

Abstract
Background
Premenstrual syndrome (PMS) is a common cause of physical, psychological and social problems in women of reproductive age. The key characteristic of PMS is the timing of symptoms, which occur only during the two weeks leading up to menstruation (the luteal phase of the menstrual cycle). Selective serotonin reuptake inhibitors (SSRIs) are increasingly used as first line therapy for PMS. SSRIs can be taken either in the luteal phase or else continuously (every day). SSRIs are generally considered to be effective for reducing premenstrual symptoms but they can cause adverse effects.
Objectives
The objective of this review was to evaluate the effectiveness and safety of SSRIs for treating premenstrual syndrome.
Search methods
Electronic searches for relevant randomised controlled trials (RCTs) were undertaken in the Cochrane Menstrual Disorders and Subfertility Group Specialised Register, Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library), MEDLINE, EMBASE, PsycINFO, and CINAHL (February 2013). Where insufficient data were presented in a report, attempts were made to contact the original authors for further details.
Selection criteria
Studies were considered in which women with a prospective diagnosis of PMS, PMDD or late luteal phase dysphoric disorder (LPDD) were randomised to receive SSRIs or placebo for the treatment of premenstrual syndrome.
Data collection and analysis
Two review authors independently selected the studies, assessed eligible studies for risk of bias, and extracted data on premenstrual symptoms and adverse effects. Studies were pooled using random‐effects models. Standardised mean differences (SMDs) with 95% confidence intervals (CIs) were calculated for premenstrual symptom scores, using separate analyses for different types of continuous data (that is end scores and change scores). Odds ratios (ORs) with 95% confidence intervals (CIs) were calculated for dichotomous outcomes. Analyses were stratified by type of drug administration (luteal or continuous) and by drug dose (low, medium, or high). We calculated the number of women who would need to be taking a moderate dose of SSRI in order to cause one additional adverse event (number needed to harm: NNH). The overall quality of the evidence for the main findings was assessed using the GRADE working group methods.
Main results
Thirty‐one RCTs were included in the review. They compared fluoxetine, paroxetine, sertraline, escitalopram and citalopram versus placebo. SSRIs reduced overall self‐rated symptoms significantly more effectively than placebo. The effect size was moderate when studies reporting end scores were pooled (for moderate dose SSRIs: SMD ‐0.65, 95% CI ‐0.46 to ‐0.84, nine studies, 1276 women; moderate heterogeneity (I2 = 58%), low quality evidence). The effect size was small when studies reporting change scores were pooled (for moderate dose SSRIs: SMD ‐0.36, 95% CI ‐0.20 to ‐0.51, four studies, 657 women; low heterogeneity (I2=29%), moderate quality evidence).
SSRIs were effective for symptom relief whether taken only in the luteal phase or continuously, with no clear evidence of a difference in effectiveness between these modes of administration. However, few studies directly compared luteal and continuous regimens and more evidence is needed on this question.
Withdrawals due to adverse effects were significantly more likely to occur in the SSRI group (moderate dose: OR 2.55, 95% CI 1.84 to 3.53, 15 studies, 2447 women; no heterogeneity (I2 = 0%), moderate quality evidence). The most common side effects associated with a moderate dose of SSRIs were nausea (NNH = 7), asthenia or decreased energy (NNH = 9), somnolence (NNH = 13), fatigue (NNH = 14), decreased libido (NNH = 14) and sweating (NNH = 14). In secondary analyses, SSRIs were effective for treating specific types of symptoms (that is psychological, physical and functional symptoms, and irritability). Adverse effects were dose‐related.
The overall quality of the evidence was low to moderate, the main weakness in the included studies being poor reporting of methods. Heterogeneity was low or absent for most outcomes, though (as noted above) there was moderate heterogeneity for one of the primary analyses.
Authors' conclusions
SSRIs are effective in reducing the symptoms of PMS, whether taken in the luteal phase only or continuously. Adverse effects are relatively frequent, the most common being nausea and asthenia. Adverse effects are dose‐dependent.
Plain language summary
Selective serotonin reuptake inhibitors (SSRIs) for premenstrual syndrome
Premenstrual syndrome (PMS) is a common cause of physical, psychological and social problems in women of reproductive age. PMS is distinguished from 'normal' premenstrual symptoms by the degree of distress and disruption it causes. Symptoms occur during the period leading up to the menstrual period and are relieved by the onset of menstruation. Common symptoms include irritability, depression, anxiety and lethargy. A clinical diagnosis of PMS requires that the symptoms are confirmed by prospective recording (that is recorded as they occur) for at least two menstrual cycles and that they cause substantial distress or impairment to daily life. It is estimated that approximately one in five women of reproductive age are affected. PMS can severely disrupt a woman's daily life and some women seek medical treatment. Researchers in The Cochrane Collaboration reviewed the evidence about the effectiveness and safety of selective serotonin reuptake inhibitors (SSRIs) for treating PMS. They examined the research up to February 2013.
The review included 31 randomised controlled trials which compared SSRIs with placebo in a total of 4372 women who were clinically diagnosed with PMS. SSRIs were found to be effective for reducing the overall symptoms of PMS and also for reducing specific types of symptoms (psychological, physical and functional symptoms, and irritability). SSRIs were usually taken for about two weeks before the start of the menstrual period (the luteal phase) or every day (continuously). Both regimens appeared to be equally effective, although more research is needed to confirm this.
Adverse effects were more common in the women taking SSRIs than in those taking placebo. The most commonly occurring side effects were nausea and decreased energy. The review authors calculated that nausea is likely to occur as a drug side effect in approximately one out of seven women with PMS taking a moderate dose of SSRIs, and lack of energy is likely to occur as a drug side effect in approximately one out of every nine women.
The overall quality of the evidence was rated as low to moderate, the main weakness being poor reporting of methods in the included studies. At least 21 of the studies received funding from pharmaceutical companies.
Summary of findings
Background
Description of the condition
Most women of reproductive age experience premenstrual symptoms that are associated with the rise and fall of ovarian sex steroids precipitated by ovulation (Rapkin 2008). Premenstrual syndrome (PMS) is distinguished from 'normal' premenstrual symptoms by the degree of distress it causes or its detrimental effect on daily functioning, or both (O'Brien 2011). The physiology of PMS is complex and the disorder is poorly understood (Freeman 2012). It may be associated with the actions of serotonin and gamma‐aminobutyric acid, which are neurotransmitters influenced by the menstrual cycle. Abnormal function or deficiency of these neurotransmitters may cause increased sensitivity to progesterone, precipitating PMS symptoms (Baker 2012).
Definitions of PMS vary, and a wide range of psychological and physical symptoms has been reported. The key characteristic of PMS is the timing of symptoms, which occur only during all or part of the two weeks leading up to menstruation (the luteal phase of the menstrual cycle). Symptoms disappear by the end of menstruation and do not recur before ovulation, giving a symptom‐free interval of at least one week. PMS is cyclical, and occurs in most menstrual cycles (O'Brien 2011). Psychological symptoms can include irritability, depression, anxiety, mood swings, a flat mood (anhedonia) and lethargy. Physical symptoms may include breast tenderness, weight gain, bloating, muscle or joint pain, headache and swelling of the extremities (hands and feet). A clinical diagnosis requires that symptoms are confirmed by prospective recording for at least two menstrual cycles and that they cause substantial distress or impairment to daily life (for example activities at home, work or school, social activities, hobbies, interpersonal relationships) (ACOG 2000; Baker 2012; Epperson 2012; O'Brien 2011). As collecting multiple, daily data points is a laborious process, most diagnoses of PMS are made based on a woman's own perception of her problem. Hence it is suggested that up to 50% of women with reported PMS do not meet the clinical criteria for the disorder (Plouffe 1993). PMS in this review is defined as symptoms meeting the clinical criteria described above.
A severe form of PMS is known as premenstrual dysphoria or premenstrual dysphoric disorder (PMDD), and was previously also known as late luteal phase dysphoric disorder (LLPDD) (O'Brien 2011). PMDD is characterised by severe symptoms occurring for a week before each menstrual period and remitting in the week after menstruation, over a period of at least a year. According to American Psychiatric Association (APA) criteria, in their Diagnostic and Statistical Manual version 4 (DSM IV), at least five of the following symptoms must occur for most of the week prior to menstruation: depression, anxiety, mood swings, irritability (at least one of these four), decreased interest in usual activities, difficulty concentrating, fatigue, appetite changes, sleep disturbance, feeling overwhelmed, and physical symptoms (APA 2000). Symptoms should remit within a few days of menstruation. This definition has been questioned because it focuses on severe psychological symptoms while placing relatively little importance on physical symptoms, and may exclude some women with debilitating symptoms that do not meet these specific criteria (O'Brien 2011).
DSM criteria are currently being updated to include PMDD as a new diagnostic category rather than (as previously) a mood disorder needing further research. Although the proposed DSM V criteria for PMDD are similar to those of DSM IV, they differ in the following respects (Epperson 2012):
symptoms must occur during the final week before menstruation, but do not need to be present most of the time;
symptoms are not required to remit within a few days of menstruation, but should improve and should be minimal (if not absent) in the week following menstruation;
mood lability and irritability are the leading two symptoms;
symptoms cause clinically significant distress or interference with activities at work or school or at home, or both (previously there was no mention of clinically significant distress or of activities at home);
PMDD symptoms are not due to an ongoing medical disorder or substance‐induced condition.
There is a wide variation in estimates of the prevalence of PMS, but it is estimated that 15% to 20% of women of reproductive age have PMS with significantly impaired functioning, and a further 3% to 8% have PMDD. Thus approximately one in five women of reproductive age are affected (Pearlstein 2007).
There is currently no haematological or biochemical test for PMS, and studies have not shown consistent differences in cyclical hormone levels. In the absence of any objective parameter to measure or diagnose PMD, clinicians and researchers rely largely on validated scales in which a woman self‐rates her symptoms. The most widely used self‐rating tool is the Daily Record of Severity of Problems (DRSP), which is a prospective scale that focuses largely on psychological rather than physical symptoms (O'Brien 2011).
Description of the intervention
Selective serotonin reuptake inhibitors (SSRIs) are a class of drugs that are believed to inhibit the absorption of serotonin, a naturally occurring chemical which acts as a messenger between brain cells (a neurotransmitter). Changing the balance of serotonin appears to improve neurotransmission and enhance mood.
SSRIs are most commonly used to treat depression and anxiety disorders, and appear to take four to eight weeks to reach clinical efficacy in these disorders (Freeman 1999). However, it has been shown that SSRIs may become effective for PMS in a matter of days, and usually within four weeks from the start of treatment (Steiner 1995). This may be due to the cyclical nature of PMS and may reflect SSRI action at a different receptor site to that involved in affective disorders (Sundblad 1997).
The rapid efficacy of SSRI treatment in PMS permits the use of intermittent dosing regimens. For treatment of PMS a relatively small dose of SSRI is generally used. Administration can be:
continuous, SSRI is taken every day throughout the menstrual cycle;
luteal or intermittent, SSRI is taken only during the luteal phase of the menstrual cycle (i.e. from estimated ovulation to menstruation). The SSRI is started about 14 days prior to expected menstruation, based on a woman's usual cycle length;
semi‐intermittent, taken every day, with a low SSRI dose during the follicular phase of the menstrual cycle and a higher dose in the luteal phase;
as required, SSRI is started at the onset of PMS symptoms and continued until the onset of menstruation.
It is suggested that avoidance of continuous use may reduce the risk of side effects of SSRIs, which can include anxiety, dizziness, insomnia, sedation, gastrointestinal disturbance, headache, loss of libido and anorgasmia (inability to achieve orgasm) (Baker 2012; Pearlstein 2002).
SSRIs are licensed for treating PMMD in the United States, but not in Europe (Ismail 2006).
Other interventions used for premenstrual symptoms include lifestyle modification (for example exercise, smoking cessation, weight management), herbal remedies (for example vitex agnus castus), calcium, vitamin C, hormones, gonadotrophin‐releasing hormone (GnRH) analogues, danazol and (rarely and as a last resort) hysterectomy with bilateral oophorectomy (Baker 2012). Uncertainty about the pathogenesis of PMS has led to many other treatments being suggested as possible therapies. It has been suggested that, as there is a substantial placebo response, a large number of uncontrolled trials have resulted in a proliferation of claims for ineffective therapies (Magos 1986).
Different PMS symptoms may have separate causes and therefore require different treatment strategies (O'Brien 2011). It is suggested that most women with severe PMS require either hormonal medication (estrogen with or without progestin) or a psychotropic medication (such as an SSRI) for symptom relief (Baker 2012). As the disorder is usually chronic, and may require treatment for 20 years or more, the long‐term effects of an intervention are important (Rapkin 2008).
Some of the interventions are the subject of other Cochrane reviews, either published or in preparation (as of April 2013), as follows.
Oral contraceptives containing drosperinone
This review found that drospirenone 3 mg plus ethinyl estradiol 20 μg may be beneficial for PMDD. It was unclear whether oral contraceptives containing drospirenone were effective for women with less severe symptoms, or were better than other oral contraceptives. A strong placebo effect was noted (Lopez 2012).
Chinese herbal medicines
This review found that there was insufficient evidence to support the use of Chinese herbal medicines for PMS (Jing 2009).
Progesterone
This review found that it was unclear whether or not progesterone is an effective treatment for PMS (Ford 2012).
Acupuncture
This review is in preparation. The protocol has been published (Yu 2005).
Vitex agnus castus
This review is in preparation. The protocol has been published (Shaw 2003).
Non‐contraceptive estrogen‐containing preparations for controlling symptoms of PMS
This review is in preparation. The protocol has been published (Naheed 2013).
How the intervention might work
Serotonin levels appear to vary during the menstrual cycle under the influence of estrogen and progesterone (Baker 2012). SSRIs may increase the amount of serotonin available for neurotransmission.
It has been noted that treatments that enhance the action of serotonin improve premenstrual irritability and low mood with a rapid onset of action, which suggests a different mechanism of action than in the treatment of depression. Neurosteroids such as progesterone metabolites may be responsible for the rapid action of SSRIs in this context (Pearlstein 2002).
Why it is important to do this review
In view of the debilitating symptoms and economic cost of PMS, and the likelihood that it will persist long‐term, it is important to confirm and quantify the effectiveness and safety of SSRIs for treating this disorder. This is an update of a Cochrane review first published in 2002.
Objectives
To determine the effectiveness and safety of SSRIs for treating premenstrual syndrome.
Methods
Criteria for considering studies for this review
Types of studies
Published and unpublished randomised controlled trials (RCTs) were eligible for inclusion. We excluded non‐randomised studies (for example studies with evidence of inadequate sequence generation such as alternate days, patient numbers) as they are associated with a high risk of bias. Crossover trials were eligible but it was planned that only data from the first phase would be included in meta‐analyses.
Types of participants
Studies of women of any age who met the medically defined diagnostic criteria for premenstrual syndrome (PMS) or premenstrual dysphoric disorder (PMDD) were eligible for inclusion. Diagnosis must have been made prior to inclusion in the trial by a general practitioner (GP), hospital clinician, or other healthcare professional. Diagnosis of PMS requires that symptoms are confirmed by prospective recording for at least two menstrual cycles and must cause substantial distress or impairment to daily life. Diagnosis of PMDD must meet established psychiatric diagnostic criteria.
Studies of women with only a self‐diagnosis of PMS were excluded.
Types of interventions
Studies of SSRIs, at any dose and in any dosing regimen for any duration longer than one menstrual cycle, versus placebo were eligible.
Trials of tricyclic antidepressants were excluded. Even when described as serotonin reuptake inhibitors, these drugs are not selective and act in a different manner to SSRIs.
Types of outcome measures
Primary
1. Self‐rated overall premenstrual symptoms, measured using a validated prospective screening tool (e.g. Moos' MDQ, Abraham's classification) or by pre‐defined medical diagnostic criteria
2. Adverse events (all adverse events, specific adverse effects, withdrawals for adverse effects)
Secondary
3. Specific symptoms of PMS: psychological, physical and functional symptoms, irritability
4. Response rate (according to how response defined in individual studies)
5. Overall withdrawals from study
Search methods for identification of studies
We searched for all published and unpublished RCTs meeting the inclusion criteria. The search was conducted without language restriction and in consultation with the Menstrual Disorders and Subfertility Group Trials Search Co‐ordinator.
Electronic searches
For the latest search (February 2013), we searched the following electronic databases, trials registers and websites: Menstrual Disorders and Subfertility Group (MDSG) Specialised Register of Controlled Trials, Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, EMBASE, and PsycINFO.
Other electronic sources of trials included: a. trials registers for ongoing and registered trials, http://www.controlled‐trials.com, http://clinicaltrials.gov/ct2/home, http://www.who.int/trialsearch/Default.aspx; b. Web of Knowledge (including the citation database Web of Science).
For versions of the review prior to 2013, we also searched http://www.clinicalstudyresults.org/ for the results of clinical trials of marketed pharmaceuticals. However, this database has not been accessible since June 2012.
See Appendix 1; Appendix 2; Appendix 3 for database search strategies.
Searching other resources
a) We handsearched the reference lists of articles retrieved by the search.
b) For the 2002 version of this review:
drug and pharmaceutical companies manufacturing SSRIs (fluoxetine: Eli Lilly; paroxetine: Smith Kline Beecham; sertraline: Invicta; fluvoxamine: Solvay; Citalopram: Du Pont) were contacted to request other published or unpublished trials;
the personal databases on PMS therapies maintained by the authors were searched for relevant articles;
the UK‐based National Association for Premenstrual Syndrome (NAPS) was also contacted for relevant articles.
c) For the 2013 version of this review:
attempts were made to contact the following drug and pharmaceutical companies manufacturing SSRIs (Lilly, GlaxoSmithKline, Pfizer, Forest Labs) to request other published or unpublished trials. However, only one company (GlaxoSmithKline) replied.
Data collection and analysis
Selection of studies
The 2013 update
For the 2013 update of this review, JM conducted an initial screen of titles and abstracts retrieved by the search and retrieved the full texts of all potentially eligible studies. Two review authors (JM and KMW) independently examined these full text articles for compliance with the inclusion criteria and selected studies eligible for inclusion. We corresponded with study investigators, as required, to clarify study eligibility. Disagreements as to study eligibility were resolved by discussion or by a third review author.
Previous versions of the review
For the original version of this review (2002), all publications identified in the search strategy were assessed by two authors (PWD and KMW) working in parallel. Selection of the trials for inclusion was performed by PWD and KW. Any disagreements were assessed by a third review author and other uncertainties regarding inclusion were resolved by contacting the primary study authors. For the 2008 update of the review, an additional 22 studies were identified by JB and JM, who independently checked the potentially eligible studies.
Data extraction and management
For the 2013 update of this review, two review authors (JM and JB) independently extracted data from the eligible studies. Any disagreements were resolved by discussion or by a third review author. Data extracted included study characteristics and effect estimates.
Where there were multiple arms in a study with a common placebo, the placebo numbers were divided as equally as possible between the arms (see footnotes in forest plots).
Where studies had multiple publications, the main trial report was used as the reference and additional details were derived from secondary papers. We corresponded with study investigators for further data on methods and results, as required.
Assessment of risk of bias in included studies
For the 2008 and 2013 updates, two review authors independently assessed the included studies for risk of bias using the Cochrane risk of bias assessment tool (Higgins 2011). This assesses: allocation (random sequence generation and allocation concealment); blinding of participants and personnel, blinding of outcome assessors; and completeness of outcome data.
For the 2013 update, new studies were also assessed for risk of selective reporting bias, which refers to the selective reporting of some outcomes (for example positive outcomes) and the failure to report others (for example adverse events), and for other potential sources of bias.
Disagreements were resolved by discussion or by a third review author. We described all judgements fully and presented the conclusions in the 'Risk of bias' table. Differences in study quality were incorporated into the interpretation of review findings by means of sensitivity analyses.
Measures of treatment effect
For continuous data (for example symptom scores), as similar outcomes were reported on different scales, we calculated the standardised mean difference (SMD) between the end scores or change scores for the control and intervention groups of each study. End scores were extracted in preference to change scores, where available, as they may be preferable for outcomes which are unstable or difficult to measure precisely (Higgins 2011). For dichotomous data (for example withdrawal rates), we used the numbers of events in the two groups to calculate Mantel‐Haenszel odds ratios (ORs). We presented 95% confidence intervals (CIs) for all outcomes. We compared the magnitude and direction of effect reported by studies with how they were presented in the review, taking account of legitimate differences.
Where there was a statistically significant difference between the two groups in the rate of adverse events, we calculated numbers needed to harm (NNH) for the moderate dose (that is an estimate of the number of women who would need to receive treatment using a moderate dose in order for one additional harm to occur).
Standard mean differences were interpreted using the following rule of thumb: 0.2 represents a small effect, 0.5 a moderate effect, and 0.8 a large effect (Cohen 1988a; Higgins 2011).
Unit of analysis issues
We planned to include only first‐phase data from crossover trials.
Dealing with missing data
The data were analysed on an intention‐to‐treat basis as far as possible and attempts were made to obtain missing data from the original trialists. Where these were unobtainable, only the available data were analysed.
Assessment of heterogeneity
We considered whether the clinical and methodological characteristics of the included studies were sufficiently similar for meta‐analysis in order to provide a clinically meaningful summary. We assessed statistical heterogeneity by the I2 statistic.
A rough guide to interpretation of I2 values is as follows (Higgins 2011):
0% to 40%, might not be important;
30% to 60%, may represent moderate heterogeneity;
50% to 90%, may represent substantial heterogeneity;
75% to 100%, considerable heterogeneity.
Assessment of reporting biases
In view of the difficulty of detecting and correcting for publication bias and other reporting biases, we aimed to minimise their potential impact by ensuring a comprehensive search for eligible studies and by being alert for duplication of data.
If there were 10 or more studies in the analysis of a primary outcome, we used a funnel plot to explore the possibility of small study effects (a tendency for estimates of the intervention effect to be more beneficial in smaller studies).
Data synthesis
If the studies were sufficiently similar, we combined the data using random‐effects models to compare SSRIs versus placebo. The primary outcome was stratified by SSRI dose (low, moderate or high, see Table 20) and by type of administration (luteal or continuous). The SMDs of end scores and change scores were analysed separately and were not pooled since the differences in the standard deviations reflect differences in the reliability of the measurements and not differences in measurement scales (Higgins 2011).
