

Abstract
AcrAB(Z)-TolC is the main drug efflux transporter complex in Escherichia coli. The extrusion of various toxic compounds depends on several drug binding sites within the trimeric AcrB transporter. Membrane-localized carboxylated substrates, like fusidic acid and hydrophobic β-lactams, access the pump via a groove between the transmembrane helices TM1 and TM2. In this report, the transport route from the initial TM1/TM2 groove binding site towards the deep binding pocket located in the periplasmic part has been addressed via molecular modelling studies followed by functional and structural characterization of several AcrB variants. We propose that membrane-embedded drugs bind initially to the TM1/TM2 groove, are oriented by the AcrB PN2 subdomain, and are subsequently transported via a PN2/PC1 interface pathway directly towards the deep binding pocket. Our work emphasizes the exploitation of multiple transport pathways by AcrB tuned to substrate physicochemical properties related to the polyspecificity of the pump.
Keywords: crystallography, drug transport, efflux, membrane protein, molecular modeling
Graphical Abstract
Introduction
In Gram-negative bacteria, Resistance Nodulation and cell Division (RND)-type efflux pumps are key players in the removal of toxic compounds, including clinically important antibiotics. The Escherichia coli AcrAB(Z)-TolC pump [1] comprises the inner membrane transporter AcrB as a homotrimeric membrane protein containing 36 transmembrane helices (TM1–12 in each monomer) and large periplasmic porter and funnel domains [2–4]. A membrane fusion protein, AcrA, is acting as a connection duct bridging AcrB and the outer membrane factor, TolC [5]. The resulting assembly forms an efficient and functional tripartite complex exporting toxic compounds localized in the periplasm or in the outer leaflet of the inner membrane [6].
Structural and functional studies indicated that the periplasmic porter domain is the substrate specificity determinant while energy transduction is mediated by proton translocation through the TM domain, energized by the electrochemical proton gradient across the inner membrane [5,7,8]. X-ray structural analysis of asymmetric trimeric AcrB revealed three different conformations of its protomers: loose (L), tight (T) and open (O), suggesting a multi-step transport cycle (functional rotation). Two main binding areas in the periplasmic porter domain were identified, the access pocket in the L conformation and the deep binding pocket in the T conformation. In addition, different channels with entrances located at the membrane/periplasm interface (Channel 1, CH1) or in the periplasm (CH2) lead to the deep binding or access pocket, respectively [3,4,7]. Recently, site-directed mutagenesis analysis revealed another substrate channel, designated CH3, whose entrance is located at the interface between periplasm and central cavity of the AcrB T protomer. This channel is connected to the deep binding pocket and it is involved in the direct transport of planar aromatic cations from the central cavity [9].
High molecular weight drugs initially bind to the access pocket in L and are presumably transported to the deep binding pocket upon conformational change to the T conformer. It has been suggested that low molecular weight drugs reach the deep binding pocket without prior binding to the access pocket in L [10]. Finally, the drug is expelled to the exit duct (comprised of an AcrA hexamer and a TolC trimer) [6], driven by a T to O transition, which is triggered by protonation of acidic residues involved in the proton translocation pathway in the AcrB TM domain [2–4,11–13]. AcrB is able to extrude a variety of structurally diverse toxins [8,14] including minocycline [2,11,12,15], doxorubicin [2,12], puromycin [6], erythromycin [11], rifampicin [11], rhodamine 6G [15], as well as bind pyranopyrimidine/pyranopyridine inhibitors [15,16]. These compounds were shown to bind in various positions at the access or deep binding sites in the periplasmic porter domain.
Substrates that are bound at the TM domain of AcrB have been reported in several previous studies. These ligand-bound AcrB structures represent the symmetric conformation of LLL protomers and were mainly identified at the central cavity of the transporter [17]. In 2016, we determined a crystal structure of AcrB/DARPin in complex with fusidic acid (FUA) bound at the TM domain, interacting with residues located at the TM1/TM2 groove (Fig. S1) [18]. From mutational analysis and substrate protection cross-linking assays, this newly identified membrane-localized binding pocket furthermore recognizes other carboxylated drugs like oxacillin (OXA), cloxacillin, dicloxacillin (DCX) and piperacillin (PIP), but not erythromycin, linezolid, novobiocin, taurocholate, deoxycholate, or rhodamine 6G [18]. Residue I337 was found to be most sensitive upon substitution, whereas substitution of V341 or H338 was less detrimental for binding and transport of the carboxylated substrates (Fig. S1). During the functional rotation cycle of the protomers, TM2 is upshifting (from T to O), and downshifting (from O to L), possibly changing the binding affinity for these drugs. The AcrB/FUA complex structure also displayed concomitant binding of an n-dodecyl-β-D-maltoside (DDM) molecule to the TM1/TM2 groove in close proximity to FUA (Fig. S1B). Previous crystal structures indicated binding of DDM to the TM7/TM8 groove, which is pseudosymmetric to the TM1/TM2 groove on the C-terminal part of each AcrB protomer [4,12] (Fig. S1C). It has been postulated that the latter groove is also a binding site for compounds that are sequestered from the outer leaflet of the inner membrane [12] and further transported via CH2 to the deep binding pocket.
In an effort to shed light on the putative transport pathway(s) of FUA and carboxylated β-lactams from the TM1/TM2 groove to the deep binding pocket of AcrB, in this work we combined structural analysis via X-ray crystallography, molecular modeling simulations, and biochemical/microbiological assays. Upon identification of new putative drug binding sites on AcrB in silico, we conducted site-directed mutagenesis and evaluated the effect of the imposed substitutions on susceptibility towards selected antibiotics. Concomitantly, we determined the AcrB structures of the functionally affected I27A, N298A, and V340A substitution variants. Our results suggest that FUA and carboxylated β-lactams are transported via the postulated PN2/PC1 down pathway [19] to the periplasmic porter domain by interaction with the PN2 subdomain loop, comprising residues N298 and L300 located just above the TM1/TM2 groove (Fig. S1B). Moreover, we propose that these antibiotics are subsequently transported towards the deep binding pocket via a PN2/PC1 subdomain interface comprising Y327 and S630. Thus, the initial binding site at the TM1/TM2 interface appears to be a hitherto unknown entrance site for sequestration and transport of carboxylated antibacterial agents by AcrB.
Results
Binding of FUA and carboxylated β-lactams at the TM1/TM2 groove
The AcrB/FUA complex X-ray structure shows binding of FUA and DDM to the TM1/TM2 groove of asymmetric AcrB [18] (Fig. S1). FUA and DDM were assigned to the binding sites on all three protomers (L, T, and O), however the binding to the T protomer was the tightest based on the electron density data. To corroborate these findings, we performed molecular mechanics and free energy calculations for FUA bound to the L, T, and O protomer, respectively, either in the presence or absence of the corresponding DDM molecules. The results (Table S1) agree with the X-ray data in that the binding free energy ΔG calculated for each of the experimental poses indicate that FUA binding to the T protomer is thermodynamically favored compared to binding to the other protomers. Furthermore, according to our calculations, FUA maintains the highest affinity to the T protomer even upon removal of DDM.
Substitution of FUA-interacting side chains and subsequent in vivo activity analysis indicated an important role for residue I337 in binding and/or transport of other carboxylated substrates, like the β-lactams DCX, OXA, and PIP, which should also bind to TM1/TM2 groove according to cross-linking protection analysis [18]. To support the implication of such experimental findings and to identify further potential binding modes for these drugs within the TM1/TM2 groove, we first performed systematic guided docking calculations of FUA, DCX, OXA, and PIP to this region. We used as a model receptor the asymmetric AcrB crystal structure (PDB ID: 5JMN), from which we removed FUA but not the DDM molecule within the TM1/TM2 groove. Ligand flexibility was considered by using different conformations extracted from molecular dynamics simulations of each antibiotic in explicit water [20] (see Materials and Methods). The binding conformation with the highest affinity of FUA, where the carboxyl moiety is oriented towards the periplasm, is consistent with the X-ray data [18] (the RMSD calculated on all non-hydrogenous atoms amounts to ~1 Å, see Fig. 1A). Furthermore, FUA binding is predicted, also according to the docking scoring function, to be stronger in the T protomer compared to the other protomers in all cases considered (Table S2). Also carboxylated β-lactams (namely DCX, OXA and PIP) were found to bind to the TM1/TM2 groove of T protomer (Fig. 1A), congruent to the observed FUA binding site (designated as binding site 1, S1) and in accordance with the cross-link protection studies [18].
Figure 1. Potential binding conformations of FUA and carboxylated β-lactams (DCX, OXA and PIP) at the S1, S2, and S3 binding sites.

Two major docking poses were selected for all sites and depicted as light and dark gray sticks. Both (A) S1 binding sites, and (B) S2 binding sites, are presented as overlay of the two selected binding modes. (C) S3 binding sites where carboxyl groups of FUA, DCX, OXA and PIP are oriented away from the PN2 subdomain. (D) S3 binding sites where carboxyl groups of FUA, DCX, OXA and PIP are oriented towards the PN2 subdomain. The highest affinity pose of FUA where the carboxyl moiety is oriented towards the periplasm (A, light gray), is consistent with the X-ray data [18] (Fig. S1B, RMSD ~1 Å). Cartoon and surface representation are colored in yellow. DDM molecules are shown as stick models (carbon = orange oxygen = red). Drug molecules are depicted as ball and stick (carbon = black or white; oxygen = red; nitrogen = blue; chlorine = green; sulfur = yellow). Residues are depicted as stick models (carbon = cyan or green).
