

Abstract
Apoptosis signal-regulating kinase 1 (ASK1) is a mitogen-activated protein kinase kinase kinase (MAPKKK) that regulates activation of the c-Jun N-terminal kinase (JNK)- and p38-stress response pathways leading to apoptosis in nucleated cells. We have previously shown that ASK1 is expressed in platelets and regulates agonist-induced platelet activation and thrombosis. However, the mechanism by which platelet agonists cause activation of ASK1 is unknown. Here, we show that in platelets agonist-induced activation of p38 is exclusively dependent on ASK1. Both thrombin and collagen were able to activate ASK1/p38. Activation of ASK1/p38 was strongly dependent on thromboxane A2 (TxA2) and ADP. Agonist-induced ASK1 activation is blocked by inhibition of phospholipase C (PLC) β/γ activity or by chelating intracellular Ca2+. Furthermore, treatment of platelets with thapsigargin or Ca2+ ionophore robustly induced ASK1/p38 activation. In addition, calcium and integrin-binding protein 1 (CIB1), a Ca2+-dependent negative regulator of ASK1, associates with ASK1 in resting platelets and is dissociated upon platelet activation by thrombin. Dissociation of CIB1 corresponds with ASK1 binding to tumor necrosis factor (TNF) receptor associated factor 6 (TRAF6) and the autophosphorylation of ASK1 Thr838 within the catalytic domain results in full activation of ASK1. Furthermore, genetic ablation of Cib1 in mice augments agonist-induced Ask1/p38 activation. Together our results suggest that in resting platelets ASK1 is bound to CIB1 at low Ca2+ concentrations. Agonist-induced platelet activation causes an increase in intracellular Ca2+ concentration that leads to the dissociation of CIB1 from ASK1, allowing for proper dimerization through ASK1 N-terminal coiled-coil (NCC) domains.
Keywords: p38, ASK1, platelets, ROS, calcium, CIB1, GPCR, TRAF6, signal transduction
Introduction
Mitogen activated protein kinases (MAPK) are a family of serine/threonine protein kinases that are central mediators of many cellular functions from proliferation to apoptosis. MAPK are activated by a kinase signaling cascade composed of MAPKKK and MAPKK; where MAPKKK activate MAPKK, which in turn activate MAPK. All three MAPK families are expressed in platelets and are activated in response to agonist stimulation. Studies of MAPK in platelets using both genetic and pharmacological methods have highlighted the importance of MAPK signaling in the regulation of hemostasis and thrombosis [1, 2].
While the signaling mechanisms linking MAPKKK to MAPK activation are conserved and well known, relatively little is known about the upstream mechanisms that regulate agonist-induced MAPK activation in platelets. Recently, we and others showed that ASK1, a MAPKKK, is expressed in platelets and that loss of Ask1 attenuated platelet functions and protected mice from thrombosis [3, 4].
In nucleated cells, ASK1 is known to mediate stress induced apoptosis [5–7]. ASK1 forms a homodimer through association with its C-terminal coiled-coil (CCC) domain. In its resting state, the ASK1 homodimer is kept inactive by binding to reduced Thioredoxin (Trx) at the N-terminal regulatory domain of ASK1, which prevents ASK1’s kinase domains from interacting through its N-terminal coiled-coil (NCC) domains [8–10]. During oxidative stress Trx is oxidized by reactive oxygen species (ROS), causing its dissociation from ASK1. Dissociation of Trx frees the N-terminal regulatory domain to bind TRAF2/6 which facilitates N-terminal dimerization of ASK1, allowing ASK1’s kinase domains to induce activation through autophosphorylation at Thr838 (Thr845 in mice), which renders ASK1 fully active [8–11]. Activated ASK1 phosphorylates its downstream targets MKK3/6 and MKK4/7 to induce p38 and JNK activation respectively [6]. However, the mechanism governing ASK1 activation in platelets remains unknown.
In the present study, we set out to determine the signaling mechanisms through which platelet agonists activate ASK1. We show that the major platelet agonists activate ASK1 through a common calcium-dependent mechanism that is independent of ROS, a preferred mechanism followed in nucleated cells. Further, we found that ASK1 is bound to CIB1, a calcium dependent negative regulator, in resting platelets, and that an agonist-induced rise in intracellular Ca2+ dissociates CIB1 from ASK1 to facilitate ASK1 activation.
Material and methods
Reagents
Dimethyl sulfoxide (DMSO), Arg-Gly-Asp-Ser (RGDS), U73122, hydrogen peroxide (H2O2), and calcium ionophore (A23187) were purchased from Sigma-Aldrich. Fluo-4-AM was purchased from Thermo-Fisher Scientific. PAR1-AP (SFLLRN) and PAR4-AP (AYPGKF) were purchased from AnaSpec. Collagen was purchased from Chronolog. Human α-thrombin was purchased from Enzyme Research. Convulxin was purchased from Pentapharm. Bisindolylmaleimide I (BIS) and 1,2-bis(2-aminophenoxy)ethane N,N,N′,N′-tetraacetic acid acetoxymethyl ester (BAPTA/AM) was purchased from Calbiochem. YM-254890 was purchased from Focus Biomolecules. Manganese (III) tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP) was purchased from Santa Cruz Biotechnology. Thapsigargin was purchased from Cayman Chemical. All other chemicals used were of analytical grade and purchased from Sigma-Aldrich.
Antibodies
Anti-ASK1 (H-300) antibody: sc7931, 1:1000; normal rabbit IgG: sc2027, was obtained from Santa Cruz Biotechnology. Following antibodies were obtained from Cell Signaling, Danvers, MA. Anti-P-p38 (Thr180/Tyr182) antibody: #9211, 1:5000; anti-p38 antibody: #9212, 1:3000; anti-P-ASK1 (Thr845) antibody: #3765, 1:200; anti-ASK1 antibody: #8662, 1:1000; anti-P-SEK/MKK4 (Thr261) antibody: #9151, 1:200; anti-SEK/MKK4 antibody: #9152, 1:200; anti-P-MKK7 (Ser271/Thr275) antibody: #4171, 1:200; anti-MKK7 antibody: #4172, 1:200; anti-P-MKK3(Ser189)/MKK6(Ser207) antibody: #9236, 1:200; anti-MKK6 antibody: #9264, 1:200; anti-MKK3 antibody: # 5674, 1:200; anti-TRAF6 antibody: #4743, 1:500; anti-Trx1 antibody: #2429, 1:100; HRP conjugated anti-rabbit IgG antibody; # 7074, 1:2000-7000; HRP conjugated anti-mouse IgG antibody: # 7076, 1:5000; HRP conjugated anti-rabbit IgG (conformation specific) antibody; #5127S, 1:12,500. Anti-TRAF6 antibody: ab137452, 1:1000; was obtained from Abcam. Additionally, the polyclonal anti-P-ASK1 (Thr838) antibody and anti-ASK1 antibody was raised against an antigenic synthetic peptide (830-AGINPCTE(p)TFTGTLQY-845) of human ASK1 by immunizing rabbits. The antisera were analyzed by ELISA to determine specificity for ASK1. A polyclonal phospho-specific antibody was affinity-purified from the antisera by Neo Scientific (Cambridge, MA). The anti-CIB1 antibody (clone 7.79; 1:200) was generated in our laboratory as described [12].
Human platelet preparation
Human whole blood was drawn, with informed consent, by venipuncture from healthy adult volunteers (irrespective of gender and race). Approval for this study was obtained from the Institutional Review Board of Thomas Jefferson University, according to the Declaration of Helsinki. Blood was collected using acidified citrate dextrose as an anticoagulant. Washed human platelets were prepared in Tyrode’s buffer to 4 x 108 platelets/mL as previously described [13]. For experiments using inhibitors, platelets were incubated at 37°C for 5 minutes (min) (for experiments involving apyrase) or 30 min (for experiments involving BAPTA/AM, U73122, aspirin, MnTMPyP, BIS, or YM-254890) before being stimulated as indicated at room temperature. For experiments using BAPTA/AM, washed platelets were prepared and maintained in calcium-free Tyrode’s buffer throughout the experiment.
Mouse platelet preparation
Both male and female 8-12-week-old WT and Cib1−/− mice of C57BL/6J genetic background were used in these studies as described previously [14, 15]. Washed murine platelet suspensions (2 x 108 platelets/mL) were prepared as previously described [3]. Approval for animal experimental studies was received from the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University.
Immunoprecipitation
Immunoprecipitation was performed as previously described [16]. Briefly, washed human platelet suspension (4 x 108 platelets/mL) was stimulated with thrombin (1U/mL) for 3 min at room temperature. Platelet suspensions were lysed by the addition of an equal volume of ice-cold 2X lysis buffer to reach a final concentration of (1% Triton X-100, 150mM NaCl, 50 mM Tris-HCl pH 7.5, 10μg/mL leupeptin, 10μg/mL aprotinin, 1mM NaF, 1mM sodium orthovanadate, and 1mM PMSF). Pre-cleared lysates were incubated with anti-ASK1 [Santa Cruz Biotechnology, 1:100] for IP of human ASK1, or normal rabbit IgG [Santa Cruz Biotechnology, 1:100] and the immunoprecipitation was performed as described [16]. Precipitate was western blotted with anti-ASK1, anti-CIB1, anti-TRAF6, and anti-Trx1 antibodies.
