

Abstract
Alterations to branched-chain keto acid (BCKA) oxidation have been implicated in a wide variety of human diseases, ranging from diabetes to cancer. Although global shifts in BCKA metabolism—evident by gene transcription, metabolite profiling, and in vivo flux analyses have been documented across various pathological conditions, the underlying biochemical mechanism(s) within the mitochondrion remain largely unknown. In vitro experiments using isolated mitochondria represent a powerful biochemical tool for elucidating the role of the mitochondrion in driving disease. Such analyses have routinely been utilized across disciplines to shed valuable insight into mitochondrial-linked pathologies. That said, few studies have attempted to model in vitro BCKA oxidation in isolated organelles. The impetus for the present study stemmed from the knowledge that complete oxidation of each of the three BCKAs involves a reaction dependent upon bicarbonate and ATP, both of which are not typically included in respiration experiments. Based on this, it was hypothesized that the inclusion of exogenous bicarbonate and stimulation of respiration using physiological shifts in ATP-free energy, rather than excess ADP, would allow for maximal BCKA-supported respiratory flux in isolated mitochondria. This hypothesis was confirmed in mitochondria from several mouse tissues, including heart, liver and skeletal muscle. What follows is a thorough characterization and validation of a novel biochemical tool for investigating BCKA metabolism in isolated mitochondria.
Introduction
Leucine, isoleucine, and valine, collectively referred to as ‘branched-chain amino acids’ (BCAAs), are essential amino acids with vital roles in cellular metabolism. One such role involves energy production through the oxidation of the BCAA carbon skeletons within the mitochondrion [1]. These metabolites include ketoisocaproate (KIC), ketomethylvalerate (KMV), and ketoisovalerate (KIV), known generally as the ‘branched-chain keto acids’ (BCKAs). The importance of BCKA oxidation in the periphery is perhaps best demonstrated by the pathophysiological consequences linked to maple syrup urine disease (MSUD), caused by inborn errors within the BCKA dehydrogenase complex (BCKDH) genes [2]. In addition to MSUD, interest in BCKA metabolism has dramatically expanded in recent years with the discovery that limitations in BCKA catabolism underlie several other human diseases. To date, dysfunctional BCKA oxidation has been demonstrated in diabetes [3,4], hepatocellular carcinoma [5], heart failure [6], and ischemia–reperfusion injury [7]. Apart from BCKDH inborn errors (i.e. MSUD), the precise biochemical lesion responsible for lower overall BCKA catabolic flux in these diseases remains unclear.
Elucidating the mechanism(s) driving stress-induced shifts in BCKA metabolism requires experimental methodologies capable of isolating the BCKA pathway and interrogating individual enzyme flux. One such tool involves in vitro analysis of isolated mitochondria. Indeed, numerous examples exist in which bioenergetic conclusions deduced from in vitro mitochondrial profiling have been paramount in the discovery, support or validation of various mitochondrial pathologies [8–12]. That said, only a handful of studies have attempted to model BCKA oxidation in isolated organelles [6,7,13–15]. In every case, evidence of respiratory control (i.e. a change in mitochondrial respiration in response to free ADP) specifically mediated by exogenous BCKAs has not been demonstrated, possibly due to insufficient optimization of the experimental conditions.
Following transamination, all BCKAs undergo decarboxylation via mitochondrial matrix-localized BCKDH. Decarboxylation by BCKDH represents the first committed step of BCKA oxidation and produces BCKA-specific short-chain acyl-CoAs (KIC > isovaleryl-CoA: KMV > methylbutyryl-CoA:KIV > isobutyryl-CoA). Each acyl-CoA is then processed via a series of keto acid-specific steps ultimately leading to the production of either acetyl-CoA and/or succinyl-CoA (Figure 1). Of note, BCKA catabolism is distinct from pyruvate or fatty acyl-CoA metabolism in that complete oxidation of each BCKA requires both bicarbonate and ATP, neither of which are traditionally present in isolated mitochondria experiments. The natural sequitur to this issue formed the guiding hypothesis herein: inclusion of bicarbonate and ATP-free energy would allow for appreciable BCKA-supported oxygen consumption. To test this hypothesis, a recently described mitochondrial diagnostics workflow was employed which leverages the creatine kinase (CK) energetic clamp to allow respiration measurements across a physiological span of ATP/ADP ratios (i.e. ATP-free energy of hydrolysis; ΔGATP) [16]. Using this methodology in the presence of exogenous bicarbonate, several novel aspects of mitochondrial BCKA metabolism were unveiled, highlighted by tissue-specific (e.g. heart, liver, skeletal muscle) differences in pathway responsivity to ATP-free energy. Specifically, in the absence of post-translation control (i.e. BCKDH phosphorylation) [17], BCKA oxidation in muscle mitochondria was found to be directly responsive to ATP-free energy, rather than the adenylate pool per se. The methodological framework outlined and validated herein serves as a blueprint for a powerful new biochemical tool for investigating BCKA oxidation. The assays outlined will likely be of use across multiple disciplines to identify and localize stress-induced lesions within the BCKA oxidation pathway.
Figure 1. Overview of BCKA oxidation.
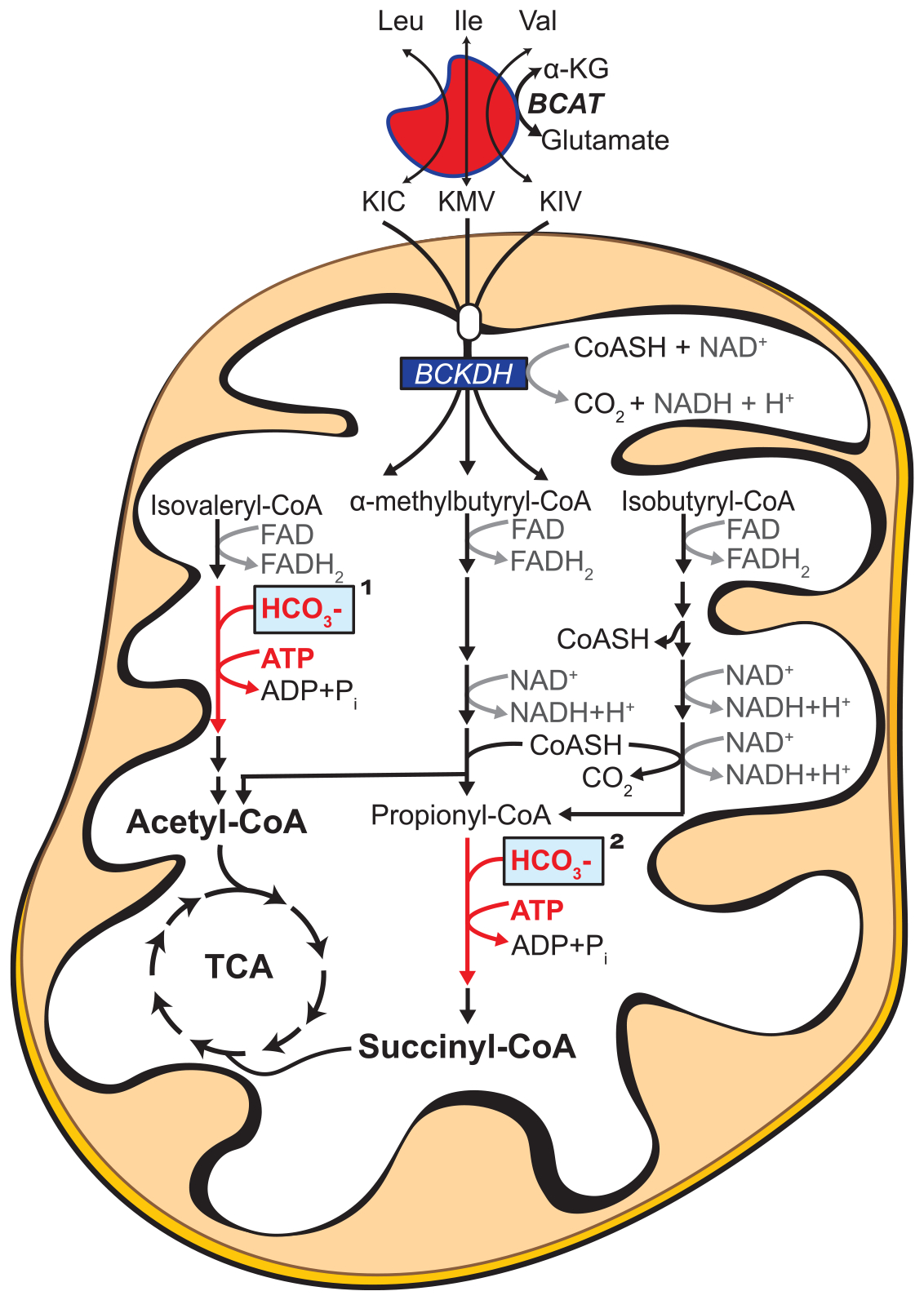
BCAA/BCKA catabolic pathway resulting in TCA cycle intermediates. Complete oxidation of all three BCKAs in isolated mitochondria requires a carboxylase reaction. This reaction is either carried out by methylcrotonyl-CoA carboxylase (KIC; reaction indicated by ‘1’ in the figure) or propionyl-CoA carboxylase (KMV and KIV; reaction indicated by ‘2’ in the figure). Both enzymes are biotin-dependent enzymes that use bicarbonate to catalyze carboxylation in an ATP-dependent reaction. Unlike biotin, which is bound to the matrix enzymes, both ATP and bicarbonate are depleted from the mitochondrial matrix during the isolation procedure.
Materials and methods
Rodent models
All animal studies were approved by the East Carolina University Institutional Animal Care and Use Committee. All experiments involving laboratory mice were carried out at the East Carolina Diabetes and Obesity Institute. C57BL/6J male mice (8 weeks old) were purchased from Jackson Laboratory (strain #000664). Unless otherwise stated, all experiments were completed using 12- to 16-week-old mice following a 2-h fast. All mice were housed in a temperature (22°C) and light-controlled (12 h light/12 h dark) room and given free access to food and water. Unless otherwise stated, mice were anesthetized with isoflurane, and the following tissues were removed for mitochondrial isolation and functional analysis: skeletal muscle (gastrocnemius, plantaris, soleus, and quadriceps), heart (complete left and right ventricles), and liver (whole). For experiments designed to inhibit the branched-chain keto acid dehydrogenase kinase (BCKDK) in vivo, C57BL/6J mice were fasted for 4 h then injected with either 3-chloro-benzo[b]thiophene-2-carboxylic acid (BT2; 40 mg/kg) [18,19] or vehicle control (DMSO). Inhibition of BCKDH kinase via BT2 has been shown to activate BCKA oxidation in vivo by promoting dephosphorylation of BCKDH [18,19]. Average injection volumes per mouse were ~25 μl. Tissues were harvested 1-h post-injection and heart mitochondria were isolated as described below.