For all compounds, two major conformations were found in S1, respectively featuring the carboxyl moiety either pointing to the lipid-periplasm interface, like in the AcrB/FUA co-crystal structure (Fig. S1B), or buried deeper into the lipid bilayer. In view of previous experimental findings [18], and considering the physicochemical properties of the compounds in a hydrophobic environment, we propose that the carboxyl moieties of FUA and carboxylated β-lactams should be located near the lipid-periplasm interface (conformations shown as light gray sticks in Fig. 1A). The same line of reason will be applied in the selection of putative binding poses at two additional sites S2 and S3, defined below.
Interestingly, predicted binding conformations of FUA and carboxylated β-lactams in S1 imply that these drugs coordinate within the TM1/TM2 groove mainly through hydrophobic interactions. More specifically, the four drug molecules considered were found to interact with I337 and V341, a result which is again consistent with the published structure of AcrB bound to FUA [18] (Fig. S1B).
We further investigated the binding of FUA and carboxylated β-lactams to the TM1/TM2 groove by means of ensemble-docking calculations [21] using again different conformations of FUA, DCX, OXA, and PIP [20] and multiple AcrB structures derived from previous [14] and new MD simulations of the wildtype transporter embedded in a model bilayer constituted of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) molecules and in the absence of any DDM molecule (Fig. 1B, 1C). The use of multiple AcrB structures appears necessary, since it is wel-lknown that even small side-chain rearrangements can drastically alter the outcome of docking calculations [22]. Considering that the X-ray structure of the AcrB/FUA complex [18] also features local rearrangements to host specifically that antibiotic and the DDM molecule, the structure at the TM1/TM2 groove might not be optimal to accommodate other putative substrates. Moreover, the DDM molecule observed in the X-ray structure might be specific for FUA binding, since no X-ray co-structures have been published thus far with other carboxylated drugs bound to this specific region. These ensemble-docking calculations identified, in addition to the S1 site described above, a binding site located more deeply into the cleft of TM1/TM2 groove region (designated as S2) (Fig. 1B) and a distal binding site at a position congruent to that of the DDM maltose moiety binding site in the T protomer of the AcrB/FUA complex structure (designated as S3) (Fig. 1C).
In S2, FUA accommodates deeply in the TM1/TM2 groove, and is in close contact with TM3 and TM4, occupying the binding site of the acyl chain of DDM at the TM1/TM2 groove region. The antibiotic is mainly coordinated by a clamp formed by residues with hydrophobic side chains such as I337, V340, and V341 located at TM2, together with I27 (TM1) and F380 (TM3) located at the opposite side of TM2. We have shown previously that I337 plays an important role in binding or transport of FUA and carboxylated β-lactams, whereas substitution of V341 to Ala does not elicit a particular phenotype against these drugs [18]. Interestingly, based on observation of the AcrB/FUA complex structure, residues I27, V340 and F380 do not show a direct interaction with FUA, but these residues interact with the acyl chain of DDM in the TM1/TM2 groove (Fig. S1B). The DDM acyl chain (occupying the S2 binding site in the X-ray structure 5JMN [18]) is indeed lined by residues I27 (TM1), I337, V341, V345 (TM2), L376, L377, F380 (TM3), I390 and M395 (TM4). Similar to FUA, in the absence of DDM also DCX, OXA, and PIP were found buried deeper in the TM1/TM2 groove, in close contact with I27, I337, V340, and F380 (Fig. 1B) and further stabilized by sparse H-bonds. Interestingly, K334 interacts with the carboxyl moiety of FUA and the carboxylated β-lactams via electrostatic interaction (conformations shown as light gray sticks in Fig. 1B), while H338 is not involved in any interaction with the four drugs considered, unlike what has been observed in the previously reported AcrB/FUA/DDM complex structure (Fig. S1B) [18].
In S3, two major binding conformations of FUA and carboxylated β-lactams were identified in silico, featuring the carboxyl moieties oriented towards the TM domain (FUATM, DCXTM, OXATM, and PIPTM) (Fig. 1C, Fig. S2A) or towards the periplasmic PN2 subdomain (hereafter FUAPN, DCXPN, OXAPN and PIPPN) (Fig. 1D, Fig. S2A). Interestingly, in both orientations FUA interacts with the same residues of the PN2 subdomain of the AcrB T protomer as for the maltose moiety of DDM, i.e. N298 and L300, as observed in the X-ray structure (Fig. 1C–D, Fig. S1B). Moreover, in this setting, FUA also interacts with residues I27, I337 and V341 buried in the TM1/TM2 groove (Fig. 1C–D). In the T protomer, residue N298 coordinates via hydrogen bonds with the carboxyl moiety of FUAPN (Fig. 1D) or the hydroxyl group of the triterpene moiety of FUATM (Fig. 1C), reminiscent of the interaction this residue makes with the maltose moiety of DDM in the crystal structure [18] (Fig. S1B). In the latter, both the N298 and D301 side chains of the T protomer form hydrogen bonds with the maltose moiety of DDM whereas this interaction is not seen between DDM and the other protomers [18]. Binding is further stabilized by hydrophobic interactions with I27, I337 and V341, which sandwich the 4-methyl-3-penten-1-yl or triterpene moiety of FUATM or FUAPN, respectively.
Importantly, N298 and L300 are identified to play a role in the binding of carboxylated β-lactams in all selected conformations in S3, regardless of the orientation of the carboxyl moieties (Fig. 1C–D). Like for FUA, there are two possibilities of drug-protein interaction with N298 via hydrogen bonding, either i) with the carboxyl moieties of DCXPN, OXAPN, and PIPPN; or ii) with isoxazol moiety of DCXTM and OXATM, or with 1-ethyl-2,3-dioxopiperazine moiety of PIPTM. Hydrophobic moieties such as the penam ring (DCX, OXA, and PIP), the isoxazol ring (DCX, and OXA), the phenyl ring (OXA, and PIP), the dichlorophenyl ring (DCX) or the 1-ethyl-2,3-dioxopiperazine ring (PIP), are clamped by I27 and I337, as well as by L300 located near N298, via hydrophobic or van der Waals interactions. After the initial binding step as seen in the crystal structure (Fig. S1B), we suggest that the carboxyl moieties of the substrates orient in a similar position as shown in Fig. 1D. On the basis of these results, we postulate that N298, L300, D301, and K334 lining the S3 site are involved in substrate capturing from the initial TM1/TM2 groove binding S1 or S2 from where these substrates are further transported towards the deep binding pocket.
Phenotypic and structural analysis of TM1/TM2 substitution variants
With a clear idea of potential interacting residues in binding conformations obtained from docking, we substituted these residues to alanine to validate their importance for antibiotic resistance by AcrB in vivo. We subjected E. coli BW25113ΔacrB harboring the plasmid-encoded single-substitution AcrB variants to LB agar plate dilution and IC50 liquid growth analysis in presence of drugs. The most differentiating phenotypes for substitutions in the S3 binding site were obtained with the N298A and L300A variants, whereas the D301A and K334A substitution variants showed no apparent impairment to confer resistance against all drugs tested (Fig. 2). N298A and L300A variants showed reduced ability to confer resistance against FUA, DCX, and OXA, and in addition, the N298A variant was impaired to confer resistance against tetraphenylphosphonium (TPP+), and PIP (Fig. 2). It appears that the pump is specifically affected by these substitutions, since both variants N298A and L300A showed wildtype activity towards erythromycin and for the latter also towards TPP+. Characterization of the phenotypes by calculating the efflux ratio clearly indicated that the N298A variant retained half of the efflux activity of wildtype AcrB for FUA, OXA and PIP, and ~60% of the wildtype efflux activity for DCX, while the L300A phenotype retained >75% efflux activity against FUA, DCX, and OXA (Table 1). Moreover, the L300A variant shows similar efflux activity for PIP as wildtype AcrB, in good agreement with the observations from the drug-LB agar plate dilution experiments (Table 1, Fig. 2). It is likely that both side chains of N298 and L300 are involved in binding and/or transport of FUA, DCX, and OXA, whereas PIP susceptibility is only affected by the N298A variant. To investigate the molecular reasons behind the prominent impact of the N298A variant of AcrB, we determined the N298A/DARPin structure at 2.8 Å, co-crystallized in complex with FUA and DDM in the T protomer. The overall structure was invariant compared to the wildtype AcrB/DARPin structure (0.30 Å RMSD of Cα), and binding of FUA to the TM1/TM2 can only be postulated for the T protomer, based on polder map calculations [23](Figure 3A, Table S3). For this AcrB variant and for the two other variants, the polder map correlation coefficients (CCs) between the experimental map and the calculated map are unequivocally indicating the presence of ligand density (Table S3). The m1-m3 CCs are comparable to that of the wildtype AcrB/FUA co-structure, which has been experimentally verified for the presence of FUA using the anomalous signal of bound 24-bromofusidic acid [18]. It is also highly unlikely that the density can be assigned to other compounds present in the crystallization buffer, since the buffer and mother liquor composition has been used for apo-AcrB crystal growth as well (identical composition, but without FUA), also by another laboratory [4] and resulted in electron densities without additional unaccounted densities. Interestingly, in the T protomer of the N298A variant, the maltoside moiety of DDM adopts an entirely different orientation compared to wildtype (Fig. 3A, Fig. S2B). In the wildtype AcrB/FUA co-structure, DDM is positioned in a perpendicular orientation to the membrane lipid plane and its maltose moiety is clamped by N298 and D301 by hydrogen bonds and interacts with L300 by hydrophobic interaction (Fig. S1). In contrast, the maltose moiety of DDM in N298A/FUA structure is tilted around ~52° towards the membrane plane as the α-D-glucose moiety is oriented parallel to the membrane lipid plane (Fig. 3A, Fig. S2B), akin to the orientation of the DDM molecules seen in the AcrB/FUA complex structures in the L and O protomers [18].