IP of mouse Ask1 followed the above protocol with the following modifications. Briefly, washed mouse platelet suspension (4 x 108 platelets/mL) was stimulated with thrombin (1U/mL) for 1 min at room temperature before being lysed with ice-cold 2X lysis buffer. Lysates were then incubated with anti-Ask1 [Cell Signaling, 0.23 μg/mL] for IP of mouse Ask1, or normal rabbit IgG [Santa Cruz Biotechnology, 0.23μg/mL] as control and the immunoprecipitation was performed as described [16]. Precipitate was western blotted with anti-Ask1 and anti-Traf6 antibodies.
Western blotting
Aliquots of washed platelet suspension in Tyrode’s buffer, either resting or stimulated with agonists, were lysed with reducing Laemmli-sample buffer at various time points. Lysates were subjected to immunoblotting as described [3]. Molecular weight markings correspond to the standard weights of molecular ladder used (Spectra BR, 26623, Thermo-Fisher Scientific).
Quantification of western blot band intensities was performed using NIH Image J software. The ratio of optical density (phospho-band/ total band or Co-IP band/ IP band) was calculated for each lane, within replicates, before plotting and statistical analysis.
Measurement of intracellular Ca2+
Intracellular Ca2+ levels were measured, as described [3]. Briefly, aliquots of washed human platelets (5x106 platelets/mL in Ca2+ free Tyrode’s buffer) were incubated with 2.5 μM Fluo-4-AM for 1 min. Platelets were then diluted to 4.17x105 platelets/mL with Tyrode’s buffer containing 1mM CaCl2 and fluorescence intensity was measured using an Accuri C6 flow cytometer (Becton Dickinson). Agonists were added following a 60 second baseline and traces recorded for the indicated time.
Statistical analysis
Statistical analysis of the data was performed using Prism 6 (GraphPad). Student’s t-test, a one-way ANOVA with Dunnett’s multiple comparisons test to the control, or a two-way ANOVA were used to determine statistical significance. P≤0.05 was regarded as statistically significant. Each experiment was repeated independently at least 3 times using platelets isolated from individual donors. Data (mean ± standard error of the mean) was plotted using Prism 6.
Results
Platelet agonists induce p38 activation
We have previously shown that ASK1 is activated by most agonists in human platelets, and in murine platelets activation of p38 is solely dependent on Ask1 [3, 4, 17]. Here, we evaluated if p38 activation corresponds to ASK1 activation in human platelets. We stimulated washed human platelets with thrombin, protease-activated receptor (PAR) 1 activating peptide (SFLLRN, PAR1-AP), PAR4 activating peptide (AYPGKF, PAR4-AP), convulxin (a GPVI specific agonist), U46619 (a TxA2 mimetic TP receptor agonist), or ADP (the P2Y1/12 receptor agonist). We found that resting platelets showed minimal phosphorylation of p38 (Thr180/Tyr182, an indicator of its activation), but stimulation with each of these agonists triggered a robust, time-dependent, but transient increase in the phosphorylation of p38, except that ADP induced a much lower amount (Fig.1A–F). These results suggest that the major platelet agonists are capable of activating ASK1/p38, making ASK1 a common signaling node in platelet activation.
Figure 1. Platelet agonists rapidly induce p38 activation.
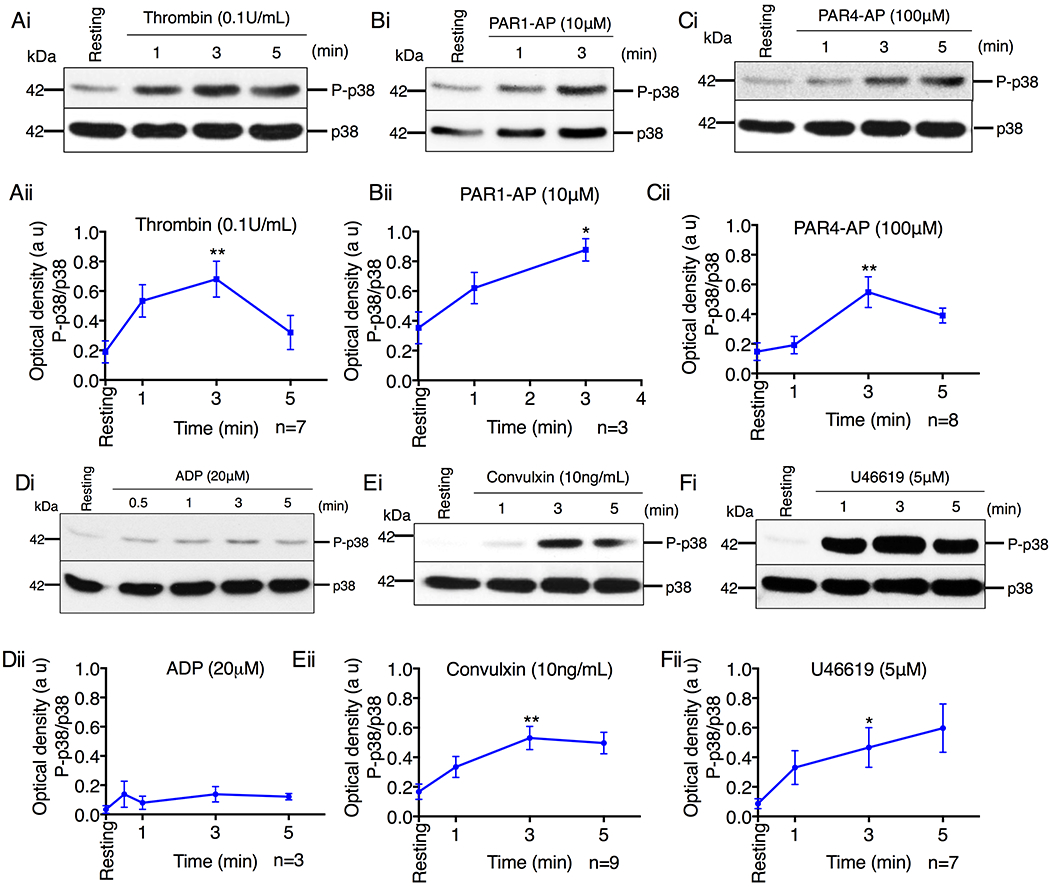
(A-F) Western blot analysis of lysates of human platelets stimulated with (A) thrombin (0.1U/mL), (B) PAR1-AP (10μM), (C) PAR4-AP (100μM), (D) ADP (20μM), (E) convulxin (10ng/mL), (F) U46619 (5μM). (i) Images of representative immunoblots of phosphorylation of p38 using a phospho-specific antibody. Blots were reprobed with anti-p38 antibody to ensure equal protein loading. (ii) Quantification of band intensities from (i). *P<0.05, **P<0.01, one-way ANOVA with Dunnett’s multiple comparisons test.
Canonical ASK1 targets are activated in human platelets
It is well known that ASK1 activates p38 and JNK in nucleated cells via activation of the intermediate MAPKKs, MKK3/4/6 and MKK4/7 respectively [5, 6, 18–22]. We therefore determined whether these kinases are activated (denoted by phosphorylation) in human platelets following agonist stimulation. Stimulation of platelets with thrombin induced rapid activation of MKK3/4/6/7, which corresponded with phosphorylation of ASK1 (Thr838), a marker of its kinase activity) and p38 (Fig. 2). These results agree with our previous report showing that Ask1 regulates thrombin induced activation of MKK3/4/6 as well as p38 activation in murine platelets [3].
Figure 2. Canonical ASK1 targets are activated in platelets.
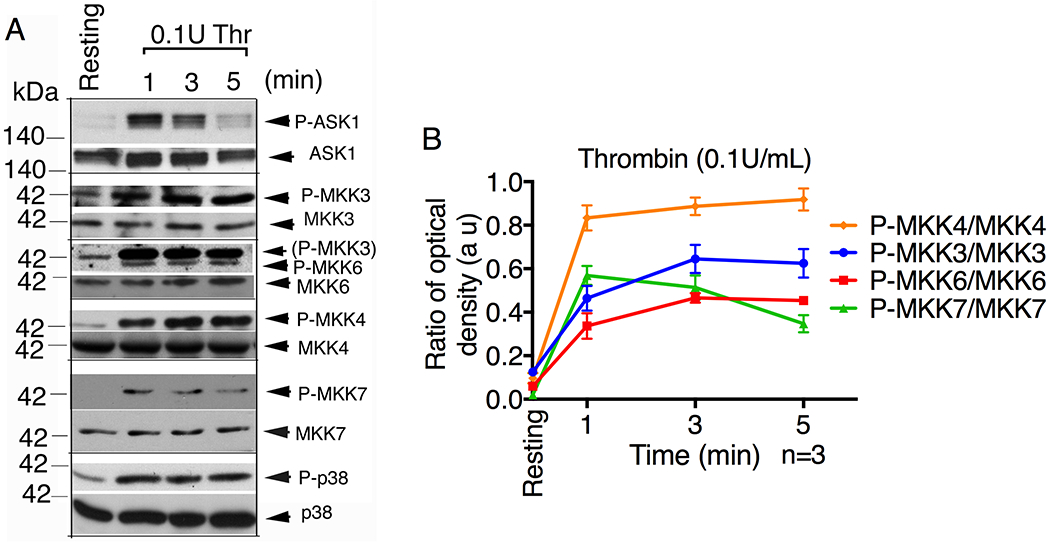
Western blot analysis of lysates of human platelets stimulated with thrombin (0.1U/mL) for the indicated time. (A) Images of representative immunoblots of phosphorylation of ASK1, MKK3, MKK4, MKK6, MKK7 and p38 using phospho-specific antibodies. Phospho-blots were reprobed for their respective total (anti-ASK1, anti-MKKs, anti-p38) antibodies to ensure equal protein loading. (B) Quantification of band intensities of each MKKs from (A).
