanoglu MS, Schommers P, Gieselmann L, et al. (2019). Polyclonal and convergent antibody response to Ebola virus vaccine rVSV-ZEBOV. Nat Med. [DOI] [PubMed] [Google Scholar]
- Ferreira FL, Bota DP, Bross A, Melot C, and Vincent JL (2001). Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA. 286(14), 1754–1758. [DOI] [PubMed] [Google Scholar]
- Fuentes S, Coyle EM, Beeler J, Golding H, and Khurana S (2016). Antigenic Fingerprinting following Primary RSV Infection in Young Children Identifies Novel Antigenic Sites and Reveals Unlinked Evolution of Human Antibody Repertoires to Fusion and Attachment Glycoproteins. PLoS Pathog. 12(4), e1005554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes S, Ravichandran S, and Khurana S (2018). Antibody Repertoire of Human Polyclonal Antibodies Against Ebola Virus Glycoprotein Generated After Deoxyribonucleic Acid and Protein Vaccination of Transchromosomal Bovines. J Infect Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group PIS, Sneller MC, Reilly C, Badio M, Bishop RJ, Eghrari AO, Moses SJ, Johnson KL, Gayedyu-Dennis D, Hensley LE, et al. (2019). A Longitudinal Study of Ebola Sequelae in Liberia. N Engl J Med. 380(10), 924–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Restrepo AM, Longini IM, Egger M, Dean NE, Edmunds WJ, Camacho A, Carroll MW, Doumbia M, Draguez B, Duraffour S, et al. (2015). Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet. 386(9996), 857–866.. [DOI] [PubMed] [Google Scholar]
- Holtsberg FW, Shulenin S, Vu H, Howell KA, Patel SJ, Gunn B, Karim M, Lai JR, Frei JC, Nyakatura EK, et al. (2015). Pan-ebolavirus and pan-filovirus mouse monoclonal antibodies: protection against Ebola and Sudan viruses. J Virol. 90:266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajihara M, Marzi A, Nakayama E, Noda T, Kuroda M, Manzoor R, Matsuno K, Feldmann H, Yoshida R, Kawaoka Y, et al. (2012). Inhibition of Marburg virus budding by nonneutralizing antibodies to the envelope glycoprotein. J Virol. 86(24), 13467–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash JC, Walters KA, Kindrachuk J, Baxter D, Scherler K, Janosko KB, Adams RD, Herbert AS, James RM, Stonier SW, et al. (2017). Longitudinal peripheral blood transcriptional analysis of a patient with severe Ebola virus disease. Sci Transl Med. 9(385). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SB, Bolay F, Kieh M, Grandits G, Badio M, Ballou R, Eckes R, Feinberg M, Follmann D, Grund B, et al. (2017). Phase 2 Placebo-Controlled Trial of Two Vaccines to Prevent Ebola in Liberia. N Engl J Med. 377(15), 1438–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Chearwae W, Castellino F, Manischewitz J, King LR, Honorkiewicz A, Rock MT, Edwards KM, Del Giudice G, Rappuoli R, et al. (2010). Vaccines with MF59 adjuvant expand the antibody repertoire to target protective sites of pandemic avian H5N1 influenza virus. Sci Transl Med. 2(15), 15ra15. [DOI] [PubMed] [Google Scholar]
- Khurana S, Fuentes S, Coyle EM, Ravichandran S, Davey RT Jr., and Beigel JH (2016). Human antibody repertoire after VSV-Ebola vaccination identifies novel targets and virus-neutralizing IgM antibodies. Nat Med. 22(12), 1439–1447. [DOI] [PubMed] [Google Scholar]
- Khurana S, Hahn M, Coyle EM, King LR, Lin TL, Treanor J, Sant A, and Golding H (2019). Repeat vaccination reduces antibody affinity maturation across different influenza vaccine platforms in humans. Nat Commun. 10(1), 3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Sasono P, Fox A, Nguyen VK, Le QM, Pham QT, Nguyen TH, Nguyen TL, Horby P, and Golding H (2011a). H5N1-SeroDetect EIA and rapid test: a novel differential diagnostic assay for serodiagnosis of H5N1 infections and surveillance. J Virol. 85(23), 12455–12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Suguitan AL Jr., Rivera Y, Simmons CP, Lanzavecchia A, Sallusto F, Manischewitz J, King LR, Subbarao K, and Golding H (2009). Antigenic fingerprinting of H5N1 avian influenza using convalescent sera and monoclonal antibodies reveals potential vaccine and diagnostic targets. PLoS Med. 6(4), e1000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Verma N, Yewdell JW, Hilbert AK, Castellino F, Lattanzi M, Del Giudice G, Rappuoli R, and Golding H (2011b). MF59 adjuvant enhances diversity and affinity of antibody-mediated immune response to pandemic influenza vaccines. Sci Transl Med. 3(85), 85ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause PR (2015). Interim results from a phase 3 Ebola vaccine study in Guinea. Lancet. 386(9996), 831–833. [DOI] [PubMed] [Google Scholar]
- Krause PR, Cavaleri M, Coleman G, and Gruber MF (2015). Approaches to demonstration of Ebola virus vaccine efficacy. Lancet Infect Dis. 15(6), 627–629. [DOI] [PubMed] [Google Scholar]
- Kupferschmidt K (2019). Successful Ebola treatments promise to tame outbreak. Science. 365(6454), 628–629. [DOI] [PubMed] [Google Scholar]
