

Abstract
During hypoxic conditions, cells undergo critical adaptive responses that include the up-regulation of hypoxia-inducible proteins (HIFs) and the induction of the unfolded protein response (UPR). While their induced signaling pathways have many distinct targets, there are some important connections as well. Despite the extensive studies on both of these signaling pathways, the exact mechanisms involved that determine survival versus apoptosis remain largely unexplained and therefore beyond therapeutic control. Here we discuss the complex relationship between the HIF and UPR signaling pathways and the importance of understanding how these pathways differ between normal and cancer cell models.
Keywords: ER-stress, Angiogenesis, Hypoxia-reoxygenation injury, Ischemia, Cell fate determination, UPRmt
This article was specially invited by the editors and represents work by leading researchers.
Introduction
Aerobic organisms employ critical control strategies to ensure proper oxygen supply through various physiological and metabolic cellular signaling networks. The inability to meet cellular oxygen demands, termed hypoxia, results in the activation of specific cellular stress responses [1, 2]. Hypoxic stress induces global gene expression changes in order to help cells adapt and survive by altering the cell’s metabolic and angiogenic pathways and restoring oxygen homeostasis [3–10]. If these repair and adaptive mechanisms fail, cells modify their gene expression profiles to induce programmed cell death [11–16]. Although active hypoxia signaling networks are necessary during embryogenesis and development [17–19], hypoxic conditions either diminish normally, or they contribute to pathological events in mature organisms [20–23].
Efficient activation of hypoxia signaling and angiogenesis is critical, for example, after stroke [24], myocardial infarction [25], and other ischemic events [26–29]. Alternatively, metabolic adaptation to low oxygen levels and the related tissue revascularization allows for the survival and progression of the majority of human tumors [30–32], and contributes to macular degeneration [33–36], glaucoma progression [37], and diabetic retinopathy [38–41]. Thus, the discovery and development of therapeutic strategies exploiting hypoxia-related cellular networks are of great interest to modern medicine, as evidenced by the awarding of the 2019 Nobel Prize in Physiology or Medicine to Drs. Semenza, Ratcliffe, Kaelin on their research into how cells detect oxygen and react to hypoxia [42–46].
The main goal of the cellular response to hypoxia is to promote cell survival and restore oxygen homeostasis. This goal, however, is accompanied by deregulation of cellular organelle changes in mitochondria and endoplasmic reticulum (ER) function that are reflected in perturbations in protein folding and trafficking [4, 47–53]. Erratic protein folding activates another specific stress response pathway, the unfolded protein response (UPR). The UPR promotes cellular survival by restoring endoplasmic and mitochondrial homeostasis through its distinct signaling networks [54–56], but if unsuccessful, the UPR induces cell death [57–59].
Although activation of the UPR supports surviving hypoxia, it can also impair cellular survival [60]. The ER, for example, is responsible for folding and maturation of transmembrane and secretory proteins [61–69] that include proangiogenic receptors and ligands such as vascular endothelial growth factor (VEGF) and erythropoietin (EPO) that are critical for hypoxia-induced angiogenesis and erythropoiesis, respectively [70–72]. Thus, although underappreciated, understanding mutual crosstalk between these stress response pathways is important for understanding and developing therapeutic interventions in cardiovascular diseases and cancer. Nevertheless, despite the extensive studies on both of these stress responses, the resulting consequences of their collective activation remain largely unexplained and are mainly limited to in vitro cell culture-based models. In this review, we summarize these two cell survival pathways and the implications of UPR involvement in the hypoxia cellular response pathway.
Hypoxia-inducible factor responses to hypoxia
The unmet cellular oxygen demand is reflected by the accumulation of functional heterodimeric α/β-subunit complexes of specific transcription factors called hypoxia-inducible factors (HIFs) [42–46]. HIFs mediate both the adaptive and apoptotic responses to hypoxia through transcriptional modulation of genes containing their specific target sequences that are termed hypoxia-response elements (HREs) [7, 73–77]. If cells are sufficiently supplied with the oxygen, the formation of active HIF complexes is inhibited by the limited availability of the alpha (α) subunits. Under normal oxygen pressure (normoxia), HIF-α subunits undergo oxygen-dependent post-translational modifications by proline-hydroxylases (PHDs) that mark these subunits for subsequent proteasomal degradation [42–46]. Furthermore, during normoxia there is another oxygen-dependent post-translational modification of α-subunits that is mediated by the factor inhibiting HIF (FIH) which impairs HIF transcriptional activity (Fig. 1) [78]. In contrast, the cellular levels of HIF-β subunits are oxygen independent [42–46]. During hypoxia, the post-translational modifications of HIF-α subunits are inhibited and lead to accumulation of the alpha subunit and the transcriptionally active HIF-αβ hetero-complexes. Despite the fact that HIF-1α is considered a major mediator of HIF signaling in higher metazoans, other tissue specific isoforms of α-subunits, HIF-2α and HIF-3α, are also known to participate in the cellular responses to hypoxia [7, 79–84].
Fig. 1.
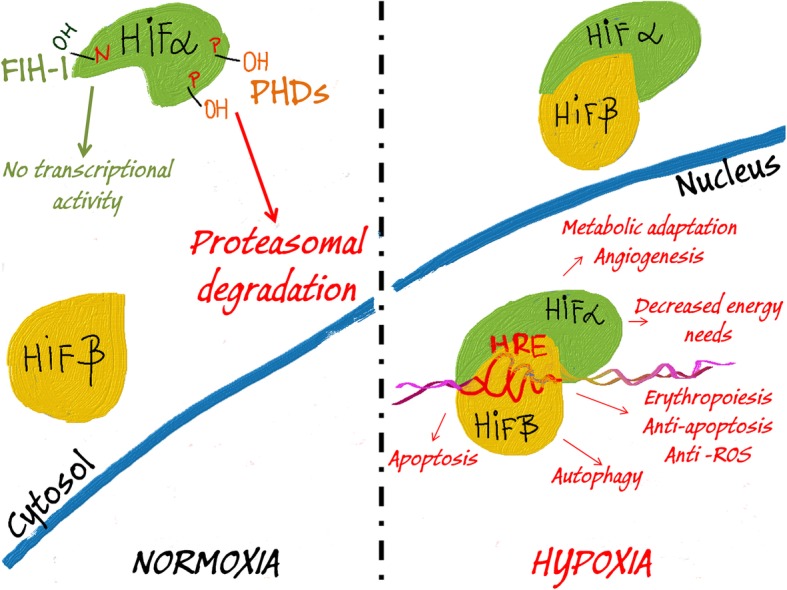
Oxygen availability regulates HIF signaling. In normoxia, proline (P) residues on HIFα subunits are hydroxylated by PHDs that marks them for proteasomal degradation. Additionally, FIH-1 mediates hydroxylation of asparagine residues (N) on HIFα to prevent HIF transcriptional activity. Hypoxia impairs the ability of PHDs and FIH-1 to hydroxylate the HIFα subunits, and thus results in the accumulation of this subunit and its heterodimerization with the stable HIFβ subunits. In the nucleus, the HIFα/β complex binds to HRE elements in the HIF target genes and governs their expression in order to adapt the cells to hypoxic conditions
The pro-survival pathway
During hypoxia, HIFs execute pro-survival transcriptomic strategies that allow cells to sustain energy levels via utilization of less efficient non-oxidative energy production. To sustain energy levels, HIFs upregulate glucose transporter genes and glycolytic enzymes, and inhibit oxidative phosphorylation (1) by preventing the conversion of pyruvate to acetyl-Co-A, (2) by reducing glucose oxidation and (3) by inhibiting β-oxidation of fatty acids [85–88]. Importantly, hypoxia-related utilization of this alternate metabolic pathway is accompanied by a HIF-mediated activation of the mechanisms that allow for a more efficient utilization of the anaerobic glycolytic pathway and that minimize its negative impact on the cell. The goal is to increase the electron transfer efficiency and to reduce reactive oxygen species (ROS) production. HIF-1 also regulates cytochrome c oxidase (COX) subunit composition to optimize the efficiency of respiration during hypoxia and to reduce ROS by promoting ROS scavenging pathways [89, 90]. Furthermore, since anaerobic glycolysis results in increased proton release, HIF-1 induces the expression of carbonic anhydrase 9 (CA-IX) and monocarboxylate transporter 4 (MCT4) to counteract acidosis by regulating intracellular pH [91, 92].
Since non-oxidative energy production of the cellular levels of ATP is less efficient than oxidative phosphorylation, HIFs activate pathways to decrease the cell’s energy needs. To accomplish this, HIFs selectively suppress translation and therefore decrease total protein production [93–95] and induce induction of autophagy [96, 97] and mitophagy [95, 98]. Notably, the mTOR pathway also reduces protein synthesis and cell growth and induces autophagy via a HIF-independent mechanism [48, 99].
In order to restore oxygen homeostasis and maintain the well-being of the endothelium, HIFs stimulate the expression of a number of angiogenic genes that include the vascular endothelial growth factor (VEGF) [9, 100], heme oxygenase-1 (HMOX1) [101], matrix metalloproteinases (MMP) 2 and 13 [102], the stem cell factor OCT-3/4 [103, 104], angiopoietin 2 (ANGPT2) [105], stromal derived factor 1 (SDF1) [106], platelet-derived growth factor B (PDGFB) [107], placental growth factor (PGF) [108], and stem cell factor (SCF )[109] and endothelial nitric oxide synthase (NOS3) [110, 111]. While HIF-induced angiogenesis ensures increased blood flow to hypoxic tissues, the oxygen caring capacity of the blood is enhanced via HIF-dependent upregulation of erythropoietin [112–114]. Importantly, to secure proper cellular iron levels that are required for the efficient erythropoiesis, HIFs adjust the expression of transferrin as well as of other genes mediating iron homeostasis [115, 116]. Furthermore, EPO supports anti-apoptotic proteins and inhibits caspase activity [117–119].
The UPR pathway responses to hypoxia
The fundamental function of the cellular response to hypoxia is surviving precarious conditions and restoring oxygen homeostasis. Hence, despite the HIF-related mechanisms to reduce the negative effects of anaerobic glycolysis and the reduced energy availability, this metabolic switch eventually disturbs cellular homeostasis. This energy deficiency limits the activity of ATP-dependent processes such as maintenance of ion homeostasis and the related redox potential, and limits protein and lipid synthesis, and post-translational protein folding capabilities due to the impaired disulfide-bond formation and ROS activity [4, 120–124]. All of these factors can disturb endoplasmic reticulum homeostasis (termed as ER stress), and lead to the accumulation of unfolded or misfolded proteins in the ER [125]. The accumulation of misfolded proteins activate another specialized stress response signaling pathway called unfolded protein response (UPR) [125]. During hypoxia, there are critical changes in mitochondrial function that lead to elevated ROS levels. Furthermore, the proper folding of mitochondria-encoded as well as the import and corresponding refolding of mitochondrial nucleus-encoded proteins are crucial for mitochondrial function [126, 127]. Hence, prolonged hypoxia will eventually result in perturbations in mitochondrial protein folding and activation of a related specific stress response mechanism termed the mitochondrial unfolded protein response (UPRmt) [126, 128–130].
The three UPR signaling pathways
Controlling ER homeostasis relies on the interplay between three signaling pathways of UPR that are initiated by three distinctive transmembrane sensors [125]. Buildup of unfolded/misfolded proteins in the ER induces a higher demand for chaperone proteins that include glucose-regulated protein 78 (GRP78 also known as BiP (binding immunoglobin protein) [57]. BiP initiates the UPR by dissociating from luminal domains of three proteins, protein kinase RNA-like endoplasmic reticulum kinase (PERK), the inositol-requiring enzyme 1α (IRE1α), and with activating transcription factor 6 (ATF6) [57]. Upon BiP release, PERK and IRE1α are activated via multimerization and trans-autophosphorylation, whereas ATF6 is translocated to the Golgi apparatus where it is proteolytically processed to a cytoplasmically soluble and active ATF6f (p50) transcription factor (Fig. 2) [131–133]. This activation cascade results in three distinctive UPR signaling pathways/axes that are mediated by the PERK, IRE1α and ATF6 sensors.
Fig. 2.