1. Classification of SSRI doses used by included studies.
| SSRI | Low dose | Moderate dose | High dose |
| Fluoxetine* | 10 mg daily | 20 mg daily | 60 mg daily |
| Sertraline* | 25‐50 mg daily | 100 ‐105 mg daily | |
| Paroxetine* | 10‐12.5 daily | 20‐30 daily | |
| Citalopram** | 20‐50 daily | ||
| Escitalopram** | 10 mg daily | 20 mg daily |
* Based on suggested doses for PMDD (Micromedex 2013)
**Based on suggested doses for depression (Micromedex 2013) as PMDD data not available
Subgroup analysis and investigation of heterogeneity
Where data were available, we conducted subgroup analyses to determine the separate evidence for the primary outcomes within the following subgroups:
administration mode (continuous versus intermittent or as required);
placebo run‐in versus non‐placebo run‐in.
If we detected substantial heterogeneity (I2 > 50%), we planned to explore possible explanations by checking the data, conducting sensitivity analyses (see below), and by examining clinical and methodological differences between the studies, to check whether there was a plausible explanation. Where there were three or more studies using the same SSRI and dose we examined whether the findings differed in subgroups using the same SSRI. We planned to take statistical heterogeneity into account when interpreting the results, especially if there was any variation in the direction of effect.
Sensitivity analysis
We conducted sensitivity analyses for the primary outcomes to determine whether the conclusions were robust to arbitrary decisions made regarding the eligibility of the studies and analysis. These analyses included consideration of whether the review conclusions would have differed if: 1. eligibility was restricted to studies without high risk of bias, defined as studies at low risk of selection bias; 2. a fixed‐effect model had been adopted.
Summary of findings table
A 'Summary of findings' table was generated using GRADEPRO software to evaluate the overall quality of the body of evidence for the main review outcomes, using GRADE working group criteria (that is study limitations (risk of bias), consistency of effect, imprecision, indirectness and publication bias). Judgements about the quality of the evidence (high, moderate or low) were justified, documented, and incorporated into reporting of results for each outcome.
Results
Description of studies
Results of the search
Searches up to 2009
Ninety‐six potentially relevant articles were retrieved in searches up to 2008. Twenty‐eight studies were identified as RCTs that used SSRIs in the management of PMS and were included.
Search update in 2013
Nineteen potentially eligible articles were retrieved in the 2013 search. Six were included as new studies (Eriksson 2008; Freeman 2010; Glaxo 1996; Glaxo 1996a; Glaxo 2001; Steiner 2008) and three (Glaxo 2002; Miller 2008; Wu 2008) were excluded. Two articles were additional publications for included studies (Freeman 2004; Landen 2007), six were the abstracts of studies already included, and two were ongoing studies (Yonkers 2007; Yonkers 2010).
In the 2009 version of the review, the 28 included studies were divided into 40 comparisons each with a separate study reference. For the 2013 update, data relating to the same study were combined under a common study reference. Three previously included studies were excluded in the update after discussion between the review authors: one (Veeninga 1990) because the participants were self‐diagnosed and did not clearly meet medically defined diagnostic criteria for PMS, and two (Sundblad 1992a; Sundblad 1993a) because the intervention was clomipramine, which is not an SSRI.
Thus, a total of 31 studies are now included (n = 4372 ), comprising 25 studies from the previous version of the review and six new studies. See the study flow diagram (Figure 1).
1.
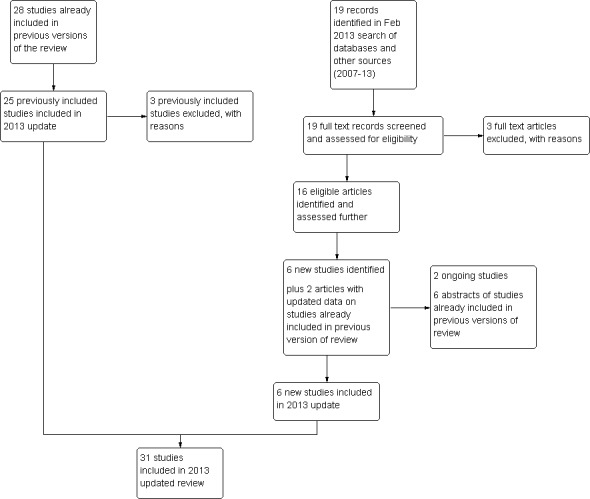
Study flow diagram.
Included studies
Study design and funding source
All studies were RCTs. Fifteen reported that they were multi‐centred (Cohen 2002; Cohen 2004; Glaxo 1996; Glaxo 1996a; Glaxo 2001; Halbreich 2002; Kornstein 2006; Landen 2007; Miner 2002; Pearlstein 1997; Pearlstein 2005; Steiner 1995; Steiner 2005; Steiner 2008; Yonkers 1997).
Most studies were of parallel‐group design but six used a crossover design (Halbreich 1997; Jermain 1999; Menkes 1992; Su 1997; Wood 1992; Young 1998). The first‐arm data (before crossover) for overall symptom reduction could be extracted for only one of these trials (Jermain 1999); the remaining crossover trials were not used in the data pooling. Where data were incomplete, all authors were contacted. However, no additional data were received.
Twenty‐one studies were solely or partially funded by pharmaceutical companies (Cohen 2002; Eriksson 1995; Eriksson 2008; Freeman 2010; Glaxo 1996; Glaxo 1996a; Glaxo 2001; Halbreich 1997; Halbreich 2002; Jermain 1999; Kornstein 2006; Landen 2007; Miner 2002; Pearlstein 1997; Pearlstein 2005, Steiner 1995; Steiner 2005; Steiner 2008; Stone 1991; Yonkers 1997) or had pharmaceutical company employees among their authors (Cohen 2004). Seven studies were funded independently (Freeman 1999; Freeman 2004; Menkes 1992; Su 1997; Wikander 1998; Wood 1992; Young 1998). The source of funding was unclear in three studies (Arrendondo 1997; Crnobaric 1998; Ozeren 1997).
Participants
Women in most of the included studies were aged from 18 to 45 years (range 18 to 49 years) where reported, though one study enrolled teenagers aged 15 to 19 years (Freeman 2010).
Most of the studies diagnosed women with PMS or PMDD by means of validated self‐rating symptom scales completed over multiple menstrual cycles, or by means of psychiatric diagnostic criteria. Measures used were:
Penn Daily Symptoms Rating Scale (Arrendondo 1997; Freeman 1999; Freeman 2004; Kornstein 2006);
Daily Record of Severity of Problems (DRSP) (Cohen 2002);
DSM III or IV diagnostic criteria for PMDD or LPDD (Cohen 2004; Crnobaric 1998; Eriksson 1995; Eriksson 2008; Glaxo 1996; Glaxo 1996a; Glaxo 2001; Halbreich 1997; Halbreich 2002; Jermain 1999; Landen 2007; Menkes 1992; Miner 2002; Ozeren 1997; Pearlstein 1997; Pearlstein 2005; Steiner 1995; Steiner 2005; Steiner 2008; Stone 1991; Wikander 1998; Wood 1992; Yonkers 1997; Young 1998);
Visual analogue scale (VAS) (Su 1997).
In most studies women were recruited from clinical settings (for example psychiatric, gynaecological or PMS outpatient clinics) or via media, television, or local newspaper advertising. Five studies provided no details of recruitment methods.
Exclusion criteria varied, but most of the studies excluded women with the following characteristics:
current or recent major or Axis 1 psychiatric diagnosis (other than PMDD);
other clinically significant disease;
recent hormonal contraceptive use;
current or planned pregnancy;
use of concurrent medication (including psychotropic drugs).
Interventions
Description of the interventions
SSRIs used were:
sertraline 50 to 150 mg (Arrendondo 1997; Freeman 1999; Freeman 2004; Halbreich 1997; Halbreich 2002; Jermain 1999; Kornstein 2006; Yonkers 1997; Young 1998);
fluoxetine 10 to 20 mg (Cohen 2002; Crnobaric 1998; Menkes 1992; Miner 2002; Ozeren 1997; Pearlstein 1997; Steiner 1995; Stone 1991; Su 1997; Wood 1992);
paroxetine 5 to 25 mg (Cohen 2004; Eriksson 1995; Glaxo 1996; Glaxo 1996a; Glaxo 2001; Landen 2007; Pearlstein 2005; Steiner 2005; Steiner 2008);
escitalopram 10 to 20 mg (Eriksson 2008; Freeman 2010);c
italopram 10 to 30 mg (Wikander 1998).
The timing regimen of the intervention varied as follows (some studies included more than one):
luteal or intermittent (Cohen 2002; Eriksson 2008; Freeman 2004; Freeman 2010; Halbreich 1997; Halbreich 2002; Jermain 1999; Kornstein 2006; Landen 2007; Miner 2002; Steiner 2005; Steiner 2008; Wikander 1998; Young 1998);
semi‐intermittent (low dose in follicular phase, higher dose in luteal phase) (Wikander 1998);
continuous (Arrendondo 1997; Cohen 2004; Crnobaric 1998; Eriksson 1995; Freeman 1999; Freeman 2004; Glaxo 1996; Glaxo 1996a; Kornstein 2006; Landen 2007; Menkes 1992; Ozeren 1997; Pearlstein 1997; Pearlstein 2005; Steiner 1995; Stone 1991; Su 1997; Wikander 1998; Wood 1992; Yonkers 1997).
The number of treatment cycles for these interventions varied from two to six.
Three studies compared luteal versus continuous (Freeman 2004; Landen 2007; Wikander 1998) or semi‐intermittent (Wikander 1998) SSRI dosing strategies. A fourth study (Kornstein 2006) included an 'as required' regimen, whereby women took an SSRI from symptom onset to menstruation. These data were not included in the review because the 'as required' regimen was administered after two cycles of luteal administration and had a duration of only one cycle, and thus did not meet our inclusion criteria.
Some of the studies had more than one active treatment arm. They included different drug doses (Cohen 2002; Cohen 2004; Eriksson 2008; Kornstein 2006; Steiner 1995; Steiner 2008; Wikander 1998) or different drug timing regimens (Freeman 2004; Landen 2007; Miner 2002; Wikander 1998) as well as a placebo group.
The following studies included a placebo run‐in period: Cohen 2002; Cohen 2004; Freeman 2004; Glaxo 1996; Glaxo 1996a; Halbreich 1997; Halbreich 2002; Miner 2002; Pearlstein 1997; Pearlstein 2005; Steiner 1995; Steiner 2005; Stone 1991; Yonkers 1997.
Review outcomes reported in the included studies
Primary review outcomes
1. Overall premenstrual symptoms
The following 15 studies reported total self‐rated symptom scores in a form that allowed pooling of the data: Cohen 2002; Cohen 2004; Eriksson 2008; Freeman 1999; Freeman 2004; Halbreich 2002; Jermain 1999; Kornstein 2006; Miner 2002; Ozeren 1997; Steiner 1995; Steiner 2005; Yonkers 1997. These studies utilised a range of self‐rated tools, which are identified by footnotes in the forest plots.
Three other studies reported self‐reported total symptom scores in a form that did not allow pooling of the data (Eriksson 2008; Freeman 2010; Glaxo 1996)
2. Adverse effects
Sixteen studies reported withdrawals due to adverse events in a form that allowed pooling of the data (Cohen 2002; Cohen 2004; Eriksson 1995; Eriksson 2008; Glaxo 2001; Halbreich 2002; Landen 2007; Ozeren 1997; Pearlstein 2005; Steiner 1995; Steiner 2005; Steiner 2008; Yonkers 1997).
The following 17 studies reported individual adverse effects in a form that allowed pooling of the data: Cohen 2002; Cohen 2004; Eriksson 1995; Eriksson 2008; Freeman 1999; Glaxo 1996a; Glaxo 2001; Halbreich 2002; Kornstein 2006; Landen 2007; Miner 2002; Pearlstein 1997; Pearlstein 2005; Steiner 1995; Steiner 2005; Steiner 2008; Stone 1991.
Secondary review outcomes
3. Specific symptoms of PMS
A wide variety of symptom measurement tools were used by the included studies, please see Table 21.
2. Outcome measures utilised by included studies.
| Outcome Measure | Studies | Description |
| Daily Symptom Report | Kornstein 2006 | 17 common PMS symptoms self rated daily on a 5‐point scale (from 0=none to 4=severe/overwhelming/unable to function). Mood (anxiety, irritability, depression, nervous tension, mood swing, feeling out of control); Behavioural ( poor coordination, insomnia, confusion/poor concentration, headache, crying, fatigue); Pain (aches, cramps, breast tenderness); Physical symptoms (food cravings, swelling); Distress. |
| Clinical Global Impression Severity of Illness (CGI‐S) | Freeman 1999, Kornstein 2006, Pearlstein 2005, Miner 2002, Steiner 2005, Yonkers 1997; Cohen 2004, Freeman, 1999, Freeman, 1999, Halbreich 2002, Pearlstein 1997, Yonkers 2006 | Clinician rated. 7‐point scale (1= not ill to 7 = extremely ill) |
| Clinical Global Improvement (CGI‐I) | Kornstein 2006, Landen 2007, Crnobaric 1998, Miner 2002, Steiner 2005, Menkes 1993, Yonkers 1997, Cohen 2004, Halbreich 1997, Freeman, 1999, Halbreich 2002, Yonkers 2006, Steiner 2008 | Clinician rated. 7‐point scale (1= very much better to 7 = very much worse) |
| Patient Global Evaluation (PGE) | Kornstein 2006, Landen 2007, Steiner 2005 a,b; Yonkers 1997, Halbreich 2002 | Self rated 7‐point ordinal scale (from 1 = very much improved to 7 = very much worse) that rates the degree of overall improvement in PMS symptoms compared with pre‐treatment baseline. Assessments were based on the past week. |
| Quality of Life Enjoyment and Satisfaction Scale | Kornstein 2006, Halbreich 2002 | Self rated 5 point ordinal scale 1 = very poor to 5 = very good. A total score computed by adding the first 14 items, dividing by 70 (maximum total score) and multiplying by 100. |
| Social Adjustment Scale Self Report (SAS‐SR) | Kornstein 2006, Yonkers 1997, Halbreich 2002 | Self rated 55‐item scale assessing work and or housework, interpersonal relationships, and social and leisure activities during the previous week. |
| Calender of Premenstrual Experience (COPE) | Young 1998, Crnobaric 1998, Ozeren 1997, Jermain 1999, Wood 1992 | Self rated 22 symptoms grouped into behavioural (14 symptoms,) and physical (8 symptoms) categories. Symptoms rated daily from 0 (none) to 3 (severe). |
| Visual Analogue Scale (VAS) | Menkes 1993 | 0‐100mm for irritability, depressed mood, increased appetite/carbohydrate cravings, breast tenderness and bloating. 0 = no complaints to 100 = maximum complaints. |
| Visual Analogue Scale (VAS) | Menkes 1992 | 0‐100mm for irritability, depressed mood, increased appetite/carbohydrate cravings, breast tenderness and bloating. 0 = no complaints to 100 = maximum complaints. |
| Visual Analogue Scale (VAS) | Landen 2007 | 0‐100mm, no details as to symptoms included in scale. |
| Visual Analogue Scale (VAS) | Steiner 1995 | 0‐100mm for tension, irritability and dysphoria with 0 being no symptoms and 100 being severe or extreme symptoms. The mean of the three scales was used determine the total psychological ‐ symptom score. |
| Visual Analogue Scale (VAS) | Pearlstein 2005, Steiner 2005, Cohen 2004; | 0‐100mm with 0 being 'not at all' and 100 being 'extreme'. Eleven symptoms were recorded irritability, tension, affective lability, depressed mood, decreased interest, difficulty concentrating, lack of energy, change in appetite, change in sleep pattern, feeling out of control and physical symptoms. VAS Mood is a composite score of irritability, tension, depressed mood and affective lability. |
| Visual Analogue Scale (VAS) | Su 1997 | A 16‐item extended version of the VAS scale. |
| Visual Analogue Scale (VAS) | Wikander 1998, Eriksson 1995 | 0‐100mm scale with 0 = no complaints and 100 = maximal complaints. Symptoms include irritability, depressed mood, tension, anxiety, increased appetite, bloating, breast tenderness. |
| Premenstrual tension scale (PMTS) ‐ observer and self rated | Landen 2007, Steiner 1995, Miner 2002, Steiner 2005, Cohen 2002, Su 1997, Yonkers 2006 | 36‐item scale completed by patient and 10‐item scale completed by therapist/clinician. Both scales rate premenstrual symptoms on a given day and the score can range from 0 to 36 indicating all symptoms present and severe. |
| Sheehan Disability Scale (SDS) | Landen 2007, Pearlstein 2005, Miner 2002, Steiner 2005, Cohen 2004 | Assesses the extent to which their symptoms affect work, social life/leisure activities and family life/home responsibilities (scale 0 = not at all impaired to 10 = cannot function). |
| Penn Daily Symptom Rating Form (DSR) | Arrendondo 1997, Freeman 2004, Freeman 1999 | Depression, feeling hopeless or guilty, anxiety/tension, mood swings, irritability/anger, decreased interest, concentration difficulties, fatigue, food cravings/increased appetite, insomnia or hypersomnia, feeling out of control/overwhelmed, poor coordination, headache, aches, swelling/bloating/weight gain, cramps and breast tenderness. Rated on a five point scale from 0‐4 (no disruption to severe disruption). Scores were calculated by adding the ratings of cycle days 5 through 10 for post menstrual scores and by adding the scores for the 6 days before menses for the premenstrual scores. |
| Subject Global Ratings of Functioning | Freeman 2004 | Depression, feeling hopeless or guilty, anxiety/tension, mood swings, irritability/anger, decreased interest, concentration difficulties, fatigue, food cravings/increased appetite, insomnia or hypersomnia, feeling out of control/overwhelmed, poor coordination, headache, aches, swelling/bloating/weight gain, cramps and breast tenderness. Rated on a five point scale from 0‐4 (no disruption to severe disruption). Scores were calculated by adding the ratings of cycle days 5 through 10 for post menstrual scores and by adding the scores for the 6 days before menses for the premenstrual scores. |
| Prospective Record of the Impact and Severity of Menstrual Symptomatology Calendar | Steiner 1995 | No details in paper. |
| Hamilton Rating Scale for Depression (HAM‐D) (HRSD) | Crnobaric 1998, Yonkers 1997, Halbreich 1997, Freeman, 1999, Halbreich 2002, Pearlstein 1997, Yonkers 2006 | No details in paper. |
| Daily Record of Severity of Problems (DRSP) | Miner 2002, Cohen 2002, Yonkers 1997, Halbreich 2002, Yonkers 2006 | Scale consisting 21 numbered items grouped into 11 categories (depressed/hopeless/worthless; tension; mood swings/ feelings hurt; irritability; less interest in activities; difficulty concentrating; lethargy; increased appetite/cravings; sleeping more/insomnia; overwhelmed/out of control; breast tenderness/bloating/headache/joint or muscle pain.). It has three additional questions measuring impairment of social functioning (at work/school/home; hobbies or social activities; relationships). Severity of each symptom is rated on a scale from 1 (not at all) to 6 (extreme). Mean score was calculated as average scores for the five most symptomatic days from six days before through to the first days of menses. Yonkers used an updated version using 24 items. |
| Daily Assessment Form (DAF) | Stone 1991 | 33‐item checklist used to assess each of the 10 symptom categories found in the DSM‐III‐R criteria. Symptoms are rated with a 6‐point rating scale ranging from 1 (none) to 6 (extreme). |
| Global Assessment Scale (GAS) | Stone 1991, Pearlstein 1997 | Self‐assessed scale with 18 summary scale scores reflecting composite ratings from 4‐14 items scored from 1(no premenstrual change) to 6 (extreme change). |
| Premenstrual Assessment Form (PAF) | Menkes 1993 | Includes irritability, low energy, mood swings, mastalgia, depression, bloating, impulsivity, abdominal pain, anxiety, food cravings. Scale of no change or worse to remitted. |
| Daily Ratings Form (DRF) | Menkes 1993 | Includes irritability, low energy, mood swings, mastalgia, depression, bloating, impulsivity, abdominal pain, anxiety, food cravings. Scale of no change or worse to remitted. |
| Daily Ratings Form (DRF) | Su 1997 | 21‐item 6‐point scale; including sadness, anxiety, irritability, mood swings, breast pain, bloating, fatigue, food cravings, impaired social and work functioning, impulsivity and global impairment, sleep and sexual interest. |
| Modified Daily Ratings Form (DRF) | Halbeich 1997 | No details in paper. |
| Beck Depression Inventory | Su 1997, Jermain 1999, Wood 1992 | 22‐item patient rated scale assessing depression. Rated on a 4‐point severity scale. |
| Stait Trait Anxiety Inventory State Form | Su 1997, Wood 1992 | No details in papers. |
| Physical symptom checklist | Su 1997 | Designed to detect the side effects of fluoxetine. |
| Profile of Mood State | Wood 1992 | No details in paper. |
| Global Ratings of Functioning and Improvement | Freeman 1999 | 5‐point rating scale using descriptors for each point ranging from 0 (none) to 4 (complete). Functioning rated for work, family life, and social activity with 0 (no disruption) to 4 (severe disruption). |
| Prospective Record of the Impact and Severity of Menstrual Symptomology calendar | Steiner 1995 | No details in paper but completed daily. |
| Quality of Life Scale (QOLS) | Freeman 1999 | Self‐reported measure of various aspects of daily living plus a global assessment of QOL over the past week. The 14 QOLS items are the summary scales of the Quality of Life Enjoyment and Satisfaction Questionnaire. |
| Premenstrual Tension Scale (PMTS‐O) | Steiner 2008 | Observer‐assessed |
3.1 Psychological symptoms
Ten studies reported psychological symptoms of PMS in a form that allowed pooling of the data (Arrendondo 1997; Cohen 2002; Freeman 1999; Glaxo 2001; Kornstein 2006; Miner 2002; Pearlstein 2005; Steiner 2005; Yonkers 1997).
3.2 Physical symptoms
Eleven studies reported physical symptoms of PMS in a form that allowed pooling of the data (Cohen 2002; Freeman 1999; Glaxo 2001; Halbreich 2002; Kornstein 2006; Miner 2002; Pearlstein 1997; Steiner 1995; Yonkers 1997).