Figure 2. Phenotypic analysis of AcrB S1, S2, and S3 substitution variants on drug agar plates.

Drug agar plate after 22 h of growth with diluted E. coli complemented with wild-type AcrB or S1 or S2 substitution variants (spotted on the plate in serial dilution, values given below the images) (A) N298A, D301A, and K334A; (B) I27A and L300A; (C) V340A and F380A; (D) M395A and M398A; (E) Western blot analysis of the AcrB variants showed no difference compared to wildtype on the expression level. Uncropped versions of the Western blots are shown in Fig. S6.
Table 1. The minimal inhibitory concentration and the growth efflux ratios for fusidic acid, oxacillin, dicloxacillin and piperacillin.
Both the growth efflux ratio and MIC (minimum inhibitory concentration) are obtained by fitting the OD600 of the MIC experiments applying the EC50 shift equation in GraphPad Prism. The efflux ratio is the EC50 of the individual AcrB substitution variants (I27A, N298A, L300A, Y327A, V340A, F332A, I335A, V340A, F380A, S630A and Q1000A) or wildtype AcrB, divided by the EC50 of inactive AcrB_D407N substitution variant. In bold are the MIC values differentiating by 2-dilution steps or more, and the EC50 values with statistically significant differences. The MIC experiments (for every substrate and clone) were repeated at least five times (biological replicates) with two technical replicates each. Indicated in parentheses are the number of biological replicates if done more than five times.
| Fusidic acid | Oxacillin | Dicloxacillin | Piperacillin | |||||
|---|---|---|---|---|---|---|---|---|
| AcrB Variants | MIC (μg ml−1) | Efflux ratio | MIC (μg ml−1) | Efflux ratio | MIC (μg ml−1) | Efflux ratio | MIC (μg ml−1) | Efflux ratio |
| D407N | 8–16 (9) | - | 2–4 (11) | - | 16 | - | 0.125–0.25 (8) | - |
| WT | 128–256 (8) | 8.64 ± 1.31 | 64–128 (9) | 20.49 ± 2.10 | 512 | 24.01 ± 4.04 | 0.50 | 2.44 ± 0.31 |
| I27A | 256 | 22.95 ± 3.48 | 256 | 51.11 ± 5.24 | 1024 | 52.19 ± 8.79 | 1.00 | 3.75 ± 0.48 |
| N298A | 64 | 4.35 ± 0.66 | 32–64 (8) | 10.31 ± 1.06 | 256 | 14.50 ± 2.44 | 0.25 | 1.30 ± 0.17 |
| L300A | 64–128 (7) | 6.56 ± 0.99 | 64 | 15.71 ± 1.61 | 512 | 21.01 ± 3.54 | 0.50 | 2.51 ± 0.32 |
| Y327A | 128–256 (7) | 13.18 ± 2.00 | 32–64 (8) | 13.06 ± 1.34 | 256 | 16.96 ± 2.86 | 0.25 | 1.75 ± 0.23 |
| F332A | 128 | 7.83 ± 1.19 | 64 (7) | 16.61 ± 1.79 | 256 | 16.93 ± 2.85 | 0.50 | 3.26 ± 0.42 |
| I335A | 128–256 (7) | 7.98 ± 1.21 | 64 (6) | 18.76 ± 1.93 | 256–512 (7) | 22.99 ± 3.87 | 0.25–0.50 (8) | 2.54 ± 0.33 |
| V340A | 64 | 4.25 ± 0.64 | 32 | 7.86 ± 0.81 | 256 | 10.86 ± 1.83 | 0.25 | 1.49 ± 0.19 |
| F380A | 64 | 4.53 ± 0.69 | 32–64 (7) | 8.60 ±0.88 | 256 | 12.03 ±2.03 | 0.25 | 1.42 ± 0.18 |
| S630A | 64–128 (8) | 5.60 ± 0.85 | 64 | 11.85 ± 1.22 | 256 | 15.56 ± 2.62 | 0.25 | 1.46 ± 0.19 |
| Q1000A | 128–256 (8) | 9.00 ± 1.36 | 64 (7) | 15.22 ± 1.56 | 256–512 (8) | 24.53 ± 4.13 | 0.50 | 3.33 ± 0.43 |
Figure 3. Fusidic acid and DDM AcrB co-structures.

Shown are fusidic acid (FUA) and dodecyl-maltoside (DDM) bound to the TM1/TM2 groove (including I27 or A27, K334, I337, H338, V340 or A340, and V341) and the PN2 subdomain (including N298 or A298, L300, and D301) of the AcrB T protomer (cartoon representation of TM1, TM2, and PN2; side chains as grey transparent surface representation). (A) AcrB/DARPins-N298A. A298 (N298 in wild-type, PN2 subdomain) is shown in orange spheres. (B) AcrB/DARPins-V340A. A340 (V340 in wild-type, TM2) is shown in orange spheres. (C) AcrB/DARPins-I27A. Residue A27 (I27 in wild-type, TM1) is shown as orange sphere. Shown in A-C are simulated annealing omit mFo-DFc electron densities (blue mesh) contoured at 2.0, 1.5, and 1.5σ, respectively for both FUA and DDM. Hydrogen bonds are shown as dashed lines. All residues are shown as sticks (in green or grey color, oxygen = red, nitrogen = blue) unless indicated otherwise below. FUA and DDM molecules are depicted as ball and stick model (carbon = black, oxygen = red). (D) Superimposition of FUA of wild-type AcrB (carbon yellow), I27A (carbon = blue), N298A (carbon = orange) and V340A (carbon = red) variant in T protomer. The DDM shown (carbon = black, oxygen = red) is from the wildtype AcrB/FUA co-structure (PDB ID: 5JMN).
While DDM is considered a substrate for AcrB, susceptibility tests are intrinsically difficult due to the very low susceptibility of E. coli towards this detergent. We could, however, clearly identify AcrB-mediated resistance on LB agar plate using an assay showing a zone of inhibition for E. coli harboring the inactive AcrB_D407N variant, whereas AcrB wildtype was able to confer lower susceptibility for the same concentration of DDM (Fig. S2C).
Subsequently, we analyzed residues I27, V340 and F380, predicted by docking to play an important role in coordination with FUA and carboxylated β-lactams, predominantly at S2 (Fig. 1B). E. coli BW25113(DE3) ΔacrB expressing acrB carrying the F380A or V340A substitution (Fig. 2C) showed increased susceptibility against DCX, OXA, and PIP, but not FUA, TPP+ and ERY on LB agar plate dilution experiments. However, in LB liquid medium and using an extended range of FUA concentrations, the F380A variant was shown to infer a higher susceptibility against FUA compared to wildtype (Table 1). We also found that cells expressing V340A were impaired in growth (compared to cells expressing the wildtype acrB gene) in liquid medium supplemented with FUA and the tested carboxylated β-lactams (Table 1). The impact of the V340A substitution on the MIC and efflux ratio for PIP (61% of wildtype AcrB ratio) is less severe than for FUA (49%), OXA (38%) and DCX (45%). No major conformational differences were observed between the crystal structure at 2.8 Å of the V340A variant in complex with DARPins and FUA, and the wildtype AcrB/DARPin/FUA complex structure (PDB ID: 5JMN), with an RMSD over Cα atoms of 0.44 Å. In the V340A structure, FUA and DDM were assigned on the basis of polder map analysis (Table S3) to two non-proteinaceous electron densities located at the TM1/TM2 groove of the T conformer (Fig. 3B). For the L and O conformer, in contrast to the wildtype AcrB/FUA/DDM complex structure, no density for DDM could be identified. Localization of FUA at the T conformer TM1/TM2 groove of the V340A variant is similar to wildtype AcrB with 0.88 Å RMSD of FUA (Fig. 3B, 3D). Two other residues, M395 and M398, located on TM4, were also considered potential influential for substrate binding and transport based on their local vicinity to V340 and F380 and their central position within the TM1/TM2 groove. However, the M395A and M398A variants revealed no specific effect for carboxylated substrates, and M398A displayed an increased susceptibility for all substrates tested (Fig. 2D).