Chemicals and reagents
Unless otherwise stated, all chemicals were purchased from Millipore-Sigma, including dipotassium phosphocreatine (Cat#237911), KIC (Cat#K0629), KMV (Cat#198978), and KIV (Cat#198978). Potassium NADP+ (Cat#AK671068) and ATP (Cat#AK54737) were purchased from Ark-Pharm. Potassium pyruvate was purchased from Combi-Blocks (QA-1116). Amplex UltraRed (AUR) and tetramethylrhodamine methyl ester (TMRM) were purchased from Thermo Fisher Scientific. The following experimental buffers were used for all assays: Buffer A—50 mM MOPS supplemented with KCl (100 mM), EGTA (1 mM), and MgSO4 (5 mM), pH 7.1; Buffer B—Buffer A, supplemented with 0.2% BSA, pH 7.1; Buffer C—105 mM MES potassium salt supplemented with KCl (30 mM), EGTA (1 mM), KH2PO4 (10 mM), MgCl2 (5 mM), and BSA (0.25%), pH 7.2. Buffer D—HEPES (20 mM), KCl (100 mM), KH2PO4 (2.5 mM), MgCl2 (2.5 mM), glycerol (1%), pH 8.0.
Mitochondrial isolation
Differential centrifugation was used to isolate mitochondria from heart, liver, and skeletal muscle. Buffers A and B were utilized in all isolations. Heart, liver, and skeletal muscle were dissected and placed in ice-cold Buffer B. Tissues were then minced, resuspended in 10 ml of Buffer B and homogenized for ~10–15 passes via a Teflon pestle and borosilicate glass vessel. The homogenate was transferred to a 15 ml tube, the volume was brought to 12 ml, and samples were centrifuged at 600×g for 10 min at 4°C. The supernatants were poured through two layers of gauze and spun at 10 000×g for 10 min at 4°C. Mitochondrial pellets were washed in 1.4 ml of Buffer A and spun at 10 000×g for 10 min at 4°C. The resulting pellets were resuspended in Buffer A. Protein content was determined using the Pierce BCA assay.
Mitochondrial respiratory control using the creatine kinase clamp
High-resolution O2 consumption measurements were conducted using the Oroboros Oxygraph-2K (Oroboros Instruments). All experiments were carried out at 37°C in a 2 ml reaction volume with continuous stirring. Steady-state oxygen consumption rates (JO2) were determined within individual experiments using a modified version of the CK energetic clamp technique [16,20,21]. In this assay, the free energy of ATP hydrolysis (ΔG′ATP, depicted throughout the manuscript as ΔGATP) can be calculated based on known amounts of creatine (Cr), phosphocreatine (PCR) and ATP in combination with excess amounts of CK and the equilibrium constant for the CK reaction (i.e. KCK). Calculation of ΔG′ATP was performed according to the following formula:
where ΔG′°ATP is the standard apparent transformed Gibbs energy (under a specified pH, ionic strength, free magnesium, and pressure), R is the gas constant (8.3145 J/kmol), and T is the temperature in Kelvin (310.15). Given that experiments were performed via sequential additions of PCr, both the ΔG′°ATP and K′CK were determined at each titration step based on the changes in buffer ionic strength and free magnesium, as previously described [16]. Although not accounted for in the present study, the presence of mitochondrial CK within the inner mitochondrial membrane of heart mitochondria may have influenced the accuracy of the calculated ΔGATP [22]. Buffer for all assays was Buffer C, supplemented with ATP (5 mM), Cr (5 mM), PCR (1 mM), and CK (20 U/ml). For all experiments, PCR final concentrations of 1, 6, 9, 15, 21, and 27 mM corresponded to ΔGATP of −12.94, −14.08, −14.34, −14.65, −14.84, and −14.99 kcal/mol. For reference, ΔGATP of −12.94 and −14.84 kcal/mol correspond to ATP/ADP ratios of ~29.5 and ~554.6, respectively. Experiments designed to test the impact of bicarbonate utilized potassium bicarbonate (2 mM). For each experiment, isolated mitochondria were added to assay buffer, followed by the addition of respiratory substrates. A complete list of all substrate combinations and inhibitors used is provided in Supplementary Table S1. Following substrate additions, sequential additions of PCR were performed to manipulate ATP-free energy. For experiments designed to test the impact of BCKAs on JO2, a 1:1:1 mixture of BCKAs (1 mM each of KIC, KMV, KIV) was compared with NaCl (3 mM). The use of NaCl for control was required as all BCKAs were provided as sodium salts. Either BCKAs or NaCl was added to mitochondria prior to the addition of each respiratory substrate.
Bicarbonate titrations
Experiments were performed in liver (0.1 mg/ml) and heart (0.05 mg/ml) mitochondria. For the hexokinase–ADP clamp, Buffer C was supplemented with Cr (5 mM), ADP (0.5 mM), glucose (5 mM), and hexokinase (HK; 1 U/ml). For the CK clamp, Buffer C was supplemented with Cr (5 mM), ATP (5 mM), PCR (21 mM), and CK (20 U/ml). The inclusion of PCR at 21 mM (i.e. ΔGATP of −14.84 kcal/mol) for the bicarbonate titrations was to ensure that ATP-free energy was not rate-limiting throughout the CK clamp experiment. Following the addition of buffer and mitochondria to the chambers, KIV was added and allowed to equilibrate. Potassium bicarbonate was then titrated in using sequential additions, resulting in concentrations of 0.1 mM, 0.2, 0.5, 1.0, 2.0, and 4.0 mM. Stable flux values were recorded at baseline and after each bicarbonate titration.
Mitochondrial membrane potential (ΔΨ) and NAD(P)H/NAD(P)+ redox
Fluorescent determination of ΔΨ and NAD(P)H/NAD(P)+ was carried out simultaneously via a QuantaMaster Spectrofluorometer (QM-400; Horiba Scientific) as previously described [16] with slight modifications. Determination of ΔΨ via TMRM was done by recording the fluorescence ratio of the following excitation/emission parameters [Ex:Em, (572:590)/(551:590)]. NAD(P)H excitation/emission parameters were 350:450. All experiments were carried out at 37°C in a 0.2 ml reaction volume with continuous stirring. Buffer for all assays was Buffer C, supplemented with Cr (5 mM), PCR (1 mM), ATP (5 mM), CK (20 U/ml), TMRM (0.2 μM), and where indicated, bicarbonate (2 mM). For each assay, isolated mitochondria were added to the assay buffer, followed by the addition of respiratory substrates and then sequential PCR additions. At the end of the protocol, potassium cyanide (10 mM) was added to induce a state of 100% reduction within the NAD(P)H/NAD (P)+ couple, followed by alamethicin (0.03 mg/ml) to eliminate ΔΨ polarization and NAD(P)H reduction. The fluorescence (Ex:Em, 350:450) signal, as well as the 572/551 ratio described above, recorded in the presence of mitochondria plus alamethicin was subtracted from all other values during the assay (Supplementary Figure S1). Final ΔΨ values are presented as the alamethicin-mediated 572/551 ratio subtracted from the observed 572/551 ratio. Final NAD(P)H/NAD(P)+ redox data are presented as the fluorescence intensity (350:450) recorded minus that recorded in the presence of alamethicin.
H2O2 emission
Mitochondrial H2O2 emission was measured fluorometrically via the AUR/horseradish peroxidase (HRP) detection system (Ex:Em, 565:600) as described previously [16], with slight modifications. Fluorescence was monitored via a QuantaMaster Spectrofluorometer (Horiba Scientific). For each experiment, resorufin fluorescence was converted to picomoles H2O2 via an H2O2 standard curve generated under identical substrate conditions as employed for each protocol. All experiments were carried out at 37°C in a 0.2 ml reaction volume. Buffer for all assays was Buffer C, supplemented with Cr (5 mM), PCR (1 mM), CK (20 U/ml), AUR (10 μM), HRP (1 U/ml), superoxide dismutase (20 U/ml), and auranofin (AF; 0.1 μM). Auranofin at 0.1 μM has been demonstrated to be specific for thioredoxin reductase inhibition [23,24]. At higher concentrations (~10 μM), auranofin inhibits glutathione peroxidase [24]. To begin, isolated mitochondria (0.1 mg/ml) were added to assay buffer, followed by the addition of respiratory substrates and then sequential PCR additions to 6 and 21 mM. A specific inhibitor of respiratory complex III (antimycin A; 5 μM) was added at the conclusion of each protocol to serve as a positive control for JH2O2 emission. The percentage of electron leak was calculated by dividing the rate of H2O2 production by the corresponding O2 consumption rate measured under similar conditions and expressed as a percentage (% Leak = JH2O2/JO2). Of note, the JH2O2 rates used in the calculation were generated in the presence of auranofin; however, the corresponding JO2 assays did not contain auranofin, as the inhibitor does not impact respiratory conductance [16].
JNADH and JNADPH production
All assays were performed using alamethicin-permeabilized (0.03 mg/ml) mitochondria suspended in Buffer A [25]. Rates of NADH and NADPH production were determined as described previously [16]. Buffer for the assays was Buffer C, supplemented with rotenone (0.005 mM) and NAD+ (2 mM) or NADP+ (2 mM). For experiments designed to assess JNADH from the pyruvate dehydrogenase complex (PDH), the α-ketoglutarate dehydrogenase complex (AKGDH), and the BCKDH, coenzyme A (0.1 mM) and thiamine pyrophosphate (0.3 mM) were included. Assay buffer (200 μl) was loaded into individual wells of a 96-well plate, followed by permeabilized mitochondria. The assay was initiated with the addition of enzymatic substrates. In the assay, NADH and NADPH were determined via autofluorescence (Ex:Em, 340:450). The following substrates were tested in parallel for each assay: pyruvate (5 mM) for PDH, AKG (10 mM) for AKGDH, KMV (5 mM) for BCKDH, glutamate (10 mM) for glutamate dehydrogenase (GDH), malate (5 mM) for NAD-linked MDH and NADP-linked malic enzyme (ME), isocitrate (5 mM) for NAD-linked IDH3 and NADP-linked IDH2. Data are presented as change in fluorescence (RFU [relative fluorescence unit]) per unit time normalized to the mitochondrial protein amount added. For experiments involving in vivo treatment with BT2, both BCKDH and PDH activity were determined under the same buffer conditions as described above; however, fluorescence measurements were made using a QuantaMaster Spectrofluorometer (QM-400; Horiba Scientific) with Ex:Em of 350:450.