3.3 Functional symptoms
Five studies reported functional symptoms of PMS in a form that allowed pooling of the data (Cohen 2002; Eriksson 2008; Kornstein 2006; Miner 2002; Yonkers 1997).
3.4 Irritability
Eight studies reported irritability in a form that allowed pooling of the data (Cohen 2002; Freeman 1999; Halbreich 2002; Pearlstein 1997; Steiner 2008; Yonkers 1997).
4. Response
Twenty‐one studies reported response rates in a form that allowed pooling of the data (Cohen 2004; Crnobaric 1998; Eriksson 1995; Eriksson 2008; Freeman 1999; Freeman 2004; Glaxo 1996; Glaxo 2001; Halbreich 2002; Kornstein 2006; Landen 2007; Ozeren 1997; Pearlstein 1997; Pearlstein 2005; Steiner 1995; Steiner 2005; Stone 1991; Wikander 1998; Yonkers 1997). The definition of response varied across studies and is defined in footnotes in the forest plots.
5. Withdrawal for any reason
Fourteen studies reported overall withdrawal rates in a form that allowed pooling of the data (Crnobaric 1998; Eriksson 2008; Freeman 1999; Glaxo 1996; Glaxo 2001; Halbreich 2002; Jermain 1999; Ozeren 1997; Steiner 2008; Stone 1991; Yonkers 1997).
For details please see Table 21 and Characteristics of included studies.
Excluded studies
Twenty‐six studies were excluded (see Characteristics of excluded studies), in most cases because they were not properly randomised, were unblinded, were not placebo controlled or did not report the comparison of interest.
Risk of bias in included studies
Summaries of the risk of bias assessment are given in Figure 2 and Figure 3.
2.
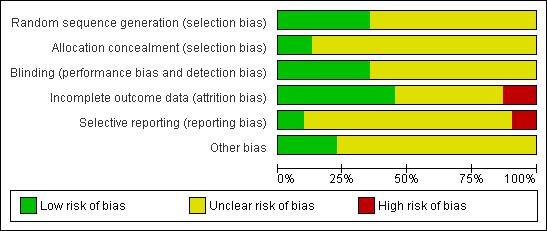
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
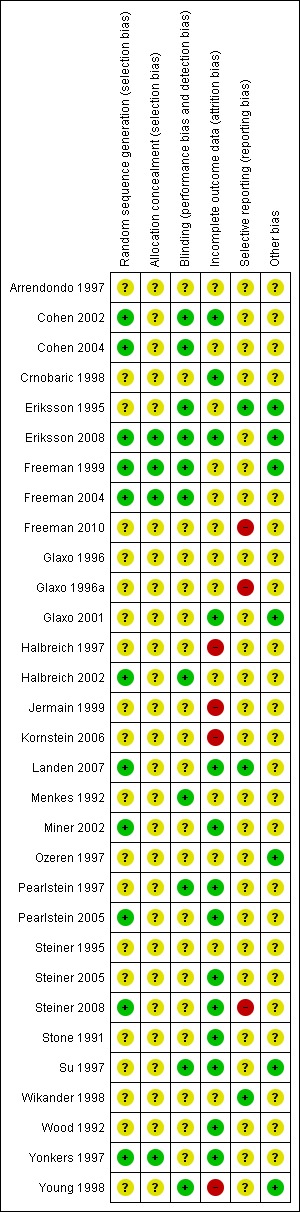
Risk of bias summary: review authors' judgments about each risk of bias item for each included study.
Allocation
Sequence generation
Eleven studies described adequate methods of randomisation and were rated as at low risk of bias related to sequence generation (Cohen 2002; Cohen 2004; Eriksson 2008; Freeman 1999; Freeman 2004; Halbreich 2002; Landen 2007; Miner 2002; Pearlstein 2005; Steiner 2008; Yonkers 1997). The other 20 studies did not clearly describe their methods and were rated as at unclear risk of bias.
Allocation concealment
Four studies described adequate methods of allocation concealment and were rated as at low risk of bias in this domain (Eriksson 2008; Freeman 1999; Freeman 2004; Yonkers 1997). The other 27 studies did not clearly describe their methods and were rated as at unclear risk of bias.
Blinding
Eleven studies reported details of double blinding and were rated as at low risk of bias in this domain (Cohen 2002; Cohen 2004; Eriksson 1995; Eriksson 2008; Freeman 1999; Freeman 2004; Freeman 2010; Menkes 1992; Pearlstein 1997; Su 1997; Young 1998). The other 20 studies were described as double blinded but provided no further details and were rated as at unclear risk of bias.
Incomplete outcome data
Fourteen studies analysed all or most women by intention to treat, and were rated as at low risk of attrition bias (Cohen 2002; Crnobaric 1998; Eriksson 2008; Glaxo 2001; Landen 2007; Miner 2002; Pearlstein 1997; Pearlstein 2005; Steiner 2005; Steiner 2008; Stone 1991; Su 1997; Wood 1992; Yonkers 1997). Four studies had data missing for over 20% of participants and were rated as at high risk of attrition bias (Halbreich 1997; Jermain 1999; Kornstein 2006; Young 1998). The other 13 studies were at unclear risk of attrition bias due to unclear reporting of numbers randomised or numbers analysed, or failure to include 10% to 20% of participants in the analysis. A number of these studies also imputed a high proportion of data.
Selective reporting
Three studies were rated as at low risk of this bias (Eriksson 1995; Landen 2007; Wikander 1998). These studies prospectively collected data on adverse effects and no potential source of selective reporting bias was identified. Three studies were rated as at high risk of this bias. These studies collected data for efficacy and adverse effects that were not fully reported or not published (Freeman 2010; Glaxo 1996a; Steiner 2008). The other 25 studies were rated as at unclear risk of selective reporting bias. Most of these studies collected data on adverse events retrospectively (often by spontaneous participant report) or did not report adverse events data in an extractable form for either comparison group.
Other potential sources of bias
No other potential source of bias was identified in seven studies, and these studies were rated as at low risk for this domain (Eriksson 1995; Eriksson 2008; Freeman 1999; Glaxo 2001; Ozeren 1997; Su 1997; Young 1998). The other 24 studies were rated as at unclear risk of other potential bias, in most cases because they excluded placebo responders from the study, were crossover studies (for which only first‐phase data were included in this review), or very few details were reported about the study design.
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings for the main comparison. SSRIs for premenstrual syndrome: all symptoms (end scores).
| SSRIs compared to placebo ‐ all symptoms (end scores) for premenstrual syndrome | ||||
| Patient or population: women with premenstrual syndrome Settings: community or outpatient Intervention: SSRIs Comparison: placebo ‐ all symptoms (end scores) | ||||
| Outcomes | Illustrative comparative risks* (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments |
|
Moderate dose SSRI versus placebo Luteal or continuous administration |
The mean score for all symptoms in the intervention groups was 0.67 standard deviations lower (0.46 to 0.84 lower) | 1276 (9 studies) | ⊕⊕⊝⊝ low1,2 | SMD ‐0.65 (95% CI ‐0.42 to ‐0.84) Symptoms were significantly less severe in the SSRI group. The size of the effect was moderate. |
|
Moderate dose SSRI versus placebo Luteal administration |
The mean score for all symptoms in the intervention groups was 0.51 standard deviations lower (0.71 to 0.31 lower) | 457 (4 studies) | ⊕⊕⊕⊝ moderate2 | SMD 0.51 (95% CI ‐0.71 to ‐0.31) Symptoms were significantly less severe in the SSRI group. The size of the effect was moderate. |
| Moderate dose SSRI versus placebo Continuous administration | The mean score for all symptoms in the intervention groups was 0.72 standard deviations lower (0.97 to 0.47 lower) | 843 (7 studies) | ⊕⊕⊝⊝ low1,2 | SMD ‐0.72 (95% CI ‐0.97 to ‐0.47) Symptoms were significantly less severe in the SSRI group. The size of the effect was moderate. |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD standardised mean difference | ||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||
1 Only 4/9 studies overall (2/4 of luteal and 3/7 of continuous administration) described adequate methods of randomisation and allocation concealment; 8/9 studies were at uncertain or high risk of attrition bias. 2 Substantial overall heterogeneity (I squared= 58%), attributable to heterogeneity in continuous administration group (I squared=68%), which included two studies with larger intervention effects. No obvious explanation (though studies used wide variety of assessment tools).
Summary of findings 2. SSRIs for premenstrual syndrome: all symptoms (change scores).
| SSRIs for premenstrual syndrome (change scores) | ||||
|
Patient or population: women with premenstrual syndrome
Settings: community or outpatient
Intervention: SSRIs Comparison: placebo | ||||
| Outcomes | Illustrative comparative risks* (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments |
|
Moderate dose SSRI versus placebo Luteal administration2 |
The mean moderate dose ssri in the intervention groups was 0.36 standard deviations lower (0.51 to 0.2 lower) | 657 (4 studies) | ⊕⊕⊕⊝ moderate1 | SMD ‐0.36 (‐0.51 to ‐0.2) Symptoms were significantly less severe in the SSRI group. The size of the effect was small. |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the ccomparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD standardised mean difference | ||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||
1 No studies described adequate methods of both randomisation and allocation concealment; one was high risk of attrition bias.
2 Change score data were not extracted for any studies of continuous administration, as all reported end scores.
Summary of findings 3. SSRIs for premenstrual syndrome: withdrawal due to adverse effects.
| SSRIs versus placebo: withdrawal due to adverse effects | ||||||
|
Patient or population: women with premenstrual syndrome
Settings: community or outpatient
Intervention: SSRIs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Placebo | SSRIs | |||||
|
Low dose SSRI versus placebo Luteal or continuous administration |
53 per 1000 | 91 per 1000 (60 to 135) | OR 1.76 (1.13 to 2.75) | 1301 (7 studies) | ⊕⊕⊕⊝ moderate1 | Withdrawal due to adverse effects was significantly more common in the SSRI groups |
|
Mod dose SSRI versus placebo Luteal or continuous administration |
45 per 1000 | 107 per 1000 (79 to 142) | OR 2.55 (1.84 to 3.53) | 2447 (15 studies) | ⊕⊕⊕⊝ moderate2 | |
|
High dose SSRIs versus placebo Continuous administration |
72 per 1000 | 457 per 1000 (207 to 1000) | RR 6.35 (2.88 to 14) | 231 (1 study) | ⊕⊕⊕⊝ moderate3 | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Only 1/4 studies reported adequate methods of randomisation and allocation concealment.
2 Only 3/15 studies described adequate methods of randomisation and allocation concealment and 7/15 were at unclear or high risk of attrition bias. 3 Single study (n=235), which did not describe adequate method of allocation concealment.
Primary outcomes
1. Total symptoms
Fifteen studies reported total self‐rated symptom scores. Nine studies reported end scores for this outcome (Cohen 2004; Eriksson 2008; Freeman 1999; Freeman 2004; Halbreich 2002; Jermain 1999; Ozeren 1997; Steiner 1995; Yonkers 1997), four only reported change scores (Cohen 2002; Kornstein 2006; Miner 2002; Steiner 2005) and one reported only descriptive data (Glaxo 1996).
1.1. Total symptoms: end scores
When effects were assessed with end scores, SSRIs reduced overall symptoms significantly more effectively than placebo, with a moderate effect size. This applied to low dose SSRIs (SMD ‐ 0.67, 95% CI ‐0.29 to ‐1.05, two studies, 301 women; I2 = 59%; Analysis 1.1), moderate dose SSRIs (SMD ‐0.65, 95% CI ‐0.46 to ‐0.84, nine studies, 1276 women; I2 = 58%; Analysis 1.2, Figure 4) and high dose SSRIs (SMD ‐0.95, 95% CI ‐ 0.58 to ‐1.31, one study, 134 women; Analysis 1.3). There was substantial heterogeneity for both pooled analyses.
1.1. Analysis.
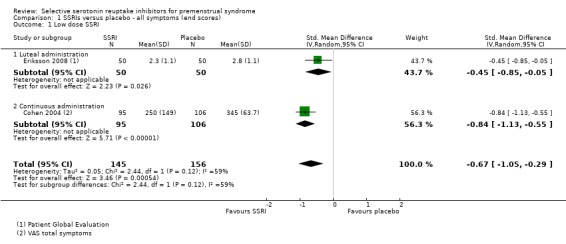
Comparison 1 SSRIs versus placebo ‐ all symptoms (end scores), Outcome 1 Low dose SSRI.
1.2. Analysis.
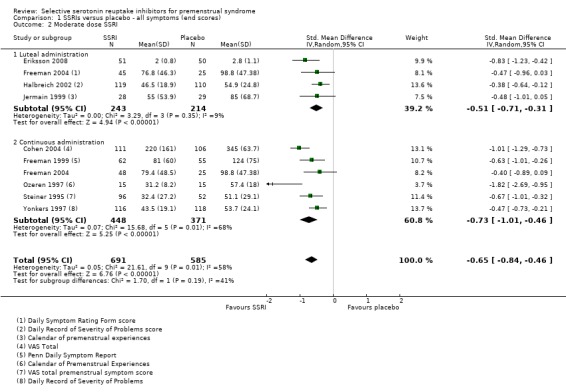
Comparison 1 SSRIs versus placebo ‐ all symptoms (end scores), Outcome 2 Moderate dose SSRI.
4.
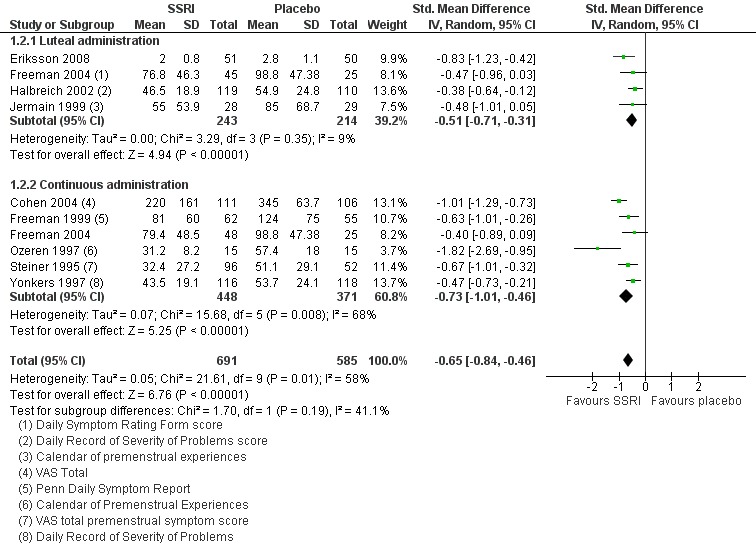
Forest plot of comparison: 1 SSRIs versus placebo ‐ all symptoms (end scores), outcome: 1.2 Moderate dose SSRI.
1.3. Analysis.
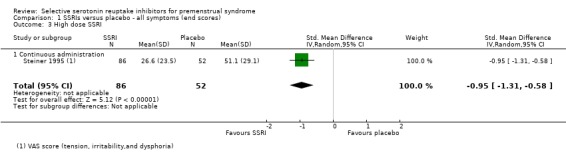
Comparison 1 SSRIs versus placebo ‐ all symptoms (end scores), Outcome 3 High dose SSRI.
The analyses were stratified by type of intervention (luteal or continuous). The effects of the intervention were non‐significantly higher in the studies of continuous SSRIs, but there may have been too few studies in each group to show a significant difference.
Five of the studies in this analysis used sertraline. When analysis was restricted to these studies, there was a significant benefit for the SSRI group with a smaller effect size and no heterogeneity (SMD 0.46, 95% CI ‐0.32 to ‐0.60, five studies, 780 women; I2 = 0%). The other studies in this analysis used three different types of SSRI and there were too few using the same type to permit subgrouping.
1.2 Total symptoms: change scores
Similarly, when effects were assessed with change scores, SSRIs reduced overall symptoms significantly more effectively than placebo. However, the effect sizes were small. This applied to both low dose SSRIs (SMD ‐0.23, 95% CI ‐0.05 to ‐0.41, four studies, 677 women; I2 = 29%; Analysis 2.1) and moderate dose SSRIs (SMD ‐ 0.36, 95% CI ‐0.20 to ‐0.51, four studies, 657 women; I2 = 0%; Analysis 2.2, Figure 5). Heterogeneity was low or absent.
2.1. Analysis.
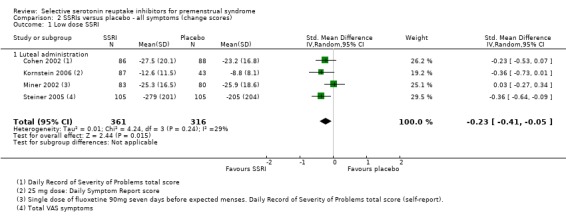
Comparison 2 SSRIs versus placebo ‐ all symptoms (change scores), Outcome 1 Low dose SSRI.
2.2. Analysis.
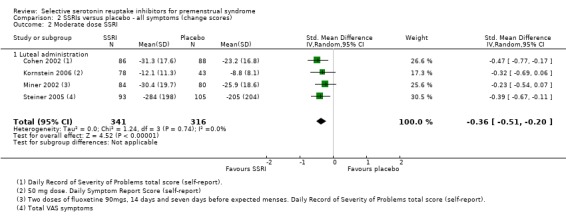
Comparison 2 SSRIs versus placebo ‐ all symptoms (change scores), Outcome 2 Moderate dose SSRI.
5.
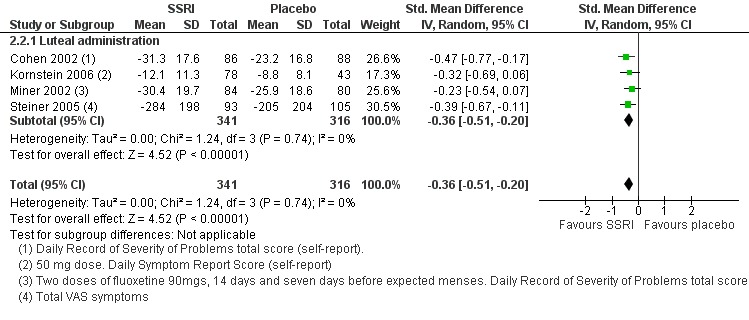
Forest plot of comparison: 2 SSRIs versus placebo ‐ all symptoms (change scores), outcome: 2.2 Moderate dose SSRI.
1.3 Total symptoms: descriptive data
Among studies reporting total self‐assessed symptoms scores in a form that was not suitable for meta‐analysis:
Eriksson 2008 reported that women taking escitalopram 10 mg or 20 mg had a significantly greater improvement in four key VAS symptoms than the placebo group;
Glaxo 1996, an unpublished study, reported no significant difference in luteal phase COPE score between paroxetine 20 mg and placebo;
Freeman 2010, an unpublished pilot study (n = 11), reported that women in both arms significantly improved from baseline, with no significant difference between the groups (which was attributed to lack of statistical power in this very small study). Penn Daily Symptom Report (DSR) scores decreased 41% in the drug arm and 28% in the placebo arm.
1.4 Sensitivity analyses
Quality: restricting analysis to the few studies that were at low risk of selection bias (Eriksson 2008; Freeman 1999; Freeman 2004; Yonkers 1997) did not materially change the main findings with regard to total symptom control.
Statistical model: use of a fixed‐effect model did not materially alter the main findings for the primary outcomes.
2. Adverse events
2.1 Withdrawals due to adverse events
Seventeen studies reported withdrawal from the study due to adverse events and reported results in a form that allowed pooling of the data. Women taking SSRIs were significantly more likely to withdraw from the study than women taking placebo. This applied to women taking low dose SSRIs (OR 1.76, 95% CI 1.13, to 2.75, seven studies, 1301 women; I2 = 0%), moderate dose SSRIs (OR 2.55, 95% CI 1.84 to 3.53, 15 studies, 2447 women; I2 = 0%; Analysis 3.2, Figure 6) and high dose SSRIs (OR 6.35, 95% CI 2.88 to 14.00, one study; Analysis 3.3). Heterogeneity was absent for these analyses (I2 = 0%).
3.2. Analysis.
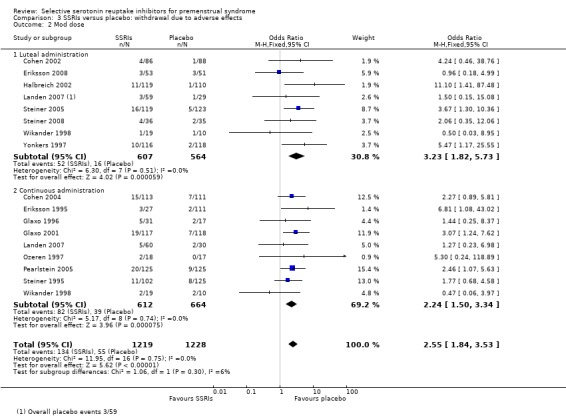
Comparison 3 SSRIs versus placebo: withdrawal due to adverse effects, Outcome 2 Mod dose.
6.
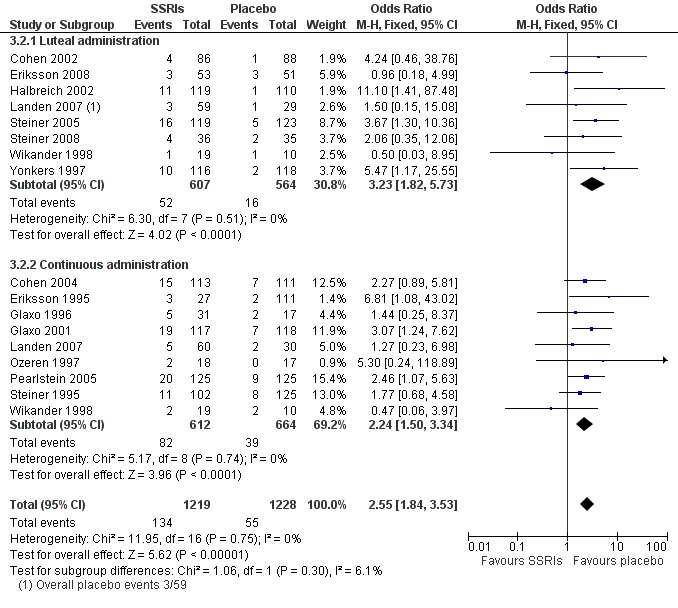
Forest plot of comparison: 4 SSRIs versus placebo: withdrawal due to adverse events, outcome: 4.2 Mod dose.