Unexpectedly, the I27A variant conferred (much) better growth compared to cells harboring wildtype AcrB on LB agar plate supplemented with carboxylated substrates, especially for DCX and OXA (Fig. 2B). Western blot analysis indicated that the increased resistance due to the I27A substitution is most likely not related to enhanced expression levels (Fig. 2E). The efflux ratio of cells comprising the I27A variant showed substantially increased resistance against FUA, DCX, OXA, and PIP, in comparison to cells harboring wildtype AcrB (activity increased with respect to wildtype AcrB by a factor of 2.7, 2.2, 2.5 and 1.5 respectively) (Table 1). Despite the clear hyperactive phenotype of the I27A variant, no major conformational differences were observed in the crystal structure of the I27A variant at 2.7 Å in complex with DARPins and FUA (0.49 Å RMSD of Cα compared to wildtype AcrB (PDB ID: 5JMN). Similar to the wildtype electron density data, the I27A variant provided two clear non-proteinaceous electron densities at the TM1/TM2 groove for the T protomer, which we assigned to FUA and DDM, respectively (Table S3, Fig. 3C, Fig. S2B). For the other protomers, no density for FUA could be observed. Isothermal titration calorimetry (ITC) experiments conducted with wildtype AcrB, and the I27A, N298A, and V340A variants indicated different binding affinities for FUA (Fig. S3). The dissociation constant (Kd) values for FUA are 527.9 ± 8.6 μM for wildtype AcrB, higher for the N298A and V340A variant (1122.3 ± 18.2 μM, and 826.5 ± 4.8 μM, respectively), but lower for the I27A variant (404.8 ± 6.6 μM). Interestingly, by comparing the volumes of the cavities in the TM1/TM2 groove for the I27A and wildtype AcrB variants (PDB_IDs: 5JMN and 6Q4O, respectively; see Materials and Methods), we found that the substitution leads to an increase of ~ 80 Å3, which corresponds roughly to the van der Waals volume difference between the Ile and Ala side chains. In tendency, this would relate to a mildly lower FUA binding affinity for the first two variants, and a slightly higher binding affinity for the latter. Within the cell, we anticipate the very hydrophobic FUA to partition into the inner membrane and reach mM concentration inside the membrane even with μM concentration in the periplasm/medium. Hence, an AcrB binding site within the membrane region does not require to bind hydrophobic compounds with high affinity as their concentration (in the membrane) will exceed the apparent Kd by far under antibiotic stress.
Validation of docking sites in the presence of a phospholipid molecule within the TM1/TM2 groove
In its physiological environment, AcrB is embedded in a biological membrane, thus it is important to assess the possibility for binding of drugs to the TM1/TM2 groove in the presence of phospholipids at this site. To this end, we performed additional ensemble docking calculations using structures of AcrB extracted from previous ([14]) as well as new μs-long MD simulations (see Materials and Methods) of the protein embedded in a model POPE bilayer and salty water solution (0.15 M KCl). Notably, the simulations did not contain DDM as observed for the crystal co-structure [18].
Before describing docking results in the presence of phospholipids at S1, it is instructive to describe the main findings from the new MD simulations, performed for the wildtype AcrB as well as for the I27A and V340A variants, which had opposite effects on the interaction of the transporter with FUA and carboxylated β-lactams. Interestingly, in the case of the wildtype protein, H-bonds and salt bridges are formed between POPE molecules and residues N298, K334 and H338, and to a minor extent with the sidechain of L28 (Table S4), in part recovering the interaction pattern between the TM1/TM2 groove and DDM seen in the X-ray structure [18]. For I27A and V340A, a stronger interaction (via formation of long-lived H-bonds) with K334 was observed, while all the other interactions weakened (Table S4). In contrast, the I27A and V340A substitutions had opposite effects on the interaction of POPE lipids with the TM1/TM2 groove, respectively decreasing (I27A) and increasing (V340A) the number of atomic contacts with respect to wildtype AcrB (Fig. S4A). This difference is mirrored in the reduced (increased) affinity of a POPE molecule towards the TM1/TM2 groove arising upon the I27A (V340A) substitution (Table S4).
To corroborate the possibility of substrate binding to the S3 site in the presence of phospholipids at the TM1/TM2 region, new ensemble-docking calculations were performed using as input “receptor” structures a set of complexes between AcrB and a POPE molecule. The pool of structures was selected by inspecting the MD trajectory for conformations having the largest contact surface with the TM1/TM2 groove and one acyl chain virtually superposed to that of the DDM molecules as seen in the X-ray structure (Fig. S4B). The docking results confirmed indeed the possibility of binding of all compounds at S3 (Fig. S4C). Unlike the binding of DCX, OXA and PIP, FUA binds further away from the TM1/TM2 groove (between the upper part of TM1 and PN2 subdomain) as compared to binding conformations of FUA at S3 without POPE molecule at TM1/TM2 groove (Fig. 1C).
In the hypothesis that binding of carboxylate drugs and POPE at S3 occurs competitively, the different interaction pattern of POPE with the TM1/TM2 groove in the I27A (V340A) AcrB variant, as well as the increased (reduced) affinity compared to the wildtype protein, could provide a rationale for the enhanced (impaired) efflux of FUA and carboxylated β-lactam substrates. In this respect, note also that the effect of the N298A mutation could be related to a decreased binding affinity of substrates towards the S3 site, which lacks in that variant an important carboxamide sidechain H-bonding partner.
Towards the deep binding pocket: Further FUA and β-lactam binding sites
The proposed pathways for drug transport through AcrB towards its distal funnel and the AcrA/TolC channels [6] all include the area defined as deep binding pocket [2–4,8–10,16]. Hence, the initial binding of FUA or carboxylated β-lactams is most likely followed by further transport steps and possibly involves other, thus far undiscovered, binding sites on a path towards the deep binding pocket. We therefore decided to conduct a final campaign of ensemble-docking calculations enlarging the docking region so as to cover all putative binding sites from S1 to the distal pocket. In this way, we identified two additional interaction spots. The first one, hereafter designated as S1’, is located at the interface between the periplasmic and TM domain of the T protomer, and lined by the upper ends of TM2, TM11 and TM12 (residues P988, L989, I991, T993, G994, A995, S997, and Q1000 from the loop between TM11 and TM12, residues F396 and V399 located on TM4, residue W634 located on the PC1 subdomain, and residues I335, E339, K342 and T343 located on TM2, juxtaposed to the S1 side (Fig. 4A)). As expected, FUA and carboxylated β-lactams are mainly surrounded by hydrophobic residues at the S1’ binding site, the most frequent residue being I335. Hydrogen bonds are found to coordinate with drugs at S1’, involving E339, K342 (located on TM2), T993, S997, and Q1000 (located on the loop between TM11 and TM12) (Fig. 4A). We selected prominent candidate residues along TM2 and the loop region between TM11 and TM12 (residues I335, E339, K342, T993, S997, and Q1000) to be substituted with Ala and tested their effect on susceptibility towards FUA and carboxylated β-lactams. All mutants were well expressed (Fig. S5A). Interestingly, E. coli BW25113ΔacrB harboring the Q1000A substitution variant showed clear susceptibility to OXA on LB agar and slight growth defect on DCX, but not in the presence of FUA and PIP. Also, the I335A substitution variant conferred slight reduced susceptibility against FUA and carboxylated β-lactams (Table 1, Fig. S5B). Our results suggest that S1’ could represent either a subsequent or an alternative uptake gate to S1/S2.
Figure 4. Potential binding conformations of FUA and carboxylated β-lactams at the S1’ and S4 docking site.

(A) S1’ binding site (upper part: carboxyl moiety of drug molecules is oriented towards TM; lower part: carboxyl moiety of drug molecules is oriented away from TM), and (B) S4 binding site (S4down: carboxyl moiety of drug molecules is oriented towards S1’; S4up: carboxyl moiety of drug molecules is oriented towards upper part of the cleft between PC1 and PN2 subdomain).Cartoon and surface representation are colored in yellow. Drug molecules are depicted as ball and stick (carbon = black or white; oxygen = red; nitrogen = blue; chlorine = green). Residues are depicted as stick models (carbon = green).