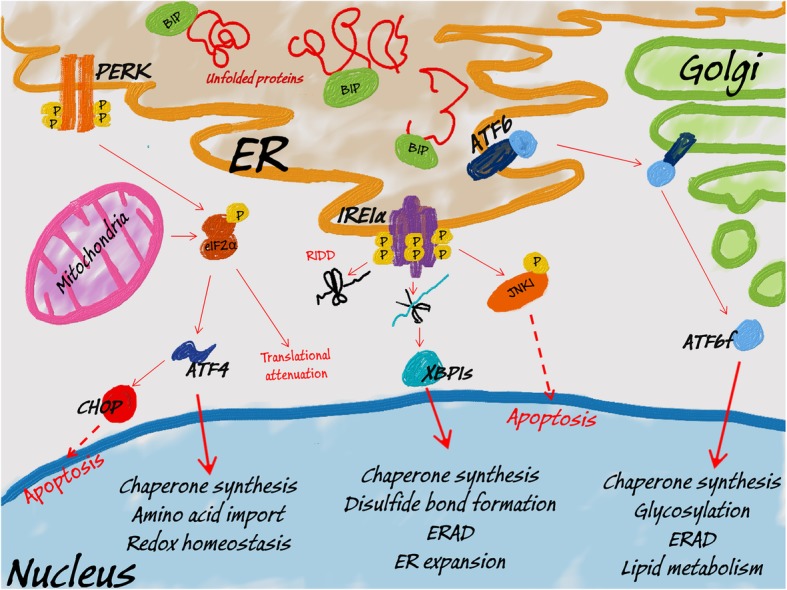
UPR and UPRmt signaling. Upon buildup of misfolded/unfolded proteins in ER, BIP is released from ER membrane to induce PERK dimerization and its subsequent autophosphorylation. Activated PERK phosphorylates the eIF2α, leading to global translation attenuation. Some transcripts, however, including ATF4 remain preferably translated. ATF4 provides the transcriptional signal to restore ER homeostasis, however, it can also induce proapoptotic CHOP. Similarly, accumulation of unfolded proteins in mitochondria leads to PERK activation and the induction of ATF4 signaling (UPRmt). Upon its dissociation from BIP, IRE1α undergoes oligomerization and autophosphorylation and thus gains endoribonuclease activity. To decrease the ER load, activated IRE1α degrades mRNAs and miRNAs (RIDD). IRE1α also performs splicing of XBP1 mRNA to release transcriptionally active XBP1s. XBP1s activates a transcriptional program to restore ER homeostasis. Alternatively, IRE1α can activate a proapoptotic kinase JNK1. Finally, BIP dissociation allows ATF6 translocation to Golgi, where cleavage of this protein results in release of transcriptionally active ATF6f. ATF6f activates a transcriptional program to restore ER homeostasis and support ERAD
Active PERK phosphorylates the alpha subunit of the eukaryotic initiation factor eIF2, and this initiates the selective translation of certain proteins and repressing the translation others during stress conditions. Some of the selected proteins include activating transcription factor 4 (ATF4), growth arrest and DNA damage inducible protein (GADD34), and CCAAT/enhancer binding homologous protein (CHOP) [57, 134, 135]. ATF4 modulates the expression of genes involved in amino acid biosynthesis, anti-oxidative responses, protein folding and in maintaining redox homeostasis [136]. Importantly, GADD34 mediates the dephosphorylation of eIF2, thus allowing the restoration of the protein synthesis upon stress recovery [137]. If the stress is persistent, ATF4 can also facilitate autophagy and stimulate transcription of the proapoptotic CHOP to induce cell death (Fig. 2) [138, 139].
Active IRE1α reduces protein synthesis through the degradation of selected mRNAs in a process referred to as regulated IRE1-dependent decay (RIDD) [140]. Notably, IRE1α endoribonuclease activity generates the active spliced isoform of the X-box binding-protein transcription factor (XBP1s) [141]. XBP1s modulates gene expression by increasing the ER’s folding capacity. XBP1s also promotes the expression of proteins that are involved in ER membrane biosynthesis, disulfide bond formation, as well as increasing the expression of chaperones and proteins involved in ER-associated degradation (EDEM) and vesicular trafficking [141–143]. Furthermore, IRE1α kinase activity activates Janus N-terminal kinase (JNK) in order to activate the inflammatory response and to promote autophagy and apoptosis [140, 144] (Fig. 2).
ATF6f, on the other hand, initiates a transcriptional program to restore ER homeostasis and that includes the induction of BIP expression, promoting protein chaperone and lipid synthesis, stimulating ER-degradation, and enhancing N-glycosylation [145, 146]. ATF6f also induces CHOP expression and thus contributes to UPR-related cell death [147, 148] (Fig. 2). Notably however, a recent report has shown that IRE1α activation can deactivate the ATF6f pathway [149].
Despite the fact that the UPR usually mediates cell death by activating the intrinsic apoptotic pathway, recent reports indicate that during unresolved ER stress, there is strong activation of the UPR that can lead to activation of programmed-necrosis pathways such as necroptosis [149–154]. Activation of these cell death pathways usually involves PERK signaling and is associated with a rapid depletion of intracellular ATP and a rapid release of ER-stored calcium [149–154]. Notably, the necroptosis pathway has been involved in modulation of both HIF-signaling and key glycolytic enzymes that include pyruvate dehydrogenase. This results in the enhancement of aerobic respiration and ROS generation, and thus can lead to impaired cellular adaptation to hypoxia [155–158]. That being said, the origins and role of necroptosis in both the UPR and the hypoxia response will require further studies.
Mitochondrial stress responses
Since mitochondria are separated from the cytosol and ER by their outer and inner membranes, they have to rely on their own stress response mechanisms for translating and folding proteins encoded in their genomes as well as refolding the imported nuclear-encoded proteins [126, 127]. In order to maintain their protein homeostasis, these organelles have a specific set of chaperones that includes heat shock protein 60 (HSP60) and LON peptidase 1 [159–161]. Notably, it has been reported that events that lead to accumulation of unfolded/misfolded proteins in the mitochondria, or in impairment of energy dependent mitochondrial protein import, or in disturbances in mitochondrial protein synthesis and folding lead to the activation of a mitochondrial UPR (UPRmt) [126, 128–130].
To recover and preserve mitochondrial function, UPRmt modulates the expression of both mitochondria and nuclear encoded genes [126, 128–130]. However, if the stress is persistent, the UPRmt can contribute to the activation of intrinsic apoptosis pathways [126, 128–130]. In C. elegans, properly functional mitochondria import and subsequently degrade the stress sensor protein called activating transcription factor associated with stress (ATFS-1) [162]. Upon stress, however, ATFS-1 import to mitochondria is impaired, and this transcription factor accumulates in nucleus and activates a transcriptional program to restore mitochondrial homeostasis through upregulation of mitochondrial chaperons and proteases as well as components of both the protein import machinery and ROS scavenger pathway [162].
Although the regulation of the mammalian UPRmt is poorly understood, it has been suggested that the import efficiency of activating transcription factors 5 and 4 (ATF5 and ATF4) can be sensors of mitochondrial protein disturbances [163, 164]. Upon stress, these transcription factors were shown to induce expression of mitochondrial chaperones and proteases. Furthermore, it has been shown that disturbances of mitochondrial protein homeostasis lead to activation of the PERK axis of the UPR, and this reduces global protein synthesis and selectively promotes expression of ATF4, ATF5 and the proapoptotic protein CHOP (Fig. 2) [126, 128–130, 165]. However, the molecular mechanisms underpinning the integrated feedback between the UPR and the UPRmt will require further study.
The crosstalk between hypoxia and UPR in cancer versus normal cell models
Despite the fact that normal endothelial cells are the main effectors of the adaptive cellular response to hypoxia, the vast majority of current research regarding this signaling pathway is from cancer cells [31, 48, 166, 167]. The mainstream reports of the interplay between hypoxia and UPR are limited to cancer models as well [71, 72, 167–171]. Importantly, cancer progression and cancer cell survival often result from the deregulation of the cell fate decision mechanisms during both hypoxia and the UPR. Although hypoxia was shown to induce all three UPR signaling axes, and given their activation could also result from cancer cell-specific adaptations, it is important that the prosurvival consequences of the UPR need to be directly compared to normal cell types.
Hypoxia-related induction of BIP expression has been reported in both cancer and endothelial cells models [50, 110, 172–176]. This suggests that hypoxia-induced perturbations in ER may increase BIP demand in both cell types and promote UPR induction. Indeed, activation of PERK signaling is also observed in both cancer and normal cells including endothelial cells, regardless of the hypoxia model applied [170, 177–182]. PERK-mediated eIF2 phosphorylation was observed in cells within minutes after exposure to acute hypoxia (below 0.1% O2), whereas this reaction rate continuously declined with increasing oxygen concentrations [177]. Furthermore, activation of the PERK axis was also reported in transient (cyclic hypoxia) models that better resemble the fluctuating oxygen availability conditions that occur in solid tumors [183–187]. Hence, it can be concluded that the hypoxia-required reduction of energy demand is partially achieved via UPR-mediated translational attenuation. Notably, this pathway was shown to be deactivated during prolonged hypoxia (16 h) as shown by dephosphorylation of eIF2 that is probably due to a negative feedback loop with GADD34 [177, 188, 189]. During prolonged hypoxia, HIF-1 signaling is only partially sustained by the HIF-2 activity during the transition from HIF-1 to HIF-2 expression [7, 76, 77]. This would suggest that the activation of PERK axis can only be modulated by the HIF-1, whereas during prolonged hypoxia, HIF-2 mediates the translational repression via an alternate mechanism [167]. However, this hypothesis will require further study. Interestingly, the PERK pathway was also shown to inhibit HIF-1α translation and thus prevent HIF-1 signaling in cancer cells [190].
Besides attenuation of protein synthesis, the PERK pathway mediated by ATF4 activates genes supporting ER and mitochondrial homeostasis [126, 128–130, 165]. Notably, however, the PERK pathway can induce cell death through CHOP accumulation [191]. Although CHOP accumulation and the potential induction of apoptotic response were observed in some hypoxia experiments (including lung endothelial cells) [192–194], this protein and mRNA levels were much lower than those observed during ER stress [177]. Inhibition of the entire PERK axis during hypoxia, however, has more drastic effects on cell survival [177]. Furthermore, hypoxic PERK activation was shown to regulate carbonic anhydrase 9 (CA9) levels and thus is important for maintaining cellular pH [195, 196]. Importantly, CHOP also directly reduces the expression of the proangiogenic endothelial nitric synthase (NOS3, eNOS) [197]. The reduction of eNOS activity during hypoxia, however, may be required to prevent the uncoupling of this enzyme and the related ROS accumulation [198, 199]. Therefore, the evaluation of CHOP’s role in hypoxic cell survival requires careful consideration and further study [200].
The activation of IRE1 axis and the role of XBP1s during hypoxia remain less clear. Despite some functionally relevant accumulation of XBP1s that supported cancer cell survival and tumor growth studies in cancers cell lines exposed to hypoxia, this effect was observed in acute and moderate hypoxia only [177, 201–209]. In contrast, impairment of XBP1 splicing under acute hypoxia was also reported [210]. Furthermore, although some potentially IRE1-related activity was observed in human pulmonary artery smooth muscle cells (PAMSCs), this did not result in direct hypoxia-induced XBP1s protein accumulation [211]. Hence, IRE1α involvement in cellular response may be very oxygen pressure- and cell type-specific and will require further studies in a wide range of primary endothelial cells [212–216]. Finally, although numerous known ATF6 transcriptional targets were shown to be elevated in some experimental models by prolonged hypoxia and ischemia [192, 217, 218], the general direct hypoxic activation of the ATF6 UPR axis has been convincingly presented [177]. Hopefully, the novel ATF6 pathway inhibitors, Ceapins [219], will be helpful in clarifying the role of this UPR branch during hypoxia.
The main function of adaptive HIF activity is with the induction of angiogenesis and erythropoiesis. The successful implementation of these cell rescue programs requires increased synthesis of proangiogenic factors (ligands and receptors) as well as increased erythropoietin production. All of these proteins fall into either the transmembrane or secretory proteins category, and as such they have to mature in the ER [52, 121, 220–223]. Hence, recovery from hypoxia absolutely depends on proper ER function [224]. Importantly, the PERK/ATF4 axis has been reported as a limiting factor for EPO production, and thus hypoxic UPR activation may limit adaptation to hypoxia [70].