3.3. Analysis.
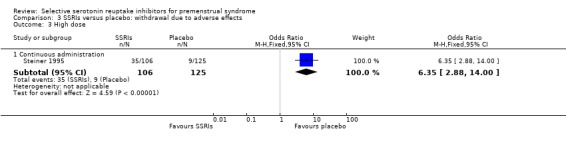
Comparison 3 SSRIs versus placebo: withdrawal due to adverse effects, Outcome 3 High dose.
2.2 Individual adverse events
Sixteen studies reported one or more individual adverse events in a form that allowed pooling of the data.
Compared to the placebo group, the SSRI group had higher rates of the following events.
Nausea (OR 3.43, 95% 2.63 to 4.47, 16 studies, 3385 women; I2 = 0%; Analysis 4.1); NNH for moderate dose of 7.
Insomnia or sleep disturbance (OR 1.84, 95% CI 1.36 to 2.47, 16 studies, 3388 women; I2 = 0%; Analysis 4.2); NNH for moderate dose of 25.
Sexual dysfunction or decreased libido (OR 2.26, 95% CI 1.54 to 3.31, 14 studies, 2847 women; I2 = 0%; Analysis 4.3); NNH for moderate dose of 14.
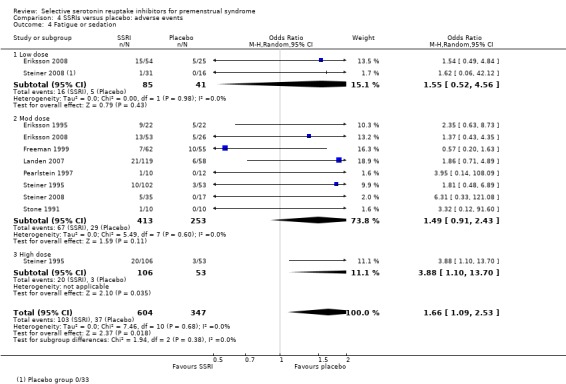
Fatigue or sedation (OR 1.66, 95% CI 1.09 to 2.53, eight studies, 951 women; I2 = 0%; Analysis 4.4); NNH for moderate dose of 14.
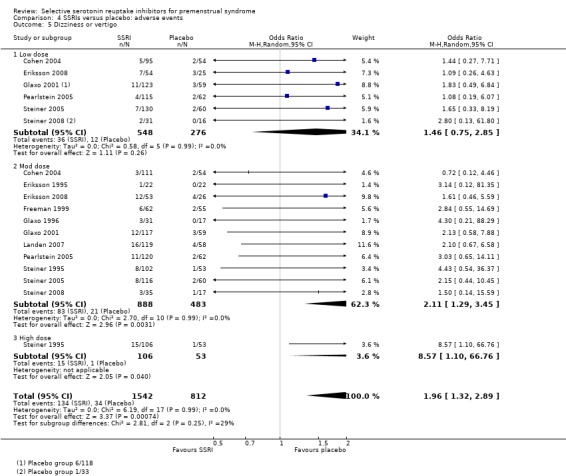
Dizziness or vertigo (OR 1.96, 95% CI 1.32 to 2.89, 11 studies, 2354 women; I2 = 0%; Analysis 4.5); NNH for moderate dose of 25.
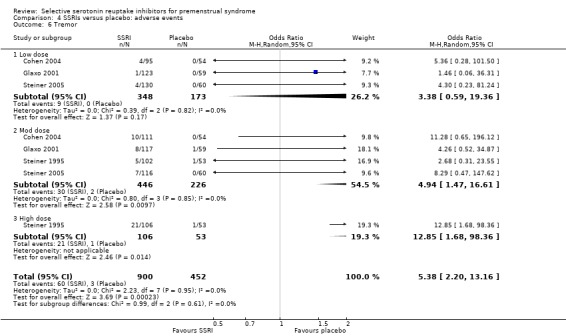
Tremor (OR 5.38, 95% CI 2.20 to 13.16, four studies, 1352 women; I2 = 0%; Analysis 4.6); NNH for moderate dose of 20.
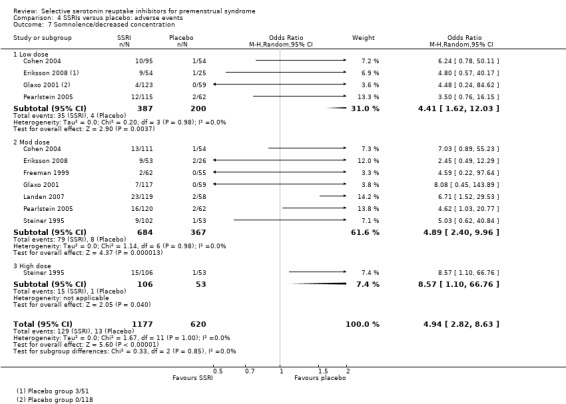
Somnolence and decreased concentration (OR 4.94, 95% CI 2.82 to 8.63, seven studies, 1797 women; I2 = 0%; Analysis 4.7); NNH for moderate dose of 13.
Sweating (OR 3.02, 95% CI 1.79 to 5.11, nine studies, 2051 women; I2 = 0%; Analysis 4.8); NNH for moderate dose of 14.
Dry mouth (OR 2.70, 95% CI 1.656 to 4.41, nine studies, 1474 women; I2 = 0%; Analysis 4.9); NNH for moderate dose of 17.
Yawning (OR 4.24, 95% CI 1.63 to 10.99, five studies, 975 women; I2 = 0%; Analysis 4.10); NNH for moderate dose of 14.
Asthenia or decreased energy (OR 3.28, 95% CI 2.16 to 4.98, seven studies, 1704 women; I2 = 0%; Analysis 4.11); NNH for moderate dose of 9.
Diarrhoea (OR 2.21, 95% CI 1.35 to 3.62, 10 studies, 2402 women; I2 = 0%; Analysis 4.12); NNH for moderate dose of 25.
Constipation (OR 2.35, 95% CI 1.04 to 5.29, six studies, 996 women; I2 = 0%; Analysis 4.13); NNH for moderate dose of 33.
4.1. Analysis.
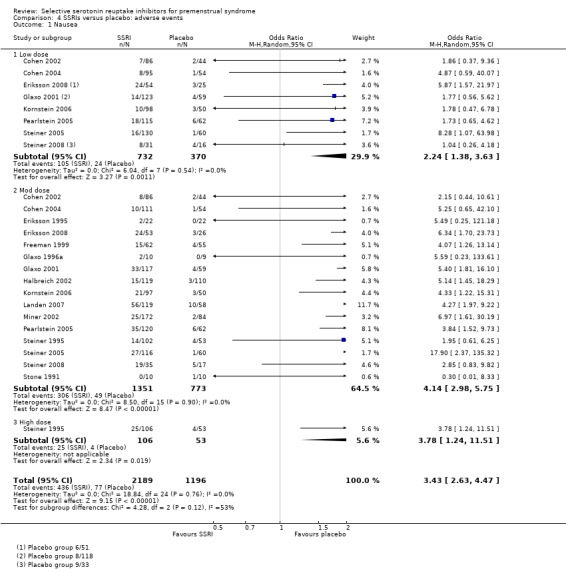
Comparison 4 SSRIs versus placebo: adverse events, Outcome 1 Nausea.
4.2. Analysis.
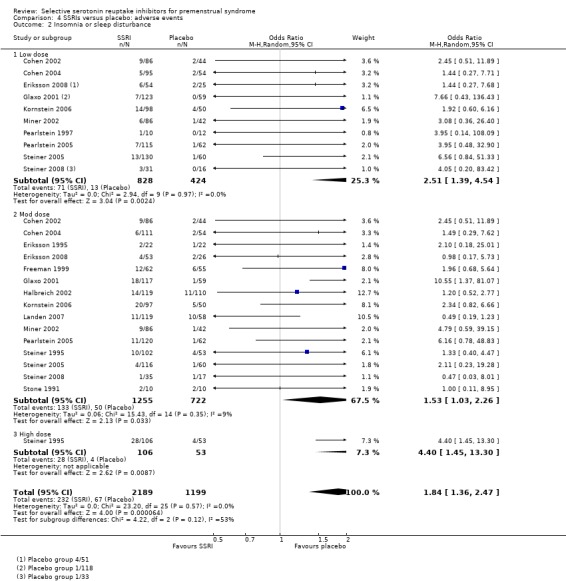
Comparison 4 SSRIs versus placebo: adverse events, Outcome 2 Insomnia or sleep disturbance.
4.3. Analysis.
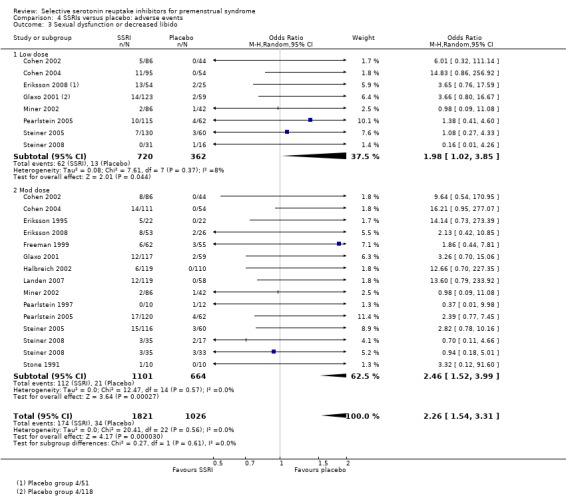
Comparison 4 SSRIs versus placebo: adverse events, Outcome 3 Sexual dysfunction or decreased libido.
4.4. Analysis.
Comparison 4 SSRIs versus placebo: adverse events, Outcome 4 Fatigue or sedation.
4.5. Analysis.
Comparison 4 SSRIs versus placebo: adverse events, Outcome 5 Dizziness or vertigo.
4.6. Analysis.
Comparison 4 SSRIs versus placebo: adverse events, Outcome 6 Tremor.
4.7. Analysis.
Comparison 4 SSRIs versus placebo: adverse events, Outcome 7 Somnolence/decreased concentration.
4.8. Analysis.
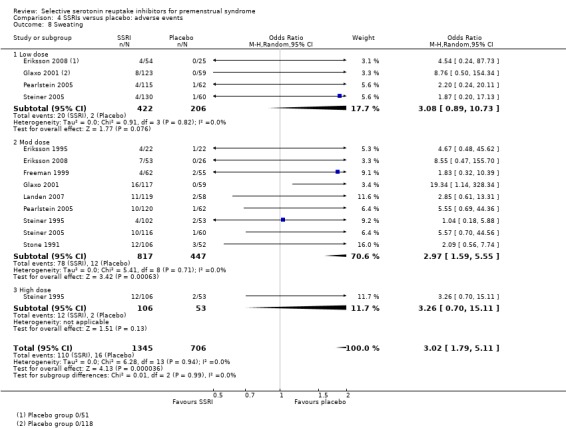
Comparison 4 SSRIs versus placebo: adverse events, Outcome 8 Sweating.
4.9. Analysis.
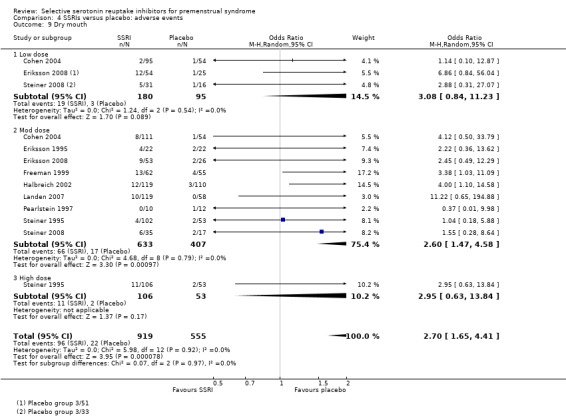
Comparison 4 SSRIs versus placebo: adverse events, Outcome 9 Dry mouth.
4.10. Analysis.
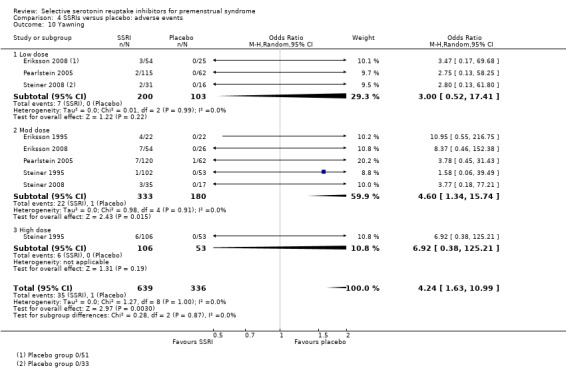
Comparison 4 SSRIs versus placebo: adverse events, Outcome 10 Yawning.
4.11. Analysis.
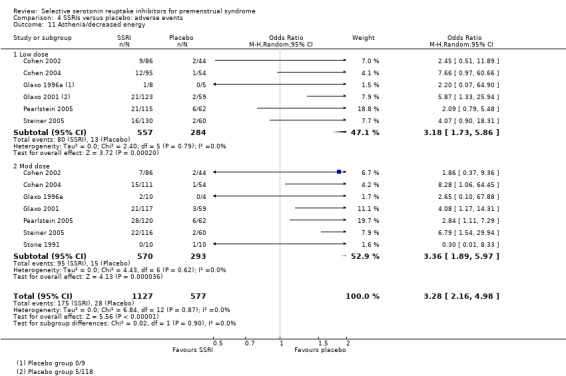
Comparison 4 SSRIs versus placebo: adverse events, Outcome 11 Asthenia/decreased energy.
4.12. Analysis.
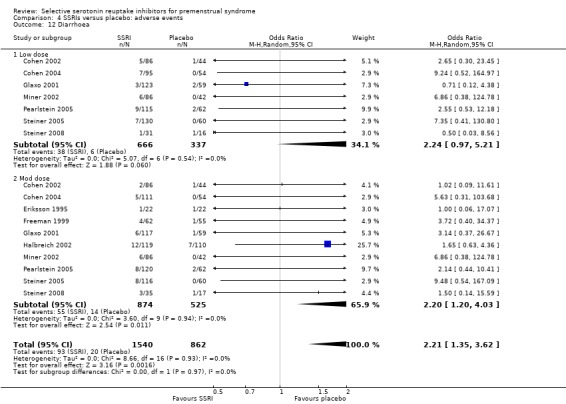
Comparison 4 SSRIs versus placebo: adverse events, Outcome 12 Diarrhoea.
4.13. Analysis.
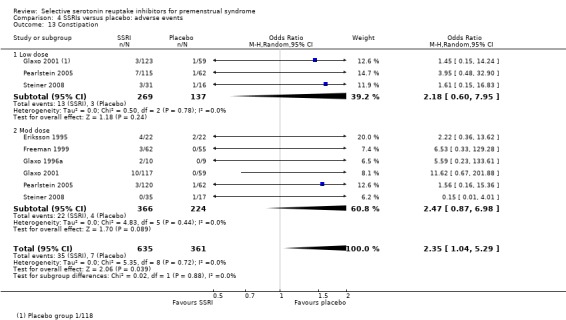
Comparison 4 SSRIs versus placebo: adverse events, Outcome 13 Constipation.
For most of these outcomes, inspection of the forest plots showed a clear dose‐response trend, with an increased risk of adverse events in women receiving a higher dose of SSRI.
Rates of the following events did not differ between the two groups.
Gastrointestinal irritability or dyspepsia (OR 2.03, 95% CI 0.78 to 5.31, five studies, 803 women; I2 = 0%; Analysis 4.14).
Headache (OR 1.13, 95% CI 0.89 to 1.42, 15 studies, 2866 women; I2 = 0%; Analysis 4.15).
Decreased appetite (OR 1.77, 95% CI 0.79 to 3.98, three studies, 433 women; I2 = 0%; Analysis 4.16).
Increased appetite (OR 1.36, 95% CI 0.34 to 5.46, three studies, 495 women; I2 = 0%; Analysis 4.17).
Anxiety (OR 1.17, 95% CI 0.45 to 302, three studies, 397 women; I2 = 0%; Analysis 4.18).
Cardiovascular symptoms (OR 3.94, 95% CI 0.89 to 17.39, three studies, 380 women; I2 = 0%; Analysis 4.19).
Respiratory disorder (OR 0.70, 95% CI 0.46 to 1.06, four studies, 1334 women; I2 = 0%; Analysis 4.20).
Sinusitis (OR 0.65, 95% CI 0.38 to 1.12, five studies, 1657 women; I2 = 0%; Analysis 4.21).
Infection (OR 1.01, 95% CI 0.60 to 1.70, four studies, 1339 women; I2 = 4%; Analysis 4.22).
4.14. Analysis.
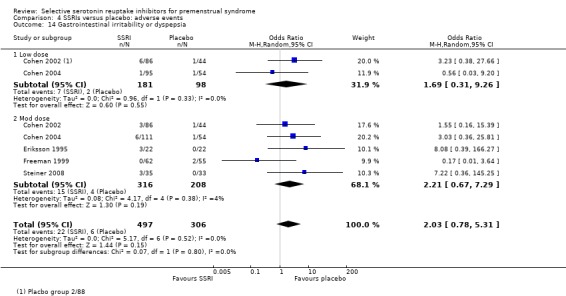
Comparison 4 SSRIs versus placebo: adverse events, Outcome 14 Gastrointestinal irritability or dyspepsia.
4.15. Analysis.
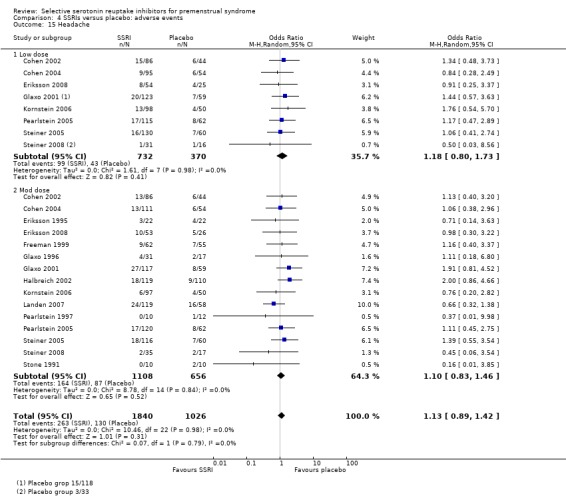
Comparison 4 SSRIs versus placebo: adverse events, Outcome 15 Headache.
4.16. Analysis.
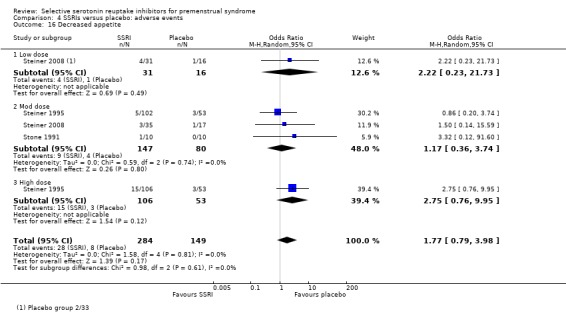
Comparison 4 SSRIs versus placebo: adverse events, Outcome 16 Decreased appetite.
4.17. Analysis.
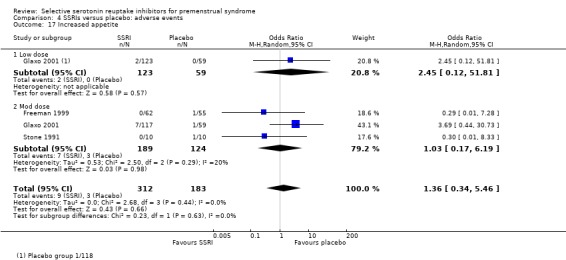
Comparison 4 SSRIs versus placebo: adverse events, Outcome 17 Increased appetite.
4.18. Analysis.
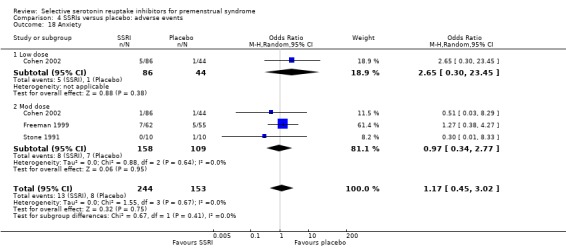
Comparison 4 SSRIs versus placebo: adverse events, Outcome 18 Anxiety.
4.19. Analysis.
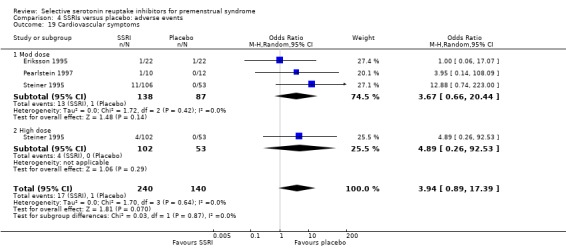
Comparison 4 SSRIs versus placebo: adverse events, Outcome 19 Cardiovascular symptoms.
4.20. Analysis.
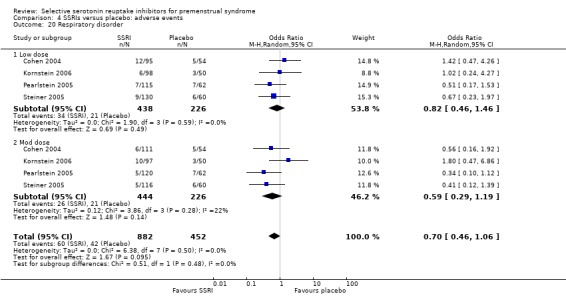
Comparison 4 SSRIs versus placebo: adverse events, Outcome 20 Respiratory disorder.
4.21. Analysis.
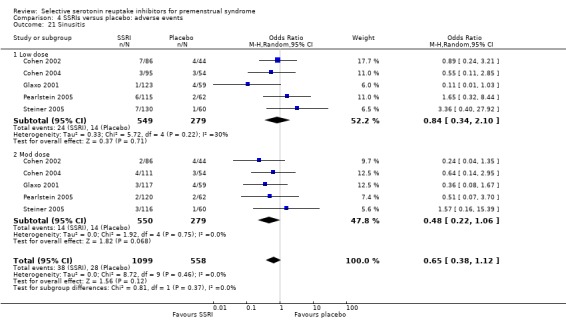
Comparison 4 SSRIs versus placebo: adverse events, Outcome 21 Sinusitis.