Another putative binding site, designated as S4, was found proximal to the deep binding pocket of the T protomer. It represents a swallow cleft between subdomains PC1 and PN2, lined by residues R307, Y325, P326, D328, P331, F332 (PN2 subdomain), and Q569, V571, E607, S608, and S630, L631, K632, D633, and W634 (PC1 subdomain) (Fig. 4B). Two major binding conformations for the substrates were observed, featuring the carboxyl moieties pointing either towards the upper part of the cleft between PC1 and PN2 subdomain (designated as S4up) or towards S1’ (designated as S4down). Similar to S1-S3 and S1’ binding sites, binding of FUA and carboxylated β-lactams to S4 is mainly coordinated by hydrophobic or van der Waals interaction, as residues P331, F332, and V571 might play a key role in drug coordination. Interestingly, F332 coordinates with all four drugs with S4down binding conformation, but not with S4up except DCX. In contrast to S1-S3 binding sites, several hydrogen bonds were mediating drug-protein interactions at S4, by residues R307, E607, S608, and S630 lining the cleft between subdomains PC1 and PN2 (Fig. 4B). Putatively interacting residues (i.e. R307, Y325, P326, Y327, D328, P331, F332, Q569, V571, E607, S608, S630, L631, K632, D633, and W634), in addition to nearby residues T329 and T330, were substituted by Ala and the single substitution AcrB variants were expressed in E. coli. All variants except W634A did express to wildtype levels, whereas the W634A variant did not express at all (Fig. S5A). Both Y327A and S630A showed reduced resistance against carboxylated β-lactams only. S630A was also impaired in FUA resistance, but Y327A was not. P326A was only showing reduced resistance against OXA, (Table 1; Fig. S5C). The calculated efflux ratio for cells expressing the S630A variant derived from growth on liquid medium was also severely reduced for FUA and the carboxylated β-lactams tested, with a retained efflux activity of 53–72% as compared to wildtype AcrB (Table 1). In addition, the F332A substitution variant conferred reduced efflux activity against FUA, OXA and DCX, but not to PIP, ERY or TPP+ (Fig. S5B). Our results indicate that F332, Y327, and S630 might play a role in direct binding to the carboxylated substrates or are part of the transport path of FUA and/or DCX, OXA, and PIP, from the initial binding site towards the deep binding pocket.
Discussion
The AcrAB-TolC efflux pump confers resistance towards an extensive set of chemically diverse compounds including antibiotics, bile salts, dyes, and solvents. One of the intriguing questions is the molecular basis of this promiscuous specificity. Previous studies [2,9,11,12,18] have shown that AcrB harbors multiple binding sites (an access pocket, a deep binding pocket, a TM1/TM2 groove binding site and a recently discovered central cavity access site to channel 3 (CH3)). These binding sites themselves exhibit a large polyspecificity on their own, and physicochemical properties like hydrophobicity and size of the drug molecule appear to be important for recognition [10,12,14,24]. The transport of substrates through the AcrB periplasmic porter domain has been subject to extensive analysis [2–4,9–12,14,19,25–27], but still remains puzzling, especially considering the path of drugs towards the deep binding pocket after the initial sequestering step.
Here, we propose a transport pathway for FUA and other carboxylated substrates within AcrB (Fig. 5). We assume that the FUA binding site as seen in the co-crystal structures [18] is the initial binding site (S1) for FUA and other carboxylated substrates, especially β-lactams, to the tight (T) protomer (Fig. 1; Fig. 5; Fig. S1B). From there we expect that these substrates are sliding either more deeply into the TM1/TM2 groove (S2, involving displacement of a phospholipid acyl chain from that region) or to the upper part of the TM1/TM2 groove (S3) (Fig. 1B–C; Fig. S4C; Fig. 5). One hypothesis would be that the TM1/TM2 groove is occupied by an acyl chain of natural lipid (Fig. S4B, Fig. S4C), which would facilitate binding of drugs to S1, like the FUA position observed in the crystal structures featuring a DDM molecule in the groove. Nevertheless, we cannot rule out the possibility that FUA and carboxylated β-lactams bind deeper inside the TM1/TM2 groove (S2) in a competitive manner with an acyl chain of detergents or phospholipids (Fig. 1B). Especially side chain V340 might be involved in the binding of substrates into this pocket. Furthermore, predicted docking poses more towards the periplasmic part of AcrB (S3) involve interactions with the tip of the PN2 subdomain especially with side chains N298 and L300, as well as I337 and I27 located at the TM1/TM2 groove (Fig. 1C). The effect of the N298A and L300A substitution on the ability of AcrB to confer resistance suggest that the PN2 subdomain is involved in transport of carboxylated substrates. We also observe an effect of the N298A substitution for TPP+, but not for erythromycin (Fig. 2). Since erythromycin is postulated to initially bind to the access pocket [11], we assume different pathways for erythromycin and carboxylated β-lactams/FUA. For TPP+, we tentatively assume yet another pathway to be involved, possibly via the CH3 channel that transports mainly cationic and aromatic drugs such as ethidium [9]. Indeed, we identified an alternative tunnel (darkgreen colored tunnel at the right-side in Fig. 5), initiating from S3 (namely at N298) and leading to deep binding pocket via the interface of central cavity and membrane plane. Notably, only the N298A variant conferred enhanced susceptibility towards TPP+, all other variants tested (in particular L300A near N298 but lining the S1/S2/S3 to S4 tunnel) displaying no noticeable effect compared to wildtype AcrB. Cells harboring the I27A substitution variant showed surprisingly a significant and specific higher resistance against FUA and β-lactam antibiotics (Fig. 2, Table 1). Notably, in the I27A/FUA co-crystal structure, the C-terminal part of TM1 is closer to FUA as in the wildtype co-structure, and FUA is closer to I337 in TM2 as well. This might indicate a tighter interaction compared to wildtype AcrB and other variants (Fig. 3D). ITC experiments showed also a slight increase in affinity for this variant, which is congruent to our other observations, although not conclusive (Fig. S3). We propose that removal of the Ile bulky side chain might enlarge the TM1/TM2 substrate binding cavity (S1 and possibly also S2 and S3) and therefore sterically facilitates the transport of these compounds; this hypothesis is supported by the cavity calculations predicting a reduced volume of the TM1/TM2 groove upon mutation. Moreover, the removal of the aliphatic Ile sidechain upon I27A substitution appears to decrease the affinity of POPE acyl chains towards the TM1/TM2 groove as compared with the wildtype AcrB (Table S4). This loss in affinity is likely related to the reduction in the contact area at the interface between POPE molecules and the TM1/TM2 binding site in the I27A variant with respect to the wildtype protein (Fig. S4A), as well as to the altered pattern of H-bonding interactions (Table S4). The reduced POPE affinity for the I27A variant and its increased affinity towards FUA would support the hypothesis that FUA/carboxylated β-lactams and POPE might bind at S2 or S3 competitively.
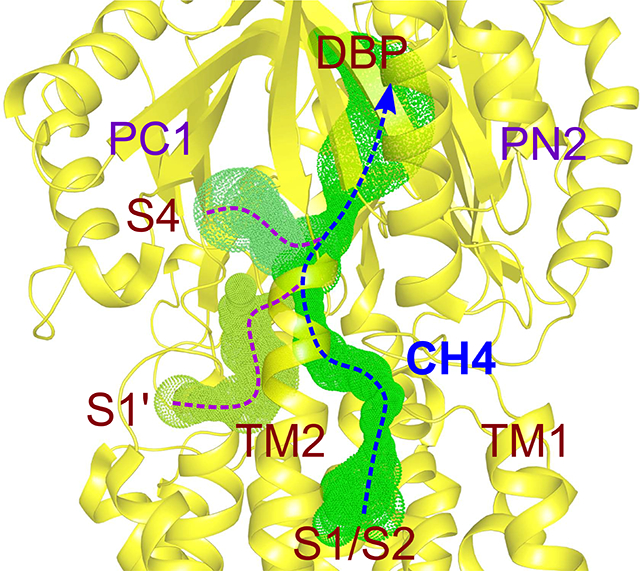
Figure 5. Postulated transport pathway of FUA and carboxylated β-lactams towards the deep binding pocket.

Tunnels (green) at the AcrB T protomer are depicted starting from initial drug binding sites S1/S2, S3, S1’ or S4 and extend towards the deep binding pocket (DBP). Residues specifically affecting AcrB activity against FUA and carboxylated β-lactams are depicted as blue stick models. Close-up view of the T protomer (cartoon representation; yellow) and indication of the transport pathways (green) starting from S1-S4, including the residues specifically affecting AcrB activity against carboxylated drugs.
Such a mechanism would also rationalize the effect of the V340A mutation conferring increased susceptibility against the compounds investigated here. Residue V340 on TM2 is located at the interface between the S1/S2 and the alternative entry gate S1’ and is predicted to play an important role in binding of substrates at S2 based on docking results. In the V340A/FUA X-ray structure, FUA binds closer to TM2 than in the complex with the wildtype protein, due to the small alanine side chain (Fig. 3D). Moreover, the interaction of POPE molecules with the TM1/TM2 site is tighter in the AcrB V340A variant than in the wildtype protein (Fig. S4A, Table S4), whereas the affinity measured for FUA via ITC was lower than wildtype (827 vs. 528 μM). In addition to V340, residue F332 (S4) located at the N-terminus of TM2 seems to play an important role in substrate binding and/or transport of these drugs. Indeed, the F332A substitution conferred reduced resistance to FUA, DCX, and OXA to certain extent, but not to PIP (Table 1).