In 2014, Karali and coworkers described the mechanism potentially linking HIF transcriptional activity with the activation of PERK, ATF6 and IRE1 pathways in human endothelial cells [225]. They reported prosurvival UPR activation in VEGF (a HIF transcriptional target) treated human umbilical vein endothelial cells (HUVECs) [226]. In these studies, the authors proposed a mechanism in which VEGF-dependent phosphorylation of vascular endothelial growth factor receptors (VGFRs) leads to phospholipase C (PLC) activation and release of ER calcium, which activates all three axes of the UPR [225]. The active UPR promotes transcriptional expression of numerous proangiogenic genes that include VEGF that can be induced directly by ATF6f, XBP1s and ATF4 [224, 227–237], and interleukin 8 that is induced by ATF4 [238, 239]. Furthermore, it has been suggested that this survival mechanism is mediated by XBP-1 through a direct interaction with HIF-1α protein [201]. Nevertheless, the extend of VEGF-related activation of UPR is rather poorly reflected in endothelial hypoxia models, and will require further study. In endothelial cells, it has been also reported that hypoxia-induced expression of very low density lipoprotein receptor (VLDLr) can contribute to the activation of the UPR cell death response [193, 194].
The UPR and UPRmt
Collectively, despite a variety of ER stress-related mechanisms reported in cancer cells, exposure to hypoxia results mainly in the activation of the PERK axis that can also be co-regulated by UPRmt (Fig. 3). Notably, however, the dramatic activation of all arms of UPR was reported in anoxia and that by definition relates to the loss of control over the cellular metabolism and energy production due to a dramatic oxygen deficit [240]. Given that limitation, such as reports are hard to interpret since they were obtained from mixed cells populations that were undergoing the anoxic necrosis and had totally lost the ability to maintain their redox homeostasis.
Fig. 3.
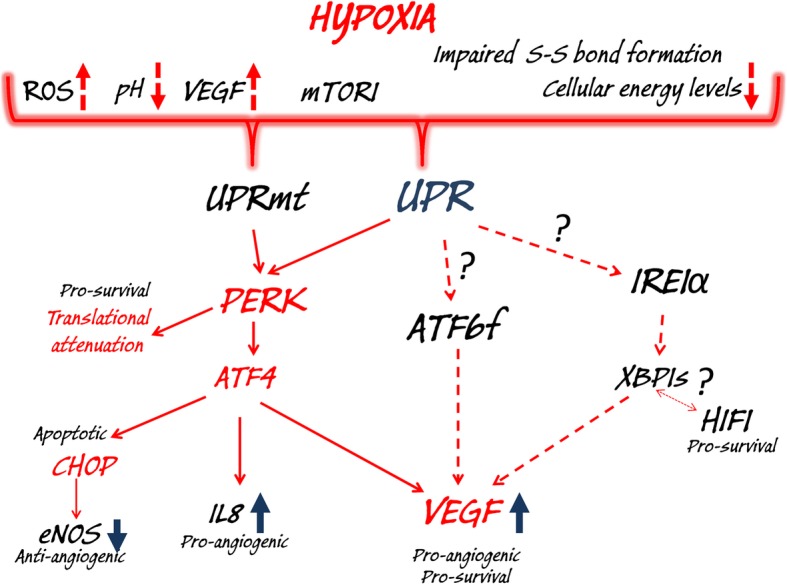
Hypoxia signaling and the related changes in cellular functions activate the UPR and UPRmt. During hypoxia, accumulation of misfolded/unfolded proteins in ER and mitochondria activate PERK signaling, and this contributes to both pro-survival (global translational arrest and induction of pro-angiogenic genes IL8 and VEGF) and apoptotic responses (induction of CHOP and inhibition of pro-angiogenic eNOS expression). Furthermore, in some models, hypoxia-related activation of ATF6 and IRE1α contributes to pro-survival and pro-angiogenic signaling. There also appears to be cooperation between XBP1s and HIF1 in pro-survival signaling
By comparison, the limited activation of the UPR and UPRmt during moderate and prolonged hypoxia suggests that the HIFs successfully prevent extensive ROS formation and alleviate stress conditions. This statement is supported by the studies that have shown that HIF-1α knockouts cells subjected to hypoxia produce lethal levels of ROS [241] as well as by numerous reports demonstrating a negative correlation between cellular ROS levels and HIF-1 stabilization [87, 90, 242–244]. Remarkably, however, cumulating evidence demonstrates that rapid re-establishment of normal oxygen levels in hypoxic cells often results in extensive ROS production and leads to cellular damage that is referred to as hypoxia-reoxygenation injury and ischemia-reperfusion injury. Although ROS accumulate in hypoxic cells, their levels are dramatically compounded by this rapid re-introduction of oxygen [90, 241, 245, 246].
Importantly, both ischemia-reperfusion injury [247–259] and hypoxia-reoxygenation injury [260–271] are also known to be accompanied by ER stress that are related to hypoxia/reoxygenation-triggered depletion of ER calcium and ROS accumulation. Indeed, an extensive UPR and UPRmt activation occurred upon rapid restoring oxygen levels in human endothelial cells, cardiomyocytes and neurons [247–271]. Although the IRE1α and ATF6 axes were activated in some models similar to their activation to hypoxia [272, 273], the PERK pathway was the common and main axis of UPR activation after hypoxia/reoxygenation. Importantly, the UPR pathway was shown to be crucial for determining cell fate during hypoxia/reoxygenation, and therefore this pathway should be considered as a potential therapeutic target for ischemic and cardiovascular diseases.
Importantly, intermittent (cyclic hypoxia) results from chronic exposure of cells to cycles of hypoxia/reoxygenation and is a basic feature of sleep apnea. Cyclic hypoxia also clearly defines the development of the majority of solid tumors, which were also shown to be accompanied by chronic ER stress [274, 275]. Nevertheless, the majority of the hypoxia-induced UPR cancer research has been performed in continuous hypoxia models that display diminished UPR activation.
Concluding remarks
The cellular response to hypoxia as well as the UPR are critical components of human pathologies and have become obvious therapeutic targets. Despite the continuous research to elucidate these complex molecular signaling pathways, however, the exact mechanisms that cells use to determine cell fate during stress remain mainly largely unexplained and beyond therapeutic control. Furthermore the current understanding of the molecular mechanisms underpinning the mutual interplay between cellular response to hypoxia and UPR remain very limited. Although hypoxia-related ER stress is well defined, the extent of the related UPR activation and its effects on angiogenesis and particularly cell survival are complicated and often contradictory. Several research cell model-related barriers need to be tested in order to gain better insight into physiological relevance of the hypoxic UPR before any type of intervention could be properly tested in vivo.
To date, the two dimensional in vitro cultures of cancer cells that are exposed to continuous hypoxia constitute the main research cell models. As mentioned, the cancer cell lines have developed specific genetic and epigenetic adaptations that allow them to adapt and survive both persistent ER-stress and hypoxia. Therefore, parallel research in matched normal, primary cells is needed to distinguish between the cancer-specific and cell line-specific signaling pathway mechanisms employed for cell fate decisions.
In solid tumors, for example, cancer cells usually benefit from endothelial angiogenesis, and thus despite sending angiogenic signals to endothelial cells, they alone not the right model for determining the extent of how hypoxia or the UPR impacts angiogenesis. Hence, the role of tumors’ endothelial cells and the development of representative models including both cancer and endothelial cells, is extremely important. Notably, the current development of high-throughput single-cell transcriptomics on organoids and 3D culture systems [276, 277] should contribute to the utilization of such a research approach.
Furthermore, solid tumors are persistently exposed to fluctuating oxygen levels (cyclic hypoxia) rather than chronic hypoxia [278–283]. Given the potent activation of the UPR during cycles of hypoxia/reoxygenation, it is plausible that majority of the cancer studies underestimate the UPR component of the response to hypoxia in these tumors. Moreover, human cells and tissues differ in their oxygen demands, and the fate of individual cells and the magnitude of the individual cellular stress responses are affected by the local levels of oxygen. This would suggest that the consequences of reoxygenation to the lower actual tissue oxygen levels (termed physoxia) should be considered as well [2].
Normal endothelial cells provide an alternative model to study hypoxia and the related UPR. The comparative analysis of endothelial cells from different vascular beds, for example, could provide novel insights into the physiological relevance of hypoxia-induced UPR. Many studies have been based on models where cells were exposed to persistent moderate continuous hypoxia (e.g., 12 or 24 h at 1% oxygen) that is physiologically irrelevant and therefore difficult to interpret.
Recent studies have identified novel post-transcriptional levels of regulation of cellular signaling pathways that depend upon the RNA interference pathway, and include mRNA modulation by microRNAs. Although, these short noncoding RNAs has been reported to be important modulators of both hypoxia and UPR [59, 77, 83, 143, 187, 284–294], their role in the interplay between these two signaling pathways remains limited. Of note in this regard, a recent report has shown that XBP1s induces antiangiogenic miR-153 during hypoxia in breast cancer cells [202, 203].
Further development of new experimental models, wide transcriptomic and proteomic approaches, as well as employment of novel specific inhibitors of UPR axes will eventually address some of these issues. Furthermore, numerous novel compounds and therapeutic strategies focusing on cancer and cardiovascular diseases are progressing through clinical trials that target either the hypoxic response or the UPR (https://clinicaltrials.gov/ct2/ and https://clinicaltrials.gov/ct2/, respectively) [294–298]. Nevertheless, the crosstalk between these stress pathways is rarely utilized in these putative therapies. For example, some of the anticancer agents are proteasome inhibitors (bortezomib and nelfinavir) that utilize the UPR pathways to decrease VEGF levels and thus directly inhibit tumor vasculature [299–302], whereas the ER chaperones inhibitor 17-AAG (geldanamycin) reduces the degree of adaptive HIF-1 signaling and thus stimulates hypoxic cell death [303, 304]. Finally, although the interplay between the hypoxia and the related UPR is just beginning to be appreciated, we are still very far from understanding their interrelated functions and therefore further research in this field will be critical for the development of future therapies.
Acknowledgments
Not applicable.
Abbreviations
- ATF4
Activating transcription factor 4
- ATF6
Activating transcription factor 6
- BiP
Also known as binding immunoglobin protein
- CHOP
CCAAT/enhancer binding homologous protein
- EDEM
ER-associated degradation
- EPO
Erythropoietin
- ER
Endoplasmic reticulum
- FIH
Factor inhibiting HIF
- GADD34
Growth arrest and DNA damage inducible protein
- GRP78
Glucose-regulated protein 78
- HIFs
Hypoxia-inducible factors
- HREs
Hypoxia-response elements
- IRE1α
The inositol-requiring enzyme 1α
- PERK
Protein kinase RNA-like endoplasmic reticulum kinase
- PHDs
Proline-hydroxylases
- RIDD
IRE1-dependent decay
- ROS
Reactive oxygen species
- UPR
Unfolded protein response
- VEGF
Vascular endothelial growth factor
Authors’ contributions
Both authors wrote, read and revised the final version of the manuscript. The authors read and approved the final manuscript.
Funding
This work has been supported by National Science Center “OPUS” and “Sonata Bis” Program under contracts UMO-2015/17/B/NZ3/01485 and 2015/18/E/NZ3/00687 (S.B.) and by the NIH P30 DK072482 grant and the Research Development Program (ROWE15R0) from the CF Foundation (J.F.C.).
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sylwia Bartoszewska, Email: sylwiabart@gumed.edu.pl.