Role of secondary agonists in ASK1/p38 activation
Given that all agonists tested were able to induce ASK1/p38 activation, we next investigated the role major secondary agonists (TxA2 and ADP, two of the major physiological agonists that induce platelet activation in vivo) play in thrombin- or collagen-induced ASK1 activation. As expected, stimulation with either thrombin or collagen was able to induce dose-dependent activation of ASK1. Activation of p38 was also dose-dependent with minimal doses of agonist being sufficient to induce ~50% p38 activation (Fig. 3A–B, Fig. S1). When pretreated with either aspirin or apyrase, to block TxA2 and ADP respectively, we found that aspirin and/or apyrase significantly attenuated both thrombin- and collagen-induced ASK1 activation (Fig. 3A–B, Fig. S1). Of these, collagen-induced ASK1 activation was more dependent on secondary agonists as ASK1/p38 activation was strongly reduced by the combination of aspirin and apyrase (Fig. 3B, Fig. S1Cii). In contrast, thrombin-induced ASK1 activation was less dependent on secondary agonists as ASK1 activation was only reduced by ~50%, compared to the untreated control. Interestingly, thrombin-induced p38 activation was not affected by pretreatment with aspirin alone, suggesting a greater dependence on ADP induced signaling for full ASK1 activation (Fig. 3A, Fig. S1Ci).
Figure 3. Secondary agonists augment ASK1 activation.
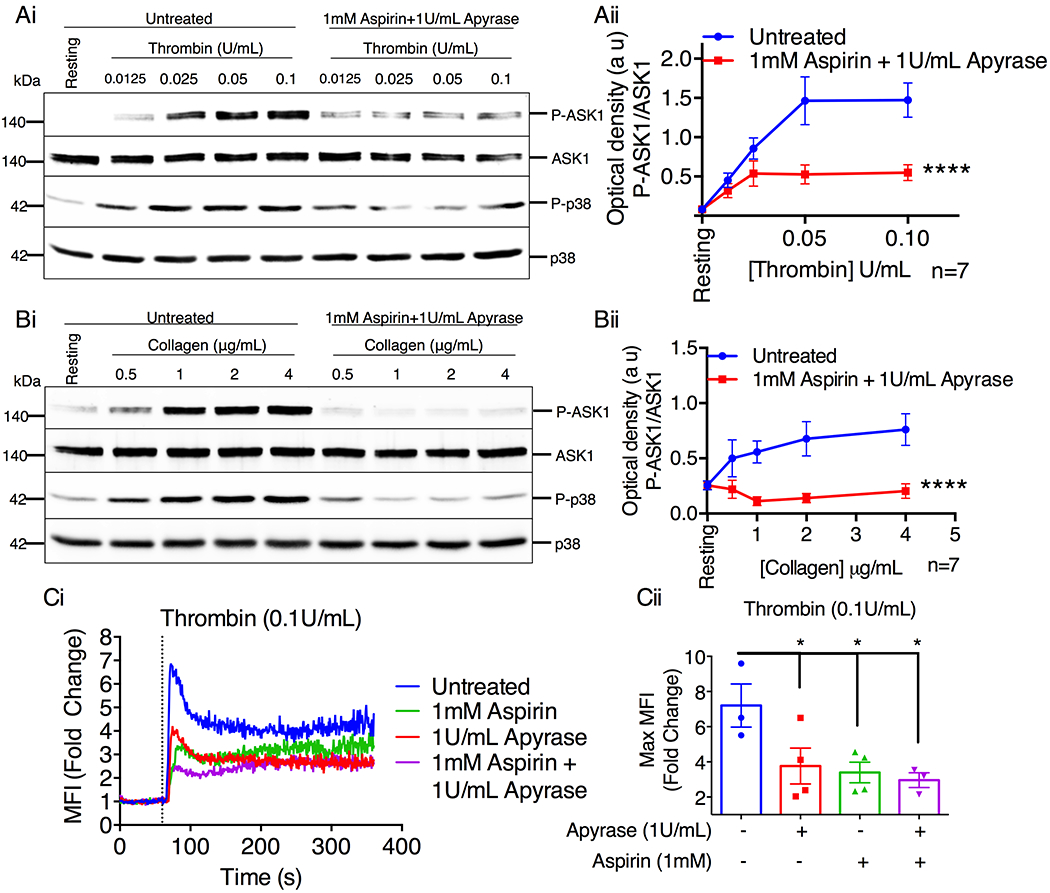
Western blot analysis of human platelets pretreated with aspirin (1mM) and apyrase (1U/mL) before being stimulated with various doses of thrombin (A) or collagen (B) for 1 and 3 min respectively. (Ai & Bi) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. (Aii & Bii) Quantification of band intensities of ASK1 from (Ai & Bi). ****P<0.0001, two-way ANOVA. (C) Intracellular Ca2+ rise was assessed by flow cytometry in washed human platelets pretreated with aspirin (1mM) and/or apyrase (1U/mL) before being stimulated with thrombin. (Ci) Representative tracings of Ca2+ rise in Fluo-4-loaded human platelets when stimulated with 0.1 U/mL thrombin. Baseline measurements were recorded before agonist addition at 60 s. Vertical dotted line denotes point of agonist addition. (Cii) Maximum fold change was assessed from 0–120 s. *P<0.05, one-way ANOVA with Dunnett’s multiple comparisons test.
To determine if intracellular signaling is perturbed by inhibition of secondary agonists, we evaluated intracellular Ca2+ rise induced by thrombin in the presence of aspirin and/or apyrase. When stimulated with 0.1U/mL of thrombin, there is robust (~7 fold) rise in intracellular Ca2+. Thrombin-induced rise in intracellular Ca2+ is reduced to ~2.5 fold by treatment with inhibitors of secondary mediators (Fig. 3Ci). These results suggest that secondary agonists play a significant role in activation of ASK1 downstream of the primary agonists, thrombin and collagen.
Platelet agonists activate ASK1 via PLCβ/γ
Platelet agonists robustly induce intracellular Ca2+ rise by first activating PLCβ or PLCγ downstream of Gαq-coupled GPCRs or ITAM receptors, respectively [23]. To determine if platelet agonists activate ASK1 via PLCβ/γ activity, we used a pan-PLC inhibitor, U73122, to inhibit agonist-induced PLC activity [24]. We chose to use the agonists PAR4-AP and convulxin as they are single receptor agonists that are representative of the major GPCR and non-GPCR platelet agonists, thrombin and collagen. We found that U73122 not only blocked PAR4-AP-induced rise in cytosolic Ca2+ concentration (Fig. 4A), but also blocked phosphorylation of ASK1 (Fig. 4B). To determine if the loss of PLC activity also affected ASK1 signaling activity, we examined agonist-induced p38 activation. We found that U73122 blocked PAR4-AP induced p38 activation (Fig. 4B). Similarly, convulxin-induced ASK1/p38 activation was completely blocked by U73122 (Fig. 4C).
Figure 4. Activation of ASK1 by platelet agonists requires PLCβ/γ activity.