Based on our findings, we propose that FUA and carboxylated β-lactam antibiotics are sequestered either from S1/S2/S3 or S1’ (a two-steps translocation from S1/S2/S3 to S1’ could be also possible (Fig. 5)). From these putative recognition sites, the next step along the translocation pathway would be the predicted site S4 within the interface between PN2 and PC1. From this site drugs could finally be transported through an internal channel leading to the deep binding pocket. Transport might occur via a transient tunnel through a conformational change of N-terminus of TM2, N-terminus of TM4, and connecting loop between TM1 and PN1, leading to the entry point of S4 binding site (Fig. 5). This hypothesis is compatible with the proposed PN2/PC1 down pathway [19], and we suggest designating this tunnel “Channel 4” (CH4), in accordance with CH1-CH3 definitions in [9]. In CH4, substitutions of Y327 and S630 to alanine within site S4 diminished the resistance against carboxylated drugs (Fig. S5C; Table 1). More importantly, Y327 and F332 reside exactly at the intersecting point of possibly three channels (channel from S1/S2, channel from S1’, and channel from S4) leading towards deep binding pocket via a transient pathway (Fig. 5). In all transport pathways discussed in this report, the access pocket is not involved in the transport of FUA and carboxylated β-lactams, and this seems compatible with the hypothesis that the access pocket is most likely not involved in the transport of low molecular mass compounds [10,16]. A network of channels similar to that reported in Fig. 5 was found in the L monomer, although the connection to the DBP is not present in this case due to the absence of this pocket (Fig. S7). As expected, no channels were found in the O monomer departing from any of the sites discussed in this report. Moreover, alanine substitutions did not affect significantly the morphology of the channels, which can be rationalized in terms of the reduced steric hindrance upon substitution introduced in a structure virtually identical to that of the wild type protein. The computational and experimental results presented here provide a first detailed insight into the structural features lining possible, previously unknown, drug transport pathways. In addition, they point to the need for additional investigations to elucidate the complete transport pathway for membrane embedded substrates, especially the route between the periplasm/outer leaflet of the inner membrane interface, and PN2/PC1 down pathway [19].
Conclusion and perspectives
This study presents an in silico inspired investigation on the RND efflux pump AcrB focused on transport pathway hypotheses verified by experimental assays. The combination of in silico prediction, functional analysis of variants and crystal structures of three variants led to the identification of multiple binding sites and transport pathways to the deep binding pocket of AcrB. The polyspecificity (binding of chemically diverse molecules, possibly in different orientations) most likely involves conformational flexibility comprising multiple side chain interactions. In silico prediction, directed-evolution with random mutagenized libraries, rather than site-directed mutagenesis, in combination with structural insights of transport-affected variants might provide further insights of intermediate transport states of the AcrB efflux pump, necessary to reconstitute the pathways of drug passage through this highly complex machinery.
Materials and Methods
Mutagenesis
Mutagenesis was performed according to ExSite protocol (Stratagene) with 5’ phosphorylated primers or QuikChange Site-directed Mutagenesis protocol (Stratagene) with pET24acrBHis [3] served as template. Mutations were verified by sequencing (Eurofins). All primers used for mutagenesis are listed in Table S5.
Drug agar plate assay
Agar dilution experiment was performed according to Wiegand et al. [27] with slight modification. Briefly, a colony of E. coli BW25113 ΔacrB [28] harboring pETacrBHis wildtype or acrB variant were grown overnight in LB medium containing 50 μg ml−1 kanamycin at 37°C/220 rpm. Dilution of the cultures to OD600 10−1–10−6 were prepared and 5 μl of each diluted cultures were spotted on an LB agar plate containing 50 μg ml−1 kanamycin, supplemented with AcrB drugs (FUA: 11 μg ml−1, TPP+: 80 mg ml−1, erythromycin: 5 μg ml−1, OXA: 20 μg ml−1, DCX: 70 μg ml−1, PIP: 0.1 μg ml−1). Plates were incubated overnight at 37°C for 22 h. Expression of AcrB or AcrB variants was tested by Western blot analysis using anti-AcrB antibody.
MIC measurement
Minimum inhibitory concentration (MIC) of various antibiotics against E. coli BW25113 ΔacrB carrying AcrB or AcrB variant was performed according to Wiegand et al. [27] with slight modification. A colony of E. coli BW25113 ΔacrB harboring pET24acrBHis wildtype or acrB variant were grown overnight in 5 ml Mueller Hinton Broth II supplemented with 50 μg ml−1 kanamycin at 37°C/220rpm. Cultures were diluted to OD600 of 0.02 and 75 μl cultures were inoculate into 75 μl of serial 2-fold substrate dilutions in LB supplemented with 50 μg ml−1 kanamycin in 96-well plates (BRANDplates pureGrade S, no. 781662). The microtiter plate was placed at 37°C for 20–22 h with 300–350 rpm (Infors HAT Multitron) and the OD600 was measured using a BioTek™ EON Microplate Spectrophotometer. The readout of the optical density measurement was used to calculate the growth inhibition curves and 50% inhibitory concentration of growth (IC50 growth) according to Iyer et al. [29] The growth efflux ratio was calculated by the difference between IC50 growth of AcrB variants (WT, I27A, N298A, L300A, Y327A, F332A, I335A V340A, F380A, S630A, and Q1000A) with AcrB inactive variant (D407N).
Overexpression, purification and crystallization of AcrB and DARPin
Purification of DARPin clone 110819 protein was previously described [4]. AcrB-I27A/DARPin, AcrB-N298A/DARPin, and AcrB-V340A/DARPin, were obtained as previously described for wildtype AcrB/DARPin [12]. AcrB variants in complex with DARPin were polished with size exclusion chromatography (Superose 6, GE Healthcare) with buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 0.03% (w/v) DDM (Glycon) and 0.05% DDAO (Anatrace). Crystallization of all variants were performed as previously described with protein concentration of 13 mg ml−1. To obtain FUA bound to AcrB-I27A/DARPin, AcrB-N298A/DARPin, and AcrB-V340A/DARPin, apo-crystals were soaked with the reservoir solution containing 4 mM FUA for 4 days.
Diffraction data collection and refinement
Datasets were collected on beam line Proxima 2A at Soleil Synchrotron, Saint-Aubin, France using a Q315r Area detector (ADSC) or Eiger X 9M detector (Dectris) with helical data collection strategy or Proxima 1 using a Pilatus 6M detector (Dectris), indexed and integrated with XDS. Structural models (starting model was the AcrB/DARPin/FUA structure without the ligands, PDB: 5JMN) were built in COOT, refined with REFMAC5 supported from CCP4 program suite [30]. Figures were prepared with PyMOL (http://www.pymol.org). Structures were validated with MolProbity. In the Ramachandran plots, 99.8% of residues were in allowed regions for all the structures reported in this manuscript (FUA bound AcrB-I27A, FUA bound AcrB-N298A, and FUA bound AcrB-V340A substitution variants). Statistics for data collection and refinement are shown in Table S5.
Isothermal Titration calorimetry (ITC)
ITC experiments were performed with a VP-ITC calorimeter (Malvern, MicroCal, Inc.), having 1.45-ml volume sample cell and a 278-μl volume syringe. The protein samples (wildtype AcrB, AcrB-I27A, AcrB-N298A, AcrB-V340A) were purified in a buffer containing 20 mM Tris-Cl, pH 7.5, 10 mM NaCl and 0.02% DDM. The syringe solution was also prepared in the same buffer and the total ionic strength was adjusted to 10 mM with NaCl. The titrations were performed under following conditions: wildtype AcrB or AcrB variant (3.5 μM) was titrated with 4 mM FUA in the syringe. The first injection was 3 μl followed by 54 further injections of 5 μl each. All titrations were performed at 25°C. The time interval between two injections was 180 s and the stirring speed was set to 307 rpm. All measurements were performed with high gain mode. Data was evaluated by using MicroCal Origin software provided by the manufacturer. The one site model was used for curve fitting. The first injection was omitted during data processing because of inaccuracy in volume associated with it. This first data point was therefore removed before analysis.
Molecular docking
All molecular docking calculations were performed using the software AutoDock Vina [31]. Protein and ligand input files were prepared with AutoDock Tools [32]. Flexibility of the ligand was taken into account indirectly by considering for each substrate 10 different configurations taken from www.dsf.unica.it/translocation/db [20]. The Iterated Local Search global optimizer (a stochastic global optimization approach) is the search algorithm implemented in AutoDock Vina [31], and was used with default parameters but for the exhaustiveness (giving a measure of the exhaustiveness of the local search), which was set to 1024 (default 8). For each docking run, we retained the top ten docking poses.
For the first docking calculations, guided towards the TM1/TM2 groove of the T protomer, the search of poses was performed within a cubic volume of 30×30×30 Å3 and centred at the centre of mass of FUA in the T protomer of the AcrB crystal structure (PDB ID: 5JMN) [18]. In this calculation, only the experimental structure of AcrB (bearing a DDM molecule within the TM1/TM2 groove) was used.
The same search space was used to perform the first and the second sets of ensemble-docking calculations, in which 13 different conformations of AcrB were used. Namely, in addition to the experimental structure, we employed 12 conformations of AcrB extracted from previous ([14]) as well as from new 1 μs long MD simulations (vide infra) of the wildtype transporter embedded in a POPE bilayer. Selected structures had the largest overlap between one alkyl chain of the POPE lipid having the tightest interaction with the TM1/TM2 groove and that of the DDM molecule in the X-ray structure (PDB ID: 5JMN) [18]. In the first set of calculations, only the structure of the protein was used as receptor, while in the second set we included, for each conformation of AcrB, also the POPE molecule having one alkyl chain virtually superposed to that of DDM.