James F. Collawn, Email: jcollawn@uab.edu
References
- 1.Connett RJ, Honig CR, Gayeski TE, Brooks GA. Defining hypoxia: a systems view of VO2, glycolysis, energetics, and intracellular PO2. J Appl Physiol (1985) 1990;68:833–842. doi: 10.1152/jappl.1990.68.3.833. [DOI] [PubMed] [Google Scholar]
- 2.McKeown SR. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol. 2014;87:20130676. doi: 10.1259/bjr.20130676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strzyz P. Hypoxia as an off switch for gene expression. Nat Rev Mol Cell Biol. 2016;17:610. doi: 10.1038/nrm.2016.119. [DOI] [PubMed] [Google Scholar]
- 4.Thiele RH. Subcellular energetics and metabolism: a cross-species framework. Anesth Analg. 2017;124:1857–1871. doi: 10.1213/ANE.0000000000001773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheaton WW, Chandel NS. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol. 2011;300:C385–C393. doi: 10.1152/ajpcell.00485.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bargiela D, Burr SP, Chinnery PF. Mitochondria and hypoxia: metabolic crosstalk in cell-fate decisions. Trends Endocrinol Metab. 2018;29:249–259. doi: 10.1016/j.tem.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Bartoszewski R, Moszynska A, Serocki M, Cabaj A, Polten A, Ochocka R, et al. Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019;33:7929–7941. doi: 10.1096/fj.201802650RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi KS, Bae MK, Jeong JW, Moon HE, Kim KW. Hypoxia-induced angiogenesis during carcinogenesis. J Biochem Mol Biol. 2003;36:120–127. doi: 10.5483/bmbrep.2003.36.1.120. [DOI] [PubMed] [Google Scholar]
- 9.Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2:1117–1133. doi: 10.1177/1947601911423654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LaManna JC, Kuo NT, Lust WD. Hypoxia-induced brain angiogenesis. Signals and consequences. Adv Exp Med Biol. 1998;454:287–293. doi: 10.1007/978-1-4615-4863-8_34. [DOI] [PubMed] [Google Scholar]
- 11.Sendoel A, Hengartner MO. Apoptotic cell death under hypoxia. Physiology (Bethesda) 2014;29:168–176. doi: 10.1152/physiol.00016.2013. [DOI] [PubMed] [Google Scholar]
- 12.Yang Z, Zhao TZ, Zou YJ, Zhang JH, Feng H. Hypoxia induces autophagic cell death through hypoxia-inducible factor 1alpha in microglia. PLoS One. 2014;9:e96509. doi: 10.1371/journal.pone.0096509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lenihan CR, Taylor CT. The impact of hypoxia on cell death pathways. Biochem Soc Trans. 2013;41:657–663. doi: 10.1042/BST20120345. [DOI] [PubMed] [Google Scholar]
- 14.Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22:177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 15.Rosenbaum DM, Michaelson M, Batter DK, Doshi P, Kessler JA. Evidence for hypoxia-induced, programmed cell death of cultured neurons. Ann Neurol. 1994;36:864–870. doi: 10.1002/ana.410360610. [DOI] [PubMed] [Google Scholar]
- 16.Rotin D, Robinson B, Tannock IF. Influence of hypoxia and an acidic environment on the metabolism and viability of cultured cells: potential implications for cell death in tumors. Cancer Res. 1986;46:2821–2826. [PubMed] [Google Scholar]
- 17.Dunwoodie SL. The role of hypoxia in development of the mammalian embryo. Dev Cell. 2009;17:755–773. doi: 10.1016/j.devcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 18.Webster WS, Abela D. The effect of hypoxia in development. Birth Defects Res C Embryo Today. 2007;81:215–228. doi: 10.1002/bdrc.20102. [DOI] [PubMed] [Google Scholar]
- 19.Giaccia AJ, Simon MC, Johnson R. The biology of hypoxia: the role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004;18:2183–2194. doi: 10.1101/gad.1243304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JW, Ko J, Ju C, Eltzschig HK. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med. 2019;51:68. doi: 10.1038/s12276-019-0235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Semenza GL, Agani F, Feldser D, Iyer N, Kotch L, Laughner E, et al. Hypoxia, HIF-1, and the pathophysiology of common human diseases. Adv Exp Med Biol. 2000;475:123–130. doi: 10.1007/0-306-46825-5_12. [DOI] [PubMed] [Google Scholar]
- 22.Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng KH, et al. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc Natl Acad Sci U S A. 2017;114:E4241–E4E50. doi: 10.1073/pnas.1621511114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi JT, Wang Z, Nuyten DS, Rodriguez EH, Schaner ME, Salim A, et al. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 2006;3:e47. doi: 10.1371/journal.pmed.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferdinand P, Roffe C. Hypoxia after stroke: a review of experimental and clinical evidence. Exp Transl Stroke Med. 2016;8:9. doi: 10.1186/s13231-016-0023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guimaraes-Camboal N, Evans SM. Redox Paradox: can hypoxia heal ischemic hearts? Dev Cell. 2016;39:392–394. doi: 10.1016/j.devcel.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semenza G. Vascular responses to ischemia regulated by hypoxia-inducible factor 1. J Vasc Res. 2011;48:27. [Google Scholar]
- 27.Semenza GL. Vascular responses to hypoxia and ischemia. Arterioscler Thromb Vasc Biol. 2010;30:648–652. doi: 10.1161/ATVBAHA.108.181644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deveza L, Choi J, Yang F. Therapeutic angiogenesis for treating cardiovascular diseases. Theranostics. 2012;2:801–814. doi: 10.7150/thno.4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Semenza GL. Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med. 2003;54:17–28. doi: 10.1146/annurev.med.54.101601.152418. [DOI] [PubMed] [Google Scholar]
- 30.Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- 31.Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83–92. doi: 10.2147/HP.S93413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Endler A, Shibasaki F. Hypoxia and angiogenesis: regulation of hypoxia-inducible factors via novel binding factors. Exp Mol Med. 2009;41:849–857. doi: 10.3858/emm.2009.41.12.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cabral T, Mello LGM, Lima LH, Polido J, Regatieri CV, Belfort R, Jr, et al. Retinal and choroidal angiogenesis: a review of new targets. Int J Retina Vitreous. 2017;3:31. doi: 10.1186/s40942-017-0084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al-Kharashi AS. Role of oxidative stress, inflammation, hypoxia and angiogenesis in the development of diabetic retinopathy. Saudi J Ophthalmol. 2018;32:318–323. doi: 10.1016/j.sjopt.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulus YM, Sodhi A. Anti-angiogenic therapy for retinal disease. Handb Exp Pharmacol. 2017;242:271–307. doi: 10.1007/164_2016_78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stone J, Itin A, Alon T, Peer J, Gnessin H, Chanling T, et al. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth-factor (Vegf) expression by neuroglia. J Neurosci. 1995;15:4738–4747. doi: 10.1523/JNEUROSCI.15-07-04738.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim M, Lee C, Payne R, Yue BYJT, Chang JH, Ying HY. Angiogenesis in glaucoma filtration surgery and neovascular glaucoma: a review. Surv Ophthalmol. 2015;60:524–535. doi: 10.1016/j.survophthal.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonn D. Blocking angiogenesis in diabetic retinopathy. Lancet. 1996;348:604. doi: 10.1016/S0140-6736(05)64809-X. [DOI] [PubMed] [Google Scholar]
- 39.Mcintosh LC, Ewen L, Forrester JV. Invitro models of angiogenesis in diabetic-retinopathy. Ophthalmic Res. 1987;19:18. [Google Scholar]
- 40.Sebag J, Mcmeel JW. Diabetic-retinopathy - pathogenesis and the role of retina-derived growth-factor in angiogenesis. Surv Ophthalmol. 1986;30:377–384. doi: 10.1016/0039-6257(86)90091-3. [DOI] [PubMed] [Google Scholar]
- 41.Frank RN. Diabetic retinopathy - clinical applications of angiogenesis research. Retinal and choroidal angiogenesis. 2008. [Google Scholar]
- 42.Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88:5680–5684. doi: 10.1073/pnas.88.13.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor-1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O-2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 45.Ivan M, Kondo K, Yang HF, Kim W, Valiando J, Ohh M, et al. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O-2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 46.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O-2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 47.Bensellam M, Maxwell EL, Chan JY, Luzuriaga J, West PK, Jonas JC, et al. Hypoxia reduces ER-to-Golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mouse beta cells. Diabetologia. 2016;59:1492–1502. doi: 10.1007/s00125-016-3947-y. [DOI] [PubMed] [Google Scholar]
- 48.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 49.Maamoun H, Benameur T, Pintus G, Munusamy S, Agouni A. Crosstalk between oxidative stress and endoplasmic reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: a focus on the protective roles of heme oxygenase (HO)-1. Front Physiol. 2019;10:70. https://www.frontiersin.org/article/10.3389/fphys.2019.00070. [DOI] [PMC free article] [PubMed]
- 50.Binet F, Sapieha P. ER stress and angiogenesis. Cell Metab. 2015;22:560–575. doi: 10.1016/j.cmet.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 51.Martinez JA, Banerjee DK. Tunicamycin inhibits angiogenesis by ER stress. Glycobiology. 2000;10:1131. [Google Scholar]
- 52.Bartoszewski R, Kroliczewski J, Piotrowski A, Jasiecka AJ, Bartoszewska S, Vecchio-Pagan B, et al. Codon bias and the folding dynamics of the cystic fibrosis transmembrane conductance regulator. Cell Mol Biol Lett. 2016;21:23. doi: 10.1186/s11658-016-0025-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartoszewski R, Rab A, Fu L, Bartoszewska S, Collawn J, Bebok Z. CFTR expression regulation by the unfolded protein response. Methods Enzymol. 2011;491:3–24. doi: 10.1016/B978-0-12-385928-0.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 55.Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–290. doi: 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karagoz GE, Acosta-Alvear D, Walter P. The unfolded protein response: detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2019;11. 10.1101/cshperspect.a033886. [DOI] [PMC free article] [PubMed]
- 57.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 58.Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- 59.Gebert M, Bartoszewska S, Janaszak-Jasiecka A, Moszynska A, Cabaj A, Kroliczewski J, et al. PIWI proteins contribute to apoptosis during the UPR in human airway epithelial cells. Sci Rep. 2018;8:16431. doi: 10.1038/s41598-018-34861-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petrillo S, Chiabrando D, Genova T, Fiorito V, Ingoglia G, Vinchi F, et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018;25:573–588. doi: 10.1038/s41418-017-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ron D. Protein folding homeostasis in the endoplasmic reticulum. FEBS J. 2011;278:23. [Google Scholar]
- 62.Naidoo N. Comprehensive biotechnology, Vol 1: scientific fundamentals of biotechnology. 2. 2011. Protein folding in the endoplasmic reticulum; pp. 217–227. [Google Scholar]
- 63.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 64.Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol. 2004;16:343–349. doi: 10.1016/j.ceb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 65.Helenius A, Tatu U, Marquardt T, Braakman I. Protein folding in the endoplasmic reticulum. In: Oka MS, Rupp RG, editors. Cell Biology and Biotechnology. New York: Springer New York; 1993.