Hydroxyacyl-CoA dehydrogenase (HADHA) activity
The assay was performed using alamethicin-permeabilized (0.03 mg/ml) mitochondria suspended in Buffer A. Buffer for the assay was Buffer D, supplemented with rotenone (0.005 mM) and NADH (0.2 mM). Assay buffer (200 μl) was loaded into individual wells of a 96-well plate, followed by permeabilized mitochondria. The assay was initiated with the addition of acetoacetyl-CoA (0.2 mM). The activity of hydroxyacyl-CoA dehydrogenase was determined via tracking the degradation in the NADH autofluorescence (Ex:Em, 340:450) signal upon acetoacetyl-CoA addition. Data are presented as change in fluorescence (RFU) per unit time normalized to the mitochondrial protein amount added.
Quantification and statistical analysis
Data are presented as mean ± SEM. Statistical analysis was performed using JMP® Pro 13 (Version 13.2). Normality of all data was assessed using a Shapiro–Wilk W test. Normally distributed data were compared using either a paired (all in vitro experiments) or unpaired (BT2 treatment experiments) Student’s t-test. Non-normal data were compared using the Wilcoxon Signed Rank test. The level of significance was set at P < 0.05. Mitochondrial diagnostics results (Figure 5) were analyzed using a data analysis toolkit developed in Python. The toolkit was written using Python 2.7 and is published on the Python Package Index at https://pypi.org/project/kfwx/ (MacOS X and Windows). Figures were generated using GraphPad Prism (Version 8.0).
Figure 5. Comparison of BCKA oxidation in skeletal muscle versus liver mitochondria.
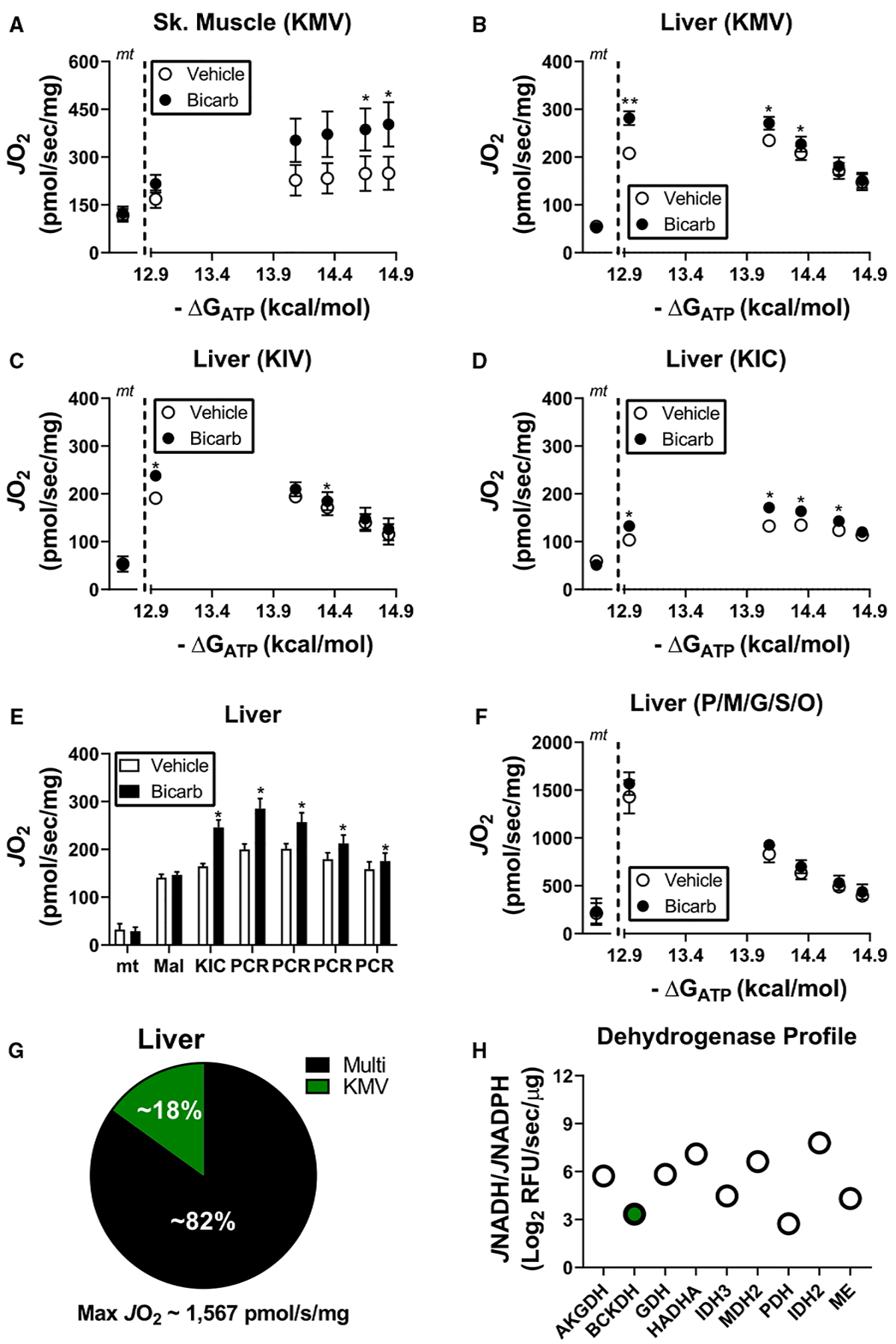
(A–F) Experiments were performed in the presence and absence of bicarbonate (2 mM). (A) Relationship between mitochondrial oxygen consumption and ATP-free energy in skeletal muscle mitochondria energized with KMV. Rates to the left of the dotted line correspond to mitochondria alone (‘mt’). (B–D) Relationship between mitochondrial oxygen consumption and ATP-free energy in liver mitochondria energized with (B) KMV, (C) KIV, or (D) KIC. Rates to the left of the dotted line correspond to mitochondria alone (‘mt’). (E) JO2 upon energization of liver mitochondria with malate and KIC across a span of ATP-free energies. Sequential PCRs in the figure correspond to final concentrations of 6, 9, 15, and 21 mM. (F) Relationship between mitochondrial oxygen consumption and ATP-free energy in liver mitochondria energized with P/M/G/S/O. Rates to the left of the dotted line correspond to mitochondria alone (‘mt’). (G) Comparison of the average maximal KMV-supported respiration in liver mitochondria versus the average maximal respiratory flux supported by P/M/G/S/O. (H) Dehydrogenase profiling in permeabilized liver mitochondria. BCKDH is highlighted in the green filled circle. Data are expressed as Log2 JNADH/JNADPH. Data are mean ± SEM, n = 4/group, *P < 0.05.
Results
BCKA oxidation in heart mitochondria requires both ATP and bicarbonate
To determine whether bicarbonate and ATP are required for BCKA oxidation, bioenergetic experiments were designed to assess mitochondrial respiration supported by saturating KMV (2 mM) in the presence and absence of potassium bicarbonate and increasing ATP-free energies of hydrolysis. Manipulation of ΔGATP was made possible through the application of a recently described mitochondrial diagnostics assay platform [16] that leverages a modified version of the CK energetic clamp [20,21] to assess respiratory control across a physiological span of ATP-free energies (−12.94 to −14.84 kcal/mol). To begin, isolated heart mitochondria were energized with KMV in the absence and presence of 2 mM bicarbonate. Despite no effect of bicarbonate at the initial ΔGATP, sequential elevations in ΔGATP (i.e. stepwise increase in ATP/ADP) revealed a dose-dependent increase in respiratory flux only in mitochondria exposed to bicarbonate (Figure 1A,B). To further investigate the influence of bicarbonate on BCKA flux, KIV-supported respiration was assessed in response to bicarbonate titration. These experiments were performed using either the CK clamp set to the highest ΔGATP (−14.84 kcal/mol) or a hexokinase–ADP clamp which maintains free ADP at 0.5 mM without allowing for appreciable ATP [26]. While the addition of bicarbonate dose-dependently increased KIV-supported JO2 with the CK clamp, this effect was completely absent from the hexokinase-clamp condition (Figure 1C,D), presumably due to insufficient ATP-free energy. Importantly, the addition of bicarbonate up to 2 mM did not impact buffer pH.
It should be noted that the CK clamp merely maintains extramitochondrial ΔGATP. As such, equilibration of ATP-free energy between the buffer and mitochondrial matrix requires a functioning phosphorylation system. To determine if matrix ATP-free energy is required for maximal BCKA flux, additional experiments were carried out in which buffer ΔGATP was set to −14.84 kcal/mol (i.e. 21 mM PCR) and KMV-supported respiration was assessed under conditions in which ATP synthesis was compromised with either oligomycin or FCCP. Although oligomycin blocks proton conductance (i.e. respiratory flux), FCCP uncouples electron transport from ATP synthesis, thus allowing for maximal KMV-dependent flux to be determined in the absence of matrix ATP-free energy. Under control conditions, KMV once again induced a robust increase in JO2 (Figure 1E). Remarkably, KMV-supported respiration was nearly eliminated in mitochondria pre-incubated with FCCP, identical with that seen with oligomycin (Figure 1E). Taken together, these experiments provide biochemical evidence that BCKA flux in cardiac mitochondria requires bicarbonate and is responsive to matrix ATP-free energy.
BCKA catabolism in cardiac mitochondria minimally contributes to total respiratory capacity
Upon establishing the CK clamp with either 1 mM (−12.94 kcal/mol) or 21 mM (−14.84 kcal/mol) PCR, creatine, and ATP are largely held constant (~5 mM), while free ADP levels drop by an order of magnitude (Figure 1F). In traditional respirometry with saturating carbon substrates, a 10-fold drop in ADP would be expected to lower respiratory flux [16,20,21]. This was indeed the case when isolated heart mitochondria were energized with saturating carbon substrates (pyruvate/malate/glutamate/succinate/octanoyl-carnitine; P/M/G/S/O; 5/2/5/5/0.2 mM) and assessed at ATP-free energies of −12.94 kcal/mol versus −14.84 kcal/mol (Figure 1G; ‘Multi’). Absolute JO2 was subsequently normalized upon the addition of FCCP (Figure 1G; ‘FCCP’), confirming that the respiratory differences between ΔGATP conditions were caused by a decrease in demand for ATP resynthesis. The traditional relationship between ATP-free energy and JO2 is reversed upon energization with KMV alone (Figure 1G; ‘KMV’). These data imply that respiratory control in the presence of KMV resides entirely with reducing equivalent supply. Thus, maximal respiration supported by KMV must be below the threshold at which respiration rate begins to respond linearly to free [ADP]. Indeed, when maximal respiration between P/M/G/S/O and KMV-energized mitochondria were compared, KMV-supported flux represented only ~3% of P/M/G/S/O flux (Figure 1H). Consistent with this, KMV alone, as compared with Pyr/M, was unable to a promote measurable reduction in the mitochondrial NAD+ pool or generate a membrane potential (ΔΨ) above that observed in mitochondria devoid of exogenous carbon (Supplementary Figure S2A–C). Increasing the mitochondrial concentration from 0.1 mg/ml to 1 mg/ml was required to observe KMV-supported NAD reduction (Supplementary Figure S2D,E). Using heart mitochondria at 1 mg/ml, KMV-supported increases in NAD(P)H/NAD(P)+ redox state were unaffected by ΔGATP (Supplementary Figure S2F), suggesting that the increase in KMV-supported JO2 in response to ΔGATP is not caused by accelerated KMV uptake across the inner mitochondrial membrane and/or BCKDH activation.