The latter set of 13 structures (AcrB+POPE molecule) was used also in the third ensemble-docking campaign, in which we adopted a 30×40×50Å3 search space centred manually to cover all the putative binding sites found in preliminary runs as well as the bottom of the distal pocket of monomer T, which allowed to detect additional binding sites on a path towards that site.
Molecular dynamics simulations
MD simulations of the wildtype, I27A and V340A variants of AcrB were carried out using AMBER18 [33]. The previously published X-ray structure (PDB ID: 5JMN) and those introduced here (PDB IDs: 6Q4N, 64QO), were used as initial conformations of wildtype, I27A and V340A variants of AcrB, respectively. Protomer specific protonation states [13] were adopted with E346 and D924 protonated in both L and T protomers while deprotonated in the O protomer of AcrB [13]. The residues D407, D408, D566 were protonated only in the O protomer of AcrB. The topology and the initial coordinate files for these apo-protein structures were created using the LEaP module of AmberTools. The proteins were successively embedded in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) bilayer patches, solvated with explicit TIP3P water model, and neutralized with the required number of randomly placed K+ ions as in previous work [14,34]. The ions count was suitably adjusted to account for an osmolarity of 0.15 M KCl. Embedding of the protein into a pre-equilibrated POPE bilayer patch was performed using the PPM server [35] and subsequently the CharmmGUI tool [36]. The lipid residue nomenclature was converted from the CHARMM to AMBER format using the charmmlipid2amber.py script provided with AmberTools. The number of lipids to be added to the central pore were obtained by dividing the approximate area of the central pore by the standard area per lipid of POPE molecules [37]. Periodic boundary conditions were used and the distance between the protein and the edge of the box was set to be at least 30 Å in each direction.
Multi-step energy minimization with a combination of steepest descent and conjugate gradient methods was carried out using the pmemd program implemented in AMBER to relax internal constrains of the systems by gradually releasing positional restraints. Following this, the systems were heated from 0 to 310 K by a 1 ns heating (0–100 K) under constant volume (NVT) followed by 5 ns of constant pressure heating (NPT) (100–310 K) with the phosphorous heads of lipids restrained along the z-axis to allow membrane merging and to bring the atmospheric pressure of the system to 1 bar. Langevin thermostat (collision frequency of 1 ps−1) was used to maintain a constant temperature, and multiple short equilibration steps of 500 ps under anisotropic pressure scaling (Berendsen barostat) in NPT conditions were performed to equilibrate the box dimensions. A time step of 2 fs was used during all these runs, while post-equilibrium MD simulations were carried out with a time step of 4 fs under constant volume conditions after hydrogen mass repartitioning [38]. The particle-mesh Ewald algorithm was used to evaluate long-range electrostatic forces with a non-bonded cutoff of 9 Å. During the MD simulations, the length of all R–H bonds was constrained with SHAKE algorithm. Coordinates were saved every 100 ps. The ff14SB version of the all-atom Amber force field [39] was used to represent the protein systems while lipid17 [37] parameters were used for the POPE bilayer. After equilibration, production MD simulations of 1 μs in length were performed for each system. Trajectory analyses were performed using the cpptraj module of AmberTools and VMD1.9.4, and graphs were plotted using xmgrace.
Structural relaxation
Molecular mechanics simulations of AcrB in complex with various ligands were performed using AMBER18 [33]. The parm14SB [39], lipid17 [37], and GAFF [40] force fields were used for the protein, the POPE bilayer, and for the substrates of AcrB (including DDM). Details on the parametrization of all substrates considered in this study were reported elsewhere [20], and the GAFF parameters are freely available at www.dsf.unica.it/translocation/db. Concerning the structural relaxation of the AcrB-ligand-(DDM/POPE) complexes (including the X-ray structure with PDB code 5JMN and the top docking poses at each site), we performed an energy minimization of each complex using a combination of steepest descent (1000 iterations) and conjugate gradient (up to 9000 iterations), as implemented in the sander module of AMBER16. Soft restraints of 1 kcal/mol/Å2 were applied to all heavy atoms of the system in order to maintain the structure near the crystallographic conformation. Structural relaxations were carried out in vacuum, and a cutoff of 99 Å was used to evaluate long-range interactions.
Binding free energy calculations
Free energy calculations were performed to assess quantitatively the relative strength of binding of FUA to the three protomers of AcrB, using both the Molecular Mechanics – Generalized Born Surface Area (MM-GBSA) and the Molecular Mechanics – Poisson-Boltzmann Surface Area (MM-PBSA) post-processing methods [41]. In these methods the binding free energy is evaluated as:
with Gcom, Grec, and Glig being the absolute free energies of complex, receptor, and ligand, respectively. According to these schemes, the free energy difference can be decomposed as:
where ΔEMM is the difference in the molecular mechanics energy, ΔGsolv is the solvation free energy, and TΔSconf is the solute conformational entropy. The first two terms were calculated with the following equations:
EMM includes the molecular mechanics energy contributed by the bonded (Ebond, Eangle, and Etorsion) and non-bonded (Evdw and Eele, calculated with no cutoff) terms of the force field. ΔGsolv is the solvation free energy, which can be modeled as the sum of an electrostatic contribution (ΔGsolv,p, evaluated using the MM-GBSA or MM-PBSA approaches) and a non-polar one (ΔGsolv,np = γΔSA + b, proportional to the difference in solvent-exposed surface area, ΔSA).
As for the MM-GBSA approach, the electrostatic solvation free energy was calculated using the implicit solvent model by Nguyen et al. [42] (igb = 8 option in AMBER) in combination with mbondi3 (for H, C, N, O, S elements) and intrinsic radii. Partial charges were taken from the AMBER/GAFF force fields, and relative dielectric constants of 1 for solute and 78.4 for the solvent (0.1 M KCl water solution) were used. The non-polar contribution is approximated by the LCPO method [43].
In the MM-PBSA approach, the electrostatic solvation free energy was evaluated by means of the linearized Poisson-Boltzmann equation, iterated up to 5000 times and using a grid spacing of 0.25 Å. The longest dimension of the rectangular finite-difference grid was set to 4 times the value of the maximum linear dimension of the solute. Since the TM1/TM2 groove is interacting with the membrane, we used in this case a relative dielectric constant of 4 for the solvent (0.1 M KCl water solution). The solute conformational entropy contribution (TΔSconf) was not evaluated [41].
Volume calculations
Volumes within the TM1/TM2 groove were computed using the cavity detetion algorithm based on a two-probe sphere method implemented in the rbcavity tool of the rDock program [44]. In particular, the binding site volume was identified within a sphere of radius 10 Å centered over the Cα of residue 27 of monomer T in both wildtype and I27A AcrB variants, using large and small probe radii of 5.0 Å and 1.5 Å, respectively. These radii were found to be optimal for our case after evaluating different combinations and checking through visual inspection their accuracy by keeping the possible inclusion of regions extending outside the cavity of interest as minimal as possible. The pathways for drug transport reported in Fig. 5 and Fig. S7 were estimated using caver 3.0 [45][https://doi.org/10.1371/journal.pcbi.1002708].
Supplementary Material
Mechanism of multi-drug transport by RND proteins such as AcrB is elusive
Structural/biophysical studies informed by in silico calculations were performed
Four binding sites for carboxylated drugs were identified at PN2/PC1 subdomain
A novel transport channel involving TM1/TM2 groove is proposed
New knowledge about the molecular mechanism of AcrB polyspecificity is provided
Acknowledgments
We acknowledge SOLEIL Synchrotron in Saint Aubin, France, for provision of synchrotron radiation facilities and we would like to thank Pierre Legrand for assistance in using beamline Proxima 1 (Proposal Number: 20150100) and Dr. Martin Savko for assistance in using beamline Proxima 2A (Proposal Number: 20160072 and 20150100). This work was supported by the German Research Foundation (SFB 807, Transport and Communication across Biological Membranes), the DFG-EXC115 (Cluster of Excellence Frankfurt–Macromolecular Complexes) and the German-Israeli Foundation (Grant Number I-1202-248.9/2012). The research leading to these results was conducted as part of the Translocation consortium (www.translocation.eu) and has received support from the Innovative Medicines Joint Undertaking under Grant Agreement Number 115525, resources that are composed of financial contribution from the European Union seventh framework program (FP7/2007-2013) and EFPIA companies in kind contribution. The work of G.M. A.A.V. and P.R. has received additional support from the National Institutes of Allergy and Infectious Diseases project number AI136799.
Footnotes
Competing interests:
The authors declare no competing interests.