- 66.Hagiwara M, Nagata K. Redox-dependent protein quality control in the endoplasmic reticulum: folding to degradation. Antioxid Redox Signal. 2012;16:1119–1128. doi: 10.1089/ars.2011.4495. [DOI] [PubMed] [Google Scholar]
- 67.Braakman I. Disulfide bond formation during protein folding in the endoplasmic reticulum. FEBS J. 2005;272:349. doi: 10.1002/j.1460-2075.1992.tb05223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Braakman I, Hebert DN. Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5(5):a013201. doi: 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bartoszewski R, Rab A, Twitty G, Stevenson L, Fortenberry J, Piotrowski A, et al. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J Biol Chem. 2008;283:12154–12165. doi: 10.1074/jbc.M707610200. [DOI] [PubMed] [Google Scholar]
- 70.Chiang CK, Nangaku M, Tanaka T, Iwawaki T, Inagi R. Endoplasmic reticulum stress signal impairs erythropoietin production: a role for ATF4. Am J Physiol Cell Physiol. 2013;304:C342–CC53. doi: 10.1152/ajpcell.00153.2012. [DOI] [PubMed] [Google Scholar]
- 71.Manalo RVM. Anastasis and the ER stress response: solving the paradox of the unfolded protein response in cancer. Med Hypotheses. 2017;109:25–27. doi: 10.1016/j.mehy.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 72.Vandewynckel YP, Laukens D, Geerts A, Bogaerts E, Paridaens A, Verhelst X, et al. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013;33:4683–4694. [PubMed] [Google Scholar]
- 73.Hu C-J, Wang L-Y, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seton-Rogers S. Hypoxia: HIF switch. Nat Rev Cancer. 2011;11:391. doi: 10.1038/nrc3074. [DOI] [PubMed] [Google Scholar]
- 75.Semenza GL. HIF-1: using two hands to flip the angiogenic switch. Cancer Metastasis Rev. 2000;19:59–65. doi: 10.1023/A:1026544214667. [DOI] [PubMed] [Google Scholar]
- 76.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37:364–372. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Serocki M, Bartoszewska S, Janaszak-Jasiecka A, Ochocka RJ, Collawn JF, Bartoszewski R. miRNAs regulate the HIF switch during hypoxia: a novel therapeutic target. Angiogenesis. 2018;21:183–202. doi: 10.1007/s10456-018-9600-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? Clin Invest. 2007;117:862–865. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loboda A, Jozkowicz A, Dulak J. HIF-1 versus HIF-2 — Is one more important than the other? Vascul Pharmacol. 2012;56:245–251. doi: 10.1016/j.vph.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 81.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang P, Yao Q, Lu L, Li Y, Chen PJ, Duan CM. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014;6:1110–1121. doi: 10.1016/j.celrep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 83.Janaszak-Jasiecka A, Bartoszewska S, Kochan K, Piotrowski A, Kalinowski L, Kamysz W, et al. miR-429 regulates the transition between Hypoxia-Inducible Factor (HIF)1A and HIF3A expression in human endothelial cells. Sci Rep. 2016;6:22775. doi: 10.1038/srep22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ravenna L, Salvatori L, Russo MA. HIF3alpha: the little we know. FEBS J. 2016;283:993–1003. doi: 10.1111/febs.13572. [DOI] [PubMed] [Google Scholar]
- 85.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 86.Huang D, Li TT, Li XH, Zhang L, Sun LC, He XP, et al. HIF-1-mediated suppression of Acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014;8:1930–1942. doi: 10.1016/j.celrep.2014.08.028. [DOI] [PubMed] [Google Scholar]
- 87.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 88.Wu PF, Peters JM, Harris RA. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem Biophys Res Commun. 2001;287:391–396. doi: 10.1006/bbrc.2001.5608. [DOI] [PubMed] [Google Scholar]
- 89.Fukuda R, Zhang HF, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 90.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med. 2010;14:771–794. doi: 10.1111/j.1582-4934.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM, et al. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009;69:358–368. doi: 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- 93.Chee NT, Lohse I, Brothers SP. mRNA-to-protein translation in hypoxia. Mol Cancer. 2019;18(1):49. doi: 10.1186/s12943-019-0968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thomas JD, Dias LM, Johannes GJ. Translational repression during chronic hypoxia is dependent on glucose levels. RNA. 2008;14:771–781. doi: 10.1261/rna.857308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fahling M. Surviving hypoxia by modulation of mRNA translation rate. J Cell Mol Med. 2009;13:2770–2779. doi: 10.1111/j.1582-4934.2009.00875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yi TF, Papadopoulos E, Hagner PR, Wagner G. Hypoxia-inducible factor-1 alpha (HIF-1 alpha) promotes cap-dependent translation of selective mRNAs through up-regulating initiation factor eIF4E1 in breast cancer cells under hypoxia conditions. J Biol Chem. 2013;288:18732–18742. doi: 10.1074/jbc.M113.471466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van den Beucken T, Magagnin MG, Jutten B, Seigneuric R, Lambin P, Koritzinsky M, et al. Translational control is a major contributor to hypoxia induced gene expression. Radiother Oncol. 2011;99:379–384. doi: 10.1016/j.radonc.2011.05.058. [DOI] [PubMed] [Google Scholar]
- 98.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15:1572–1581. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 100.Buchler P, Reber HA, Buchler M, Shrinkante S, Buchler MW, Friess H, et al. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas. 2003;26:56–64. doi: 10.1097/00006676-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 101.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem. 1997;272:5375–5381. doi: 10.1074/jbc.272.9.5375. [DOI] [PubMed] [Google Scholar]
- 102.Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1 alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Koh MY, Lemos R, Jr, Liu X, Powis G. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011;71:4015–4027. doi: 10.1158/0008-5472.CAN-10-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Covello KL, Kehler J, Yu HW, Gordan JD, Arsham AM, Hu CJ, et al. HIF-2 alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pichiule P, Chavez JC, LaManna JC. Hypoxic regulation of angiopoietin-2 expression in endothelial cells. J Biol Chem. 2004;279:12171–12180. doi: 10.1074/jbc.M305146200. [DOI] [PubMed] [Google Scholar]
- 106.Hitchon C, Wong K, Ma GP, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46:2587–2597. doi: 10.1002/art.10520. [DOI] [PubMed] [Google Scholar]
- 107.Schito L, Rey S, Tafani M, Zhang HF, Wong CCL, Russo A, et al. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proc Natl Acad Sci U S A. 2012;109:E2707–E2E16. doi: 10.1073/pnas.1214019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Adelman DM, Gertsenstein M, Nagy A, Simon MC, Maltepe E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000;14:3191–3203. doi: 10.1101/gad.853700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Han Z-B, Ren H, Zhao H, Chi Y, Chen K, Zhou B, et al. Hypoxia-inducible factor (HIF)-1α directly enhances the transcriptional activity of stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF) Carcinogenesis. 2008;29:1853–1861. doi: 10.1093/carcin/bgn066. [DOI] [PubMed] [Google Scholar]
- 110.Kalinowski L, Janaszak-Jasiecka A, Siekierzycka A, Bartoszewska S, Wozniak M, Lejnowski D, et al. Posttranscriptional and transcriptional regulation of endothelial nitric-oxide synthase during hypoxia: the role of microRNAs. Cell Mol Biol Lett. 2016;21:16. doi: 10.1186/s11658-016-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37:4587–4602. doi: 10.1093/nar/gkp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013;27:41–53. doi: 10.1016/j.blre.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kirito K. Regulation of erythropoiesis by hypoxia inducible factors (HIFs) Rinsho Ketsueki. 2011;52:368–375. [PubMed] [Google Scholar]
- 114.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/MCB.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009;9:152–164. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tramontano AF, Muniyappa R, Black AD, Blendea MC, Cohen I, Deng LL, et al. Erythropoietin protects cardiac myocytes from hypoxia-induced apoptosis through an Akt-dependent pathway. Biochem Biophys Res Commun. 2003;308:990–994. doi: 10.1016/S0006-291X(03)01503-1. [DOI] [PubMed] [Google Scholar]
- 118.Wu Y, Shang Y, Sun SG, Liang HF, Liu RG. Erythropoietin prevents PC12 cells from 1-methyl-4-phenylpyridinium ion-induced apoptosis via the Akt/GSK-3 beta/caspase-3 mediated signaling pathway. Apoptosis. 2007;12:1365–1375. doi: 10.1007/s10495-007-0065-9. [DOI] [PubMed] [Google Scholar]
- 119.Arcasoy MO. The non-haematopoietic biological effects of erythropoietin. Br J Haematol. 2008;141:14–31. doi: 10.1111/j.1365-2141.2008.07014.x. [DOI] [PubMed] [Google Scholar]
- 120.Koritzinsky M, Levitin F, van den Beucken T, Rumantir RA, Harding NJ, Chu KC, et al. Two phases of disulfide bond formation have differing requirements for oxygen. J Cell Biol. 2013;203:615–627. doi: 10.1083/jcb.201307185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.May D, Itin A, Gal O, Kalinski H, Feinstein E, Keshet E. Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene. 2005;24:1011–1020. doi: 10.1038/sj.onc.1208325. [DOI] [PubMed] [Google Scholar]
- 122.Arnould T, Michiels C, Alexandre I, Remacle J. Effect of hypoxia upon intracellular calcium-concentration of human endothelial-cells. J Cell Physiol. 1992;152:215–221. doi: 10.1002/jcp.1041520127. [DOI] [PubMed] [Google Scholar]
- 123.Dorner AJ, Wasley LC, Kaufman RJ. Protein dissociation from Grp78 and secretion are blocked by depletion of cellular Atp levels. Proc Natl Acad Sci U S A. 1990;87:7429–7432. doi: 10.1073/pnas.87.19.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bartoszewski R, Matalon S, Collawn JF. Ion channels of the lung and their role in disease pathogenesis. Am J Physiol Lung Cell Mol Physiol. 2017;313:L859–LL72. doi: 10.1152/ajplung.00285.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2019;286:241–278. doi: 10.1111/febs.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Melber A, Haynes CM. UPRmt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018;28:281–295. doi: 10.1038/cr.2018.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Martin J. Molecular chaperones and mitochondrial protein folding. J Bioenerg Biomembr. 1997;29:35–43. doi: 10.1023/A:1022407705182. [DOI] [PubMed] [Google Scholar]
- 128.Kueh HY, Niethammer P, Mitchison TJ. Maintenance of mitochondrial oxygen homeostasis by cosubstrate compensation. Biophys J. 2013;104:1338–1348. doi: 10.1016/j.bpj.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. 2018;19:109–120. doi: 10.1038/nrm.2017.110. [DOI] [PubMed] [Google Scholar]
- 130.Munch C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018;16(1):81. doi: 10.1186/s12915-018-0548-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 132.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/S1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 133.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gonen N, Sabath N, Burge CB, Shalgi R. Widespread PERK-dependent repression of ER targets in response to ER stress. Sci Rep. 2019;9(1):4330. doi: 10.1038/s41598-019-38705-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Han J, Backa SH, Hur J, Lin YH, Gildersleeve R, Shan JX, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–48+. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rutkowski DT, Kaufman RJ. All roads lead to ATF4. Dev Cell. 2003;4:442–444. doi: 10.1016/S1534-5807(03)00100-X. [DOI] [PubMed] [Google Scholar]
- 137.Novoa I, Zeng HQ, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2 alpha. J Cell Biol. 2001;153:1011–1021. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2003;11:381. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 139.B'chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2 alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–7699. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Han D, Lerner AG, Vande Walle L, Upton JP, Xu WH, Hagen A, et al. IRE1 alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 142.Bartoszewska S, Cabaj A, Dabrowski M, Collawn JF, Bartoszewski R. miR-34c-5p modulates X-box-binding protein 1 (XBP1) expression during the adaptive phase of the unfolded protein response. FASEB J. 2019;33:11541–11554. doi: 10.1096/fj.201900600RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bartoszewski R, Brewer JW, Rab A, Crossman DK, Bartoszewska S, Kapoor N, et al. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J Biol Chem. 2011;286:41862–41870. doi: 10.1074/jbc.M111.304956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Urano F, Wang XZ, Bertolotti A, Zhang YH, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 145.Li MQ, Baumeister P, Roy B, Phan T, Foti D, Luo SZ, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol Cell Biol. 2000;20:5096–5106. doi: 10.1128/MCB.20.14.5096-5106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang KZ, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- 147.Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283:2640–2652. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- 148.Bartoszewski R, Gebert M, Janaszak-Jasiecka A, Cabaj A, Kroliczewski J, Bartoszewska S, et al. Genome-wide mRNA profiling identifies RCAN1 and GADD45A as regulators of the transitional switch from survival to apoptosis during ER stress. FEBS J. 2019. 10.1111/febs.15195. [DOI] [PubMed]
- 149.Walter F, O'Brien A, Concannon CG, Dussmann H, Prehn JHM. ER stress signaling has an activating transcription factor 6 (ATF6)-dependent “off-switch”. J Biol Chem. 2018;293:18270–18284. doi: 10.1074/jbc.RA118.002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Saveljeva S, Mc Laughlin SL, Vandenabeele P, Samali A, Bertrand MJ. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015;6:e1587. doi: 10.1038/cddis.2014.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Livezey M, Huang R, Hergenrother PJ, Shapiro DJ. Strong and sustained activation of the anticipatory unfolded protein response induces necrotic cell death. Cell Death Differ. 2018;25:1796–1807. doi: 10.1038/s41418-018-0143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shirjang S, Mansoori B, Asghari S, Duijf PHG, Mohammadi A, Gjerstorff M, et al. MicroRNAs in cancer cell death pathways: apoptosis and necroptosis. Free Radic Biol Med. 2019;139:1–15. doi: 10.1016/j.freeradbiomed.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 153.Kishino A, Hayashi K, Maeda M, Jike T, Hidai C, Nomura Y, et al. Caspase-8 regulates endoplasmic reticulum stress-induced necroptosis independent of the apoptosis pathway in auditory cells. Int J Mol Sc. 2019;20. 10.3390/ijms20235896. [DOI] [PMC free article] [PubMed]