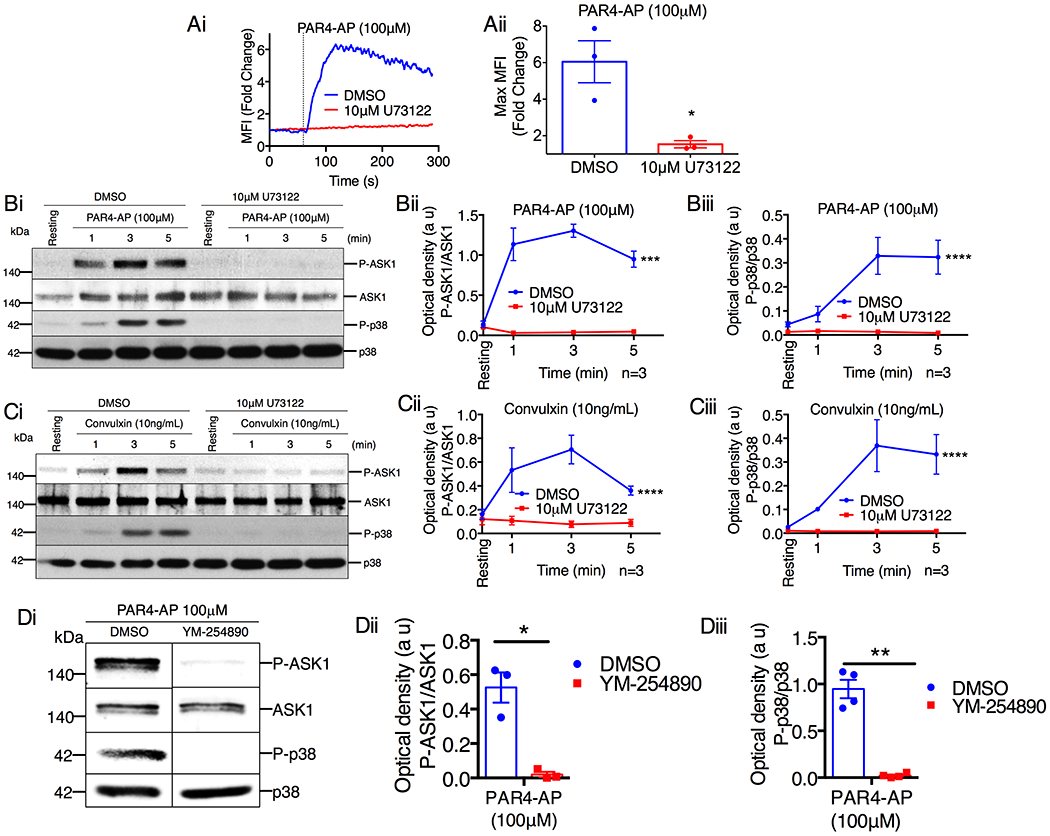
(A) Fluo-4-loaded washed human platelets pretreated with DMSO or the pan-PLC inhibitor U73122 (10μM) were activated with PAR4-AP and intracellular Ca2+ rise was measured. (Ai) Representative tracing of PAR4-AP-induced Ca2+ rise in platelets. Vertical dotted line denotes point of agonist addition. (Aii) Quantification of the maximum Ca2+ rise from Ai, *P<0.05, Student’s t-test. (B-C) Western blot analysis of human platelets pretreated with either DMSO or U73122 (10μM) before being stimulated with (B) PAR4-AP (100μM) or (C) convulxin (10ng/mL). (Bi & Ci) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. Quantification of the band intensities of (Bii & Cii) P-ASK1 and (Biii & Ciii) P-p38 from (Bi & Ci), ***P<0.001, ****P<0.0001, two-way ANOVA. (D) Western blot analysis of human platelets pretreated with either DMSO or YM-254890 (1μM) before being stimulated with PAR4-AP (100μM) for 3 min. (Di) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. Quantification of the band intensity of (Dii) P-ASK1 and (Diii) P-p38 from (Di); *P<0.05, **P<0.01, Student’s t-test.
To determine if PAR4-AP activates ASK1 via the Gαq/PLCβ signaling axis, we examined ASK1/p38 activation in platelets pretreated with a Gαq inhibitor, YM-254890. YM-254890 abolished PAR4-AP-induced ASK1/p38 activation (Fig. 4D). Taken together, these data suggest that both GPCR and ITAM receptors stimulate ASK1 activity via PLCβ/γ.
ASK1 is activated by an increase in intracellular Ca2+
PLC activity not only leads to increased cytosolic Ca2+ concentration, but also to an increase in protein kinase C (PKC) activity via generation of diacylglycerol (DAG). It is therefore possible that PKC may be responsible for activation of ASK1. To test this possibility, we pretreated platelets with bisindolylmaleimide (BIS), a pan PKC inhibitor, and activated them with thrombin. We found that inhibition of PKC had no effect on ASK1 activation induced by thrombin (Fig. 5A). To determine if ASK1 is activated by increased cytosolic Ca2+ concentration alone, we used calcium ionophore, A23187, which is known to increase cytosolic Ca2+ concentration. We found that A23187 induced rapid activation of ASK1 and p38 (Fig. 5B) suggesting that intracellular Ca2+ rise alone can activate ASK1.
Figure 5. Ca2+ influx activates ASK1 in platelets.
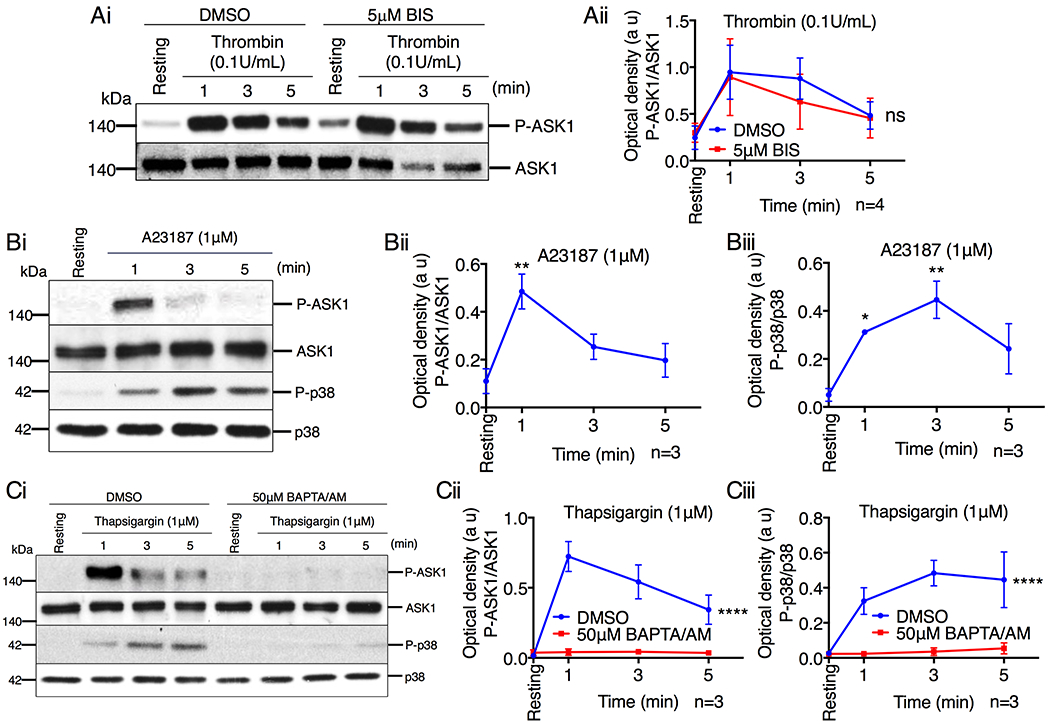
(A) Western blot analysis of human platelets pretreated with either DMSO or BIS (5μM), and stimulated with thrombin (0.1U/mL). (Ai) Images of representative immunoblots of phosphorylation of ASK1 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 antibodies to ensure equal protein loading. (Aii) Quantification of the band intensity of P-ASK1 from (Ai); Not significant (ns), two-way ANOVA. (B) Representative western blot images of human platelets stimulated with A23187 (1μM). (Bi) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. Quantification of the band intensity of (Bii) P-ASK1 and (Biii) P-p38 from (Bi), *P<0.05, **P<0.01, one-way ANOVA with Dunnett’s multiple comparisons test. (C) Western blot analysis of human platelets pretreated with either DMSO or BAPTA/AM (50μM) and stimulated with thapsigargin (1μM). (Ci) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. Quantification of the band intensity of (Cii) P-ASK1 and (Ciii) P-p38 from (Ci), ****P<0.0001, two-way ANOVA.
Both Ca2+ entry from the extracellular milieu and release of Ca2+ from internal stores can contribute to a rise in cytosolic Ca2+. To determine if Ca2+ from intracellular stores leads to ASK1 activation, we treated washed human platelets with thapsigargin in Ca2+ free buffer. We found that thapsigargin induced robust phosphorylation of both ASK1 and p38 (Fig. 5C). Additionally, pretreating platelets with the Ca2+ chelator BAPTA/AM blocked thapsigargin-induced activation of ASK1 and p38 (Fig. 5C).
To determine if platelet agonists activate ASK1 via elevation of intracellular Ca2+ levels, platelets in Ca2+ free buffer were pretreated with BAPTA/AM and stimulated with either PAR4-AP or convulxin. We found that both PAR4-AP- and convulxin-induced phosphorylation of ASK1 and p38 was blocked by chelating Ca2+ (Fig. 6, A & B). Taken together, our results suggest that agonist-induced increase in cytosolic Ca2+ concentration is responsible for the activation of ASK1.
Figure 6. Platelet agonists activate ASK1 in a Ca2+-dependent manner.