4.22. Analysis.
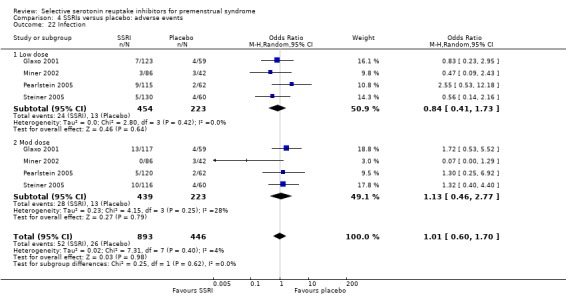
Comparison 4 SSRIs versus placebo: adverse events, Outcome 22 Infection.
Overall, heterogeneity was absent or minimal for analyses of adverse events (I2 = 0% to 4%).
Other side effects mentioned in one or more studies were breast tenderness (Stone 1991), numbness (Eriksson 1995), rash (Pearlstein 1997), trauma (Cohen 2004), vomiting (Pearlstein 2005), genital disorders (Pearlstein 2005), flu syndrome (Cohen 2004; Steiner 2008), back pain (Cohen 2002; Steiner 2008), pharyngitis and rhinitis (Cohen 2002), visual disturbance (Eriksson 1995; Steiner 1995), pain (Cohen 2002; Miner 2002), accidental injury (Cohen 2002) and abdominal pain (Glaxo 1996a). There was no significant difference between the groups for any of these outcomes, and the reported event rates were very low.
Secondary outcomes
3. Specific symptoms of PMS
3.1 Psychological symptoms
Eleven studies reported psychological symptom scores, either as end scores (five studies) or as change scores (four studies).
When psychological symptoms were assessed with end scores, SSRIs reduced symptoms significantly more effectively than placebo. Low dose SSRIs were associated with a small effect size (SMD ‐0.38, 95% CI ‐20.0 to ‐0.57, three studies, 470 women; I2 = 0%; Analysis 5.1) and moderate dose SSRIs with a moderate effect size (SMD ‐0.51, 95% CI ‐0.37 to ‐0.65, five studies, 795 women; I2 = 0%; Analysis 5.2 ). This applied to both luteal and continuous administration. Heterogeneity was absent.
5.1. Analysis.
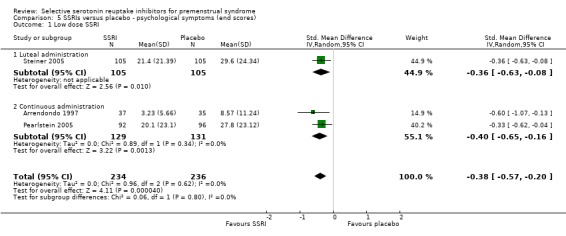
Comparison 5 SSRIs versus placebo ‐ psychological symptoms (end scores), Outcome 1 Low dose SSRI.
5.2. Analysis.
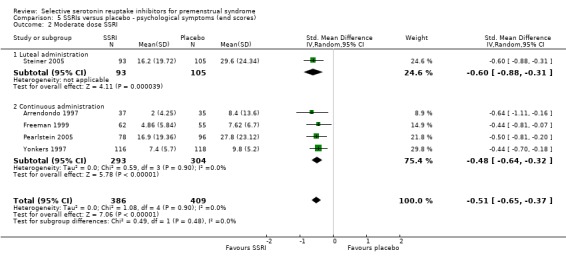
Comparison 5 SSRIs versus placebo ‐ psychological symptoms (end scores), Outcome 2 Moderate dose SSRI.
When psychological symptoms were assessed with change scores, SSRIs reduced symptoms significantly more effectively than placebo, with a small effect size. This applied to both low dose SSRIs (SMD ‐0.26, 95% CI ‐0.11 to ‐0.42, four studies, 683 women; I2 = 5%; Analysis 6.1) and moderate dose SSRIs (SMD ‐0.30, 95% CI ‐0.11 to ‐0.48, four studies, 681 women; I2 = 32%; Analysis 6.2). There were few studies in this analysis, and findings were not significant when studies of luteal administration were considered separately. Overall, statistical heterogeneity was low to moderate.
6.1. Analysis.
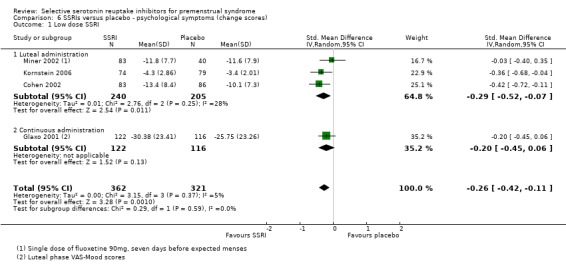
Comparison 6 SSRIs versus placebo ‐ psychological symptoms (change scores), Outcome 1 Low dose SSRI.
6.2. Analysis.
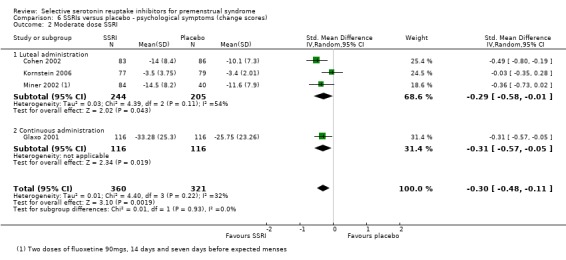
Comparison 6 SSRIs versus placebo ‐ psychological symptoms (change scores), Outcome 2 Moderate dose SSRI.
3.2 Physical symptoms
Nine studies reported physical symptom scores, either as end scores (five studies) or as change scores (four studies).
When physical symptoms were assessed with end scores, there were no data for low dose SSRIs. Moderate dose SSRIs reduced physical symptoms significantly more than placebo, with a small effect size and moderate heterogeneity (SMD ‐0.43, 95% CI ‐0.21 to ‐0.65, five studies, 781 women; I2 = 50%; Analysis 7.1). The heterogeneity was attributable to differences in type of administration. In the single study of luteal administration there was no significant difference between moderate dose SSRIs and placebo for this outcome (OR ‐0.13, 95% CI ‐0.40 to 0.13, one study, 219 women), while in the studies of continuous administration there was a significant benefit for the SSRI group, of moderate effect size (OR ‐0.52, 95% CI ‐0.69 to ‐0.3, four studies, 562 women; I2 = 0%). High dose SSRIs were associated with a moderate effect size (SMD ‐0.56, 95% CI ‐0.26 to ‐0.86, one study, 179 women; Analysis 7.2).
7.1. Analysis.
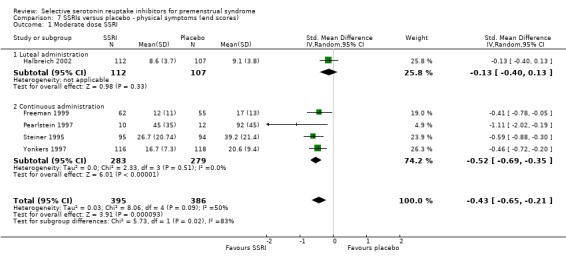
Comparison 7 SSRIs versus placebo ‐ physical symptoms (end scores), Outcome 1 Moderate dose SSRI.
7.2. Analysis.
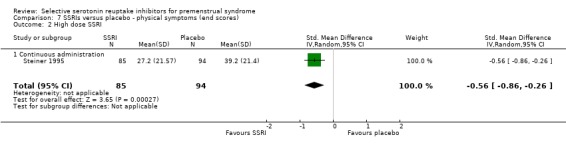
Comparison 7 SSRIs versus placebo ‐ physical symptoms (end scores), Outcome 2 High dose SSRI.
When physical symptoms were assessed with change scores, SSRIs reduced symptoms significantly more effectively than placebo, with a small effect size. This applied to both low dose SSRIs (SMD ‐0.17, 95% CI ‐0.03 to ‐0.31, four studies, 752 women; I2 = 0%; Analysis 8.1) and moderate dose SSRIs (SMD ‐0.27, 95% CI ‐0.10 to ‐0.44, four studies, 742 women; I2 = 27%; Analysis 8.2). When the results were reported by type of administration, findings were no longer statistically significant except for moderate dose luteal administration. Overall, heterogeneity was absent or low.
8.1. Analysis.
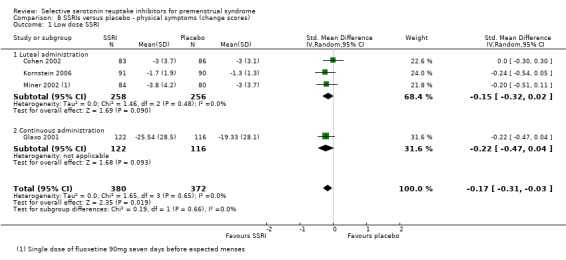
Comparison 8 SSRIs versus placebo ‐ physical symptoms (change scores), Outcome 1 Low dose SSRI.
8.2. Analysis.
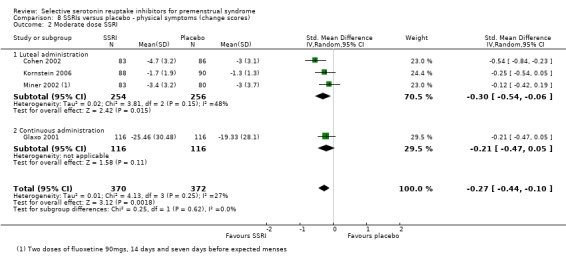
Comparison 8 SSRIs versus placebo ‐ physical symptoms (change scores), Outcome 2 Moderate dose SSRI.
3.3 Functional symptoms
Five studies reported functional symptom scores, reported either as end scores (two studies) or as change scores (three studies).
When functional symptoms were assessed with end scores, no significant difference was found between SSRIs and placebo for low dose SSRIs (SMD ‐0.34, 95% CI 0.05 to ‐0.74, one study, 100 women; Analysis 9.1). Moderate dose SSRIs were associated with significantly reduced functional symptoms, with a moderate effect size (SMD ‐0.71, 95% CI ‐0.49 to ‐0.93, two studies, 334 women; I2 = 0%; Analysis 9.2).
9.1. Analysis.
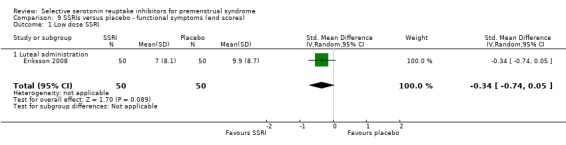
Comparison 9 SSRIs versus placebo ‐ functional symptoms (end scores), Outcome 1 Low dose SSRI.
9.2. Analysis.
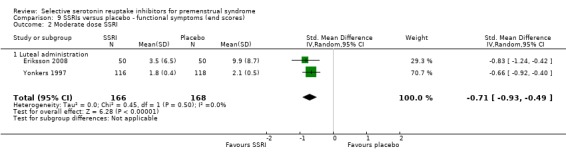
Comparison 9 SSRIs versus placebo ‐ functional symptoms (end scores), Outcome 2 Moderate dose SSRI.
When functional symptoms were assessed with change scores, low dose SSRIs were of significant benefit compared to placebo, with a small effect size (SMD ‐0.22, 95% CI ‐0.05 to ‐0.40, three studies, 514 women; I2 = 0%; Analysis 10.1). There was no significant difference between moderate dose SSRIs and placebo for this outcome (SMD ‐0.13, 95% CI 0.11 to ‐0.37, three studies, 467 women; I2 = 40%; Analysis 10.2). Heterogeneity was absent for the analysis of low dose SSRIs, and moderate for the analysis of moderate dose SSRIs.
10.1. Analysis.
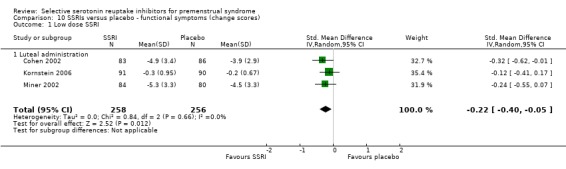
Comparison 10 SSRIs versus placebo ‐ functional symptoms (change scores), Outcome 1 Low dose SSRI.
10.2. Analysis.
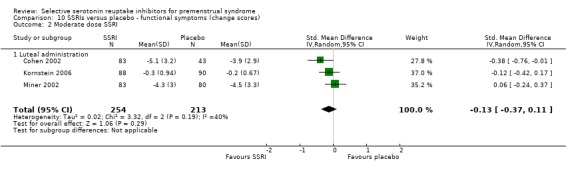
Comparison 10 SSRIs versus placebo ‐ functional symptoms (change scores), Outcome 2 Moderate dose SSRI.
3.4 Irritability
Eight studies reported irritability scores, reported either as end scores (seven studies) or as change scores (one study).
When irritability was assessed with end scores, SSRIs reduced symptoms significantly more than placebo, with a moderate effect size. This applied to both low dose SSRIs (SMD ‐0.57, 95% CI ‐0.02 to ‐0.57, one study, 53 women; Analysis 11.1) and moderate dose SSRIs (SMD ‐0.56, 95% CI ‐0.40 to ‐0.72, five studies, 655 women; I2 = 0%; Analysis 11.2). Heterogeneity was absent.
11.1. Analysis.
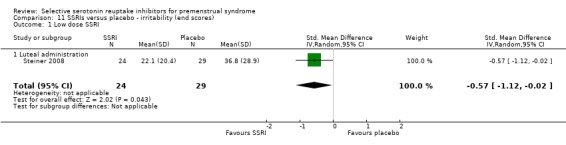
Comparison 11 SSRIs versus placebo ‐ irritability (end scores), Outcome 1 Low dose SSRI.
11.2. Analysis.
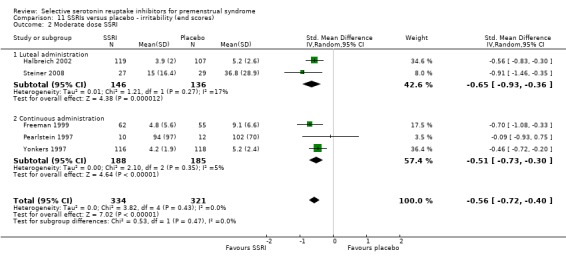
Comparison 11 SSRIs versus placebo ‐ irritability (end scores), Outcome 2 Moderate dose SSRI.
Similarly, when irritability was assessed with change scores, SSRIs reduced symptoms significantly more than placebo. Low dose SSRIs were associated with a small effect size (SMD ‐0.39, 95% CI ‐0.09 to ‐0.70, one study, 169 women; Analysis 12.1) and moderate dose SSRIs were associated with a moderate effect size (SMD ‐0.50, 95% CI ‐0.19 to ‐0.80, one study, 169 women; Analysis 12.2).
12.1. Analysis.
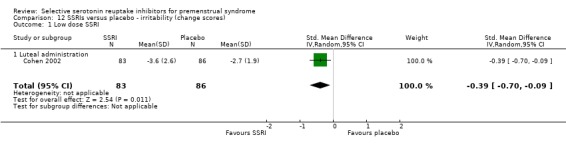
Comparison 12 SSRIs versus placebo ‐ irritability (change scores), Outcome 1 Low dose SSRI.
12.2. Analysis.
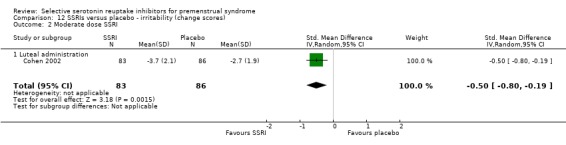
Comparison 12 SSRIs versus placebo ‐ irritability (change scores), Outcome 2 Moderate dose SSRI.
4. Response rates
Nineteen studies reported response rates.
A treatment response was significantly more likely in women taking SSRIs than in those taking placebo. This applied to low dose SSRIs (OR 1.78, 95% CI 1.41 to 2.25, six studies, 1243 women; I2 = 0%; Analysis 13.1), moderate dose SSRIs (OR 2.75, 95% CI 2.20 to 3.44, 19 studies, 2647 women; I2 = 33%; Analysis 13.2) and high dose SSRIs (OR 3.44, 95% CI 1.86 to 6.34, one study, 211 women; Analysis 13.3). Heterogeneity was absent or fairly low. The response rate was relatively low in the single study of semi‐intermittent administration (Wikander 1998) (OR 1.50, 95% CI 0.24 to 9.38, one study, 40 women).
13.1. Analysis.
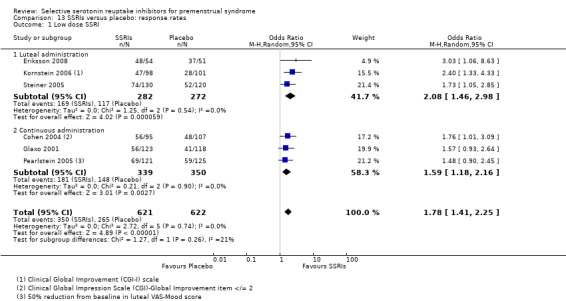
Comparison 13 SSRIs versus placebo: response rates, Outcome 1 Low dose SSRI.
13.2. Analysis.
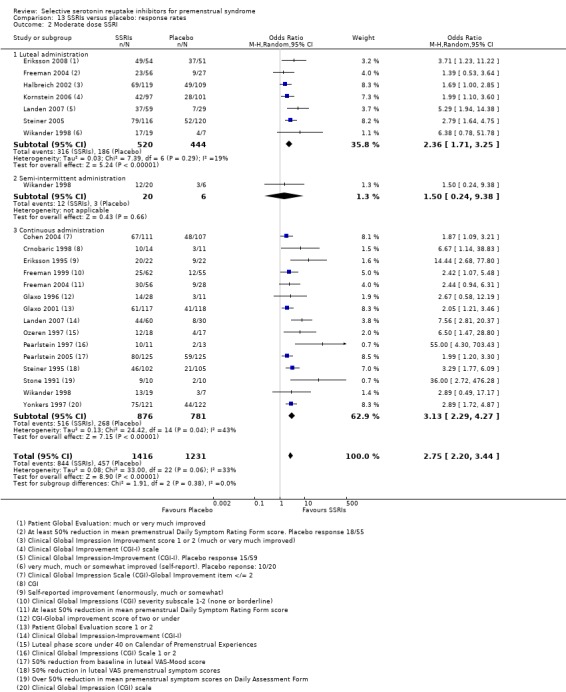
Comparison 13 SSRIs versus placebo: response rates, Outcome 2 Moderate dose SSRI.
13.3. Analysis.
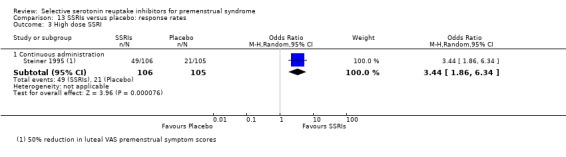
Comparison 13 SSRIs versus placebo: response rates, Outcome 3 High dose SSRI.
5. Study withdrawal for any reason
Twelve studies reported this outcome.
There was no significant difference between the SSRI and placebo groups in the overall study withdrawal rate. This applied both to low dose SSRIs (OR 1.03, 95% CI 0.45 to 2.35, three studies, 385 women; I2 = 53%; Analysis 14.1) and to moderate dose SSRIs (OR 0.97, 95% CI 0.73 to1.28, 12 studies, 1217 women; I2 = 0%; Analysis 14.2). There was high heterogeneity for the analysis of low dose SSRIs, attributable to a high dropout rate in the placebo group in one study.
14.1. Analysis.
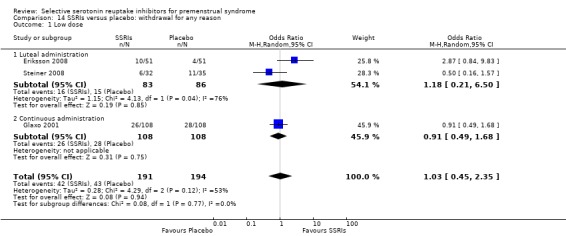
Comparison 14 SSRIs versus placebo: withdrawal for any reason, Outcome 1 Low dose.
14.2. Analysis.
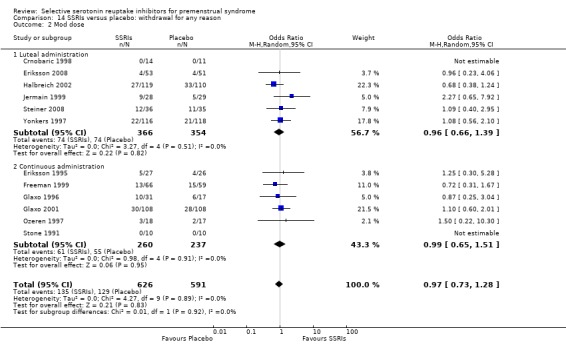
Comparison 14 SSRIs versus placebo: withdrawal for any reason, Outcome 2 Mod dose.
Subgroup analyses
1. Studies with no placebo run‐in
Analysis was restricted to studies without a placebo run‐in (Eriksson 2008; Freeman 1999; Jermain 1999; Ozeren 1997) for the primary outcome with the largest number of included studies (Analysis 1.2). SSRIs were associated with a significant improvement in overall symptoms compared to placebo, with a large effect size (OR ‐0.82, 95% CI ‐0.43 to ‐1.21, four studies, 305 women; I2 = 59%). However, as in the main analysis there was substantial heterogeneity (I2 = 59%).
No studies directly compared a placebo run‐in versus no placebo run‐in.
2. Luteal versus continuous administration
Both luteal and continuous administration of SSRIs were effective in symptom reduction compared to placebo. Three studies (Freeman 2004; Landen 2007; Wikander 1998) compared luteal phase versus continuous administration.
Total symptom scores
There was no significant difference between the two groups in symptom end scores (‐0.04, 95% CI ‐0.39 to 0.31, two studies, 128 women; I2 = 0%) or in response rate (OR 0.82, 95% CI 0.37 to 1.80, three studies, 269 women; I2 = 50%). There was moderate heterogeneity for the analysis of response rates, possibly due to use of differing definitions of response.