Data availability: Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes PDB 6Q4N (AcrB-V340A/DARPin in complex with fusidic acid), 6Q4O (AcrB-I27A/DARPin in complex with fusidic acid), and 6Q4P (AcrB-N298A/DARPin in complex with fusidic acid).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Li X-Z, Plésiat P, Nikaido H, The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria, Clin. Microbiol. Rev 28 (2015) 337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A, Crystal structures of a multidrug transporter reveal a functionally rotating mechanism, Nature 443 (2006) 173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- [3].Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM, Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism, Science 313 (2006) 1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- [4].Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG, Grutter MG, Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors, PLoS Biol. 5 (2007) e7. doi:06-PLBI-RA-1517R2 doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Du D, van Veen HW, Murakami S, Pos KM, Luisi BF, Structure, mechanism and cooperation of bacterial multidrug transporters, Curr. Opin. Struct. Biol 33 (2015) 76–91. doi: 10.1016/j.sbi.2015.07.015. [DOI] [PubMed] [Google Scholar]
- [6].Wang Z, Fan G, Hryc CF, Blaza JN, Serysheva II, Schmid MF, Chiu W, Luisi BF, Du D, An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump, Elife 6 (2017) 1–19. doi: 10.7554/eLife.24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pos KM, Drug transport mechanism of the AcrB efflux pump, Biochim Biophys Acta. 1794 (2009) 782–793. doi: 10.1016/j.bbapap.2008.12.015 [DOI] [PubMed] [Google Scholar]
- [8].Ruggerone P, Murakami S, Pos KM, V Vargiu A, Pos KM, V Vargiu A, RND Efflux Pumps: Structural Information Translated into Function and Inhibition Mechanisms, Curr. Top. Med. Chem 13 (2013) 3079–100. doi: 10.2174/15680266113136660220. [DOI] [PubMed] [Google Scholar]
- [9].Zwama M, Yamasaki S, Nakashima R, Sakurai K, Nishino K, Yamaguchi A, Multiple entry pathways within the efflux transporter AcrB contribute to multidrug recognition, Nat. Commun 9 (2018). doi: 10.1038/s41467-017-02493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yamaguchi A, Nakashima R, Sakurai K, Structural basis of RND-type multidrug exporters, Front. Microbiol 6 (2015) 327. doi: 10.3389/fmicb.2015.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A, Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket, Nature 480 (2011) 565–569. doi: 10.1038/nature10641. [DOI] [PubMed] [Google Scholar]
- [12].Eicher T, Cha HJ, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, V Kern W, Verrey F, Grütter MG, Diederichs K, Pos KM, Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop, Proc Natl Acad Sci U S A. 109 (2012) 5687–5692. doi: 10.1073/pnas.1114944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eicher T, Seeger MA, Anselmi C, Zhou W, Brandstätter L, Verrey F, Diederichs K, Faraldo-Gómez JD, Pos KM, Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB, Elife 3 (2014) e03145. doi: 10.7554/eLife.03145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ramaswamy VK, V Vargiu A, Malloci G, Dreier J, Ruggerone P, Molecular Rationale behind the Differential Substrate Specificity of Bacterial RND Multi-Drug Transporters, Sci. Rep 7 (2017) 1–18. doi: 10.1038/s41598-017-08747-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sjuts H, V Vargiu A, Kwasny SM, Nguyen ST, Kim H-S, Ding X, Ornik AR, Ruggerone P, Bowlin TL, Nikaido H, Pos KM, Opperman TJ, Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivatives, Proc. Natl. Acad. Sci. U. S. A 113 (2016) 3509–3514. doi: 10.1073/pnas.1602472113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, Onodera Y, Nishino K, Yamaguchi A, Structural basis for the inhibition of bacterial multidrug exporters, Nature. 500 (2013) 102–106. doi: 10.1038/nature12300. [DOI] [PubMed] [Google Scholar]
- [17].Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE Jr., D.E. Koshland, Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump, Science (80-. ). 300 (2003) 976–980. doi: 10.1126/science.1083137300/5621/976. [DOI] [PubMed] [Google Scholar]
- [18].Oswald C, Tam H-K, Pos KM, Transport of lipophilic carboxylates is mediated by transmembrane helix 2 in multidrug transporter AcrB, Nat. Commun 7 (2016) 13819. doi: 10.1038/ncomms13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yao X-QQ, Kimura N, Murakami S, Takada S, Drug Uptake Pathways of Multidrug Transporter AcrB Studied by Molecular Simulations and Site-Directed Mutagenesis Experiments, J. Am. Chem. Soc 135 (2013) 7474–85. doi: 10.1021/ja310548h. [DOI] [PubMed] [Google Scholar]
- [20].Malloci G, Vargiu A, Serra G, Bosin A, Ruggerone P, Ceccarelli M, A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds, Molecules 20 (2015) 13997–14021. doi: 10.3390/molecules200813997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang S-Y, Zou X, Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking, Proteins 66 (2006) 399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- [22].Du X, Li Y, Xia Y-L, Ai S-M, Liang J, Sang P, Ji X-L, Liu S-Q, Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods, Int. J. Mol. Sci 17 (2016) 144. doi: 10.3390/ijms17020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liebschner D, V Afonine P, Moriarty NW, Poon BK, V Sobolev O, Terwilliger TC, Adams PD, Polder maps: improving OMIT maps by excluding bulk solvent, Acta Crystallogr. Sect. D, Struct. Biol 73 (2017) 148–157. doi: 10.1107/S2059798316018210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].V Vargiu A, Nikaido H, Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 20637–42. doi: 10.1073/pnas.1218348109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zuo Z, Wang B, Weng J, Wang W, Stepwise substrate translocation mechanism revealed by free energy calculations of doxorubicin in the multidrug transporter AcrB, Sci. Rep 5 (2015) 13905. doi: 10.1038/srep13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vargiu AV, Ramaswamy VK, Malvacio I, Malloci G, Kleinekathöfer U, Ruggerone P, Water-mediated interactions enable smooth substrate transport in a bacterial efflux pump, Biochim. Biophys. Acta - Gen. Subj 1862 (2018) 836–845. doi: 10.1016/j.bbagen.2018.01.010. [DOI] [PubMed] [Google Scholar]
- [27].Wiegand I, Hilpert K, Hancock REW, Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances, Nat. Protoc 3 (2008) 163–75. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- [28].Seeger MA, von Ballmoos C, Eicher T, Brandstätter L, Verrey F, Diederichs K, Pos KM, Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB, Nature Struct. Mol. Biol 15 (2008) 199–205. doi: 10.1038/nsmb.1379. [DOI] [PubMed] [Google Scholar]
- [29].Iyer R, Ferrari A, Rijnbrand R, Erwin AL, A fluorescent microplate assay quantifies bacterial efflux and demonstrates two distinct compound binding sites in AcrB, Antimicrob. Agents Chemother. 59 (2015) 2388–97. doi: 10.1128/AAC.05112-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Overview of the CCP4 suite and current developments, Acta Crystallogr. D. Biol. Crystallogr 67 (2011) 235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Trott O, Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem 31 (2010) 455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem 30 (2009) 2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].D.M.Y. and Case PAKDA, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III, Cruzeiro VWD, Darden TA, Duke RE, Ghoreishi D, Gilson MK, Gohlke H, Goetz AW, Greene D, Harris R, Homeyer N, Izadi S, Kovalenko A, Kurtzman T, Lee TS, LeGra S, AMBER 2018. University of California San Francisco, (2018). [Google Scholar]
- [34].Atzori A, Malviya VN, Malloci G, Dreier J, Pos KM, Vargiu AV, Ruggerone P, Identification and characterization of carbapenem binding sites within the RND-transporter AcrB, Biochim. Biophys. Acta - Biomembr 1861 (2019) 62–74. doi: 10.1016/j.bbamem.2018.10.012. [DOI] [PubMed] [Google Scholar]
- [35].Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL, OPM database and PPM web server: resources for positioning of proteins in membranes, Nucleic Acids Res. 40 (2012) D370–6. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jo S, Kim T, Iyer VG, Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM, J. Comput. Chem. 29 (2008) 1859–65. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- [37].IR Gould RW, Skjevik AA, Dickson CJ, Madej BD, Lipid17: A Comprehensive AMBER Force Field for the Simulation of Zwitterionic and Anionic Lipids, in preparation (2019). [Google Scholar]
- [38].Hopkins CW, Le Grand S, Walker RC, Roitberg AE, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning, J. Chem. Theory Comput. 11 (2015) 1864–74. doi: 10.1021/ct5010406. [DOI] [PubMed] [Google Scholar]
- [39].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput 11 (2015) 3696–713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA, Development and testing of a general amber force field, J. Comput. Chem 25 (2004) 1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- [41].Genheden S, Ryde U, The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert Opin. Drug Discov. 10 (2015) 449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nguyen H, Roe DR, Simmerling C, Improved Generalized Born Solvent Model Parameters for Protein Simulations, J. Chem. Theory Comput 9 (2013) 2020–2034. doi: 10.1021/ct3010485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Weiser J, Shenkin PS, Still WC, Approximate solvent-accessible surface areas from tetrahedrally directed neighbor densities, Biopolymers 50 (1999) 373–380. doi:. [DOI] [PubMed] [Google Scholar]
- [44].Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P, Barril X, Hubbard RE, Morley SD, rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids, PLoS Comput. Biol 10 (2014) e1003571 10.1371/journal.pcbi.1003571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chovancova E, Pavelka A, Benes P, Strnad O, Brezovsky J, Kozlikova B, Gora A,. Sustr V, Klvana M, Medek P, Biedermannova L, Sochor J, Damborsky J, CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput Biol 8 (2012) e1002708. doi: 10.1371/journal.pcbi.1002708 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.