Dehydrogenase profiling in permeabilized heart mitochondria corroborated the respirometry findings as BCKDH flux was found to represent ~2% of maximal NAD-linked capacity (Figure 1I). Respirometry analysis and dehydrogenase profiling agree with a recent report demonstrating that BCKA contribution to the TCA cycle is <1% in mouse heart [19]. Together, these data indicate that the BCKA oxidation pathway minimally contributes to maximal catabolic flux in mouse heart.
The inclusion of bicarbonate and ATP permit full BCKA oxidation to either acetyl-CoA or succinyl-CoA
Having established that bicarbonate and ATP are required for KMV oxidation, the impact of bicarbonate was next assessed in cardiac mitochondria energized with P/M/G/S/O or the BCKA corresponding to valine (KIV) or leucine (KIC) metabolism. Although bicarbonate did not impact respiratory sensitivity in the presence of P/M/G/S/O (Figure 2A), KIV-supported JO2 was dose-dependently increased across the ΔGATP range (Figure 2B), similar to KMV-supported respiration (Figure 1B). Interestingly, KIC only marginally increased JO2 above that of mitochondria alone and was unresponsive to ΔGATP (Figure 2C). KIC oxidation produces acetyl-CoA exclusively, raising the possibility that acetyl-CoA build up could be inhibiting KIC-supported respiration, similar to pyruvate-supported JO2 [27]. To test this hypothesis, KIC-supported JO2 was assessed in the presence of high carnitine or following pre-treatment with malate. The presence of excess carnitine provides a substrate for carnitine acetyltransferase (CrAT), thus relieving acetyl-CoA-mediated control of respiration [27,28]. Malate relieves acetyl-CoA buildup by serving as a source of oxaloacetate (acetyl-CoA condenses with oxaloacetate to form citrate). The presence of both carnitine and malate increased KIC-supported respiratory flux in a bicarbonate-dependent manner (Figure 2D). These findings confirm that in the presence of bicarbonate and ATP-free energy, in vitro metabolism of KIC is constrained through accumulation of its TCA cycle end product, acetyl-CoA.
Figure 2. BCKA oxidation in heart mitochondria requires both bicarbonate and physiological ATP-free energy of hydrolysis.
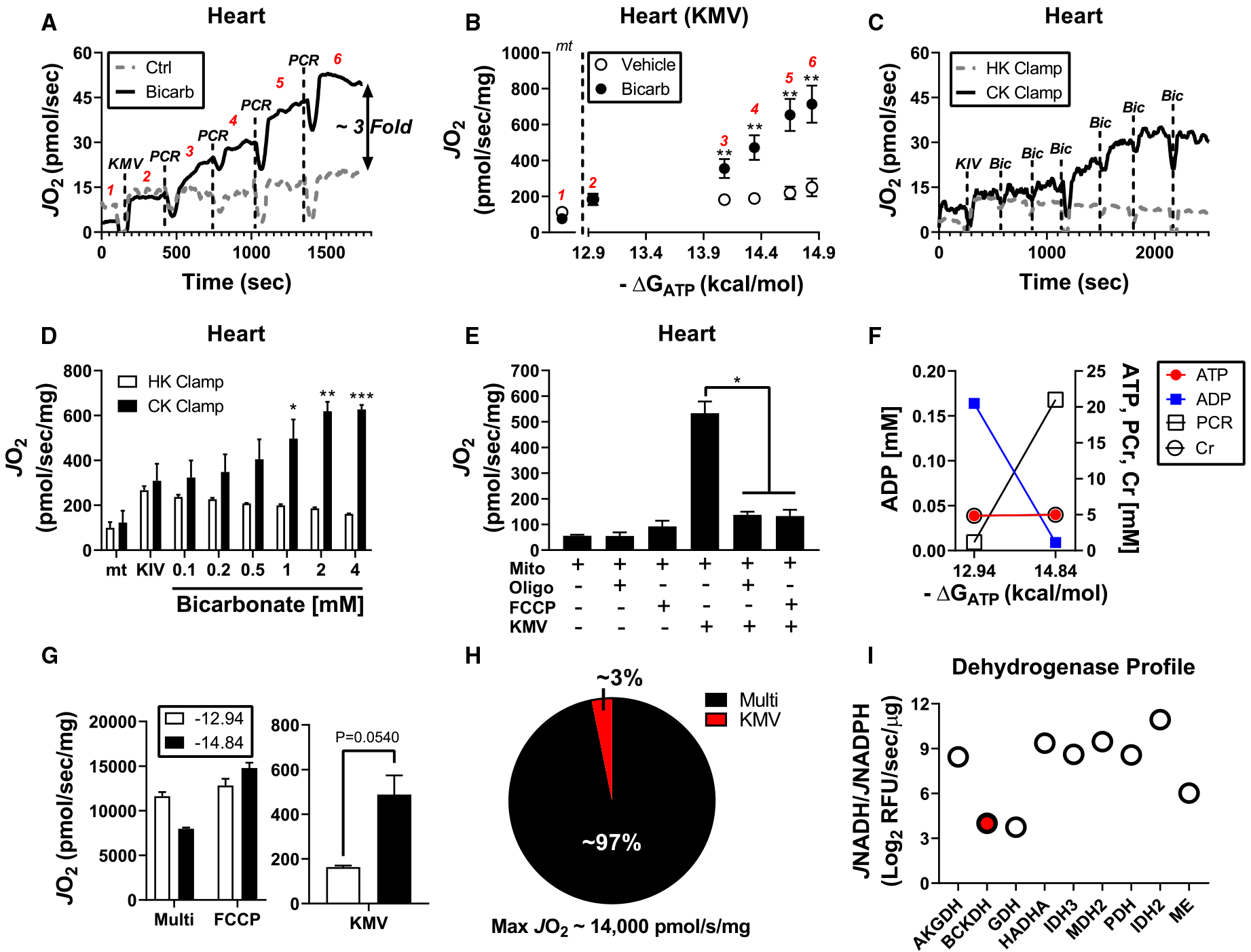
Isolated heart mitochondria were used for all experiments. (A) Representative trace of JO2 during a phosphocreatine (PCR) titration in the absence and presence of 2 mM bicarbonate. (B) Quantification of data from (A) depicting the relationship between JO2 and ΔGATP in mitochondria energized with KMV in the presence and absence of bicarbonate. Rates to the left of the dotted line correspond to mitochondria alone (‘mt’). (C) Representative trace of JO2 during a bicarbonate titration in the presence of the CK or hexokinase clamp. (D) JO2 quantification of the bicarbonate titration from the data depicted in (C). (E) Mitochondrial JO2 without exogenous carbon substrates or saturating KMV in the absence and presence of either FCCP (2 μM) or oligomycin (Oligo; 2 μM). (F) Calculated changed in ATP, ADP, PCR, and Cr at different ΔGATP (−12.94 kcal/mol vs. −14.84 kcal/mol). (G) Heart mitochondria incubated at ΔGATP of −12.94 kcal/mol versus. −14.84 kcal/mol and energized with P/M/G/S/O (5/2/5/5/0.2 mM; ‘Multi’), P/M/G/S/O plus FCCP (2 μM; ‘FCCP’) or KMV alone (‘KMV’). (H) Comparison of KMV-supported respiration versus maximal respiratory flux (G compare ‘FCCP’ to ‘KMV’ at ΔGATP of −14.84 kcal/mol). (I) Dehydrogenase profiling in permeabilized cardiac mitochondria. BCKDH is highlighted in the red filled circle. Data are expressed as Log2 JNADH/JNADPH. Data are mean ± SEM, n = 3–7/group, *P < 0.05, **P < 0.001, ***P < 0.0001.
Oxidation of KIV produces succinyl-CoA exclusively, whereas KMV oxidation can produce either acetyl-CoA or succinyl-CoA (Figure 1). To determine if the combination of bicarbonate and ATP-free energy permits complete metabolism of KMV to succinyl-CoA, KMV-supported JO2 was assessed under high ΔGATP followed by the addition of the complex I inhibitor rotenone or the succinate dehydrogenase (SDH) inhibitor malonate. Both rotenone and malonate eliminated KMV-dependent flux (Figure 2E,F). Importantly, JO2 was restored following the addition of 1 mM succinate only in the plus rotenone condition, thus confirming SDH inhibition by malonate (Figure 2F; ‘Succ’). Malonate did not impact respiration supported by KIC in heart or liver mitochondria (Supplementary Figure S3A,B). Complete inhibition of KMV-dependent flux by malonate confirms that KMV metabolism under the experimental conditions employed herein proceeds at least to the level of succinate. Metabolite exchange with buffer phosphate across the inner mitochondrial membrane could restrict dehydrogenase activity downstream of SDH.
Mitochondrial BCKA oxidation contributes to electron leak
Given that BCKDH is a known source of mitochondrial superoxide [29], experiments were designed to assess the peroxide emitting potential of the BCKA oxidation pathway. Mitochondrial H2O2 emission was determined in isolated cardiac mitochondria exposed to bicarbonate and energized with either KMV or P/M/G/S/O across a span of ΔGATP (−12.94, −14.08, −14.84 kcal/mol). Antimycin A was included at the end of the protocol as a positive control for H2O2 emission. Although absolute H2O2 emission was lower in the presence of KMV compared with P/M/G/S/O (Figure 2G), normalization to the corresponding respiration rates (i.e. JH2O2/JO2 = % electron leak) revealed percent electron leak to be 10-fold higher in KMV-energized mitochondria (Figure 2H). Interestingly, % electron leak supported by KMV decreased by ~3-fold across the ΔGATP span, suggesting that electron leak is minimal when BCKA catabolism is maximal (Figure 2G; compare ‘KMV’ mean at −12.94 vs. −14.84 kcal/mol). Potential sources of electron leak within the BCKA oxidation pathway include BCKDH, electron transfer flavoprotein (ETF), and ETF dehydrogenase (ETFDH) [30].