Adverse events
Only one study (Landen 2007) compared side effects in women using luteal phase versus continuous SSRIs. There was no significant difference between the two groups in overall adverse events nor in the most commonly reported events (nausea, fatigue, headache, and somnolence), but significantly fewer women taking luteal SSRIs reported decreased libido (OR 0.18, 95% CI 0.04 to 0.84, one study, 118 women; Analysis 15.3).
15.3. Analysis.
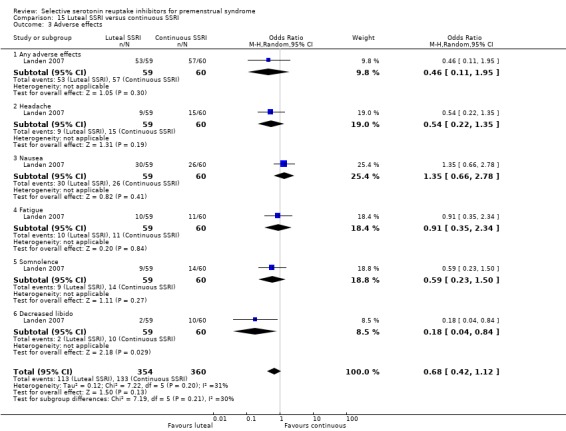
Comparison 15 Luteal SSRI versus continuous SSRI, Outcome 3 Adverse effects.
3. Semi‐intermittent versus luteal or continuous administration
Only one study (Wikander 1998) included a group receiving semi‐intermittent SSRIs. This type of administration was compared with luteal and continuous regimens. Only 39 women were included in each of these comparisons. Total symptom scores were not reported in this study.
Withdrawal due to adverse events
There was no significant difference for this outcome between the semi‐intermittent group and either the luteal group (OR 3.18, 95% CI 0.30 to 33.58, one study, 39 women) or the continuous group (OR 1.50, 95% CI 0.22 to 10.14, one study, 39 women) (Analysis 16.2).
16.2. Analysis.
Comparison 16 Semi‐intermittent versus other regimens, Outcome 2 Withdrawal due to adverse effects.
Response rate
The response rate was significantly lower in the semi‐intermittent group than in the luteal group (OR 0.18, 95% CI 0.03 to 0.98, one study, 39 women) but did not differ significantly between the semi‐intermittent and the continuous group (OR 0.69, 95% CI 0.19 to 2.59, one study, 39 women) (Analysis 16.1).
16.1. Analysis.
Comparison 16 Semi‐intermittent versus other regimens, Outcome 1 Response rate.
Assessment of reporting bias
A funnel plot (Figure 7) was constructed for Analysis 1.2, which was the primary efficacy outcome with the largest number of studies (n = 9). It was not suggestive of publication bias.
7.
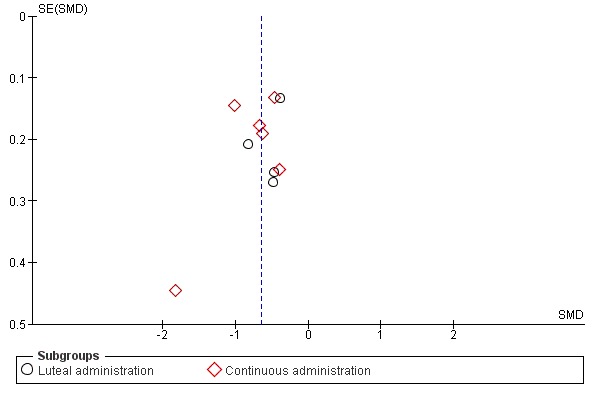
Funnel plot of comparison: 1 SSRIs versus placebo ‐ all symptoms (end scores), outcome: 1.2 Moderate dose SSRI.
Discussion
Summary of main results
Effectiveness of SSRIs
The findings of this review indicate that SSRIs are effective for reducing the symptoms of premenstrual syndrome (PMS). This applies both to overall symptoms and also to specific types of symptom (that is psychological, physical, functional symptoms and irritability). Most of the data related to a moderate dose of SSRIs, taken either in the luteal phase only or continuously. Findings were fairly consistent regardless of the dose and timing of the SSRIs. Effect sizes were small or moderate for most outcomes. All the SSRIs tested appeared effective.
There was consistent evidence that even low doses of SSRIs were effective, although moderate doses generally were associated with a larger effect size and a higher response rate.
Adverse events
A wide range of side effects were reported in women taking SSRIs. When numbers needed to harm (NNH) were calculated (that is the number of women who would need to take a drug in order for one women to be likely to experience an event over and above her underlying risk), the most common side effects associated with a moderate dose of SSRI were nausea (NNH = 7), asthenia (NNH = 9), somnolence (NNH = 13), fatigue (NNH = 14), decreased libido (NNH = 14) and sweating (NNH = 14).
For most of these adverse events there was a clear dose‐response trend, with an increased risk of adverse events in women receiving a higher dose of SSRI. Adverse events were very common in the only study that used a high dose of SSRI.
Effectiveness and adverse events associated with different types of administration
Effectiveness
Only three of the included studies compared different types of SSRI administration, and they included only 269 participants. No significant difference in effectiveness was found between luteal and continuous administration for most outcomes, though there was substantial heterogeneity for the analysis of response rates (I2 = 50%).
It has been suggested that intermittent SSRI administration may be as effective as continuous administration with respect to symptoms such as irritability and mood swings, but may be less effective for somatic symptoms (Eriksson 2008). Our analysis of end scores for physical symptoms (Analysis 7.1) provides some indirect support for this suggestion. However, direct comparisons between administration regimes are required and currently there are insufficient data to draw any conclusions. This is an area that could be further explored.
Adverse effects
Only one study compared the rates of individual adverse effects associated with different types of administration, and the difference between luteal and continuous administration was significant only for decreased libido (favouring luteal administration). It seems likely that the risk of adverse effects will be minimised by use of the lowest effective dose and an intermittent dosing regimen.
Overall findings
Overall, these findings support the use of SSRIs as an effective and reasonably tolerable first line treatment for PMS.
Overall completeness and applicability of evidence
The review included 31 studies and the findings were generally consistent with regard to both efficacy and safety outcomes. There was very little direct evidence about the relative effectiveness and tolerability of luteal versus continuous drug regimens.
There was some indication that heterogeneity for the primary outcome might relate to differences in effectiveness between SSRI types. However, there were too few studies of specific SSRIs to determine whether any particular SSRI appeared substantially more effective than any other, and subgroup analyses require very cautious interpretation. Direct head‐to‐head comparisons of different SSRIs would help determine their relative effectiveness and we propose that future updates of this review include such comparisons.
Several of the trials included in this analysis had a single‐blind placebo run‐in stage to exclude placebo responders, and exclusion of placebo responders may yield estimates of effect that differ from findings in an unselected population. A subgroup analysis of studies without placebo run‐in protocols was carried out, which found a large effect size in the SSRI group, suggesting that the benefit of SSRIs may be stronger in an unselected population than in placebo responders. However subgroup findings require very cautious interpretation and, moreover, there was substantial heterogeneity for this analysis (I2 = 59%).
Studies have reported a high risk of relapse following cessation of treatment (Freeman 2009; Pearlstein 1994; Sundblad 1997). The risk appears to be highest in women with severe symptoms at baseline and in those who do not achieve remission, and it has been suggested that these factors be considered when determining the optimum duration of treatment (Freeman 2009).
Quality of the evidence
Overall, the study quality was poor, with only four studies reporting acceptable methods of randomisation and allocation concealment and nine studies at high risk of either attrition bias or selective reporting bias. For most studies the risk of bias was unclear, largely due to poor reporting of methods. The overall quality of the evidence for the main findings was graded as low to moderate using the GRADE criteria, due to the suboptimal quality of most of the included studies and moderate heterogeneity (I2 = 59%) in one of the primary analyses (Analysis 1.2). See Table 1; Table 2; Table 3.
Potential biases in the review process
Efforts were made to retrieve all eligible studies. However, it is likely that some unpublished studies were not retrieved. This is mainly because efforts to contact pharmaceutical companies, in 2013, were unsuccessful in most cases; automated responses were received but no information was sent. GlaxoSmithKline have recently undertaken to make unpublished data available (Glaxo 2012), and hence we were able to add to this review three unpublished studies on paroxetine dating from 1996. This raises the possibility that there may be other unpublished data that we are unable to access.
The choice of end scores (where reported) rather than change scores had the potential to influence effect estimates for the primary outcome. We considered including an additional analysis pooling all studies that reported change scores for the primary outcome, but we found that this allowed only two additional studies to be included in the analysis of change scores for moderate dose SSRIs. Other studies did not report effect estimates in a form which permitted the standard deviations of the change score to be extracted or calculated.
Agreements and disagreements with other studies or reviews
A systematic review of behavioural and pharmacological interventions for PMS (Kleinstauber 2012) included 14 RCTs of SSRIs versus no treatment, wait list or placebo, and found that SSRIs were associated with a small to moderate benefit for PMS symptoms. Similarly, an earlier systematic review (Halbreich 2008), which included 14 placebo‐controlled RCTs of SSRIs for PMS, concluded that even though SSRIs are effective for many women with PMDD, alternative targeted treatments need to be developed since around 40% of women fail to respond to SSRIs.
A systematic review of 19 RCTs (Shah 2008) also found SSRIs effective for treating PMS and PMDD compared to placebo (OR 0.40, 95% 0.31 to 0.51), with substantial heterogeneity (I2 = 66%). In subgroup analysis, luteal dosing was found to be less effective than continuous administration (OR 0.55, 95% CI 0.45 to 0.68; OR 0.28, 95% CI 0.18 to 0.42, respectively).
The current review found no convincing evidence of a significant difference in effectiveness between luteal and continuous dosing (Analysis 15.1; Analysis 15.2). The difference in findings between the current review and Shah 2008 may possibly be due to differences in data management. In Shah 2008, with the exception of Veeninga 1990 (which was excluded from the current review), the three studies with the lowest effect estimates were studies of luteal phase SSRIs in which change scores had been converted to end scores by the review authors. In the current review, a) studies reporting end scores tended to report higher effect estimates than those reporting change scores, b) all studies of continuous administration reported end scores, and c) studies reporting end scores were not pooled with those reporting change scores.
15.1. Analysis.
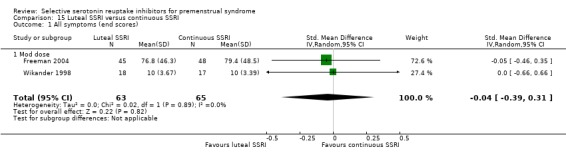
Comparison 15 Luteal SSRI versus continuous SSRI, Outcome 1 All symptoms (end scores).
15.2. Analysis.
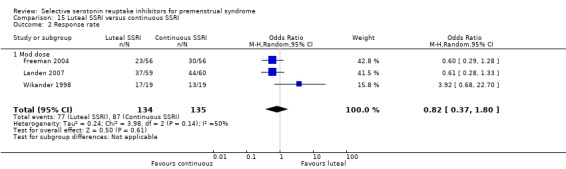
Comparison 15 Luteal SSRI versus continuous SSRI, Outcome 2 Response rate.
A small (n = 36) open‐label RCT (Wu 2008) comparing intermittent versus continuous paroxetine found that luteal and continuous administration were both effective for treating symptoms of PMDD. Effects were maintained over six months of follow‐up and did not differ significantly between the groups. This study was not included in the current review because there was no placebo group.
As the authors of Shah 2008 suggest, more head‐to‐head trials of luteal versus continuous dosing strategies are needed to provide conclusive evidence on this point. Such studies should report both effectiveness and adverse effects.
A subgroup analysis by type of SSRI in Shah 2008 found no significant difference in effectiveness between the most commonly used SSRIs (citalopram, fluoxetine, paroxetine and sertraline).
Authors' conclusions
Implications for practice.
There is now very convincing evidence to support the use of SSRIs for reducing the symptoms of PMS. All doses appear effective, but there is a dose‐response effect that applies both to symptom reduction and to adverse events. High doses of SSRIs may be intolerable. This review suggests that both intermittent and continuous dosing regimens are effective in reducing symptoms.
In most instances the choice of SSRI dose and type of administration could be based on an individual woman's preference and modified according to the effectiveness and tolerability of the chosen regimen.
Implications for research.
Future research should focus on direct comparisons between different SSRI administration regimens, including their safety, tolerability and effectiveness for overall symptoms and for specific symptom types over long‐term follow‐up. Head‐to‐head comparisons of the effectiveness and tolerability of different SSRIs may also be useful, and such comparisons should be included in future updates of this review. A multiple treatment meta‐analysis (network meta analysis or mixed treatment comparisons) should also be considered for this topic. Primary studies should report full statistical information with standard deviations for all continuous outcomes to facilitate their inclusion in meta‐analysis.
What's new
| Date | Event | Description |
|---|---|---|
| 23 May 2013 | New citation required but conclusions have not changed | The additional evidence did not change the conclusions of this review. |
| 20 February 2013 | New search has been performed | Added six new studies: Eriksson 2008, Freeman 2010, Glaxo 1996, Glaxo 1996a, Glaxo 2001, Steiner 2008. Some studies were split into multiple comparisons in earlier versions of the review, each with a separate study reference; such studies have been combined and referenced as a single study in this update. Excluded three previously included studies: Sundblad 1992, Sundblad 1993, Veeninga 1990. |
History
Protocol first published: Issue 1, 1999 Review first published: Issue 4, 2002
| Date | Event | Description |
|---|---|---|
| 11 February 2009 | New citation required but conclusions have not changed | Review updated May 2008 |
| 4 May 2008 | New search has been performed | New studies identified and major update completed |
| 31 March 2008 | Amended | Converted to new review format. |
| 28 February 2002 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
This review first appeared in the Lancet (Lancet 2000;356:1131‐6). The Lancet version includes more RCTs, partly because the Cochrane statistical reviewers excluded five RCTs of crossover design from the meta‐analysis of this review. The authors would like to acknowledge the contribution of K Wyatt and P Dimmock who were involved in the original review.
Appendices
Appendix 1. MEDLINE search strategy
1 Premenstrual Syndrome/
2 premenstrua$.tw.
3 pre‐menstrua$.tw.
4 late luteal.tw.
5 luteal phase.tw.
6 (luteal adj5 symptom$).tw.
7 (PMS or PMD or PMDD or LLPDD).tw.
8 or/1‐7
9 exp serotonin uptake inhibitors/ or amoxapine/ or citalopram/ or clomipramine/ or fenfluramine/ or fluoxetine/ or fluvoxamine/ or norfenfluramine/ or paroxetine/ or sertraline/ or trazodone/ or zimeldine/
10 (serotonin adj5 inhibitor$).tw.
11 (amoxapine or citalopram or clomipramine or fenfluramine or fluoxetine or fluvoxamine or norfenfluramine or paroxetine or sertraline or trazodone or zimeldine).tw.
12 SSRI.tw.
13 (5‐hydroxytryptamine adj5 inhibitor$).tw.
14 (5‐ht adj5 inhibitor$).tw.
15 or/9‐14
16 8 and 15
17 randomised controlled trial.pt.
18 controlled clinical trial.pt.
19 Randomized Controlled Trials/
20 Random allocation/
21 Double‐blind method/
22 Single‐blind method/
23 or/17‐22
24 clinical trial.pt.
25 exp clinical trials/
26 (clin$ adj25 trial$).ti,ab,sh.
27 ((singl$ or doubl$ or tripl$ or trebl$) adj25 (blind$ or mask$)).ti,ab,sh.
28 Placebos/
29 placebo$.ti,ab,sh.
30 random$.ti,ab,sh.
31 Research design/
32 or/24‐31
33 animal/ not (human/ and animal/)
34 23 or 32
35 34 not 33
36 16 and 35
37 (2002$ or 2003$ or 2004$ or 2005$ or 2006$ or 2007$ or 2008$).ed.
38 36 and 37
Appendix 2. EMBASE search strategy
1 premenstrua$.tw. 2 pre‐menstrua$.tw. 3 late luteal.tw. 4 luteal phase.tw. 5 (luteal adj5 symptom$).tw. 6 (PMS or PMD or PMDD or LLPDD).tw. 7 premenstrual dysphoric disorder/ or premenstrual syndrome/ 8 exp Serotonin Uptake Inhibitor/ 9 (serotonin adj5 inhibitor$).tw. 10 (amoxapine or citalopram or clomipramine or fenfluramine or fluoxetine or fluvoxamine or norfenfluramine or paroxetine or sertraline or trazodone or zimeldine).tw. 11 SSRI.tw. 12 (5‐hydroxytryptamine adj5 inhibitor$).tw. 13 (5‐ht adj5 inhibitor$).tw. 14 or/1‐7 15 or/8‐13 16 14 and 15 17 Controlled study/ or randomized controlled trial/ 18 double blind procedure/ 19 single blind procedure/ 20 crossover procedure/ 21 drug comparison/ 22 placebo/ 23 random$.ti,ab,hw,tn,mf. 24 latin square.ti,ab,hw,tn,mf. 25 crossover.ti,ab,hw,tn,mf. 26 cross‐over.ti,ab,hw,tn,mf. 27 placebo$.ti,ab,hw,tn,mf. 28 ((doubl$ or singl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).ti,ab,hw,tn,mf. 29 (comparative adj5 trial$).ti,ab,hw,tn,mf. 30 (clinical adj5 trial$).ti,ab,hw,tn,mf. 31 or/17‐30 32 nonhuman/ 33 animal/ not (human/ and animal/) 34 or/32‐33 35 31 not 34 36 16 and 35 37 (2012$ or 2013$).em. 38 36 and 37
Appendix 3. PsycINFO search strategy
atabase: PsycINFO <1806 to February Week 1 2013> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 exp Premenstrual Syndrome/ 2 premenstrua$.tw. 3 pre‐menstrua$.tw. 4 late luteal.tw. 5 luteal phase.tw. 6 (luteal adj5 symptom$).tw. 7 (PMS or PMD or PMDD or LLPDD).tw. 8 (Dysphoric adj2 disorder$).tw. 9 exp Premenstrual Dysphoric Disorder/ 10 or/1‐9 11 exp Serotonin Reuptake Inhibitors/ or exp Antidepressant Drugs/ 12 exp Citalopram/ 13 exp Chlorimipramine/ 14 exp Fenfluramine/ 15 exp Fluoxetine/ 16 exp Fluvoxamine/ 17 exp Paroxetine/ 18 exp Sertraline/ 19 exp Trazodone/ 20 exp Zimeldine/ 21 (serotonin adj5 inhibitor$).tw. 22 (amoxapine or citalopram or clomipramine or fenfluramine or fluoxetine or fluvoxamine or norfenfluramine or paroxetine or sertraline or trazodone or zimeldine).tw. 23 SSRI$.tw. 24 (5‐hydroxytryptamine adj5 inhibitor$).tw. 25 (5‐ht adj5 inhibitor$).tw. 26 or/11‐25 27 10 and 26 28 random.tw. 29 control.tw. 30 double‐blind.tw. 31 clinical trials/ 32 placebo/ 33 exp Treatment/ 34 or/28‐33 35 27 and 34
Data and analyses
Comparison 1. SSRIs versus placebo ‐ all symptoms (end scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 2 | 301 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.05, ‐0.29] |
| 1.1 Luteal administration | 1 | 100 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.45 [‐0.85, ‐0.05] |
| 1.2 Continuous administration | 1 | 201 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.84 [‐1.13, ‐0.55] |
| 2 Moderate dose SSRI | 9 | 1276 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐0.84, ‐0.46] |
| 2.1 Luteal administration | 4 | 457 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐0.71, ‐0.31] |
| 2.2 Continuous administration | 6 | 819 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.73 [‐1.01, ‐0.46] |
| 3 High dose SSRI | 1 | 138 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.95 [‐1.31, ‐0.58] |
| 3.1 Continuous administration | 1 | 138 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.95 [‐1.31, ‐0.58] |
Comparison 2. SSRIs versus placebo ‐ all symptoms (change scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 4 | 677 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.41, ‐0.05] |
| 1.1 Luteal administration | 4 | 677 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.41, ‐0.05] |
| 2 Moderate dose SSRI | 4 | 657 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.51, ‐0.20] |
| 2.1 Luteal administration | 4 | 657 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.51, ‐0.20] |
Comparison 3. SSRIs versus placebo: withdrawal due to adverse effects.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose | 7 | 1301 | Odds Ratio (M‐H, Random, 95% CI) | 1.76 [1.13, 2.75] |
| 1.1 Luteal administration | 4 | 600 | Odds Ratio (M‐H, Random, 95% CI) | 2.21 [1.05, 4.65] |
| 1.2 Continuous administration | 3 | 701 | Odds Ratio (M‐H, Random, 95% CI) | 1.56 [0.90, 2.71] |
| 2 Mod dose | 15 | 2447 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.55 [1.84, 3.53] |
| 2.1 Luteal administration | 8 | 1171 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.23 [1.82, 5.73] |
| 2.2 Continuous administration | 9 | 1276 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.24 [1.50, 3.34] |
| 3 High dose | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Continuous administration | 1 | 231 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.35 [2.88, 14.00] |
3.1. Analysis.
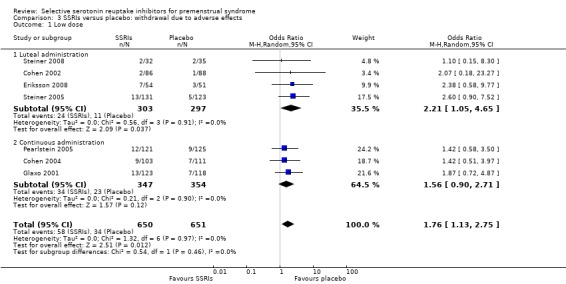
Comparison 3 SSRIs versus placebo: withdrawal due to adverse effects, Outcome 1 Low dose.