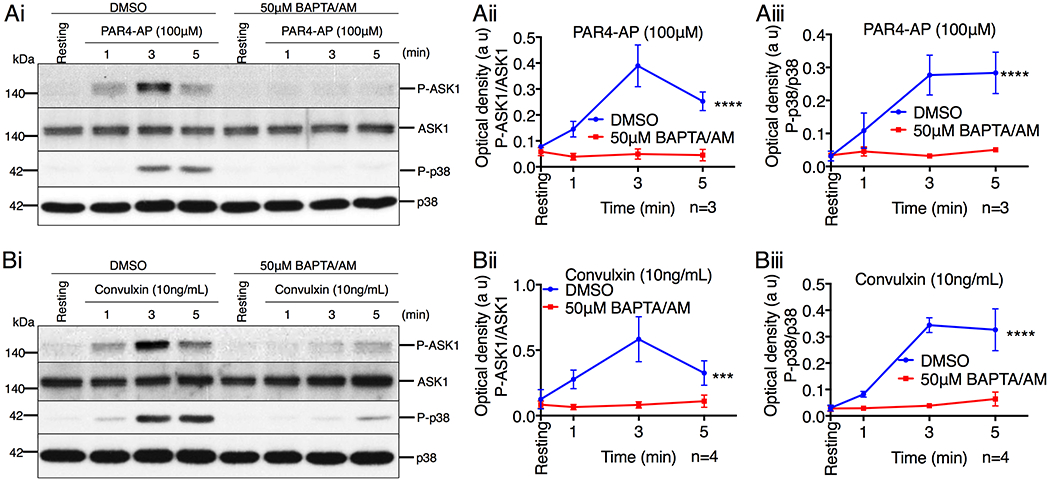
(A & B) Western blot analysis of human platelets pretreated with either DMSO or BAPTA/AM (50μM) before being stimulated with (A) PAR4-AP (100μM) or (B) convulxin (10ng/mL). (Ai & Bi) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. Quantification of the band intensities of (Aii & Bii) P-ASK1 and (Aiii & Biii) P-p38 from (Ai & Bi), ***P<0.001, ****P<0.0001, two-way ANOVA.
H2O2 induces ASK1 activation via a Ca2+ dependent mechanism
ASK1 is known to be activated primarily by ROS in nucleated cells [10, 25]. To determine if ROS can activate ASK1 in platelets, platelets were stimulated with the ROS donor H2O2. We found that H2O2 dose-dependently induced phosphorylation of ASK1/p38 (Fig. 7A). However, we found that H2O2 only induced a marginal increase in the intracellular Ca2+ levels at these concentrations (Fig. 7B). To determine if this marginal increase in intracellular Ca2+ is responsible for H2O2-induced activation of ASK1, we stimulated platelets with H2O2 in the presence or absence of extracellular Ca2+ and BAPTA/AM. While removal of extracellular Ca2+ did not affect H2O2 induced ASK1 activation, we found that chelating intracellular Ca2+, by the addition of BAPTA/AM, attenuated H2O2 induced ASK1 activation (Fig. 7C). Strikingly, while BAPTA/AM was able to block 50μM H2O2 induced ASK1 activation, 100μM of H2O2 was still able to induce a noticeable amount of ASK1 activation (Fig. 7C). These data suggest that H2O2 induces ASK1 activation primarily via a Ca2+ dependent mechanism.
Figure 7. Platelet agonists activate ASK1 via a ROS independent mechanism.
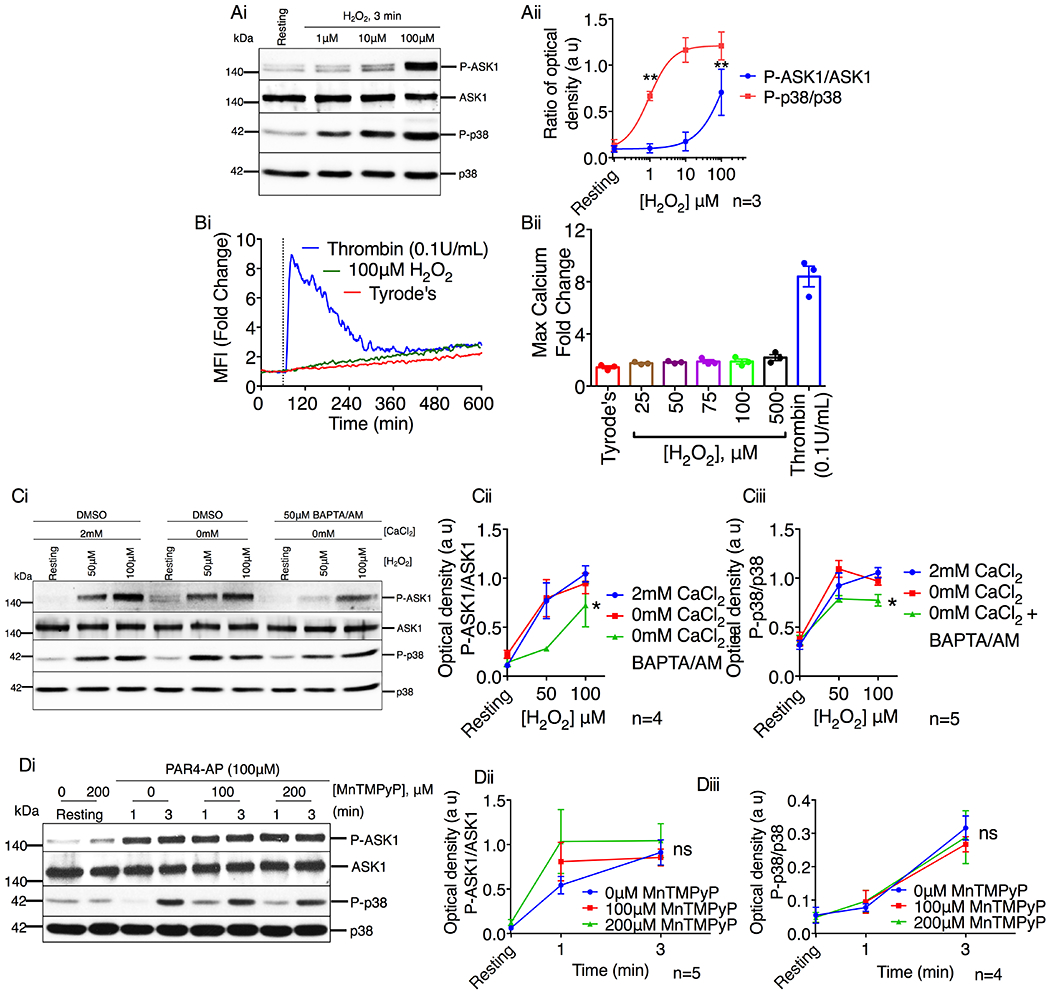
(A) Western blot analysis of human platelets stimulated with H2O2 (1–100μM). (Ai) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. (Aii) Quantification of the band intensities of P-ASK1 and P-p38 from (Ai). **P<0.01, one-way ANOVA with Dunnett’s multiple comparisons test. (B) Intracellular Ca2+ rise was assessed by flow cytometry in washed human platelets stimulated with thrombin or H2O2. (Bi) Representative tracing of thrombin (0.1U/mL) or H2O2 (100μM) induced intracellular Ca2+ rise in washed human platelets. (Bii) Quantification of the maximum mean fluorescence intensity (fold change). *P<0.05, one-way ANOVA with Dunnett’s multiple comparisons test. (C) Western blot analysis of human platelets in the presence or absence of extracellular Ca2+ and pretreated with either DMSO or BAPTA/AM (50μM) before being stimulated with H2O2 (50 & 100μM). (Ci) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. Quantification of the band intensities of (Cii) P-ASK1 and (Ciii) P-p38 from (Ci), *P<0.05, two-way ANOVA. (D) Western blot analysis of human platelets pretreated with vehicle or MnTMPyP (100 & 200μM) and stimulated with PAR4-AP (100μM). (Di) Images of representative immunoblots of phosphorylation of ASK1 and p38 using phospho-specific antibodies. Blots were reprobed with anti-ASK1 and anti-p38 antibodies to ensure equal protein loading. Quantification of the band intensities of (Dii) P-ASK1 and (Diii) P-p38 from (Di), Non significant (ns), Two-way ANOVA.
Since our data shows that high concentrations of H2O2 are capable of activating ASK1 in platelets independent of intracellular Ca2+, it is possible that platelet agonists may also activate ASK1 via a ROS generating mechanism. To determine if platelet agonists activate ASK1 by increasing ROS production, we pretreated platelets with a superoxide dismutase mimetic compound, MnTMPyP, to scavenge ROS [26] prior to stimulation with PAR4-AP, an agonist known to induce ROS generation in platelets [27]. We found that PAR4-AP-induced phosphorylation of both ASK1 and p38 was not affected by scavenging ROS (Fig. 7D). Taken together, these data suggest that PAR4-AP activates ASK1 via a ROS-independent mechanism.