In vivo inhibition of the branched-chain dehydrogenase kinase does not impact maximal BCKA oxidation in heart mitochondria
Recent in vivo analysis of BCKA oxidation in mouse heart suggested that maximal BCKA flux is constrained in unstressed C57BL/6J mice via inhibitory phosphorylation of BCKDH [19]. To determine if inhibitory phosphorylation of BCKDH limits KMV-supported respiration, short-term fasted C57BL/6J mice were injected with the BCKDH kinase inhibitor BT2 [18,19] (40 mg/kg) or vehicle control. Mice were euthanized 1 h post-injection and isolated mitochondria were prepared from heart tissue (Figure 3A). Assessment of BCKDH activity in permeabilized mitochondria revealed a ~2-fold increase in JNADH in mitochondria prepared from BT2-treated mice, consistent with inhibition of BCKDH kinase in vivo (Figure 3B,C). Importantly, this increase in JNADH in response to KMV was specific to BCKDH, as no differences were observed in pyruvate dehydrogenase (PDH) flux (Figure 3C). Despite the evidence of BCKDH activation in vivo, KMV-supported respiration in heart mitochondria was completely unaffected by BT2 treatment (Figure 3D). These data imply that the phosphorylation status of BCKDH is reversible in intact isolated organelles, likely through exposure to high BCKA concentrations and/or physiological ATP-free energy [31,32]. Moreover, the increase in JO2 in response to increasing ΔGATP cannot be explained by gradual BCKDH dephosphorylation.
Figure 3. Detailed characterization of BCKA metabolism in cardiac mitochondria.
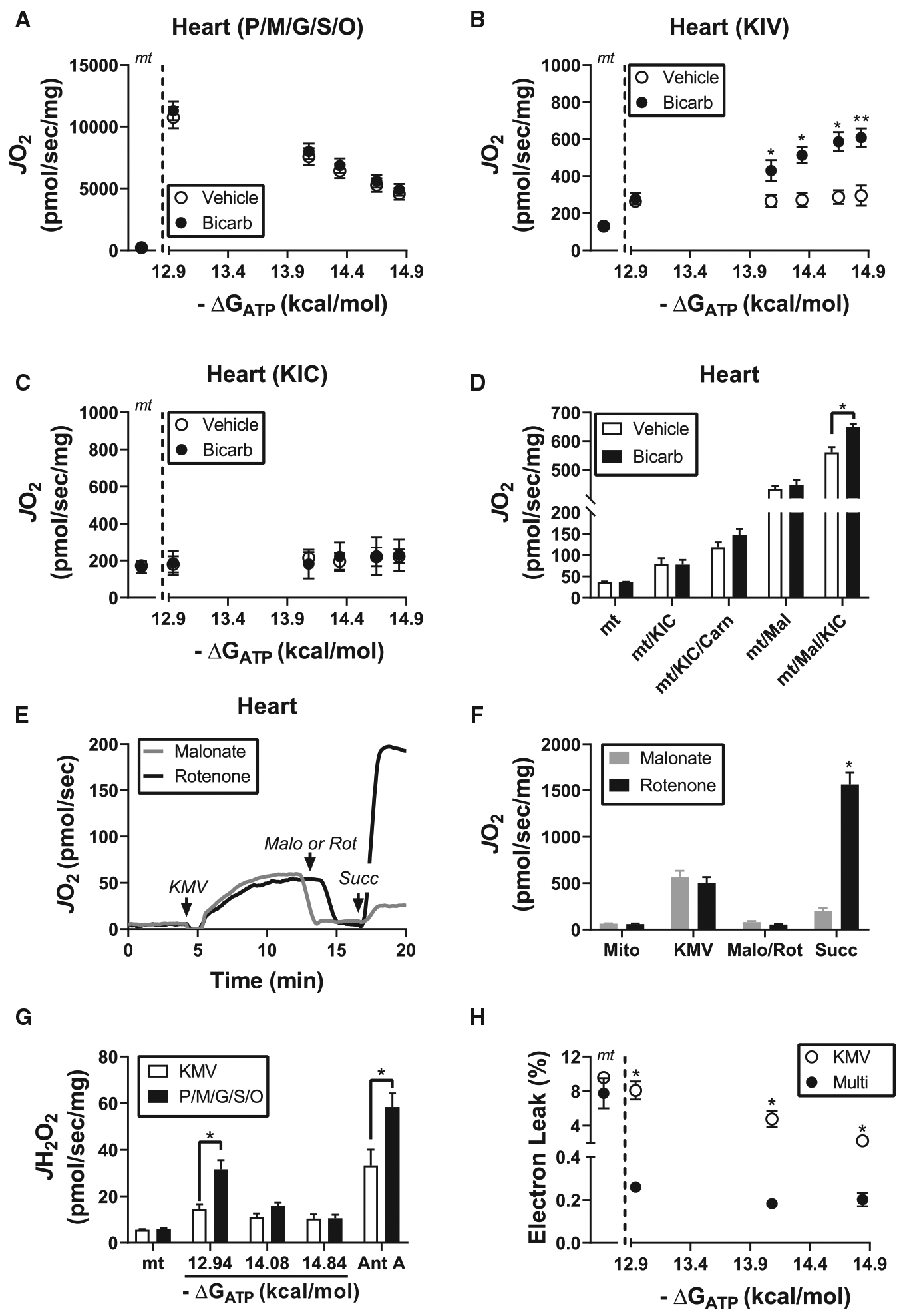
Isolated heart mitochondria were used for all experiments. All bicarbonate conditions were done in the presence of 2 mM bicarbonate. (A) Relationship between mitochondrial oxygen consumption and ATP-free energy in heart mitochondria energized with P/M/G/S/O. Rates to the left of the dotted line correspond to mitochondria alone (‘mt’). (B,C) Relationship between mitochondrial oxygen consumption and ATP-free energy in the presence of (B) KIV or (C) KIC. Rates to the left of the dotted line correspond to mitochondria alone (‘mt’). (D) Mitochondrial ΔGATP was set to −14.84 kcal/mol and JO2 was assessed under the following substrate combinations: mitochondria alone (‘mt’), KIC (‘mt/KIC’), KIC plus 5 mM carnitine (‘mt/KIC/Carn’), malate (‘mt/Mal’) and malate plus KIC (‘mt/Mal/KIC’). (E) Representative JO2 trace depicting KMV-supported flux followed by the sequential additions of either rotenone (5 μM) or malonate (‘Malo’; 5 mM) and then succinate (1 mM). Of note, mitochondrial ΔGATP was set to −14.84 kcal/mol and bicarbonate was present during the experiment. (F) JO2 quantification of the experiment in (E). (G) H2O2 production in heart mitochondria in the presence of either KMV or P/M/G/S/O assessed at various ATP-free energies. Antimycin A (5 μM) was added at the end of the protocol to serve as a positive control. (H) Relationship between electron leak (% Leak = JH2O2/JO2) and ΔGATP for both substrate conditions. Data are mean ± SEM, n = 3–4/group, *P < 0.05, **P < 0.001.
Tissue-specific differences in mitochondrial BCKA oxidation
To investigate BCKA oxidation across tissues, mitochondria were isolated from skeletal muscle and liver of C57BL/6J mice. Respiration supported by KMV was dependent on ATP-free energy and bicarbonate in skeletal muscle mitochondria, similar to that seen in cardiac mitochondria (Figure 4A). In liver mitochondria, bicarbonate once again increased respiration supported by each of the three BCKAs (KMV, Figure 4B; KIV, Figure 4C; KIC, Figure 4D) as well as the combination of malate and KIC (Figure 4E), consistent with bicarbonate being required for maximal BCKA flux in vitro. The inclusion of bicarbonate did not impact respiratory sensitivity in the presence of P/M/G/S/O (Figure 4F).
Figure 4. Inhibition of BCKDH kinase in vivo does not impact maximal BCKA-supported mitochondrial respiration.
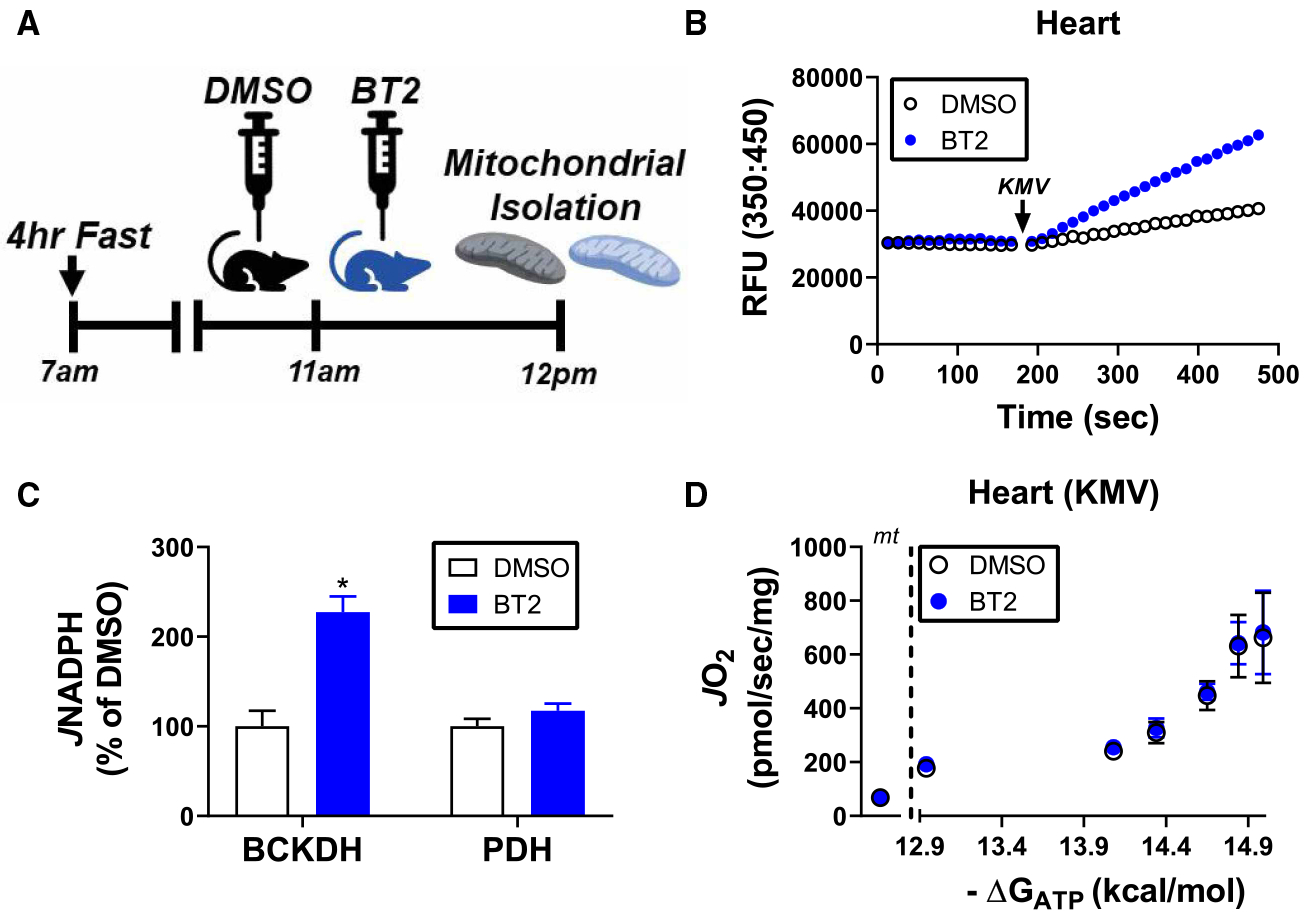
(A) Schematic of experimental timeline with BT2. (B) Representative fluorescent trace depicting KMV-supported JNADH in alamethicin-permeabilized heart mitochondria from mice treated with DMSO or BT2. (C) Quantification of BCKDH and PDH activity from heart mitochondria of mice treated with DMSO or BT2. Data are expressed as a percentage of activity from DMSO-treated mice. (D) Relationship between JO2 and ΔGATP for KMV-energized heart mitochondria from DMSO and BT2-treated mice. Rates to the left of the dotted line correspond to mitochondria alone (‘mt’). Data are mean ± SEM, n = 4/group, *P < 0.05.