Comparison 4. SSRIs versus placebo: adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Nausea | 16 | 3385 | Odds Ratio (M‐H, Random, 95% CI) | 3.43 [2.63, 4.47] |
| 1.1 Low dose | 8 | 1102 | Odds Ratio (M‐H, Random, 95% CI) | 2.24 [1.38, 3.63] |
| 1.2 Mod dose | 16 | 2124 | Odds Ratio (M‐H, Random, 95% CI) | 4.14 [2.98, 5.75] |
| 1.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 3.78 [1.24, 11.51] |
| 2 Insomnia or sleep disturbance | 16 | 3388 | Odds Ratio (M‐H, Random, 95% CI) | 1.84 [1.36, 2.47] |
| 2.1 Low dose | 10 | 1252 | Odds Ratio (M‐H, Random, 95% CI) | 2.51 [1.39, 4.54] |
| 2.2 Mod dose | 15 | 1977 | Odds Ratio (M‐H, Random, 95% CI) | 1.53 [1.03, 2.26] |
| 2.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 4.40 [1.45, 13.30] |
| 3 Sexual dysfunction or decreased libido | 14 | 2847 | Odds Ratio (M‐H, Random, 95% CI) | 2.26 [1.54, 3.31] |
| 3.1 Low dose | 8 | 1082 | Odds Ratio (M‐H, Random, 95% CI) | 1.98 [1.02, 3.85] |
| 3.2 Mod dose | 14 | 1765 | Odds Ratio (M‐H, Random, 95% CI) | 2.46 [1.52, 3.99] |
| 4 Fatigue or sedation | 8 | 951 | Odds Ratio (M‐H, Random, 95% CI) | 1.66 [1.09, 2.53] |
| 4.1 Low dose | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [0.52, 4.56] |
| 4.2 Mod dose | 8 | 666 | Odds Ratio (M‐H, Random, 95% CI) | 1.49 [0.91, 2.43] |
| 4.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 3.88 [1.10, 13.70] |
| 5 Dizziness or vertigo | 11 | 2354 | Odds Ratio (M‐H, Random, 95% CI) | 1.96 [1.32, 2.89] |
| 5.1 Low dose | 6 | 824 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [0.75, 2.85] |
| 5.2 Mod dose | 11 | 1371 | Odds Ratio (M‐H, Random, 95% CI) | 2.11 [1.29, 3.45] |
| 5.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 8.57 [1.10, 66.76] |
| 6 Tremor | 4 | 1352 | Odds Ratio (M‐H, Random, 95% CI) | 5.38 [2.20, 13.16] |
| 6.1 Low dose | 3 | 521 | Odds Ratio (M‐H, Random, 95% CI) | 3.38 [0.59, 19.36] |
| 6.2 Mod dose | 4 | 672 | Odds Ratio (M‐H, Random, 95% CI) | 4.94 [1.47, 16.61] |
| 6.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 12.85 [1.68, 98.36] |
| 7 Somnolence/decreased concentration | 7 | 1797 | Odds Ratio (M‐H, Random, 95% CI) | 4.94 [2.82, 8.63] |
| 7.1 Low dose | 4 | 587 | Odds Ratio (M‐H, Random, 95% CI) | 4.41 [1.62, 12.03] |
| 7.2 Mod dose | 7 | 1051 | Odds Ratio (M‐H, Random, 95% CI) | 4.89 [2.40, 9.96] |
| 7.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 8.57 [1.10, 66.76] |
| 8 Sweating | 9 | 2051 | Odds Ratio (M‐H, Random, 95% CI) | 3.02 [1.79, 5.11] |
| 8.1 Low dose | 4 | 628 | Odds Ratio (M‐H, Random, 95% CI) | 3.08 [0.89, 10.73] |
| 8.2 Mod dose | 9 | 1264 | Odds Ratio (M‐H, Random, 95% CI) | 2.97 [1.59, 5.55] |
| 8.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 3.26 [0.70, 15.11] |
| 9 Dry mouth | 9 | 1474 | Odds Ratio (M‐H, Random, 95% CI) | 2.70 [1.65, 4.41] |
| 9.1 Low dose | 3 | 275 | Odds Ratio (M‐H, Random, 95% CI) | 3.08 [0.84, 11.23] |
| 9.2 Mod dose | 9 | 1040 | Odds Ratio (M‐H, Random, 95% CI) | 2.60 [1.47, 4.58] |
| 9.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 2.95 [0.63, 13.84] |
| 10 Yawning | 5 | 975 | Odds Ratio (M‐H, Random, 95% CI) | 4.24 [1.63, 10.99] |
| 10.1 Low dose | 3 | 303 | Odds Ratio (M‐H, Random, 95% CI) | 3.00 [0.52, 17.41] |
| 10.2 Mod dose | 5 | 513 | Odds Ratio (M‐H, Random, 95% CI) | 4.60 [1.34, 15.74] |
| 10.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 6.92 [0.38, 125.21] |
| 11 Asthenia/decreased energy | 7 | 1704 | Odds Ratio (M‐H, Random, 95% CI) | 3.28 [2.16, 4.98] |
| 11.1 Low dose | 6 | 841 | Odds Ratio (M‐H, Random, 95% CI) | 3.18 [1.73, 5.86] |
| 11.2 Mod dose | 7 | 863 | Odds Ratio (M‐H, Random, 95% CI) | 3.36 [1.89, 5.97] |
| 12 Diarrhoea | 10 | 2402 | Odds Ratio (M‐H, Random, 95% CI) | 2.21 [1.35, 3.62] |
| 12.1 Low dose | 7 | 1003 | Odds Ratio (M‐H, Random, 95% CI) | 2.24 [0.97, 5.21] |
| 12.2 Mod dose | 10 | 1399 | Odds Ratio (M‐H, Random, 95% CI) | 2.20 [1.20, 4.03] |
| 13 Constipation | 6 | 996 | Odds Ratio (M‐H, Random, 95% CI) | 2.35 [1.04, 5.29] |
| 13.1 Low dose | 3 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 2.18 [0.60, 7.95] |
| 13.2 Mod dose | 6 | 590 | Odds Ratio (M‐H, Random, 95% CI) | 2.47 [0.87, 6.98] |
| 14 Gastrointestinal irritability or dyspepsia | 5 | 803 | Odds Ratio (M‐H, Random, 95% CI) | 2.03 [0.78, 5.31] |
| 14.1 Low dose | 2 | 279 | Odds Ratio (M‐H, Random, 95% CI) | 1.69 [0.31, 9.26] |
| 14.2 Mod dose | 5 | 524 | Odds Ratio (M‐H, Random, 95% CI) | 2.21 [0.67, 7.29] |
| 15 Headache | 15 | 2866 | Odds Ratio (M‐H, Random, 95% CI) | 1.13 [0.89, 1.42] |
| 15.1 Low dose | 8 | 1102 | Odds Ratio (M‐H, Random, 95% CI) | 1.18 [0.80, 1.73] |
| 15.2 Mod dose | 15 | 1764 | Odds Ratio (M‐H, Random, 95% CI) | 1.10 [0.83, 1.46] |
| 16 Decreased appetite | 3 | 433 | Odds Ratio (M‐H, Random, 95% CI) | 1.77 [0.79, 3.98] |
| 16.1 Low dose | 1 | 47 | Odds Ratio (M‐H, Random, 95% CI) | 2.22 [0.23, 21.73] |
| 16.2 Mod dose | 3 | 227 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.36, 3.74] |
| 16.3 High dose | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 2.75 [0.76, 9.95] |
| 17 Increased appetite | 3 | 495 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.34, 5.46] |
| 17.1 Low dose | 1 | 182 | Odds Ratio (M‐H, Random, 95% CI) | 2.45 [0.12, 51.81] |
| 17.2 Mod dose | 3 | 313 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.17, 6.19] |
| 18 Anxiety | 3 | 397 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.45, 3.02] |
| 18.1 Low dose | 1 | 130 | Odds Ratio (M‐H, Random, 95% CI) | 2.65 [0.30, 23.45] |
| 18.2 Mod dose | 3 | 267 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.34, 2.77] |
| 19 Cardiovascular symptoms | 3 | 380 | Odds Ratio (M‐H, Random, 95% CI) | 3.94 [0.89, 17.39] |
| 19.1 Mod dose | 3 | 225 | Odds Ratio (M‐H, Random, 95% CI) | 3.67 [0.66, 20.44] |
| 19.2 High dose | 1 | 155 | Odds Ratio (M‐H, Random, 95% CI) | 4.89 [0.26, 92.53] |
| 20 Respiratory disorder | 4 | 1334 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.46, 1.06] |
| 20.1 Low dose | 4 | 664 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.46, 1.46] |
| 20.2 Mod dose | 4 | 670 | Odds Ratio (M‐H, Random, 95% CI) | 0.59 [0.29, 1.19] |
| 21 Sinusitis | 5 | 1657 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.38, 1.12] |
| 21.1 Low dose | 5 | 828 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.34, 2.10] |
| 21.2 Mod dose | 5 | 829 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.22, 1.06] |
| 22 Infection | 4 | 1339 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.60, 1.70] |
| 22.1 Low dose | 4 | 677 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.41, 1.73] |
| 22.2 Mod dose | 4 | 662 | Odds Ratio (M‐H, Random, 95% CI) | 1.13 [0.46, 2.77] |
Comparison 5. SSRIs versus placebo ‐ psychological symptoms (end scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 3 | 470 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.38 [‐0.57, ‐0.20] |
| 1.1 Luteal administration | 1 | 210 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.63, ‐0.08] |
| 1.2 Continuous administration | 2 | 260 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.65, ‐0.16] |
| 2 Moderate dose SSRI | 5 | 795 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐0.65, ‐0.37] |
| 2.1 Luteal administration | 1 | 198 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐0.88, ‐0.31] |
| 2.2 Continuous administration | 4 | 597 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.48 [‐0.64, ‐0.32] |
Comparison 6. SSRIs versus placebo ‐ psychological symptoms (change scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 4 | 683 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.42, ‐0.11] |
| 1.1 Luteal administration | 3 | 445 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.52, ‐0.07] |
| 1.2 Continuous administration | 1 | 238 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.45, 0.06] |
| 2 Moderate dose SSRI | 4 | 681 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.48, ‐0.11] |
| 2.1 Luteal administration | 3 | 449 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.58, ‐0.01] |
| 2.2 Continuous administration | 1 | 232 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.57, ‐0.05] |
Comparison 7. SSRIs versus placebo ‐ physical symptoms (end scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Moderate dose SSRI | 5 | 781 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐0.65, ‐0.21] |
| 1.1 Luteal administration | 1 | 219 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.40, 0.13] |
| 1.2 Continuous administration | 4 | 562 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.52 [‐0.69, ‐0.35] |
| 2 High dose SSRI | 1 | 179 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.56 [‐0.86, ‐0.26] |
| 2.1 Continuous administration | 1 | 179 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.56 [‐0.86, ‐0.26] |
Comparison 8. SSRIs versus placebo ‐ physical symptoms (change scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 4 | 752 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.31, ‐0.03] |
| 1.1 Luteal administration | 3 | 514 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.32, 0.02] |
| 1.2 Continuous administration | 1 | 238 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.47, 0.04] |
| 2 Moderate dose SSRI | 4 | 742 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.44, ‐0.10] |
| 2.1 Luteal administration | 3 | 510 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.54, ‐0.06] |
| 2.2 Continuous administration | 1 | 232 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.47, 0.05] |
Comparison 9. SSRIs versus placebo ‐ functional symptoms (end scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 1 | 100 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.34 [‐0.74, 0.05] |
| 1.1 Luteal administration | 1 | 100 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.34 [‐0.74, 0.05] |
| 2 Moderate dose SSRI | 2 | 334 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.71 [‐0.93, ‐0.49] |
| 2.1 Luteal administration | 2 | 334 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.71 [‐0.93, ‐0.49] |
Comparison 10. SSRIs versus placebo ‐ functional symptoms (change scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 3 | 514 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.40, ‐0.05] |
| 1.1 Luteal administration | 3 | 514 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.40, ‐0.05] |
| 2 Moderate dose SSRI | 3 | 467 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.37, 0.11] |
| 2.1 Luteal administration | 3 | 467 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.37, 0.11] |
Comparison 11. SSRIs versus placebo ‐ irritability (end scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 1 | 53 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.57 [‐1.12, ‐0.02] |
| 1.1 Luteal administration | 1 | 53 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.57 [‐1.12, ‐0.02] |
| 2 Moderate dose SSRI | 5 | 655 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.56 [‐0.72, ‐0.40] |
| 2.1 Luteal administration | 2 | 282 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐0.93, ‐0.36] |
| 2.2 Continuous administration | 3 | 373 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐0.73, ‐0.30] |
Comparison 12. SSRIs versus placebo ‐ irritability (change scores).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 1 | 169 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐0.70, ‐0.09] |
| 1.1 Luteal administration | 1 | 169 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐0.70, ‐0.09] |
| 2 Moderate dose SSRI | 1 | 169 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐0.80, ‐0.19] |
| 2.1 Luteal administration | 1 | 169 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐0.80, ‐0.19] |
Comparison 13. SSRIs versus placebo: response rates.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose SSRI | 6 | 1243 | Odds Ratio (M‐H, Random, 95% CI) | 1.78 [1.41, 2.25] |
| 1.1 Luteal administration | 3 | 554 | Odds Ratio (M‐H, Random, 95% CI) | 2.08 [1.46, 2.98] |
| 1.2 Continuous administration | 3 | 689 | Odds Ratio (M‐H, Random, 95% CI) | 1.59 [1.18, 2.16] |
| 2 Moderate dose SSRI | 19 | 2647 | Odds Ratio (M‐H, Random, 95% CI) | 2.75 [2.20, 3.44] |
| 2.1 Luteal administration | 7 | 964 | Odds Ratio (M‐H, Random, 95% CI) | 2.36 [1.71, 3.25] |
| 2.2 Semi‐intermittent administration | 1 | 26 | Odds Ratio (M‐H, Random, 95% CI) | 1.5 [0.24, 9.38] |
| 2.3 Continuous administration | 15 | 1657 | Odds Ratio (M‐H, Random, 95% CI) | 3.13 [2.29, 4.27] |
| 3 High dose SSRI | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Continuous administration | 1 | 211 | Odds Ratio (M‐H, Random, 95% CI) | 3.44 [1.86, 6.34] |
Comparison 14. SSRIs versus placebo: withdrawal for any reason.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Low dose | 3 | 385 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.45, 2.35] |
| 1.1 Luteal administration | 2 | 169 | Odds Ratio (M‐H, Random, 95% CI) | 1.18 [0.21, 6.50] |
| 1.2 Continuous administration | 1 | 216 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.49, 1.68] |
| 2 Mod dose | 12 | 1217 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.73, 1.28] |
| 2.1 Luteal administration | 6 | 720 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.66, 1.39] |
| 2.2 Continuous administration | 6 | 497 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.65, 1.51] |
Comparison 15. Luteal SSRI versus continuous SSRI.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All symptoms (end scores) | 2 | 128 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.39, 0.31] |
| 1.1 Mod dose | 2 | 128 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.39, 0.31] |
| 2 Response rate | 3 | 269 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.37, 1.80] |
| 2.1 Mod dose | 3 | 269 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.37, 1.80] |
| 3 Adverse effects | 1 | 714 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.42, 1.12] |
| 3.1 Any adverse effects | 1 | 119 | Odds Ratio (M‐H, Random, 95% CI) | 0.46 [0.11, 1.95] |
| 3.2 Headache | 1 | 119 | Odds Ratio (M‐H, Random, 95% CI) | 0.54 [0.22, 1.35] |
| 3.3 Nausea | 1 | 119 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.66, 2.78] |
| 3.4 Fatigue | 1 | 119 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.35, 2.34] |
| 3.5 Somnolence | 1 | 119 | Odds Ratio (M‐H, Random, 95% CI) | 0.59 [0.23, 1.50] |
| 3.6 Decreased libido | 1 | 119 | Odds Ratio (M‐H, Random, 95% CI) | 0.18 [0.04, 0.84] |
Comparison 16. Semi‐intermittent versus other regimens.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Semi‐intermittent versus luteal | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Semi‐intermittent versus continuous | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Withdrawal due to adverse effects | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Semi‐intermittent versus continuous | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Semi‐intermittent versus luteal | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Arrendondo 1997.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel two‐arm trial | |
| Participants | Country: USA Site: No details Recruitment: 72 women no other details Refer to table of bias for details of attrition Inclusion: Meeting defined criteria for PMS using Penn Daily Symptom Report (DSR) for two cycles. Regular menstrual cycles (22‐35 days), general good health Exclusion: No current major psychiatric diagnoses as assessed by Structured Clinical Interview (SCID) No explicit exclusion criteria noted |
|
| Interventions | Screening: Screening for two cycles Placebo run‐in: None Interventions: Placebo (n=35) versus sertraline 50mg administered orally (n=37) Duration: Treatment administered for three cycles Timing of administration: No details of when in the cycle this was first administered Summary measures: Data presented as individual treatment cycles. Review has used mean data across all three cycles |
|
| Outcomes | Penn Daily Self Rating Symptoms (DSR). Depression and food craving scores. Depression score reported in current review | |
| Notes | Only available as abstract Daily recording of symptoms Measured depression and food cravings No details of ITT or power calculation No details of funding source |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States randomised. Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | States double blind. No further details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 72 women randomised, number analysed not reported |
| Selective reporting (reporting bias) | Unclear risk | Adverse effects not reported |
| Other bias | Unclear risk | Few details available as not published in full text |
Cohen 2002.
| Methods | Randomised, double‐blind, placebo‐controlled, 3‐arm parallel study Study flow: 1276 women screened for study, 260 randomised: 86 to fluoxetine 10mg (77 completed), 86 to fluoxetine 20mg (66 completed); 88 to placebo (75 completed) |
|
| Participants | Country:USA Site: Multi‐centre (19 investigators at 20 sites) Mean age placebo group 35.6±4.9 years, fluoxetine 10mg group 37.2±5.1 years, fluoxetine 20mg group 35.1±5.3 years Inclusion: PMDD: 18 to 45 years, regular menstrual cycles (23 to 35 days). Two consecutive cycles in which scores averaged 3.0 or more each for five of the eleven DRSP items during the luteal phase, and their scores averaged 2.5 or less for each of the same items during the follicular week; and if they showed 50% or more increase from follicular to luteal scores for these items; and if at least twice during the defined luteal period, they had scores of 4 or more on any of the three functional items Exclusion: Having Axis 1 disorder other than PMDD, History of Axis 1 pathology occurring within the past six months (exception of specific phobias), women using hormonal contraception |
|
| Interventions | Screening: Two screening cycles Placebo run in: One cycle single blind placebo run‐in Interventions: 1. Fluoxetine 10mg administered orally daily during the luteal phase for three cycles (n=86) versus placebo administered orally daily during the luteal phase for three cycles (n=88) followed by one cycle single blind placebo run‐out 2. Fluoxetine 20mg administered orally daily during the luteal phase for three cycles (n=86) versus placebo administered orally daily during the luteal phase for three cycles (n=88) followed by one cycle single blind placebo run‐out Duration: Three cycles of treatment Timing of administration: Medication administered 14 days before the expected date of the next menses and until the first day of active bleeding Summary measures: For change scores no details as to whether this is an average over treatment or endpoint score. For absolute scores data presented for baseline and three cycles of treatment. Review authors calculated average over the three cycles presented |
|
| Outcomes | Hand‐held electronic diary used to record Daily Record of Severity of Problems (DRSP) ‐ self‐rated Rating Scale for Premenstrual Tension Clinician rated Adverse events Arizona Sexual Experience Scale |
|
| Notes | Daily recording of symptoms luteal score calculated using scores from the five most symptomatic days occurring from six days before menses to the first day of menses Funded by Eli‐Lilly |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer generated randomisation code that was stratified by investigative site" |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | States double blind, with placebo "identical in appearance" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 84% of women randomised (218/260) completed study; 97% (252/260) included in primary analysis Reasons for dropouts as follows: fluoxetine 10 mg: 9 of 86 dropped out (2 adverse events, 2 patient decision, 5 protocol requirement), fluoxetine 20 mg: 22 of 86 dropped out (4 adverse events, 2 lack of efficacy, 5 patient decision, 1 physician decision, 3 protocol requirement, 7 loss to follow‐up). Placebo: 13 of 88 dropped out (1 adverse event, 3 lack of efficacy, 3 patient decision, 1 physician decision, 2 protocol requirement, 3 lost to follow up) |
| Selective reporting (reporting bias) | Unclear risk | States "The incidence of treatment‐emergent adverse events during placebo or fluoxetine therapy was compared using Fisher exact test". Adverse events not a clearly pre stated outcome and does not state whether adverse event data collected prospectively |
| Other bias | Unclear risk | Exclusion of placebo responders |
Cohen 2004.