- 154.Ding BX, Parmigiani A, Divakaruni AS, Archer K, Murphy AN, Budanov AV. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci Rep. 2016;6:22538. doi: 10.1038/srep22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cheng SB, Nakashima A, Huber WJ, Davis S, Banerjee S, Huang ZP, et al. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019;10(12):1–5. doi: 10.1038/s41419-019-2162-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol. 2018;20:186–197. doi: 10.1038/s41556-017-0022-y. [DOI] [PubMed] [Google Scholar]
- 157.Qiu XF, Zhang YY, Han JH. RIP3 is an upregulator of aerobic metabolism and the enhanced respiration by necrosomal RIP3 feeds back on necrosome to promote necroptosis. Cell Death Differ. 2018;25:821–824. doi: 10.1038/s41418-018-0075-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fulda S. Alternative cell death pathways and cell metabolism. Int J Cell Biol. 2013;2013:463637. doi: 10.1155/2013/463637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gibellini L, Losi L, De Biasi S, Nasi M, Lo Tartaro D, Pecorini S, et al. LonP1 differently modulates mitochondrial function and bioenergetics of primary versus metastatic colon cancer cells. Front Oncol. 2018;8:254. doi: 10.3389/fonc.2018.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Peter B, Waddington CL, Olahova M, Sommerville EW, Hopton S, Pyle A, et al. Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum Mol Genet. 2018;27:1743–1753. doi: 10.1093/hmg/ddy080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cheng MY, Hartl FU, Martin J, Pollock RA, Kalousek F, Neupert W, et al. Mitochondrial heat-shock protein Hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 1989;337:620–625. doi: 10.1038/337620a0. [DOI] [PubMed] [Google Scholar]
- 162.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Quiros PM, Prado MA, Zamboni N, D'Amico D, Williams RW, Finley D, et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol. 2017;216:2027–2045. doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol. 2016;26:2037–2043. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2 alpha kinase GCN-2. PLoS Genet. 2012;8(6):e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2:213–219. doi: 10.2147/vhrm.2006.2.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Koumenis C, Wouters BG. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. 2006;4:423–436. doi: 10.1158/1541-7786.MCR-06-0150. [DOI] [PubMed] [Google Scholar]
- 168.Moenner M, Pluquet O, Bouchecareilh M, Chevet E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007;67:10631–10634. doi: 10.1158/0008-5472.CAN-07-1705. [DOI] [PubMed] [Google Scholar]
- 169.Mujcic H, Rzymski T, Rouschop KMA, Koritzinsky M, Milani M, Harris AL, et al. Hypoxic activation of the unfolded protein response (UPR) induces expression of the metastasis-associated gene LAMP3. Radiother Oncol. 2009;92:450–459. doi: 10.1016/j.radonc.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 170.Wang YG, Alam GN, Ning Y, Visioli F, Dong ZH, Nor JE, et al. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012;72:5396–5406. doi: 10.1158/0008-5472.CAN-12-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rouschop KMA, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sun LL, Chen CM, Zhang J, Wang J, Yang CZ, Lin LZ. Glucose-regulated protein 78 signaling regulates hypoxia-induced epithelial-mesenchymal transition in A549 cells. Front Oncol. 2019;9:137. doi: 10.3389/fonc.2019.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Song MS, Park YK, Lee JH, Park K. Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumor cells through a protein kinase C-epsilon/ERK/AP-1 signaling cascade. Cancer Res. 2001;61:8322–8330. [PubMed] [Google Scholar]
- 174.Koong AC, Auger EA, Chen EY, Giaccia AJ. The regulation of Grp78 and messenger-Rna levels by hypoxia is modulated by protein-kinase-C activators and inhibitors. Radiat Res. 1994;138:S60–SS3. doi: 10.2307/3578763. [DOI] [PubMed] [Google Scholar]
- 175.Raiter A, Weiss C, Bechor Z, Ben-Dor I, Battler A, Kaplan B, et al. Activation of GRP78 on endothelial cell membranes by an ADAM15-derived peptide induces angiogenesis. J Vasc Res. 2010;47:399–411. doi: 10.1159/000281580. [DOI] [PubMed] [Google Scholar]
- 176.Koong AC, Chen EY, Lee AS, Brown JM, Giaccia AJ. Increased cytotoxicity of chronic hypoxic cells by molecular inhibition of Grp78 induction. Int J Radiat Oncol Biol Phys. 1994;28:661–666. doi: 10.1016/0360-3016(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 177.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Blais JD, Filipenko V, Bi MX, Harding HP, Ron D, Koumenis C, et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol. 2004;24:7469–7482. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Scheuner D, Song BB, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/S1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 180.Liu LP, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ye J, Koumenis C. ATF4, an ER stress and hypoxia-inducible transcription factor and its potential role in hypoxia tolerance and tumorigenesis. Curr Mol Med. 2009;9:411–416. doi: 10.2174/156652409788167096. [DOI] [PubMed] [Google Scholar]
- 182.Bensellam M, Maxwell E, Jonas JC, Chan J, Laybutt DR. Hypoxia induces beta cell death by inhibiting the adaptive UPR. Diabetologia. 2015;58:S235. doi: 10.1007/s00125-015-3503-1. [DOI] [PubMed] [Google Scholar]
- 183.Rouschop KM, Dubois LJ, Keulers TG, van den Beucken T, Lambin P, Bussink J, et al. PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc Natl Acad Sci U S A. 2013;110:4622–4627. doi: 10.1073/pnas.1210633110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mujcic H, Nagelkerke A, Rouschop KMA, Chung S, Chaudary N, Span PN, et al. Hypoxic activation of the PERK/eIF2 alpha arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin Cancer Res. 2013;19:6126–6137. doi: 10.1158/1078-0432.CCR-13-0526. [DOI] [PubMed] [Google Scholar]
- 185.Saxena K, Jolly MK. Acute vs. chronic vs. cyclic hypoxia: their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules. 2019;9. 10.3390/biom9080339. [DOI] [PMC free article] [PubMed]
- 186.Chen A, Sceneay J, Godde N, Kinwel T, Ham S, Thompson EW, et al. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene. 2018;37:4214–4225. doi: 10.1038/s41388-018-0259-3. [DOI] [PubMed] [Google Scholar]
- 187.Kochan-Jamrozy K, Kroliczewski J, Moszynska A, Collawn JF, Bartoszewski R. miRNA networks modulate human endothelial cell adaptation to cyclic hypoxia. Cell Signal. 2019;54:150–160. doi: 10.1016/j.cellsig.2018.11.020. [DOI] [PubMed] [Google Scholar]
- 188.Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J. 1999;339:135–141. [PMC free article] [PubMed] [Google Scholar]
- 189.Wolfgang CD, Chen BPC, Martindale JL, Holbrook NJ, Hai T. gadd153/Chop10, a potential target gene of the transcriptional repressor ATF3. Mol Cell Biol. 1997;17:6700–6707. doi: 10.1128/MCB.17.11.6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ivanova IG, Park CV, Yemm AI, Kenneth NS. PERK/eIF2 alpha signaling inhibits HIF-induced gene expression during the unfolded protein response via YB1-dependent regulation of HIF1 alpha translation. Nucleic Acids Res. 2018;46:3878–3890. doi: 10.1093/nar/gky127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2 alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. 2016;16:533–544. doi: 10.2174/1566524016666160523143937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Delbrel E, Soumare A, Naguez A, Label R, Bernard O, Bruhat A, et al. HIF-1 alpha triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci Rep. 2018;8(1):1–4. doi: 10.1038/s41598-018-36063-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yang D, Gao LL, Wang TF, Qiao ZD, Liang YJ, Zhang P. Hypoxia triggers endothelial endoplasmic reticulum stress and apoptosis via induction of VLDL receptor. FEBS Lett. 2014;588:4448–4456. doi: 10.1016/j.febslet.2014.09.046. [DOI] [PubMed] [Google Scholar]
- 194.Xie P, Duan YC, Guo XZ, Hu LN, Yu MH. SalA attenuates hypoxia-induced endothelial endoplasmic reticulum stress and apoptosis via down-regulation of VLDL receptor expression. Cell Physiol Biochem. 2015;35:17–28. doi: 10.1159/000369671. [DOI] [PubMed] [Google Scholar]
- 195.van den Beucken T, Koritzinsky M, Niessen H, Dubois L, Savelkouls K, Mujcic H, et al. Hypoxia-induced expression of carbonic anhydrase 9 is dependent on the unfolded protein response. J Biol Chem. 2009;284:24204–24212. doi: 10.1074/jbc.M109.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dong L, Krewson EA, Yang LV. Acidosis activates endoplasmic reticulum stress pathways through GPR4 in human vascular endothelial cells. Int J Mol Sci. 2017;18. 10.3390/ijms18020278. [DOI] [PMC free article] [PubMed]
- 197.Loinard C, Zouggari Y, Rueda P, Ramkhelawon B, Cochain C, Vilar J, et al. C/EBP homologous protein-10 (CHOP-10) limits postnatal neovascularization through control of endothelial nitric oxide synthase gene expression. Circulation. 2012;125:1014–U126. doi: 10.1161/CIRCULATIONAHA.111.041830. [DOI] [PubMed] [Google Scholar]
- 198.De Pascali F, Hemann C, Samons K, Chen CA, Zweier JL. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S-glutathionylation. Biochemistry. 2014;53:3679–3688. doi: 10.1021/bi500076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Janaszak-Jasiecka A, Siekierzycka A, Bartoszewska S, Serocki M, Dobrucki LW, Collawn JF, et al. eNOS expression and NO release during hypoxia is inhibited by miR-200b in human endothelial cells. Angiogenesis. 2018;21:711–724. doi: 10.1007/s10456-018-9620-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Badran M, Abuyassin B, Golbidi S, Ayas N, Laher I. Uncoupling of vascular nitric oxide synthase caused by intermittent hypoxia. Oxid Med Cell Longev. 2016:2354870. 10.1155/2016/2354870. [DOI] [PMC free article] [PubMed]
- 201.Xia ZX, Wu SY, Wei X, Liao YF, Yi P, Liu Y, et al. Hypoxic ER stress suppresses beta-catenin expression and promotes cooperation between the transcription factors XBP1 and HIF1 alpha for cell survival. J Biol Chem. 2019;294:13811–13821. doi: 10.1074/jbc.RA119.008353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Liang HC, Xiao J, Zhou ZM, Wu J, Ge F, Li ZC, Zhang HL, Sun J, Li FB, Liu R, Chen CS. Hypoxia induces miR-153 through the IRE1 alpha-XBP1 pathway to fine-tune the HIF1 alpha/VEGFA axis in breast cancer angiogenesis. Oncogene. 2018;37:1961-75. [DOI] [PMC free article] [PubMed]
- 203.Liang HC, Xiao J, Zhou ZM, Wu J, Ge F, Li ZC, et al. Hypoxia induces miR-153 through the IRE1 alpha-XBP1 pathway to fine tune the HIF1 alpha/VEGFA axis in breast cancer angiogenesis. Oncogene. 2018;37:1961–1975. doi: 10.1038/s41388-017-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chen X, Iliopoulos D, Zhang Q, Tang QZ, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1 alpha pathway. Nature. 2014;508:103–10+. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Xu XD, Qimuge AD, Wang HL, Xing C, Gu Y, Liu SS, et al. IRE1 alpha/XBP1s branch of UPR links HIF1 alpha activation to mediate ANGII-dependent endothelial dysfunction under particulate matter (PM) 2.5 exposure. Sci Rep. 2017;7(1):13507. doi: 10.1038/s41598-017-13156-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Romero-Ramirez L, Cao HB, Nelson D, Hammond E, Lee AH, Yoshida H, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–5947. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 207.Romero L, Cao H, Hammond E, Giaccia AJ, Le QT, Koong AC. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Int J Radiat Oncol Biol Phys. 2004;60:S192–S1S3. doi: 10.1016/j.ijrobp.2004.06.130. [DOI] [PubMed] [Google Scholar]
- 208.Bouchecareilh M, Chevet E, Bikfalvi A, Moenner M, Drogat B, Auguste P, et al. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-A expression and contributes to angiogenesis and tumor growth in vivo. Bull Du Cancer. 2007;94:S283–S2S4. doi: 10.1158/0008-5472.CAN-06-3235. [DOI] [PubMed] [Google Scholar]
- 209.Drogat B, Auguste P, Nguyen DT, Bouchecareilh M, Pineau R, Nalbantoglu J, et al. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-a expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. 2007;67:6700–6707. doi: 10.1158/0008-5472.CAN-06-3235. [DOI] [PubMed] [Google Scholar]
- 210.Karar J, Dolt KS, Pasha MAQ. Endoplasmic reticulum stress response in murine kidney exposed to acute hypobaric hypoxia. FEBS Lett. 2008;582:2521–2526. doi: 10.1016/j.febslet.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 211.Cao X, He Y, Li X, Xu Y, Liu X. The IRE1α-XBP1 pathway function in hypoxia-induced pulmonary vascular remodeling, is upregulated by quercetin, inhibits apoptosis and partially reverses the effect of quercetin in PASMCs. Am J Transl Res. 2019;11:641–654. [PMC free article] [PubMed] [Google Scholar]
- 212.Cojocari D, Vellanki RN, Sit B, Uehling D, Koritzinsky M, Wouters BG. New small molecule inhibitors of UPR activation demonstrate that PERK, but not IRE1 alpha signaling is essential for promoting adaptation and survival to hypoxia. Radiother Oncol. 2013;108:541–547. doi: 10.1016/j.radonc.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 213.Duan QL, Chen C, Yang L, Li N, Gong W, Li S, et al. MicroRNA regulation of unfolded protein response transcription factor XBP1 in the progression of cardiac hypertrophy and heart failure in vivo. J Transl Med. 2015;13:363. doi: 10.1186/s12967-015-0725-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Brewer JW, Jackson KP, Lee EH, Smith KM. The unfolded protein response, microRNA-214, and expression of the transcription factor XBP1. J Immunol. 2018;200(1). Abstract: 48.11. http://apps.webofknowledge.com/InboundService.do?customersID=ResearchSoft&mode=FullRecord&IsProductCode=Yes&product=WOS&Init=Yes&Func=Frame&DestFail=http%3A%2F%2Fwww.webofknowledge.com&action=retrieve&SrcApp=EndNote&SrcAuth=ResearchSoft&SID=C5C8j8MtCI2Q1PjgT6B&UT=WOS%3A000459977700213.