Except for a small increase in KIC-supported JO2 between ΔGATP of −12.94 and −14.08 kcal/mol (Figure 4E), increasing ATP-free energy via PCR titration induced a drop in BCKA-supported respiration in liver mitochondria. These findings differ from those observed in heart and skeletal muscle and suggest that BCKA oxidation in liver mitochondria is sufficient to support respiratory control. Consistent with this, KMV-supported respiration was calculated to represent ~18% of maximal respiratory flux in liver mitochondria (Figure 4G), considerably higher than that observed in cardiac mitochondria (Figure 1H). Dehydrogenase profiling of permeabilized liver mitochondria demonstrated that BCKDH flux represents ~7% of maximal NAD flux (Figure 4H). Observed differences in BCKA respiratory flux across heart, skeletal muscle, and liver mitochondria are entirely consistent with prior metabolic studies implicating hepatocyte metabolism in whole-body BCKA homeostasis [33–35].
Mitochondrial BCKA oxidation specifically lowers glutamate- and α-ketoglutarate (AKG)-supported respiratory control
Alterations in BCKA oxidation have been linked to numerous human diseases ranging from diabetes [3] to cancer [5]. In addition, recent work has linked defective BCKA oxidation in cardiac mitochondria to heart failure [6] and ischemia–reperfusion injury [7]. In both cases, in vitro experiments determined that BCKAs impaired mitochondrial respiration, reportedly via specific inhibition of pyruvate utilization [7]. Given that these previous studies did not assess BCKA-induced shifts in JO2 in the context of bicarbonate and physiological ATP-free energy, it was hypothesized that BCKAs may differentially impact respiratory control across ATP/ADP energetic demands. To test this, isolated heart mitochondria were exposed to BCKAs (KIC/KIV/KMV, 1 mM each) or vehicle control (NaCl) and interrogated using a recently described mitochondrial diagnostic workflow [16]. In this system, mitochondrial JO2 and the corresponding free energies controlling flux (ΔΨ and NAD(P)H/NAD(P)+ redox state) are assayed in parallel across a span of ATP-free energies. These experiments were conducted in the presence of various substrate combinations (pyruvate/malate, Pyr/M; octanoyl-carnitine/malate, Oct/M; glutamate/malate, G/M; succinate/rotenone, S/R; and AKG), each of which activates a unique subset of mitochondrial dehydrogenases and respiratory complexes. Potassium bicarbonate was present for all experiments. Compared with control, mitochondrial respiration was slightly elevated in the presence of BCKAs prior to the addition of each substrate combination (Figure 5A; ‘mt’), consistent with BCKA oxidation. Although BCKAs did not influence S/R-supported respiratory flux, all NAD-linked substrate combinations were partially lower in the presence of BCKAs at the lowest ATP-free energy, as well as in response to FCCP addition (Figure 5A; ‘−12.94 kcal/mol’ and ‘FCCP’). Interestingly, respiratory sensitivity calculated from the linear change in JO2 for a given ΔGATP shift (−14.08 to −14.84 kcal/mol) revealed BCKA-induced impairments exclusively for G/M and AKG energized mitochondria (Figure 5D). These data suggest that although high BCKA concentrations generally impinge on maximal NAD-linked flux, respiratory impacts across physiological ΔGATP induced by BCKAs are more pronounced with respect to glutamate/AKG metabolism.
Parallel measurements of membrane potential (ΔΨ) and NAD(P)H/NAD(P)+ redox state allow for differences in respiratory control to be localized into one of three distinct energy transduction control nodes: (1) ‘ATP Synthesis’, (2) Electron Transport System (‘ETS’), and (3) ‘Matrix Dehydrogenases’. Although ΔΨ was largely unaffected by BCKAs, slight depolarization was evident in the presence of both Pyr/M and Oct/M at ΔGATP of −12.94 kcal/mol (Figure 5B). Membrane potential depolarization was accompanied by NAD(P)H/NAD(P)+ oxidation across all substrate combinations (Figure 5C), consistent with the source of respiratory resistance induced by BCKAs being localized to the matrix dehydrogenases. To validate this potential mechanism, dehydrogenase activity was directly assessed in permeabilized heart mitochondria exposed to identical BCKA concentrations as those used for the respirometry experiments. While slight inhibition was evident for both NADP-linked isocitrate dehydrogenase (IDH2) and NAD-linked PDH, both GDH and AKGDH were starkly inhibited by BCKAs (Figure 5E). Taken together, these findings provide biochemical evidence that under physiological energy constraints BCKAs impinge on AKG/glutamate oxidation within heart mitochondria, primarily via inhibition of AKGDH and GDH (Figure 6). Such findings agree well with previous reports demonstrating specific inhibition of AKGDH by BCKAs [36].
Figure 6. Comprehensive assessment of heart mitochondrial respiratory control in the presence of BCKAs.
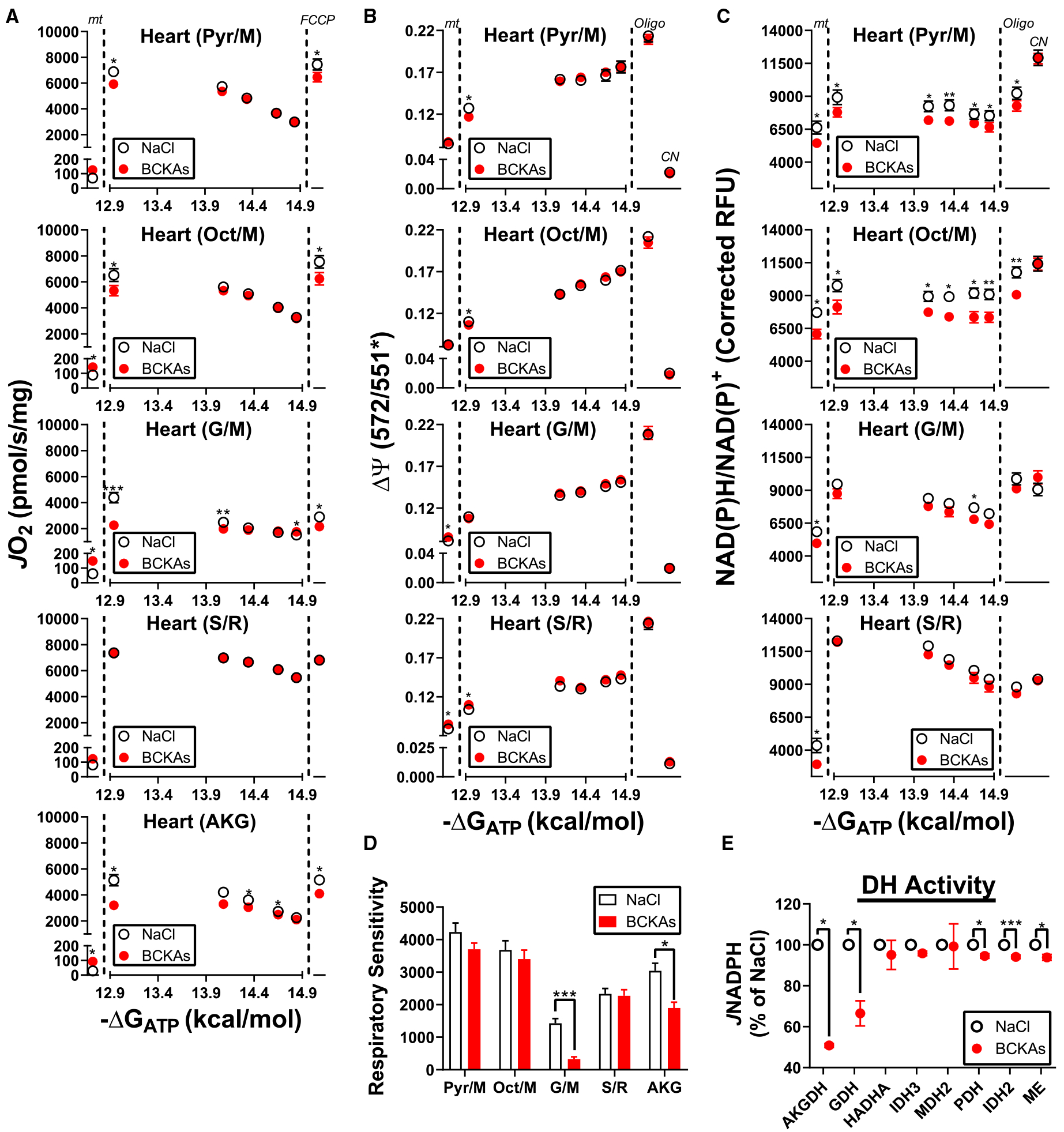
All experiments were performed in isolated heart mitochondria in the presence of either NaCl (3 mM) or a BCKA mixture (1 mM:1 mM:1 mM of KIC/KMV/KIV). (A) Relationship between JO2 and ATP-free energy in mitochondria energized with differing substrates (Pyr/M, Oct/M, G/M, S/R, AKG). Rates to the left of the first dotted line correspond to mitochondria alone (‘mt’). Rates to the right of the second dotted line represent JO2 following the addition of FCCP (2 μM) at the end of the PCR titration (‘FCCP’). (B) Relationship between mitochondrial membrane potential (ΔΨ) and ATP-free energy in mitochondria energized with varying substrates (Pyr/M, Oct/M, G/M, S/R). Values to the left of the first dotted line correspond to mitochondria alone (‘mt’). Values to the right of the second dotted line represent ΔΨ following the addition of oligomycin (‘Oligo’; 5 μM) and cyanide (‘CN’; 10 mM) at the end of the PCR titration. (C) Relationship between NAD(P)H/NAD(P)+ redox and ATP-free energy under the same conditions as in (B). (D) Respiratory sensitivity for each substrate combination determined from the linear change in JO2 as a function of ΔGATP (−14.08 kcal/mol to −14.84 kcal/mol). (E) Dehydrogenase activity in permeabilized heart mitochondria exposed to NaCl or BCKAs. Data are expressed as a percentage of the average NaCl rate for each enzyme. Data are mean ± SEM, n = 6–8/group. *P < 0.05, **P < 0.001, ***P < 0.0001.