CIB1 regulates thrombin-induced ASK1 activation in platelets
Having conclusively shown that platelet agonists activate ASK1 exclusively by increasing intracellular Ca2+, we next investigated the mechanism involved in this activation. Canonically, the ASK1 homodimer is kept inactive by binding to reduced Trx, which prevents binding of TRAF2/6 and subsequent ASK1 activation [8–11]. We therefore investigated if Trx associated with ASK1 in platelets. Interestingly, despite being highly expressed in platelets, we could not detect Trx in the co-precipitate when ASK1 was immunoprecipitated (IPed) from resting or thrombin activated platelets suggesting that Trx is not bound to ASK1 and therefore unlikely to be the endogenous inhibitor of ASK1 in platelets (Fig. 8A).
Figure 8. CIB1, but not Trx, associates with ASK1 in resting platelets.
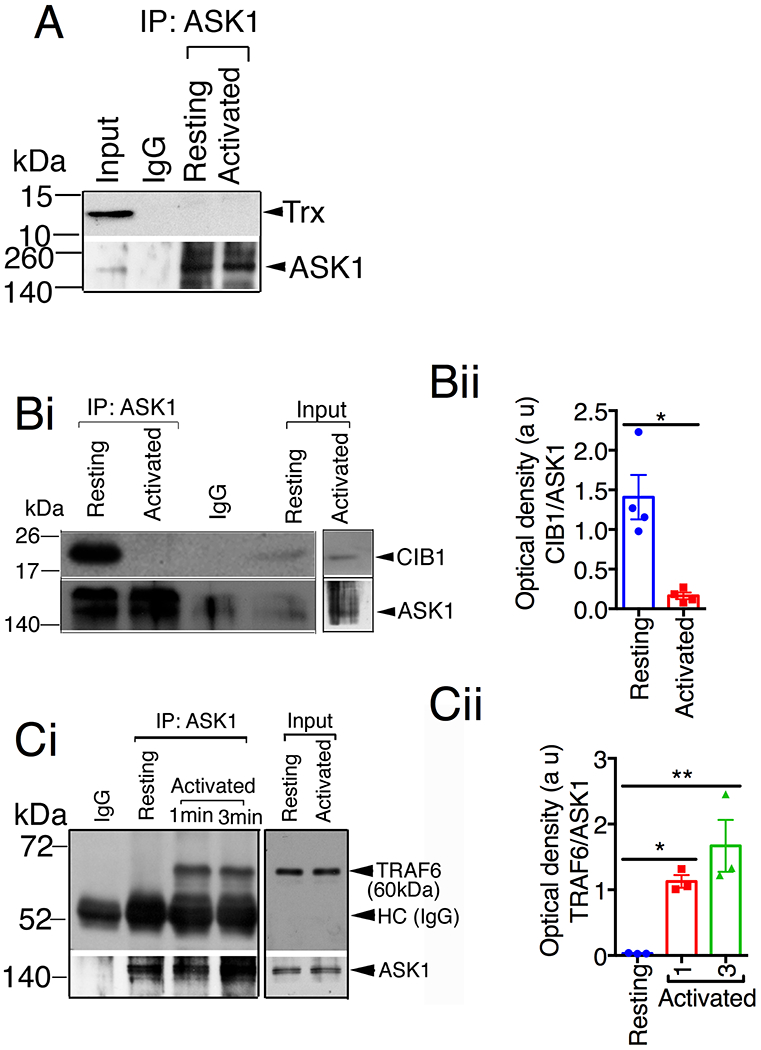
(A-C) Representative western blot images of immunoprecipitate of anti-ASK1 or normal rabbit IgG antibodies from resting and thrombin (1U/mL, used to elicit full dissociation of CIB1 from ASK1) stimulated platelet lysates and blotted for (A) Trx, (Bi) CIB1, and (Ci) TRAF6. Input is 5% of lysates used for immunoprecipitation. Blots were reprobed with anti-ASK1 antibody to ensure equal loading. (Bii) Quantification of the band intensity from (Bi); *P<0.05, Student’s t-test. (Cii) Quantification of the band intensity from (Ci); *P<0.05, **P<0.01, one-way ANOVA with Dunnett’s multiple comparisons test.
Apart from the ROS sensitive regulator Trx, CIB1is a major regulator of platelet function and is known to inhibit ASK1 in neuronal cells [8, 28]. We therefore asked if CIB1 binds to ASK1 in resting platelets. When ASK1 was IPed from resting human platelet lysates, we observed that CIB1 co-IPed with ASK1; however, following stimulation with thrombin, CIB1 no longer co-IPed with ASK1, suggesting that CIB1, which was bound to ASK1 in resting platelets, dissociated from ASK1 upon platelet activation (Fig. 8B). Consistent with this finding when ASK1 was IPed from resting human platelet lysates, we found that the positive regulator TRAF6 was absent in the ASK1 immunoprecipitate, but was readily co-IPed with ASK1 upon platelet activation by thrombin (Fig. 8C) suggesting that TRAF6 associates with ASK1 upon platelet activation.
Consistent with the above findings, one would expect ASK1 to be prone to activation in the absence of CIB1. In fact, enhanced ASK1 activation was detected in cells in which CIB1 was knocked down [28]. To test if the lack of Cib1 augmented Ask1 activation in murine platelets, we investigated thrombin induced Ask1 activation in platelets isolated from WT and Cib1−/− mice. RGDS was used to block outside-in signaling. We found that thrombin-stimulated Cib1 null platelets displayed a significant augmentation of Ask1 and p38 phosphorylation in response to thrombin, suggesting that Ask1 activation was enhanced in the absence of Cib1 in platelets (Fig. 9A&B). The augmentation of Ask1 and p38 that we observed was not due to the increased levels of Ask1 protein in Cib1 null platelets, since Ask1 protein levels were comparable between WT and Cib1−/− platelets (Fig. 9Ai). Furthermore, neither Ask1 nor p38 was significantly hyperphosphorylated in resting Cib1 null platelets, suggesting that TRAF2/6 is not freely available to bind to ASK1 prior to platelet activation (Fig. 9A&B). When we investigated the augmentation of Ask1 activation more closely, we found that more Traf6 co-IPed from thrombin stimulated mouse platelet lysates when compared to resting controls. Furthermore, when we investigated this association in Cib1−/− platelets, we found that the loss of Cib1 corresponded to an ~2-fold increased association between Ask1 and Traf6 when Ask1 was IPed from WT and Cib1−/− platelet lysates (Fig. 9C). Additionally, when we inhibited PLCβ-signaling using U73122, we found that U73122 was able to significantly attenuate thrombin induced recruitment of Traf6 to Ask1 (Fig. 9D).
Figure 9. ASK1 activation is negatively regulated by CIB1 in platelets.
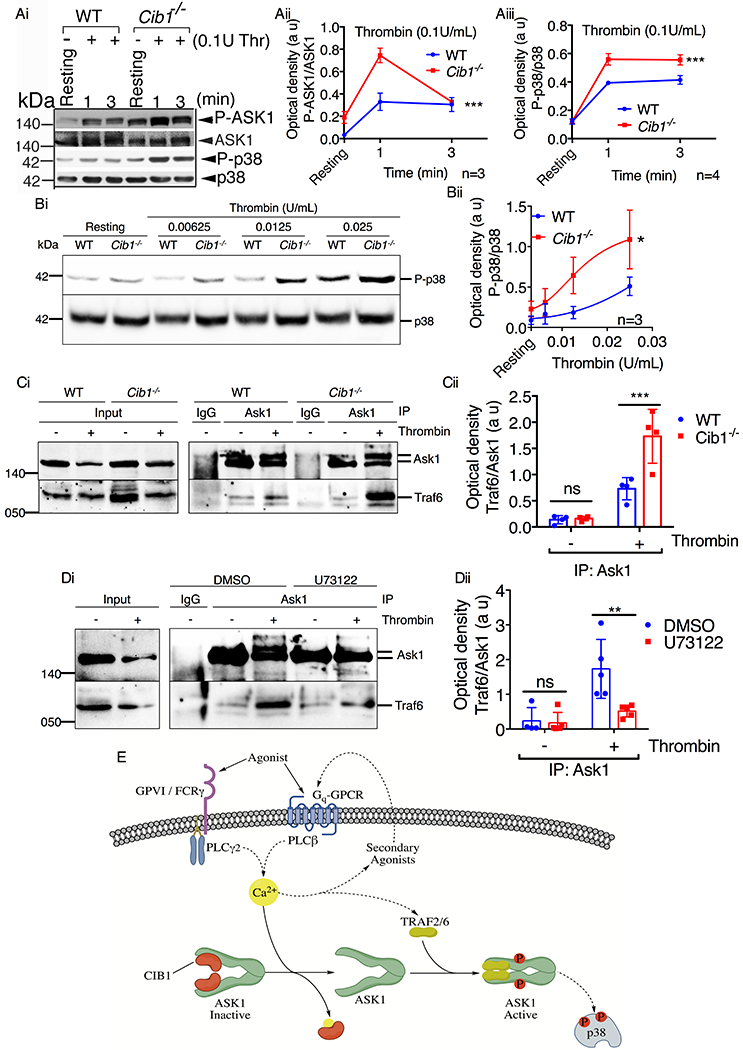
(A & B) Western blot analysis of washed murine platelets isolated from WT or Cib1−/− mice, pretreated with 200μM RGDS (5 min) and stimulated with either (A) thrombin (0.1U/mL) for 1 and 3 min or (B) thrombin (0.00625-0.025U/mL) for 3 min. Images of representative immunoblots of phosphorylation of (Ai) ASK1 and (Ai & Bi) p38 using phospho-specific antibodies. Blots were reprobed with (Ai) anti-ASK1 and (Ai & Bi) anti-p38 antibodies to ensure equal protein loading. Quantification of the band intensities of (Aii) P-ASK1 and (Aiii & Bii) P-p38 from (Ai & Bi) respectively, *P<0.05, ***P<0.001, two-way ANOVA. (C) Representative western blot images of immunoprecipitate of anti-Ask1 or normal rabbit IgG antibodies from resting and thrombin (1U/mL) stimulated platelet lysates and blotted for (Ci) Traf6. Blots were reprobed with anti-Ask1 antibody to ensure equal loading. (Cii) Quantification of the band intensity from (Ci); Non significant (ns), ***P<0.001, two-way ANOVA. (D) Representative western blot images of immunoprecipitate of anti-Ask1 or normal rabbit IgG antibodies from resting and thrombin (1U/mL) stimulated platelet lysates pretreated with either DMSO or U73122 (10μM) and blotted for (Di) Traf6. Blots were reprobed with anti-Ask1 antibody to ensure equal loading. (Dii) Quantification of the band intensity from (Di); Non significant (ns), **P<0.01, two-way ANOVA. (E) Schematic representation of the mechanism of agonist-induced ASK1 activation in platelets. In resting platelets (low Ca2+ levels) CIB1 is bound to ASK1 keeping it inactive. Platelet agonists, via their respective receptors, initiate PLCβ/γ activation causing an increase in intracellular Ca2+ concentration. Increased Ca2+ binds ASK1-bound CIB1 and dissociates it from ASK1. Free of its negative regulatory element, ASK1 then enters an inactive, intermediary state. From this intermediary state, ASK1 is then able to bind its positive regulatory elements TRAF2/6 to facilitate N-terminal dimerization and activation of ASK1 in platelets.