- 215.Chitnis N, Pytel D, Diehl JA. UPR-inducible miRNAs contribute to stressful situations. Trends Biochem Sci. 2013;38:447–452. doi: 10.1016/j.tibs.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Maurel M, Chevet E. Endoplasmic reticulum stress signaling: the microRNA connection. Am J Physiol Cell Physiol. 2013;304:C1117–C1C26. doi: 10.1152/ajpcell.00061.2013. [DOI] [PubMed] [Google Scholar]
- 217.Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. 2009;284:29735–29745. doi: 10.1074/jbc.M109.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Doroudgar S, Thuerauf DJ, Marcinko MM, Glembotski CC. Simulated Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response in cultured cardiac myocytes. Circ Res. 2008;103:E69–E70. doi: 10.1074/jbc.M109.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Gallagher CM, Walter P. Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. eLife. 2016;5:e11880. doi: 10.7554/eLife.11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sakaki K, Kaufman RJ. Interaction between quality control systems for ER protein folding and RNA biogenesis. Worm. 2013;2:e23005. doi: 10.4161/worm.23005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol. 2012;4:a015438. doi: 10.1101/cshperspect.a015438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 223.Listowski MA, Heger E, Boguslawska DM, Machnicka B, Kuliczkowski K, Leluk J, et al. microRNAs: fine tuning of erythropoiesis. Cell Mol Biol Lett. 2013;18:34–46. doi: 10.2478/s11658-012-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Pereira ER, Frudd K, Awad W, Hendershot LM. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF) J Biol Chem. 2014;289:3352–3364. doi: 10.1074/jbc.M113.507194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell. 2014;54:559–572. doi: 10.1016/j.molcel.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 226.Urra H, Hetz C. A novel ER stress-independent function of the UPR in angiogenesis. Mol Cell. 2014;54:542–544. doi: 10.1016/j.molcel.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 227.Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, et al. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS One. 2010;5:e9575. doi: 10.1371/journal.pone.0009575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Pereira ER, Liao N, Neale GA, Hendershot LM. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS One. 2010;5. 10.1371/journal.pone.0012521. [DOI] [PMC free article] [PubMed]
- 229.Roybal CN, Hunsaker LA, Barbash O, Vander Jagt DL, Abcouwer SF. The oxidative stressor arsenite activates vascular endothelial growth factor mRNA transcription by an ATF4-dependent mechanism. J Biol Chem. 2005;280:20331–20339. doi: 10.1074/jbc.M411275200. [DOI] [PubMed] [Google Scholar]
- 230.Kyriakakis E, Philippova M, Joshi MB, Pfaff D, Bochkov V, Afonyushkin T, et al. T-cadherin attenuates the PERK branch of the unfolded protein response and protects vascular endothelial cells from endoplasmic reticulum stress-induced apoptosis. Cell Signal. 2010;22:1308–1316. doi: 10.1016/j.cellsig.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 231.Afonyushkin T, Oskolkova OV, Philippova M, Resink TJ, Erne P, Binder BR, et al. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler Thromb Vasc Biol. 2010;30:1007–1013. doi: 10.1161/ATVBAHA.110.204354. [DOI] [PubMed] [Google Scholar]
- 232.Liu L, Qi X, Chen Z, Shaw L, Cai J, Smith LH, et al. Targeting the IRE1alpha/XBP1 and ATF6 arms of the unfolded protein response enhances VEGF blockade to prevent retinal and choroidal neovascularization. Am J Pathol. 2013;182:1412–1424. doi: 10.1016/j.ajpath.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Longchamp A, Mirabella T, Arduini A, MacArthur MR, Das A, Trevino-Villarreal JH, et al. Amino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S production. Cell. 2018;173:117–29.e14. doi: 10.1016/j.cell.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Terashima J, Tachikawa C, Kudo K, Habano W, Ozawa S. An aryl hydrocarbon receptor induces VEGF expression through ATF4 under glucose deprivation in HepG2. BMC Mol Biol. 2013;14:27. doi: 10.1186/1471-2199-14-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Pollreisz A, Afonyushkin T, Oskolkova OV, Gruber F, Bochkov VN, Schmidt-Erfurth U. Retinal pigment epithelium cells produce VEGF in response to oxidized phospholipids through mechanisms involving ATF4 and protein kinase CK2. Exp Eye Res. 2013;116:177–184. doi: 10.1016/j.exer.2013.08.021. [DOI] [PubMed] [Google Scholar]
- 236.Chai L, Ling K, He X, Yang R. Expression of ATF4 and VEGF in chorionic villus tissue in early spontaneous abortion. Eur J Obstet Gynecol Reprod Biol. 2013;170:434–438. doi: 10.1016/j.ejogrb.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 237.Oskolkova OV, Afonyushkin T, Leitner A, von Schlieffen E, Gargalovic PS, Lusis AJ, et al. ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood. 2008;112:330–339. doi: 10.1182/blood-2007-09-112870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 239.Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, et al. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci U S A. 2006;103:12741–12746. doi: 10.1073/pnas.0605457103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bruder L. Anoxia. In: Goldstein S, Naglieri JA, editors. Encyclopedia of child behavior and development. Boston: Springer US; 2011. pp. 108–109. [Google Scholar]
- 241.Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta. 2011;1813:1263–1268. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Sanjuan-Pla A, Cervera AM, Apostolova N, Garcia-Bou R, Victor VM, Murphy MP, et al. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1 alpha. FEBS Lett. 2005;579:2669–2674. doi: 10.1016/j.febslet.2005.03.088. [DOI] [PubMed] [Google Scholar]
- 243.Guzy RD, Hoyos B, Robin E, Chen H, Liu LP, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 244.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 245.Zweier JL. Measurement of superoxide-derived free-radicals in the reperfused heart - evidence for a free-radical mechanism of reperfusion injury. J Biol Chem. 1988;263:1353–1357. [PubMed] [Google Scholar]
- 246.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen-free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem. 1993;268:18532–18541. [PubMed] [Google Scholar]
- 247.Imarisio C, Alchera E, Revanna CB, Valente G, Follenzi A, Trisolini E, et al. Oxidative and ER stress-dependent ASK1 activation in steatotic hepatocytes and Kupffer cells sensitizes mice fatty liver to ischemia/reperfusion injury. Free Radic Biol Med. 2017;112:141–148. doi: 10.1016/j.freeradbiomed.2017.07.020. [DOI] [PubMed] [Google Scholar]
- 248.Van Kooten C, Pacchiarotta T, van der Pol P, de Fijter J, Schlagwein N, van Gijlswijk D, et al. ER stress and loss of GRP78 expression provides a link between renal ischemia/reperfusion injury and the urinary metabolome. Am J Transplant. 2016;16:638. [Google Scholar]
- 249.Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, et al. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am J Transplant. 2014;14:1552–1561. doi: 10.1111/ajt.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Gao F, Shen X, Lu T, Liu J, Busuttil RW, Kupiec-Weglinski JW, et al. IL-23 in liver ischemia/reperfusion injury (IRI): a synergy between ER stress and TLR4 activation. Am J Transplant. 2012;12:223. [Google Scholar]
- 251.Balachandran P, Dubray BJ, Upadhya GA, Jia J, Anderson C, Chapman WD. ER stress is an important mediator of ischemia reperfusion injury in hepatocytes isolated from steatotic livers. Am J Transplant. 2011;11:503. [Google Scholar]
- 252.Kaser A, Tomczak M, Blumberg RS. “ER stress(ed out)!”: paneth cells and ischemia-reperfusion injury of the small intestine. Gastroenterology. 2011;140:393–396. doi: 10.1053/j.gastro.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ren F, Liu J, Gao F, Shen XD, Busuttil RW, Kupiec-Weglinski JW, et al. Endoplasmic reticulum (ER) stress modulates tissue inflammatory responses and its implication in liver ischemia/reperfusion injury (IRI) Liver Transpl. 2010;16:S100. doi: 10.1097/TP.0b013e318259d38e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Vilatoba M, Eckstein C, Ringland S, Bilbao G, Thompson A, Eckhoff DE, et al. Sodium 4-phenylbutyrate (PBA) protects against liver ischemia reperfusion injury (I/R-injury) by inhibition of endoplasmic reticulum (ER)-stress mediated apoptosis. Am J Transplant. 2005;5:536. doi: 10.1016/j.surg.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 255.Ricca L, Lecorche E, Hamelin J, Balducci G, Azoulay D, Lemoine A. The unfolded protein response (Upr) can participate to the liver ischemic postconditioning protection against ischemia/reperfusion (I/R) injury via the modulation of Nf-Kb/Chop/Il-1 beta signaling pathway. Transpl Int. 2014;27:15. doi: 10.1111/tri.12310_2. [DOI] [Google Scholar]
- 256.Zhang CC, He SQ, Li YM, Li F, Liu ZB, Liu J, et al. Bisoprolol protects myocardium cells against ischemia/reperfusion injury by attenuating unfolded protein response in rats. Sci Rep. 2017;7:11859. doi: 10.1038/s41598-017-12366-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Le Pape S, Dimitrova E, Hannaert P, Konovalov A, Volmer R, Ron D, et al. Polynomial algebra reveals diverging roles of the unfolded protein response in endothelial cells during ischemia-reperfusion injury. FEBS Lett. 2014;588:3062–3067. doi: 10.1016/j.febslet.2014.05.065. [DOI] [PubMed] [Google Scholar]
- 258.Kim H, Zhao J, Lee D, Bai X, Cypel M, Keshavjee S, et al. Protein kinase C delta-mediated unfolded protein response and necrotic cell death contributes to ischemia-reperfusion induced injury in lung transplantation. J Heart Lung Transplant. 2014;33:S83. doi: 10.1016/j.healun.2014.01.257. [DOI] [Google Scholar]
- 259.Wang ZV, Deng YF, Gao NG, Pedrozo Z, Li DL, Tan W, Liang N, Lehrman MA, Rothermel BA, Lee AH, et al. The unfolded protein response directly activates the hexosamine biosynthetic pathway to protect the heart from ischemia/reperfusion injury. Circulation. 2013;128(22). Abstract: 11565.