Discussion
As an alternative to tracer analysis [19] and/or RNA sequencing [5], experiments in isolated mitochondria represent a powerful model system for comprehensively interrogating BCKA flux. That said, only a handful of studies have attempted to measure BCKA-supported respiration in isolated organelles [6,7,13–15]. In the majority of cases, BCKA-supported JO2 was not definitively demonstrated to be higher than that supported by mitochondria alone or mitochondria plus malate. Moreover, evidence for respiratory control (i.e. a change in JO2 in response to a change in free [ADP]) in the presence of BCKAs was lacking. This difficulty likely originated from omission of bicarbonate from the experimental media, as well as the use of bolus ADP, rather than physiological ATP/ADP, as the stimulus for increasing respiratory flux. Given that full BCKA oxidation requires bicarbonate and ATP, it was hypothesized that the inclusion of potassium bicarbonate in the respiratory buffer in conjunction with physiological ATP-free energy would stimulate maximal BCKA-supported JO2 in isolated mitochondria. This was indeed found to be the case across three murine organs (e.g. heart, liver, skeletal muscle). The contributions of BCKA oxidation to total respiratory capacity as well as the sensitivity to ΔGATP were found to be distinct across tissues. Remarkably, although flux was minimal at low ATP/ADP, BCKA-supported respiration in heart and skeletal muscle mitochondria increased over 3-fold across a physiological ΔGATP span. This increase in JO2 cannot be explained by ΔGATP-dependent changes in BCKA uptake across the inner mitochondrial membrane (Supplementary Figure S2F) or activation of BCKDH (Figure 3A–D). Collectivity, these data imply that in addition to post-translation control of BCKDH (i.e. phosphorylation) [17], BCKA oxidation in muscle mitochondria is exquisitely responsive to matrix ATP-free energy.
Mammalian mitochondria can receive reducing equivalents from each of the three canonical macronutrients: carbohydrates (e.g. glucose-derived pyruvate), fatty acids (activated to fatty acyl-CoA), and amino acids (e.g. BCKAs). Unlike pyruvate and acyl-CoA, BCKA oxidation encompasses an ATP-dependent reaction. In the case of KMV and KIV, this reaction involves the ATP/bicarbonate-dependent conversion of propionyl-CoA to D-methylmalonyl-CoA via propionyl-CoA carboxylase. A similar reaction occurs in the KIC oxidation pathway where methylcrotonyl-CoA is converted to methylglutaconyl-CoA. Aside from extreme nutrient deprivation (e.g. extreme endurance exercise and/or starvation), mitochondrial respiratory flux is principally governed by the intracellular demand for ATP regeneration. That is, any drop in ΔGATP is counter-balanced by reciprocal activation of catabolic flux (e.g. TCA cycle and β-oxidation) that increases respiration to restore ATP-free energy. The responsivity of these catabolic pathways to ΔGATP is largely mediated by NAD-linked dehydrogenases that are exquisitely sensitive to NADH/NAD+ redox balance. In the present study, BCKA-supported respiration in murine mammalian mitochondria, particularly from skeletal muscle and heart, was found to be highly responsive to increasing ΔGATP. These data provide biochemical evidence that maximal BCKA oxidation in peripheral tissues may occur under conditions of high BCKA availability and low energetic demand (i.e. fed state conditions). In support of this notion, recent in vivo flux analysis demonstrated increased BCKA contribution to the TCA cycle pool in skeletal muscle and heart in response to physiologic hyperinsulinemia [19]. Indeed, previous findings suggest that the primary function of BCKA oxidation in muscle involves anaplerotic replenishment of TCA cycle intermediates to support β-oxidation flux [37]. Additional investigation is required to elucidate if ATP-free energy is indeed rate-limiting for BCKA catabolism in vivo, particularly with respect to KIV and KMV oxidation.
Compared with muscle, BCKA oxidation in liver mitochondria was far less sensitive to both exogenous bicarbonate and ΔGATP. Given that bicarbonate is produced during BCKA decarboxylation, it is possible that higher BCKDH catalytic flux in liver mitochondria provides an endogenous source of matrix-localized bicarbonate to fuel sub-maximal BCKA oxidation. Across the three tissues interrogated, liver mitochondria were the only tissue in which BCKA-mediated respiratory control was observed. These data imply that in contrast with muscle mitochondria, the BCKA oxidation pathway in liver can substantially contribute to overall hepatic energy production. In support of this, whole-body BCKA homeostasis is restored in MSUD patients following hepatic transplantation [35].
Multiple reports exist linking increased mitochondrial BCKA exposure to impaired mitochondrial respiration [6,7,14,15]. Importantly, these studies did not assess BCKA-induced shifts in JO2 in the context of bicarbonate and physiological ATP-free energy, nor were the inhibitory effects of BCKAs assessed across multiple substrate combinations. The present study leveraged a recently described mitochondrial diagnostics workflow to unbiasedly assess the impact of BCKAs on cardiac mitochondrial respiratory control [16]. Exposure to BCKAs generally depressed maximal JO2 for all NAD-linked substrate combinations. Parallel determinations of ΔΨ and NAD(P) H/NAD(P)+ redox localized the source of BCKA-induced inhibition to the matrix dehydrogenases. Interestingly, respiratory sensitivity across a physiological span of ΔGATP was depressed by BCKAs only in the presence of G/M and AKG. Direct interrogation of dehydrogenase flux corroborated the respirometry data, evident by pronounced inhibition of both AKGDH and GDH by BCKAs. Although previous studies have documented BCKA-induced inhibition of AKGDH [15,36], loss of GDH activity may be mediated via BCKA-dependent competition with BCAA aminotransferase (BCAT) and/or reductive amination of exogenous BCKAs by GDH directly [38]. With respect to AKGDH inhibition, previous work has demonstrated that KIC is more potent than KMV or KIV [15]. Taken together, these findings suggest that the primary bioenergetic consequences of compromised BCKA metabolism likely involve limitations in AKG and/or glutamate metabolism.
In aggregate, the current study provides a framework for comprehensive interrogation of BCKA metabolism in isolated mitochondria. To illustrate the utility of this novel methodology, consider that a recent report demonstrated using RNA sequencing that several common laboratory mouse strains carry mild inborn errors of metabolism within the BCKA oxidation pathway [39]. Leveraging the assays described herein, straightforward experiments could easily be designed and carried out to validate the functional impact of these inborn errors across organs. Of course, similar methodologies could also be applied to elucidate the functional impact of stress and disease on BCKA metabolism, a critical open question across multiple disciplines.
Supplementary Material
Figure 7. Summary diagnostics of BCKA-mediated shifts in heart respiratory control.
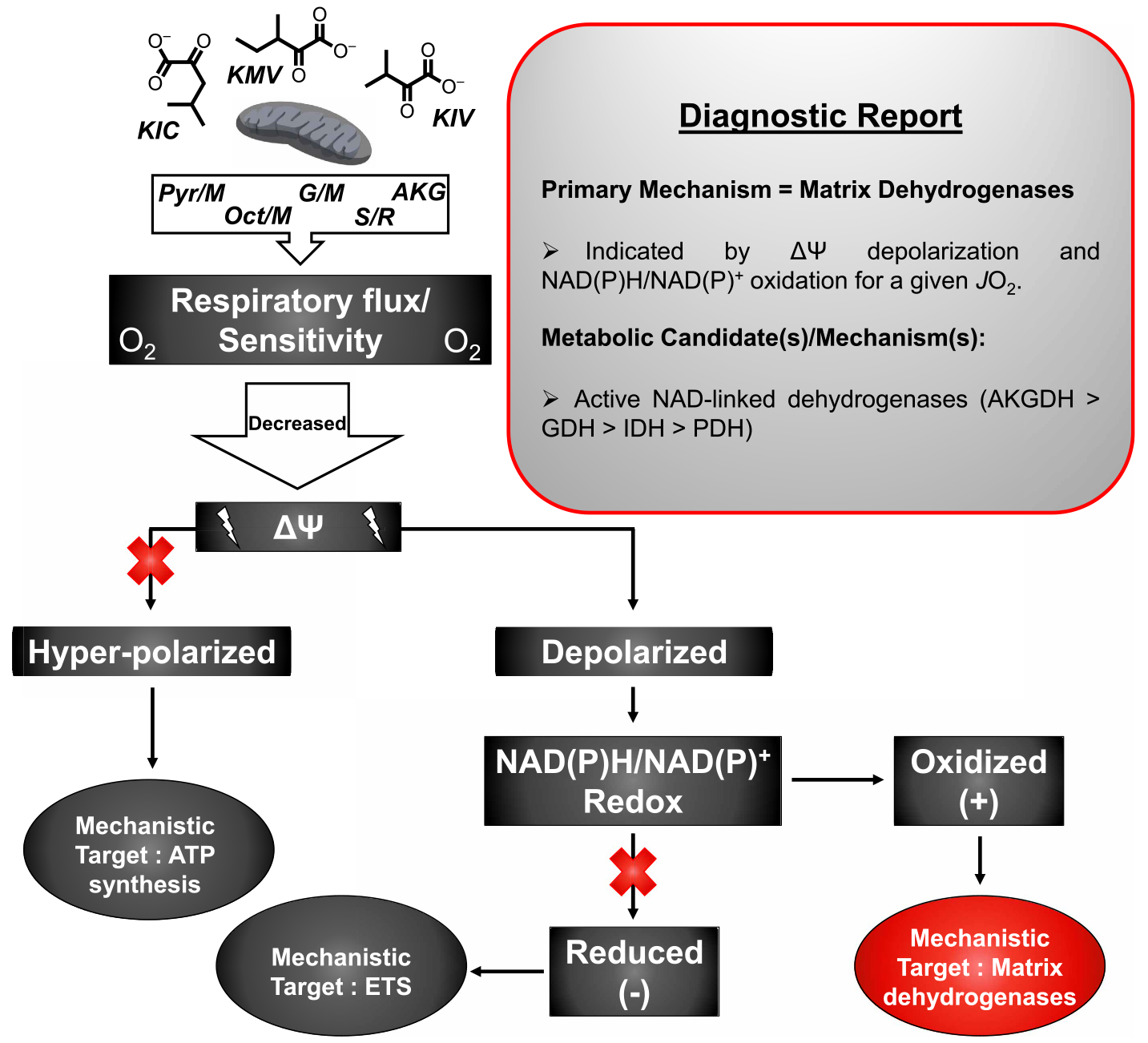
Diagnostic tree diagram depicting the primary bioenergetic mechanism by which BCKAs lower heart mitochondrial respiration. Starting with loss of respiratory flux or sensitivity, potential primary mechanisms are depicted in the dark grey-filled ovals (‘ATP synthesis’, ‘ETS’) with the BCKA-specific mechanism indicated in red (‘Matrix Dehydrogenases’). The experimental evidence in support of a ‘Matrix Dehydrogenases’ mechanism is provided in the ‘Diagnostic Report’ box.