- Lehrer AT, Wong TS, Lieberman MM, Johns L, Medina L, Feldmann F, Feldmann H, and Marzi A (2019). Recombinant subunit vaccines protect guinea pigs from lethal Ebola virus challenge. Vaccine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LL, Suscovich TJ, Fortune SM, and Alter G (2018). Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol. 18(1), 46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara JJ, Han Z, Ruthel G, Freedman BD, and Harty RN (2015). The multifunctional Ebola virus VP40 matrix protein is a promising therapeutic target. Future Virol. 10(5), 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi A, Engelmann F, Feldmann F, Haberthur K, Shupert WL, Brining D, Scott DP, Geisbert TW, Kawaoka Y, Katze MG, et al. (2013). Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc Natl Acad Sci U S A. 110(5), 1893–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi A, Robertson SJ, Haddock E, Feldmann F, Hanley PW, Scott DP, Strong JE, Kobinger G, Best SM, and Feldmann H (2015). EBOLA VACCINE. VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Science. 349(6249), 739–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matassov D, Marzi A, Latham T, Xu R, Ota-Setlik A, Feldmann F, Geisbert JB, Mire CE, Hamm S, Nowak B, et al. (2015). Vaccination With a Highly Attenuated Recombinant Vesicular Stomatitis Virus Vector Protects Against Challenge With a Lethal Dose of Ebola Virus. J Infect Dis. 212 Suppl 2, S443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy AK, Harmon JR, Flietstra TD, Campbell S, Mehta AK, Kraft CS, Lyon MG, Varkey JB, Ribner BS, Kratochvil CJ, et al. (2016). Kinetic Analysis of Biomarkers in a Cohort of US Patients With Ebola Virus Disease. Clin Infect Dis. 63(4), 460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith W, Rutledge R, Fakhry SM, Emery S, and Kromhout-Schiro S (1998). The conundrum of the Glasgow Coma Scale in intubated patients: a linear regression prediction of the Glasgow verbal score from the Glasgow eye and motor scores. J Trauma. 44(5), 839–844. [DOI] [PubMed] [Google Scholar]
- Milligan ID, Gibani MM, Sewell R, Clutterbuck EA, Campbell D, Plested E, Nuthall E, Voysey M, Silva-Reyes L, McElrath MJ, et al. (2016). Safety and Immunogenicity of Novel Adenovirus Type 26- and Modified Vaccinia Ankara-Vectored Ebola Vaccines: A Randomized Clinical Trial. JAMA. 315(15), 1610–1623. [DOI] [PubMed] [Google Scholar]
- Murin CD, Bruhn JF, Bornholdt ZA, Copps J, Stanfield R, and Ward AB (2018). Structural Basis of Pan-Ebolavirus Neutralization by an Antibody Targeting the Glycoprotein Fusion Loop. Cell Rep. 24(10), 2723–2732 e2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampling T, Ewer K, Bowyer G, Wright D, Imoukhuede EB, Payne R, Hartnell F, Gibani M, Bliss C, Minhinnick A, et al. (2015). A Monovalent Chimpanzee Adenovirus Ebola Vaccine - Preliminary Report. N Engl J Med.. [Google Scholar]
- Ravichandran S, Hahn M, Belaunzaran-Zamudio PF, Ramos-Castaneda J, Najera-Cancino G, Caballero-Sosa S, Navarro-Fuentes KR, Ruiz-Palacios G, Golding H, Beigel JH, et al. (2019). Differential human antibody repertoires following Zika infection and the implications for serodiagnostics and disease outcome. Nat Commun. 10(1), 1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regules JA, Beigel JH, Paolino KM, Voell J, Castellano AR, Munoz P, Moon JE, Ruck RC, Bennett JW, Twomey PS, et al. (2015). A Recombinant Vesicular Stomatitis Virus Ebola Vaccine - Preliminary Report. N Engl J Med. DOI: 10.1056/NEJMoa1414216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphire EO, Schendel SL, Fusco ML, Gangavarapu K, Gunn BM, Wec AZ, Halfmann PJ, Brannan JM, Herbert AS, Qiu X, et al. (2018). Systematic Analysis of Monoclonal Antibodies against Ebola Virus GP Defines Features that Contribute to Protection. Cell. 174(4), 938–952 e913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DE, Page M, Mattiuzzo G, Hassall M, Dougall T, Rigsby P, Stone L, and Minor P (2017). Comparison of platform technologies for assaying antibody to Ebola virus. Vaccine. 35(9), 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JA, Bray M, Bakken R, and Hart MK (2001). Vaccine potential of Ebola virus VP24, VP30, VP35, and VP40 proteins. Virology. 286(2), 384–390. [DOI] [PubMed] [Google Scholar]
- Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, and Hart MK (2000). Epitopes involved in antibody-mediated protection from Ebola virus. Science. 287(5458), 1664–1666. [DOI] [PubMed] [Google Scholar]
- Wong G, Richardson JS, Pillet S, Patel A, Qiu X, Alimonti J, Hogan J, Zhang Y, Takada A, Feldmann H, et al. (2012). Immune parameters correlate with protection against ebola virus infection in rodents and nonhuman primates. Sci Transl Med. 4(158), 158ra146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamayoshi S, and Kawaoka Y (2007). Mapping of a region of Ebola virus VP40 that is important in the production of virus-like particles. J Infect Dis. 196 Suppl 2, S291–295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.