- 260.Li YP, Wang SL, Liu B, Tang L, Kuang RR, Wang XB, et al. Sulforaphane prevents rat cardiomyocytes from hypoxia/reoxygenation injury in vitro via activating SIRT1 and subsequently inhibiting ER stress. Acta Pharmacol Sin. 2016;37:344–353. doi: 10.1038/aps.2015.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Xu JQ, Hu HX, Chen B, Yue RC, Zhou Z, Liu Y, et al. Lycopene protects against hypoxia/reoxygenation injury by alleviating ER stress induced apoptosis in neonatal mouse cardiomyocytes. PLoS One. 2015;10(8):e0136443. doi: 10.1371/journal.pone.0136443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Guan GP, Yang L, Huang WY, Zhang J, Zhang PH, Yu H, et al. Mechanism of interactions between endoplasmic reticulum stress and autophagy in hypoxia/reoxygenation-induced injury of H9c2 cardiomyocytes. Mol Med Rep. 2019;20:350–358. doi: 10.3892/mmr.2019.10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Xing J, Xu H, Liu CB, Wei ZL, Wang ZH, Zhao L, et al. Melatonin ameliorates endoplasmic reticulum stress in N2a neuroblastoma cell hypoxia-reoxygenation injury by activating the AMPK-Pak2 pathway. Cell Stress Chaperones. 2019;24:621–633. doi: 10.1007/s12192-019-00994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Li T, Chen LL, Yu YY, Yang BB, Li PY, Tan XQ. Resveratrol alleviates hypoxia/reoxygenation injury-induced mitochondrial oxidative stress in cardiomyocytes. Mol Med Rep. 2019;19:2774–2780. doi: 10.3892/mmr.2019.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Deng TM, Wang YH, Wang CC, Yan H. FABP4 silencing ameliorates hypoxia reoxygenation injury through the attenuation of endoplasmic reticulum stress-mediated apoptosis by activating PI3K/Akt pathway. Life Sci. 2019;224:149–156. doi: 10.1016/j.lfs.2019.03.046. [DOI] [PubMed] [Google Scholar]
- 266.Sun MY, Ma DS, Zhao S, Wang L, Ma CY, Bai Y. Salidroside mitigates hypoxia/reoxygenation injury by alleviating endoplasmic reticulum stress-induced apoptosis in H9c2 cardiomyocytes. Mol Med Rep. 2018;18:3760–3768. doi: 10.3892/mmr.2018.9403. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 267.Xu YX, Wang WT, Jin KK, Zhu QF, Lin HZ, Xie MY, et al. Perillyl alcohol protects human renal tubular epithelial cells from hypoxia/reoxygenation injury via inhibition of ROS, endoplasmic reticulum stress and activation of PI3K/Akt/eNOS pathway. Biomed Pharmacother. 2017;95:662–669. doi: 10.1016/j.biopha.2017.08.129. [DOI] [PubMed] [Google Scholar]
- 268.Lei X, Zhang S, Hu HX, Yue RC, Wang H, Chen HY, et al. Lycopene protects cardiomyocytes from hypoxia/reoxygenation injury via attenuating endoplasmic reticulum stress. J Am Coll Cardiol. 2014;64:C88–CC9. doi: 10.1016/j.jacc.2014.06.413. [DOI] [Google Scholar]
- 269.Wu XD, Zhang ZY, Sun S, Li YZ, Wang XR, Zhu XQ, et al. Hypoxic preconditioning protects microvascular endothelial cells against hypoxia/reoxygenation injury by attenuating endoplasmic reticulum stress. Apoptosis. 2013;18:85–98. doi: 10.1007/s10495-012-0766-6. [DOI] [PubMed] [Google Scholar]
- 270.Samarasinghe DA, Tapner M, Farrell GC. Role of oxidative stress in hypoxia-reoxygenation injury to cultured rat hepatic sinusoidal endothelial cells. Hepatology. 2000;31:160–165. doi: 10.1002/hep.510310124. [DOI] [PubMed] [Google Scholar]
- 271.Samarasinghe DA, Farrell GC. Role of redox stress in hypoxia-reoxygenation injury to hepatic sinusoidal endothelial cells. Hepatology. 1996;24:444. doi: 10.1002/hep.510240541. [DOI] [PubMed] [Google Scholar]
- 272.Tang JY, Jin P, He Q, Lu LH, Ma JP, Gao WL, et al. Naringenin ameliorates hypoxia/reoxygenation-induced endoplasmic reticulum stress-mediated apoptosis in H9c2 myocardial cells: involvement in ATF6, IRE1 alpha and PERK signaling activation. Mol Cell Biochem. 2017;424:111–122. doi: 10.1007/s11010-016-2848-1. [DOI] [PubMed] [Google Scholar]
- 273.Jia WK, Jian Z, Li JW, Luo L, Zhao L, Zhou Y, et al. Upregulated ATF6 contributes to chronic intermittent hypoxia-afforded protection against myocardial ischemia/reperfusion injury. Int J Mol Med. 2016;37:1199–1208. doi: 10.3892/ijmm.2016.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Song S, Tan J, Miao Y, Sun Z, Zhang Q. Intermittent-hypoxia-induced autophagy activation through the ER-stress-related PERK/eIF2alpha/ATF4 pathway is a protective response to pancreatic beta-cell apoptosis. Cell Physiol Biochem. 2018;51:2955–2971. doi: 10.1159/000496047. [DOI] [PubMed] [Google Scholar]
- 275.Yang YY, Shang J, Liu HG. Role of endoplasmic reticular stress in aortic endothelial apoptosis induced by intermittent/persistent hypoxia. Chin Med J. 2013;126:4517–4523. doi: 10.1097/00029330-201309050-00044. [DOI] [PubMed] [Google Scholar]
- 276.Brazovskaja A, Treutlein B, Camp JG. High-throughput single-cell transcriptomics on organoids. Curr Opin Biotechnol. 2019;55:167–171. doi: 10.1016/j.copbio.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 277.Tekin H, Simmons S, Cummings B, Gao LY, Adiconis X, Hession CC, et al. Effects of 3D culturing conditions on the transcriptomic profile of stem-cell-derived neurons. Nat Biomed Eng. 2018;2:540–554. doi: 10.1038/s41551-018-0219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Dewhirst MW, Cao YT, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Almendros I, Martinez-Garcia MA, Campos-Rodriguez F, Riveiro-Falkenbach E, Rodriguez-Peralto JL, Nagore E, et al. Intermittent hypoxia is associated with high hypoxia inducible factor-1 alpha but not high vascular endothelial growth factor cell expression in tumors of cutaneous melanoma patients. Front Neurol. 2018;9:272. doi: 10.3389/fneur.2018.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Yoon DW, So D, Min S, Kim J, Lee M, Khalmuratova R, et al. Accelerated tumor growth under intermittent hypoxia is associated with hypoxia-inducible factor-1-dependent adaptive responses to hypoxia. Oncotarget. 2017;8:61592–61603. doi: 10.18632/oncotarget.18644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Yoon DW, Min S, Kim Y, Kim JH, Lee GY, Lee M, et al. Intermittent hypoxia promotes tumor growth in azoxymethane and dextran sodium sulfate-induced colon carcinogenesis mouse model. Sleep Med. 2017;40:E356–E3E7. [Google Scholar]
- 282.Martinive P, Defresne F, Bouzin C, Saliez J, Lair F, Gregoire V, et al. Preconditioning of the tumor vasculature and tumor cells by intermittent hypoxia: implications for anti-cancer therapies. Acta Clin Belg. 2008;63:55. doi: 10.1158/0008-5472.CAN-06-2056. [DOI] [PubMed] [Google Scholar]
- 283.Franko AJ. Evidence against acute-hypoxia caused by intermittent blood-flow in Emt6 and Lewis lung-tumors. Radiat Res. 1981;87:457. [Google Scholar]
- 284.Bartoszewski R, Sikorski AF. Editorial focus: entering into the non-coding RNA era. Cell Mol Biol Lett. 2018;23:45. doi: 10.1186/s11658-018-0111-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Bartoszewska S, Kamysz W, Jakiela B, Sanak M, Kroliczewski J. Bebok Z, et al: miR-200b downregulates CFTR during hypoxia in human lung epithelial cells. Cell Mol Biol Lett. 2017;22:23. doi: 10.1186/s11658-017-0054-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Bartoszewski R, Serocki M, Janaszak-Jasiecka A, Bartoszewska S, Kochan-Jamrozy K, Piotrowski A, et al. miR-200b downregulates Kruppel Like Factor 2 (KLF2) during acute hypoxia in human endothelial cells. Eur J Cell Biol. 2017;96:758–766. doi: 10.1016/j.ejcb.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Bartoszewska S, Kochan K, Piotrowski A, Kamysz W, Ochocka RJ, Collawn JF, et al. The hypoxia-inducible miR-429 regulates hypoxia-inducible factor-1alpha expression in human endothelial cells through a negative feedback loop. FASEB J. 2015;29:1467–1479. doi: 10.1096/fj.14-267054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Bartoszewska S, Kochan K, Madanecki P, Piotrowski A, Ochocka R, Collawn JF, et al. Regulation of the unfolded protein response by microRNAs. Cell Mol Biol Lett. 2013;18:555–578. doi: 10.2478/s11658-013-0106-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Madanecki P, Kapoor N, Bebok Z, Ochocka R, Collawn JF, Bartoszewski R. Regulation of angiogenesis by hypoxia: the role of microRNA. Cell Mol Biol Lett. 2013;18:47–57. doi: 10.2478/s11658-012-0037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Sun SM, Xuan FJ, Ge XP, Zhu J, Zhang WX. Dynamic mRNA and miRNA expression analysis in response to hypoxia and reoxygenation in the blunt snout bream (Megalobrama amblycephala) Sci Rep. 2017;7(1):12846. doi: 10.1038/s41598-017-12537-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Rupaimoole R, Wu SY, Pradeep S, Ivan C, Pecot CV, Gharpure KM, et al. Hypoxia-mediated downregulation of miRNA biogenesis promotes tumour progression. Nat Commun. 2014;5:5202. doi: 10.1038/ncomms6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Rupaimoole R, Ivan C, Pecot C, Wu S, Pradeep S, Zand B, Nagaraja A, Gharpure K, Dalton H, Sadaoui N, et al. Hypoxia is a master regulator of Drosha- and Dicer-dependent miRNA biogenesis in cancer. Cancer Res. 2014;74(19). Abstract: 3532. 10.1158/1538-7445.AM2014-3532.
- 293.Zhao RB, Qian LJ, Jiang L. miRNA-dependent cross-talk between VEGF and Ang-2 in hypoxia-induced microvascular dysfunction. Biochem Biophys Res Commun. 2014;452:428–435. doi: 10.1016/j.bbrc.2014.08.096. [DOI] [PubMed] [Google Scholar]
- 294.Hua Z, Lv Q, Ye WB, Wong CKA, Cai GP, Gu DY, et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006;1(1):e116. doi: 10.1371/journal.pone.0000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Tharmalingham H, Hoskin P. Clinical trials targeting hypoxia. Br J Radiol. 2018. 10.1259/bjr.20170966:20170966. [DOI] [PMC free article] [PubMed]
- 296.Hetz C, Axten JM, Patterson JB. Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol. 2019;15:764–775. doi: 10.1038/s41589-019-0326-2. [DOI] [PubMed] [Google Scholar]
- 297.Rivas A, Vidal RL, Hetz C. Targeting the unfolded protein response for disease intervention. Expert Opin Ther Targets. 2015;19:1203–1218. doi: 10.1517/14728222.2015.1053869. [DOI] [PubMed] [Google Scholar]
- 298.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 299.Sharma A, Arambula JF, Koo S, Kumar R, Singh H, Sessler JL, et al. Hypoxia-targeted drug delivery. Chem Soc Rev. 2019;48:771–813. doi: 10.1039/C8CS00304A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Birle DC, Hedley DW. Suppression of the hypoxia-inducible factor-1 response in cervical carcinoma xenografts by proteasome inhibitors. Cancer Res. 2007;67:1735–1743. doi: 10.1158/0008-5472.CAN-06-2722. [DOI] [PubMed] [Google Scholar]
- 301.Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006;66:184–191. doi: 10.1158/0008-5472.CAN-05-1195. [DOI] [PubMed] [Google Scholar]
- 302.Pore N, Gupta AK, Cerniglia GJ, Maity A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia. 2006;8:889–895. doi: 10.1593/neo.06535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Pore N, Gupta AK, Cerniglia GJ, Jiang Z, Bernhard EJ, Evans SM, et al. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Cancer Res. 2006;66:9252–9259. doi: 10.1158/0008-5472.CAN-06-1239. [DOI] [PubMed] [Google Scholar]
- 304.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem. 2002;277:29936–29944. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.