Taken together these data suggest that CIB1 constitutes a major negative regulatory element bound to ASK1 in resting platelets. Our data further suggest that following agonist stimulation, increased intracellular Ca2+ causes CIB1 to dissociate from and facilitates TRAF6 to associate with ASK1, triggering ASK1 activation.
Discussion
Platelets express all members of the MAPK family. It has been well-documented that MAPKs are robustly activated downstream of most physiological platelet agonists [29–31]. However, little is known about the signaling events that are responsible for the activation of MAPKs downstream of each agonist receptor activation. We have previously shown that MKKs, kinases immediately upstream of MAPKs, are expressed in murine platelets and are activated by platelet agonists [3]. Furthermore, ASK1 is rapidly activated during platelet activation and acts as a key regulator of platelet function [3]. Activation of platelets from Ask1−/− mice with thrombin, collagen, or convulxin failed to induce any activation of MKK3/4/6 and p38, without affecting MKK7 and JNK phosphorylation suggesting that activation of p38 by agonists is entirely dependent on ASK1 and can be used as an endogenous marker of ASK1 signaling activity in platelets [3]. In this study, we elucidated the mechanisms governing agonist-induced ASK1 activation in platelets. Based on our results herein, we propose the following mechanism of ASK1 activation in platelets (Fig. 9E). In resting platelets, at conditions of low intracellular Ca2+ concentrations, CIB1 binds to the N-terminal regulatory domains of ASK1 keeping ASK1 inactive. Following agonist stimulation, receptors on the platelet surface activate the PLCβ/γ signaling pathways to increase the concentration of cytosolic Ca2+, resulting in the dissociation of CIB1 from ASK1. Free of its negative regulatory element, ASK1 is then able to bind its positive regulatory elements TRAF2/6 to facilitate N-terminal dimerization and activation of ASK1 via autophosphorylation in platelets [8, 9, 28].
ASK1 is shown to be activated in a ROS-dependent manner [25]. We observed that the ROS donor H2O2 could only activate ASK1 in platelets at relatively high concentrations. Interestingly, when Ca2+ was chelated, we found that H2O2-induced activation of ASK1 was significantly attenuated. This finding supports the notion that intracellular Ca2+ rise predominantly mediates H2O2-induced ASK1 activation in platelets, and ROS-dependent activation may be a minor contributor. Further, while platelets are known to generate ROS in response to physiological agonist stimulation [26, 32, 33], agonist-induced ASK1 activation was independent of ROS. A finding supported by the observation that thrombin, but not homocysteine (a ROS dependent agonist), induced p38 phosphorylation was not blocked by the thiol-reducing agent, N-acetyl cysteine [34]. Overall, our findings suggest that ROS-dependent activation of ASK1 may only occur in environments with high concentrations of ROS, such as those encountered at atherosclerotic plaques [35].
While Trx represents the most well characterized regulatory mechanism of ASK1 activation in nucleated cells, our results suggest that Ca2+-dependent activation of ASK1 is the most predominant in platelets. Several independent mechanisms have been described through which Ca2+ can regulate ASK1 activation [36]. In neuronal cells it was found that CIB1 binds to ASK1’s N-terminal regulatory domain preventing ASK1 from interacting through its NCC domains [8–10]. Additionally, calcium/calmodulin-dependent protein kinase II (CAMKII) was shown to regulate ASK1 phosphorylation on Thr838 in spinal astrocytes [37]. And it was also revealed that the PP5 binding protein S100A1 prevents the interaction between PP5 and ASK1 in a Ca2+-dependent manner [38].
Increased cytosolic Ca2+ occurs downstream of both GPCR and ITAM receptors via activation of PLCβ and PLCγ respectively [23, 39, 40]. It is therefore plausible that CIB1 is the Ca2+ sensitive regulator of agonist-induced ASK1 activation in platelets. Immunoprecipitation experiments clearly suggested that ASK1 and CIB1 associate in resting platelets and are dissociated upon platelet activation, an observation consistent with the findings seen in nucleated cells [8, 28]. While we cannot definitely rule out the possibility that other Ca2+ dependent regulatory mechanisms are not at play, our results strongly suggest that CIB1 is a major regulator of ASK1 activation in platelets.
Our finding of enhanced agonist-induced Ask1 activation in the absence of Cib1 supports the negative role of Cib1 in the activation of Ask1. This is opposite to the known positive role played by CIB1 in platelet integrin αIIbβ3-dependent outside-in signaling [13, 15, 41]. To avoid potential complications, we used RGDS to block outside-in signaling in WT platelets. Therefore, it is clear that in resting platelets, Ca2+-depleted CIB1 acts as a negative regulator of Ask1, and upon binding to Ca2+ it supports integrin outside-in signaling by physically associating with integrin αIIb tail.
In this study, we uniquely found that physiological agonists selectively activate platelet ASK1 only by inducing the release of Ca2+, and not via ROS, in a CIB1-dependent manner. Our report is the first of its kind to document these findings that ASK1 is regulated in a Ca2+-dependent manner downstream of both GPCRs and non-GPCR type receptors in platelets stimulated with physiological agonists. These findings have strong implications on the basic biology of ASK1 activation and are of critical importance in the development of ASK1 inhibitors for the treatment of cardiovascular disease.
Supplementary Material
Acknowledgements:
Authors will like to thank Dr. Leslie Parise (University of North Carolina at Chapel Hill) for providing Cib1−/− mice. We would also like to thank Dr. Michael Whitley and Joanne Vesci for their help in preparing washed human platelets used in some experiments.
Funding
This work was supported by an AHA Predoctoral Fellowship (18PRE33990382) to PP and a National Heart, Lung, and Blood Institute (2R01 HL113188-05), the American Heart Association Grant-in-Aid Award (#13GRNT16380023), and an American Society of Hematology Bridge Award to UN.
Abbreviations
- ASK1
apoptosis signal-regulating kinase 1
- MAPKKK
mitogen-activated protein kinase kinase kinase
- JNK
c-Jun N-terminal kinase
- PLC
phospholipase C
- CIB1
calcium and integrin-binding protein 1
- TxA2
thromboxane A2
- ROS
reactive oxygen species
- NCC
N-terminal coiled-coil
- TNF
tumor necrosis factor
- ERK
extracellular signal-regulating kinase
- GPCR
G-protein coupled receptor
- ITAM
immunoreceptor tyrosine-based activation motif
- PKC
protein kinase C
- DAG
diacylglycerol
- PAR
protease-activated receptor
- BIS
bisindolylmaleimide
- MnTMPyP
manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin
- BAPTA-AM
1,2-Bis(2-aminophynoxy)ethane-N,N,N’,N’-tetraacetic acid tetrakis(acetoxymethyl ester)
- Trx
thioredoxin
- IP
immunoprecipitation
- IPed
immunoprecipitated
- PMSF
phenylmethylsulfonyl fluoride
- TRAF
TNF receptor associated factor.