Funding
C.E.P. is supported by an American Association of Immunologists Careers in Immunology Fellowship Award.
Abbreviations
- ADP
adenosine diphosphate
- AF
auranofin
- AKGDH
α-ketoglutarate dehydrogenase complex
- ATP
adenosine triphosphate
- AUR
Amplex UltraRed
- BCAAs
branched-chain amino acids
- BCAT
BCAA aminotransferase
- BCKAs
branched-chain keto acids
- BCKDH
BCKA dehydrogenase complex
- CK
creatine kinase
- Cr
creatine
- CrAT
carnitine acetyltransferase
- ETF
electron transfer flavoprotein
- ETFDH
ETF dehydrogenase
- ETS
electron transport system
- GDH
glutamate dehydrogenase
- HK
hexokinase
- HRP
horseradish peroxidase
- JO2
oxygen consumption rate
- KIC
ketoisocaproate
- KIV
ketoisovalerate
- KMV
ketomethylvalerate
- ME
malic enzyme
- MSUD
maple syrup urine disease
- PCR
phosphocreatine
- PDH
pyruvate dehydrogenase complex
- SDH
succinate dehydrogenase
- TMRM
tetramethylrhodamine methyl ester
- ΔGATP
ATP-free energy of hydrolysis
- ΔΨ
mitochondrial membrane potential
Footnotes
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Harper AE, Miller RH and Block KP (1984) Branched-chain amino acid metabolism. Annu. Rev. Nutr 4, 409–454 10.1146/annurev.nu.04.070184.002205 [DOI] [PubMed] [Google Scholar]
- 2.Burrage LC, Nagamani SCS, Campeau PM and Lee BH (2014) Branched-chain amino acid metabolism: from rare Mendelian diseases to more common disorders. Hum. Mol. Genet 23, R1–R8 10.1093/hmg/ddu123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF et al. (2009) A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9, 311–326 10.1016/j.cmet.2009.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T et al. (2016) Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: a Mendelian randomisation analysis. PLoS Med. 13, e1002179 10.1371/journal.pmed.1002179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ericksen RE, Lim SL, McDonnell E, Shuen WH, Vadiveloo M, White PJ et al. (2019) Loss of BCAA catabolism during carcinogenesis enhances mTORC1 activity and promotes tumor development and progression. Cell Metab. 29, 1151–1165.e6 10.1016/j.cmet.2018.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z et al. (2016) Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 133, 2038–2049 10.1161/CIRCULATIONAHA.115.020226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li T, Zhang Z, Kolwicz SC, Abell L, Roe ND, Kim M et al. (2017) Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. 25, 374–385 10.1016/j.cmet.2016.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crupi AN, Nunnelee JS, Taylor DJ, Thomas A, Vit J-P, Riera CE et al. (2018) Oxidative muscles have better mitochondrial homeostasis than glycolytic muscles throughout life and maintain mitochondrial function during aging. Aging 10, 3327–3352 10.18632/aging.101643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin C-T et al. (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest 119, 573–581 10.1172/JCI37048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes MC, Ramos S V, Turnbull PC, Edgett BA, Huber JS, Polidovitch N et al. (2019) Impairments in left ventricular mitochondrial bioenergetics precede overt cardiac dysfunction and remodelling in Duchenne muscular dystrophy. J. Physiol 1–16 10.1113/JP277306 [DOI] [PubMed] [Google Scholar]
- 11.Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z et al. (2016) Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl Acad. Sci. U.S.A 113, 4320–4325 10.1073/pnas.1519858113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M et al. (2015) SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes 64, 3081–3092 10.2337/db14-1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP et al. (2016) Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol 12, 15–21 10.1038/nchembio.1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amaral AU, Leipnitz G, Fernandes CG, Seminotti B, Schuck PF and Wajner M (2010) α-Ketoisocaproic acid and leucine provoke mitochondrial bioenergetic dysfunction in rat brain. Brain Res. 1324, 75–84 10.1016/j.brainres.2010.02.018 [DOI] [PubMed] [Google Scholar]
- 15.Shestopalov AI and Kristal BS (2007) Branched chain keto-acids exert biphasic effects on α-ketoglutarate-stimulated respiration in intact rat liver mitochondria. Neurochem. Res 32, 947–951 10.1007/s11064-007-9291-3 [DOI] [PubMed] [Google Scholar]
- 16.Fisher-Wellman KH, Davidson MT, Narowski TM, Te LC, Koves TR and Muoio DM (2018) Mitochondrial diagnostics: a multiplexed assay platform for comprehensive assessment of mitochondrial energy fluxes. Cell Rep. 24, 3593–3606.e10 10.1016/j.celrep.2018.08.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris RA, Hawes JW, Popov KM, Zhao Y, Shimomura Y, Sato J et al. (1997) Studies on the regulation of the mitochondrial α-ketoacid dehydrogenase complexes and their kinases. Adv. Enzyme Regul 37, 271–293 10.1016/S0065-2571(96)00009-X [DOI] [PubMed] [Google Scholar]
- 18.White PJ, McGarrah RW, Grimsrud PA, Tso SC, Yang WH, Haldeman JM et al. (2018) The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 27, 1281–1293.e7 10.1016/j.cmet.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neinast MD, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ et al. (2018) Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab. 29, 417–429.e4 10.1016/j.cmet.2018.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glancy B, Willis WT, Chess DJ and Balaban RS (2013) Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52, 2793–2809 10.1021/bi3015983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messer JI, Jackman MR, Willis WT, Kavazis AN, Smuder AJ, Min K et al. (2004) Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. Am. J. Physiol. Cell Physiol 286, C565–C572 10.1152/ajpcell.00146.2003 [DOI] [PubMed] [Google Scholar]
- 22.Saks VA, Kuznetsov A,V, Kupriyanov V,V, Miceli M,V and Jacobus WE (1985) Creatine kinase of rat heart mitochondria. The demonstration of functional coupling to oxidative phosphorylation in an inner membrane-matrix preparation. J. Biol. Chem 260, 7757–7764 [PubMed] [Google Scholar]
- 23.Stanley BA, Sivakumaran V, Shi S, McDonald I, Lloyd D, Watson WH et al. (2011) Thioredoxin reductase-2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J. Biol. Chem 286, 33669–33677 10.1074/jbc.M111.284612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radenkovic F, Holland O, Vanderlelie JJ and Perkins AV (2017) Selective inhibition of endogenous antioxidants with Auranofin causes mitochondrial oxidative stress which can be countered by selenium supplementation. Biochem. Pharmacol 146, 42–52 10.1016/j.bcp.2017.09.009 [DOI] [PubMed] [Google Scholar]
- 25.Gostimskaya IS, Grivennikova VG, Zharova TV, Bakeeva LE and Vinogradov AD (2003) In situ assay of the intramitochondrial enzymes: use of alamethicin for permeabilization of mitochondria. Anal. Biochem 313, 46–52 10.1016/S0003-2697(02)00534-1 [DOI] [PubMed] [Google Scholar]
- 26.Lark DS, Torres MJ, Lin C-T, Ryan TE, Anderson EJ and Neufer PD (2016) Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am. J. Physiol. Cell Physiol 311, C239–C245 10.1152/ajpcell.00124.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher-Wellman KH, Lin C-T, Ryan TE, Reese LR, Gilliam LAA, Cathey BL et al. (2015) Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem. J 467, 271–280 10.1042/BJ20141447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, Debalsi KL et al. (2012) Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 15, 764–777 10.1016/j.cmet.2012.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinlan CL, Goncalves RLS, Hey-Mogensen M, Yadava N, Bunik VI and Brand MD (2014) The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem 289, 8312–8325 10.1074/jbc.M113.545301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL and Brand MD (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312 10.1016/j.redox.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami T, Matsuo M, Shimizu A and Shimomura Y (2005) Dissociation of branched-chain α-keto acid dehydrogenase kinase (BDK) from branched-chain α-keto acid dehydrogenase complex (BCKDC) by BDK inhibitors. J. Nutr. Sci. Vitaminol 51, 48–50 10.3177/jnsv.51.48 [DOI] [PubMed] [Google Scholar]
- 32.Pawelczyk T and Olson MS (1992) Regulation of pyruvate dehydrogenase kinase activity from pig kidney cortex. Biochem. J 288, 369–373 10.1042/bj2880369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin AC, Fasshauer M, Filatova N, Grundell LA, Zielinski E, Zhou JY et al. (2014) Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab. 20, 898–909 10.1016/j.cmet.2014.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakai N, Kobayashi R, Popov KM, Harris RA and Shimomura Y (2000) Determination of branched-chain α-keto acid dehydrogenase activity state and branched-chain α-keto acid dehydrogenase kinase activity and protein in mammalian tissues. 324, 48–62 10.1016/S0076-6879(00)24218-3 [DOI] [PubMed] [Google Scholar]
- 35.Mazariegos G V, Morton DH, Sindhi R, Soltys K, Nayyar N, Bond G et al. (2012) Liver transplantation for classical maple syrup urine disease: long-term follow-up in 37 patients and comparative united network for organ sharing experience. J. Pediatr 160, 116–121.e1 10.1016/j.jpeds.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson RH and Singer TP (1983) Inactivation of the 2-ketoglutarate and pyruvate dehydrogenase complexes of beef heart by branched chain keto acids. J. Biol. Chem 258, 1857–1865 [PubMed] [Google Scholar]
- 37.Lerin C, Goldfine AB, Boes T, Liu M, Kasif S, Dreyfuss JM et al. (2016) Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol. Metab 5, 926–936 10.1016/j.molmet.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drotman RB, Featherston WR and Freedland RA (1972) Reductive amination of branched chain keto acids by glutamate dehydrogenase from several animal sources. Comp. Biochem. Physiol. B 41, 171–179 10.1016/0305-0491(72)90020-X [DOI] [PubMed] [Google Scholar]
- 39.Leandro J, Violante S, Argmann CA, Hagen J, Dodatko T, Bender A et al. (2019) Mild inborn errors of metabolism in commonly used inbred mouse strains. Mol. Genet. Metab 126, 388–396 10.1016/j.ymgme.2019.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.