Footnotes
Declaration of interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Adam F, Kauskot A, Rosa JP and Bryckaert M (2008) Mitogen-activated protein kinases in hemostasis and thrombosis. Journal of thrombosis and haemostasis : JTH 6, 2007–2016 [DOI] [PubMed] [Google Scholar]
- 2.Adam F, Kauskot A, Nurden P, Sulpice E, Hoylaerts MF, Davis RJ, Rosa JP and Bryckaert M (2010) Platelet JNK1 is involved in secretion and thrombus formation. Blood. 115, 4083–4092 [DOI] [PubMed] [Google Scholar]
- 3.Naik MU, Patel P, Derstine R, Turaga R, Chen X, Golla K, Neeves KB, Ichijo H and Naik UP (2017) Ask1 regulates murine platelet granule secretion, thromboxane A2 generation, and thrombus formation. Blood. 129, 1197–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamiyama M, Shirai T, Tamura S, Suzuki-Inoue K, Ehata S, Takahashi K, Miyazono K, Hayakawa Y, Sato T, Takeda K, Naguro I and Ichijo H (2017) ASK1 facilitates tumor metastasis through phosphorylation of an ADP receptor P2Y12 in platelets. Cell Death Differ. 24, 2066–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T and Ichijo H (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO reports. 2, 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K and Gotoh Y (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science (New York, N.Y.). 275, 90–94 [DOI] [PubMed] [Google Scholar]
- 7.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A and Ichijo H (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes & Devlopment. 16, 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon KW, Cho JH, Lee JK, Kang YH, Chae JS, Kim YM, Kim J, Kim EK, Kim SE, Baik JH, Naik UP, Cho SG and Choi EJ (2009) CIB1 functions as a Ca(2+)-sensitive modulator of stress-induced signaling by targeting ASK1. Proceedings of the National Academy of Sciences of the United States of America. 106, 17389–17394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujino G, Noguchi T, Matsuzawa A, Yamauchi S, Saitoh M, Takeda K and Ichijo H (2007) Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell. Biol 27, 8152–8163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K and Ichijo H (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17, 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tobiume K, Saitoh M and Ichijo H (2002) Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol. 191, 95–104 [DOI] [PubMed] [Google Scholar]
- 12.Naik UP, Patel PM and Parise LV (1997) Identification of a novel calcium-binding protein that interacts with the integrin alphaIIb cytoplasmic domain. J. Biol. Chem 272, 4651–4654 [DOI] [PubMed] [Google Scholar]
- 13.Naik UP and Naik MU (2003) Association of CIB with GPIIb/IIIa during outside-in signaling is required for platelet spreading on fibrinogen. Blood. 102, 1355–1362 [DOI] [PubMed] [Google Scholar]
- 14.Yuan W, Leisner TM, McFadden AW, Clark S, Hiller S, Maeda N, O’Brien DA and Parise LV (2006) CIB1 is essential for mouse spermatogenesis. Mol. Cell. Biol 26, 8507–8514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naik MU, Nigam A, Manrai P, Millili P, Czymmek K, Sullivan M and Naik UP (2009) CIB1 deficiency results in impaired thrombosis: the potential role of CIB1 in outside-in signaling through integrin alpha IIb beta 3. Journal of thrombosis and haemostasis : JTH 7, 1906–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naik MU, Naik TU, Summer R and Naik UP (2017) Binding of CIB1 to the alphaIIb tail of alphaIIbbeta3 is required for FAK recruitment and activation in platelets. PLoS One. 12, e0176602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel P, Golla K and Naik UP (2018) PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway: comment. Journal of thrombosis and haemostasis : JTH. 16, 1901–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeda K, Noguchi T, Naguro I and Ichijo H (2008) Apoptosis signal-regulating kinase 1 in stress and immune response. Annu. Rev. Pharmacol. Toxicol 48, 199–225 [DOI] [PubMed] [Google Scholar]
- 19.Enslen H, Raingeaud J and Davis RJ (1998) Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem 273, 1741–1748 [DOI] [PubMed] [Google Scholar]
- 20.Alonso G, Ambrosino C, Jones M and Nebreda AR (2000) Differential activation of p38 mitogen-activated protein kinase isoforms depending on signal strength. J. Biol. Chem 275, 40641–40648 [DOI] [PubMed] [Google Scholar]
- 21.Doza NY, Cuenda A, Thomas GM, Cohen P and Nebreda AR . (1995) Activation of the MAP kinase homologue RK requires the phosphorylation of Thr-180 and Tyr-182 and both residues are phosphorylated in chemically stressed KB cells. FEBS Lett. 364, 223–228 [DOI] [PubMed] [Google Scholar]
- 22.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA and Davis RJ . (2003) Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 17, 1969–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varga-Szabo D, Braun A and Nieswandt B (2009) Calcium signaling in platelets. Journal of thrombosis and haemostasis : JTH. 7, 1057–1066 [DOI] [PubMed] [Google Scholar]
- 24.Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ and Bunting S (1990) Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J. Pharmacol. Exp. Ther 255, 756–768 [PubMed] [Google Scholar]
- 25.Shiizaki S, Naguro I and Ichijo H (2013) Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Adv Biol Regul. 53, 135–144 [DOI] [PubMed] [Google Scholar]
- 26.Begonja AJ, Gambaryan S, Geiger J, Aktas B, Pozgajova M, Nieswandt B and Walter U (2005) Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood. 106, 2757–2760 [DOI] [PubMed] [Google Scholar]
- 27.Carrim N, Arthur JF, Hamilton JR, Gardiner EE, Andrews RK, Moran N, Berndt MC and Metharom P (2015) Thrombin-induced reactive oxygen species generation in platelets: A novel role for protease-activated receptor 4 and GPIbalpha. Redox biology. 6, 640–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon KW, Yang HS, Kim YM, Kim Y, Kang S, Sun W, Naik UP, Parise LV and Choi EJ (2017) CIB1 protects against MPTP-induced neurotoxicity through inhibiting ASK1. Sci Rep. 7, 12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luber K and Siess W (1994) Integrin-dependent protein dephosphorylation on tyrosine induced by activation of the thrombin receptor in human platelets. Cell. Signal 6, 279–284 [DOI] [PubMed] [Google Scholar]
- 30.Papkoff J, Chen RH, Blenis J and Forsman J (1994) p42 mitogen-activated protein kinase and p90 ribosomal S6 kinase are selectively phosphorylated and activated during thrombin-induced platelet activation and aggregation. Mol. Cell. Biol 14, 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bugaud F, Nadal-Wollbold F, Levy-Toledano S, Rosa JP and Bryckaert M (1999) Regulation of c-jun-NH2 terminal kinase and extracellular-signal regulated kinase in human platelets. Blood. 94, 3800–3805 [PubMed] [Google Scholar]
- 32.Delaney MK, Kim K, Estevez B, Xu Z, Stojanovic-Terpo A, Shen B, Ushio-Fukai M, Cho J and Du X (2016) Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler Thromb Vasc Biol. 36, 846–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang JY, Min JH, Chae YH, Baek JY, Wang SB, Park SJ, Oh GT, Lee SH, Ho YS and Chang TS (2014) Reactive oxygen species play a critical role in collagen-induced platelet activation via SHP-2 oxidation. Antioxid. Redox Signal. 20, 2528–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leoncini G, Bruzzese D and Signorello MG (2006) Activation of p38 MAPKinase/cPLA2 pathway in homocysteine-treated platelets. Journal of thrombosis and haemostasis : JTH. 4, 209–216 [DOI] [PubMed] [Google Scholar]
- 35.Yang X, Li Y, Li Y, Ren X, Zhang X, Hu D, Gao Y, Xing Y and Shang H (2017) Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front Physiol. 8, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishida T, Hattori K and Watanabe K (2017) The regulatory and signaling mechanisms of the ASK family. Adv Biol Regul. 66, 2–22 [DOI] [PubMed] [Google Scholar]
- 37.Liu G, Zhao J, Chang Z and Guo G (2013) CaMKII activates ASK1 to induce apoptosis of spinal astrocytes under oxygen-glucose deprivation. Cell. Mol. Neurobiol 33, 543–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamaguchi F, Umeda Y, Shimamoto S, Tsuchiya M, Tokumitsu H, Tokuda M and Kobayashi R (2012) S100 proteins modulate protein phosphatase 5 function: a link between CA2+ signal transduction and protein dephosphorylation. J. Biol. Chem 287, 13787–13798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki-Inoue K, Inoue O, Frampton J and Watson SP (2003) Murine GPVI stimulates weak integrin activation in PLCgamma2−/− platelets: involvement of PLCgamma1 and PI3-kinase. Blood. 102, 1367–1373 [DOI] [PubMed] [Google Scholar]
- 40.Lian L, Wang Y, Draznin J, Eslin D, Bennett JS, Poncz M, Wu D and Abrams CS (2005) The relative role of PLCbeta and PI3Kgamma in platelet activation. Blood. 106, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naik MU and Naik UP (2003) Calcium-and integrin-binding protein regulates focal adhesion kinase activity during platelet spreading on immobilized fibrinogen. Blood. 102, 3629–3636 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.