| Methods | Randomised, double‐blind, placebo‐controlled, 3‐arm parallel study Study flow: Recruitment, 1751 women screened for study, 327 randomised, 103 to 12.5 mg paroxetine (70 completed, 95 analysed),113 to paroxetine 25 mgs (72 completed, 111 analysed), 110 to placebo (79 completed, 107 analysed) |
|
| Participants | Country: USA and Canada Site: Multi‐centre. Recruited from 43 outpatient centres Inclusion: PMDD. Women aged 18‐45 years with regular menstrual cycles (22 to 35 days) meeting diagnostic criteria for PMDD using DSM‐IV. Having symptoms of PMDD in at least 9 of 12 menstrual cycles over previous year. Onset of severe symptoms in the luteal phase subsided in the follicular phase on the four core symptoms of PMDD. Required to show a 200% worsening on one core symptom or a 100% worsening on two or more core symptoms during the luteal phase relative to their follicular phase score. A baseline Clinical Global Impression severity of illness (CGI‐S) score of ≥3. Exclusion: Presence of a primary psychiatric disorder (Axis 1 using DSM‐IV) except specific phobias in the previous 6 months; gynaecological or other clinically significant diseases, significant depressive symptoms during the follicular phase or current use of medication for PMDD symptoms. |
|
| Interventions | Screening: Two reference cycle for screening Placebo run‐in: Single blind placebo for one cycle. An additional cycle was available to patient who met all entry criteria before the first reference cycle but failed to achieve the predefined severity of core PMDD symptoms after a period of symptom tracking Interventions: Paroxetine CR 12.5 mg or 25mg, versus placebo for three treatment cycles administered daily Duration: Three treatment cycles Timing of administration: Study visits began within the first three days of menses Summary measures: Data was based on the study end point using the LOCF |
|
| Outcomes | Patient rated VAS ‐ Mood (irritability, tension, depressed mood and affective lability); Clinical Global Impression of Severity (CGI‐S); Clinical Global Impression of disease Improvement (CGI‐I); Patient rated Sheehan Disability Scale (SDS); Adverse events | |
| Notes | Funding source not stated, but some of investigators employed by GlaxoSmithKline | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer‐generated randomisation code" |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "All the participants in the study, including those administering the study visits and those assessing the outcomes, were blinded to the study assignment" ..."similar appearing placebo" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | As only 68% of women randomised completed the trial (221/327) a large number of data were imputed Text states "Fourteen randomized patients were excluded from all efficacy and safety analyses" ‐ no further explanation. Text also states "For all efficacy measures, the primary conclusions were based on the TC3 study end point using the last observation carried forward (LOCF) approach to handle missing data", but results for response rates include only 91% of "ITT population" and only 87% of those randomised |
| Selective reporting (reporting bias) | Unclear risk | Adverse events not a clearly prestated outcome and does not state whether adverse event data collected prospectively |
| Other bias | Unclear risk | Apparent exclusion of placebo responders |
Crnobaric 1998.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel study | |
| Participants | Country: Yugoslavia Site: No details in abstract Recruitment: 25 women randomised. Age 18‐40 years (mean 29.5). See Table of bias for details of attrition Inclusion: Aged 18 to 40 with regular menstrual cycles. Meeting DSM‐IV criteria for PMDD Exclusion: Current history of major depressive disorder, current mental disorder |
|
| Interventions | Screening: Two cycle screening Placebo run in: None Interventions: Fluoxetine 20mg (n=14) versus placebo (n=11) taken daily Duration: Two cycles of treatment Timing of administration: No details as to when in the cycle medication was commenced Summary measures: No details as to whether the data was the mean of two treatment cycles or the last cycle data |
|
| Outcomes | Clinical Global Impression Scale (CGI); Hamilton Rating Scale of Depression (HAM‐D); Calendar of Premenstrual Experience (COPE) | |
| Notes | No details of funding source | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States randomised. Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 25 women randomised. No attrition |
| Selective reporting (reporting bias) | Unclear risk | Three symptom scoring questionnaires used but results reported for only two |
| Other bias | Unclear risk | Few details available as not published in full text |
Eriksson 1995.
| Methods | Randomised double‐blind placebo‐controlled 3‐parallel arms (paroxetine versus maprotiline versus placebo) study | |
| Participants | Country: Sweden Site: No details Recruitment: Recruited via newspaper advertisements followed by telephone and then structured interview. 171 women recruited, 81 randomised, 65 completed (22 paroxetine, 21 maprotiline (not reported on in this review), 22 placebo). Refer to Table of bias for details of attrition. Mean age 37.5 years. Inclusion: Marked irritability and/or depressed mood starting regularly around ovulation or during two weeks preceding menstrual bleeding and terminating within a few days of onset of menstruation. Meeting DSM‐III‐R criteria for LLPDD. Increase of over 100% in either irritability or depressed mood (or both) during the premenstrual phase as compared to the postmenstrual phase, mean premenstrual irritability rating or depressed mood exceeding 20mm. Exclusion: Previous or ongoing psychiatric illness, ongoing major depressive disorder or dysthymic disorder, major depressive disorder or dysthymic disorder less than 2 years from time of interview,ongoing medication for somatic or psychiatric illness (excluding casual analgesics), use of oral contraceptives, ongoing alcohol abuse, ongoing or planned pregnancy, under 18 years of age, previous treatment with antidepressants for premenstrual complaints. |
|
| Interventions | Screening: Two screening cycles Placebo run‐in: None Intervention: 10‐30 mg paroxetine every day for 3 cycles (n=27) versus placebo (n=26) Duration: Three cycles of medication Timing of administration: Continuous; treatment started on the first day of menstruation Summary measures: Medians presented for three treatment cycles |
|
| Outcomes | Visual analogue scales (VAS); Self reported global assessment of improvement (Enormously improved to enormously deteriorated); Adverse events | |
| Notes | Daily symptom rating Funding from Swedish Medical Research Council, NovoNordisk and Ciba AB, Sweden. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States randomised. Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind (patients and investigators unaware of allocation until end of study) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 171 recruited. 81 randomised to three arms, two of which used in analysis here: n=53 44/53 women included in analysis (83%) Paroxetine n=27 lost five subjects during the study, three because of side effects, one due to pregnancy and one because of irregular menstruation Placebo n= 26 lost four women during the study, two due to side effects, one due to onset of another illness and one due to protocol violation |
| Selective reporting (reporting bias) | Low risk | Data on adverse events prospectively collected. No evidence of selective reporting identified |
| Other bias | Low risk | No other potential bias identified |
Eriksson 2008.
| Methods | Randomised, double‐blind, placebo‐controlled trial with 3 arms | |
| Participants | Country: Sweden Site: University clinic Recruitment: Newspaper advertisements, phone interview Inclusion: Women aged at least 18, meeting DSM‐IV A‐C criteria for PMDD (confirmed at interview), with regular menstrual cycles and using adequate non hormonal contraception. Exclusion: Axis 1 psychiatric disorder, psychotic disorder, major depressive disorder of less than 2 years, Montgomery‐Asber depression scores at baseline of over 15, use of hormonal contraception, recurrent severe headache or migraine, regular use of a drug that might influence PMDD symptoms or interact with trial medication, use of psychoactive drug within 2 weeks of screening visit or of fluoxetine within 5 weeks of visit. Mean age 36‐38 years |
|
| Interventions | Screening: 2‐3 screening cycles confirming cyclicity of symptoms Placebo run‐in: no Intervention: 1. 10 mg escitalopram (n=54) 2. 20 mg escitalopram (n=53) 3. placebo (n=51) for 3 cycles Timing of administration: luteal phase only ‐ taken from estimated day of ovulation to first full day of menstruation Summary measures: change scores(VAS) and end scores (PGE) |
|
| Outcomes | Sum self‐assessed VAS scores of 4 key symptoms (irritability, tension, affective lability, depressed mood) ‐ primary outcome. Patient Global Evaluation scores reported in review (all symptom ‐ end scores) Clinician rated symptom scale (PMTS‐O) Function ‐ assessed with Sheehan Disability Scale Clinician‐rated global severity (CGI‐S) and global improvement (CGI‐I) Response rate (a priori defined as 50 decrease in VAS symptoms or in irritability, or self‐rating as much improved (PGE) or clinician‐rated as by CGI‐I) Patient ‐assessed "usefulness of medication" using Patient Global Evaluation (PGE) |
|
| Notes | Funded by pharmaceutical company H Lundbeck A/S VAS data presented in graphical form: emailed author to request effects estimates and SDs ‐ no reply received |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Code generated by sponsor, details unknown to investigators |
| Allocation concealment (selection bias) | Low risk | Sequential enrolment, details unknown to investigators |
| Blinding (performance bias and detection bias) All outcomes | Low risk | All study personnel and participants blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 158 women randomised; seven excluded from analysis (incomplete baseline assessment, concomitant use of another antidepressant) 151/158 analysed (96%) by intention to treat with last observation‐carried‐forward |
| Selective reporting (reporting bias) | Unclear risk | Adverse events not prospectively reported |
| Other bias | Low risk | Groups had similar prognostic factors at baseline |
Freeman 1999.
| Methods | Randomised, double‐blind placebo‐controlled, 3‐arm parallel trial. (sertraline versus desipramine versus placebo). Only sertraline and placebo reported in this review | |
| Participants | Country: USA Site: Premenstrual Syndrome Program, University of Pennsylvania, Philadelphia Recruitment: 278 eligible women. 125 randomised to sertraline (n=66) or placebo (n=59) Inclusion: 18‐45 years, regular menstrual cycles (22‐35 days), ovulating, experiencing distressing premenstrual symptoms for at least six months, reporting moderate to severe impairment in work, family life or social activity on subject global ratings, meeting PMS criteria on DSR ratings, good health, no major psychiatric (DSM‐IV) diagnosis within past year. Exclusion: use of psychotropic drugs that could not be discontinued during the study period, all other prescription and non‐prescriptions drugs for PMS, pregnancy, lactation, hysterectomy, symptomatic endometriosis, irregular menstrual cycles, not using medically approved contraception, serious health problems, any major axis 1 psychiatric diagnosis, including major depression, current or within the past year, risk of suicide and alcohol or drug abuse within the past year. |
|
| Interventions | Placebo run‐in: None Intervention: 50mg ‐150mg sertraline administered orally continuously (average 105mg) for 3 cycles versus placebo administered orally for 3 cycles (a third study arm received desipramine) Timing of administration: Drug commenced on day 1 of the menstrual cycle Summary measures: Data for LOCF for all patients with treatment data and also for all completers |
|
| Outcomes | Daily Symptom Report Hamilton Scale for Depression Clinical Global Impression Scale | |
| Notes | Daily symptom rating Drug provided by Pfizer Inc NY, Funding from National Institute of Child Health and Human Development | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised via computer‐generated number tables |
| Allocation concealment (selection bias) | Low risk | Allocation by a technician with no clinical contact |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind, medication prepared in pharmacy in identical capsules |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 97/125 of women randomised completed treatment (78%), but all women randomised were included in efficacy analysis by last‐observation‐carried‐forward. 13/66 dropped out of the sertraline group and 15/59 from the placebo group. Reasons for withdrawal not reported by treatment group, but text states that reasons for discontinuation did not differ significantly between the sertraline and placebo groups |
| Selective reporting (reporting bias) | Unclear risk | Adverse events recorded retrospectively at follow‐up visits |
| Other bias | Low risk | No other potential bias identified |
Freeman 2004.
| Methods | Randomised double‐blind, placebo‐controlled, 3‐arm parallel study (intermittent or continuous dosing or placebo) Study flow: 167 women were randomised: 56 to intermittent (35 completed, 45 analysed), 56 to continuous (40 completed, 48 analysed), 55 to placebo (43 completed, 50 analysed). Refer to table of bias for details of attrition |
|
| Participants | Country: USA Site: University based Premenstrual Syndrome Program Inclusion: PMS or PMDD. Age 18‐45 years, regular menstrual cycles of 22‐35 days, positive urine test for ovulation, persistent premenstrual symptoms for at least 6 months, global report of at least moderate to severe impairment in work, family life or social activity; general good health; no major psychiatric diagnosis within past year, meeting stated PMS criteria based on prospective daily rating of symptoms. Required a total premenstrual Daily Symptom Rating Score >/80 and an increase of >/50% over the postmenstrual score for the mean of the three screen cycles and for the single blind placebo cycle. Mean age continuous dosing 34.5±6.2, placebo group 33.4±6.5 years. Exclusion: Any major Axis 1 psychiatric diagnosis, including major depression, currently or within previous year as assessed by SCID; use of psychotropic medications that could not be discontinued for the duration of the study, use of any prescription, non‐prescription, herbal or other therapies for PMS, pregnancy, lactation, hysterectomy, symptomatic endometriosis, irregular menstrual cycles, not using medically approved contraception, serious health problems, suicide risk, alcohol or drug abuse within the past year. |
|
| Interventions | Screening: Two screening cycles followed by one single blind placebo run in cycle Interventions: 1. 50mg sertraline taken orally until day 2 menses (n=56) 2. 50 ‐100mg of sertraline taken throughout cycle orally (n=56) (mean dosage during trial was 75mg/day +/‐25) for three cycles versus 3. placebo taken orally (n=55) for three cycles In the absence of improvement dose could be increased to 100mg sertraline or 2 tablets (sertraline or placebo) Timing of administration: Bottle A started on Day 3 of menses, switching to bottle B at 14 days before the expected date of menses and continuing through day 2 of menses Summary measures: Data is presented as end of treatment, LOCF, mean values Timing of administration: Bottle A started on Day 3 of menses, switching to bottle B at 14 days before the expected date of menses and continuing through day 2 of menses Summary measures: Data presented at end of treatment, LOCF, mean values |
|
| Outcomes | Daily Symptom Rating Form Subject Global Ratings of Functioning |
|
| Notes | Daily symptom rating Funding: National Institute of Child Health and Human Development. Drugs provided by Pfizer Inc. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation using random number tables |
| Allocation concealment (selection bias) | Low risk | Allocation conducted by a technician at the beginning of the study with no clinical contact |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind, medication taken from Bottle A and Bottle B on the same day of the menstrual cycle whatever the allocation. "Sertraline and placebo tablets were identical in appearance" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 29% of women (49/167) did not complete treatment, including 24/167 (14%) who provided no outcome data. The remaining 86% were included in analysis, using last‐observation‐carried‐forward Dropout rate variable across groups. Reasons for dropouts as follows: intermittent: 21/56 (adverse event =5, lack of efficacy =2, withdrawn consent = 8, medical problem =2, non‐compliance = 1, loss to follow up = 3); continuous: 16/56 (adverse event = 7, withdrawn consent = 4, medical problem =2, non ‐compliance =1, loss to follow up = 2); placebo 12/56 (adverse event =1, lack of efficacy =1, withdrawal of consent =5, noncompliance = 2, loss to follow‐up = 3) |
| Selective reporting (reporting bias) | Unclear risk | Adverse events not a predefined outcome and not all prospectively recorded ‐ AEs were recorded at each visit, plus women had a medication checklist to report at home any side effects experienced "while taking tablets from Bottle A" (i.e. non‐luteal phase) to identify discontinuation‐related symptoms. Individual adverse effects not reported |
| Other bias | Unclear risk | Exclusion of placebo responders |
Freeman 2010.
| Methods | Randomised double‐blind controlled trial 11 participants recruited |
|
| Participants | Country: USA Site: University of Pennsylvania Recruitment: Inclusion: Age 15‐19 years, regular menstrual cycles, in good health, using contraception if sexually active, evidence of ovulation, meets symptom criteria for PMS Exclusion: Being treated for PMS, using medications that may compromise the study drug, pregnancy or intending pregnancy, current or history of major psychiatric diagnosis, alcohol or drug abuse, sensitivity to study drug |
|
| Interventions | Screening: No Placebo run in: No Intervention: Escitalopram 10 mg daily, increasing to 20 mg if unimproved and not precluded by side effects Timing of administration: Luteal phase (approx day 14 to day 2) Placebo‐controlled Summary measures: Outcomes not published |
|
| Outcomes | Sheehan Disability Scale, Steiner Premenstrual Tension Scale, Patient Global Evaluation of Improvement, Subject Satisfaction Questionnaire, Clinical Global Impressions score | |
| Notes | Email from primary investigator to JM 10.7.2012: The study was not submitted for publication due to the small sample size. The final report of the data analysis is on file at Forest Laboratories, Inc. Eleven subjects were randomized to double‐blind treatment. Both arms significantly improved from baseline, but drug did not differ from placebo due to lack of statistical power in this very small study. Penn Daily Symptom Report (DSR) scores decreased 41% in the drug arm and 28% in the placebo arm. There were no serious AEs. E Freeman | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double‐blinded ‐ no further details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Described as double‐blinded ‐ no further details |
| Selective reporting (reporting bias) | High risk | Outcome data not published |
| Other bias | Unclear risk | Not assessable as results unpublished |
Glaxo 1996.
| Methods | Randomised double‐blind parallel‐group placebo‐controlled study 2:1 randomisation drug: placebo 48 women randomised (planned for 126); 31 to drug and 17 to placebo, 32/48 completed study, 43/48 analysed |
|
| Participants | Country: UK Site: seven centres Recruitment: Inclusion: Women with premenstrual dysphoric disorder, aged 18 to 45 years, regular menstrual cycle of previous 6 months and meeting DSM‐IV criteria for PDD Exclusion: Meeting DSM‐IV criteria for depression, severe clinically significant co‐existing conditions |
|
| Interventions | Screening: Placebo run‐in: Yes ‐ for one month Intervention: Paroxetine 20 mg daily versus placebo Timing of administration: Continuous for 4 cycles Summary measures: Change scores |
|
| Outcomes | Calendar of Premenstrual Experience (COPE); change in subsets of COPE, Beck Depression Inventory score, proportion of women with Clinical Global Impression Severity of Illness score and global Improvement score at defined cut‐off points | |
| Notes | COPE total scores given as mean and range, no SDs | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double‐blinded ‐ no further details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 43/48 analysed (90%) |
| Selective reporting (reporting bias) | Unclear risk | Adverse events recorded retrospectively at follow‐up visits |
| Other bias | Unclear risk | Exclusion of placebo responders |
Glaxo 1996a.
| Methods | Randomised double‐blind parallel‐group placebo‐controlled study 38 women randomised (planned for 126); 29 to drug and 9 to placebo, |
|
| Participants | Country: Austria, Canada, France, Begium, Germany, Holland, Sweden, Italy, Ireland Site: Recruitment: Inclusion: PMDD by DSM IV. Age 18‐45 years, regular menstrual cycles, symptoms in 10/12 previous cycle, Mean VAS score of at least 40 mm for 3 of 4 key psychological symptoms (depressed mood, irritability, tension, mood swings) in luteal phase and no more than 20mm in follicular phase. Baseline luteal phase CGI severity of illness score at least 3 Exclusion: Major depressive episode, schizophrenia, mania, suicide risk, use of other therapy |
|
| Interventions | Screening: 2 cycles Placebo run in: Yes 1 cycle Intervention: Paroxetine 5 mg, 10mg or 20 mg versus placebo Timing of administration: Continuous (for first 4 cycles) Summary measures: Only AE data presented |
|
| Outcomes | Safety (no other outcomes measured due to early termination) | |
| Notes | Study terminated early due to problems with electronic diary that it was planned to use for recording outcomes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All 38 randomised included in analysis for AEs. Terminated early, planned sample size was 248 |
| Selective reporting (reporting bias) | High risk | Terminated early, none of planned outcomes reported except AEs |
| Other bias | Unclear risk | Very small (n=11) |
Glaxo 2001.
| Methods | Randomised double‐blind parallel‐group placebo‐controlled study 361 women randomised, 274 completed, 358 analysed |
|
| Participants | Country: Europe and South Africa (46 centres) Recruitment: not stated Inclusion: Women aged 18‐45 years with regular menstrual cycles, dig nosed with PMDD according to DSM IV criteria, PMDD of at least one year, with symptoms in 9/12 previous cycles Exclusion: not stated |
|
| Interventions | Screening: 2 cycles Placebo run in: No Intervention: Paroxetine 12.5 mg, 25mg, or placebo Timing of administration: Continuous Summary measures: Change score |
|
| Outcomes | Primary: Change in VAS mood score at treatment cycle 3 Secondary: Nine other efficacy measures on various scales Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double‐blinded ‐ no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 358/361 analysed (99%) |
| Selective reporting (reporting bias) | Unclear risk | Unclear whether adverse events data prospectively collected. Unpublished except on funder's website |
| Other bias | Low risk | No other potential bias identified |
Halbreich 1997.
| Methods | Crossover trial 2X2 cycles | |
| Participants | Country: USA Site: Not stated Recruitment: Women recruited by advertisement in local newspapers and posted notices. Over 60 screened by structured telephone interview, 32 women interviewed, Twenty seven eligible for study; fifteen entered single blind phase. 11 were randomised, 8 completed. Refer to Table of bias for details of attrition. Inclusion: Age 24 to 45 years, regular menstrual cycles (25 to 34 days), not meeting criteria for DSM‐IV major diagnoses for at least 6 months, meeting DSM‐IV criteria for PMDD and criteria for dysphoric PMS. Physically healthy and not taking any medications. Confirmed PMDD symptoms during late luteal phase and no symptoms during mid follicular phase. |
|
| Interventions | Screening: Two screening cycles Placebo run in : Single blind drug run‐in Intervention: 100mg sertraline orally for 2 cycles versus placebo orally for 2 cycles luteal phase only Timing of administration: Intervention administered fourteen days before expected onset of menses or for full cycle, unclear as to which day commenced Summary measures: Data from both arms pooled |
|
| Outcomes | Clinical Global Impression ‐ Improvement Scale (CGI‐I) Hamilton Rating Scale for Depression (HAM‐D)modified Daily Rating Form (DRF) | |
| Notes | Not included in analysis due to cross‐over design. No pre‐crossover data reported Only responders to drug randomised Symptoms rated daily Funded by Pfizer |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States randomised. Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no details |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 11 were randomised, 8 completed (72%) 2 withdrawals due to side effects and one due to pregnancy: all dropped out during placebo phase |
| Selective reporting (reporting bias) | Unclear risk | Adverse events data not collected prospectively |
| Other bias | Unclear risk | Only responders to drug randomised. Review includes only first phase data from crossover trial |
Halbreich 2002.
| Methods | Randomised, double‐blind, 2‐arm parallel study. | |
| Participants | Country: USA and Canada Site: Multi‐centre study in 14 psychiatric and gynaecological outpatient clinics Recruitment: 907 screened, 281 women randomised and 221 completed study. Refer to Table of bias for details of attrition.Women recruited by advertisements in media and by referrals.Mean age 35.9+/‐5.4 and 36.5+/‐4.8 years Inclusion: Age 24 to 45 years, regular menstrual cycles (24 to 36 days), two year self reported history of PMDD, meeting DSM‐IV criteria for PMDD based on 2 cycles prospective screening using DRSP. Mean luteal phase score during 5 most symptomatic days to be at least 75% higher than mean mid follicular phase score. Also a marked level of functional impairment for a minimum of 2 premenstrual days. Required to have at least one of the following symptoms rated as moderate or greater in severity for at least two days during the late luteal phase: depression, irritability, anxiety/tension, or mood lability as well as at least four additional DSM‐IV criterion symptoms of PMDD. Exclusion: Follicular phase Hamilton Rating Scale of Depression score >10, use of oral contraceptives or other hormonal preparations within 2 months before screening, current or lifetime diagnosis of psychiatric disorder, current (or past 6 month) of major depressive disorder (other than PMDD), panic disorder, generalised anxiety disorder, posttraumatic stress disorder or eating disorder, > 38 years having luteinizing hormone levels >38U/L or follicle stimulating hormone levels > 20U/L, hysterectomy or failure to demonstrate ovulation in screening cycles, failure to respond to two or more adequate trials of antidepressants to treat their PMDD, current use of psychotropic medication. |
|
| Interventions | Screening: Two screening cycles Placebo run in: One cycle single blind placebo run‐in Intervention: Sertraline 50‐100mg given orally for 3 cycles during luteal phase only (n=142) versus placebo given orally for 3 cycles during luteal phase (n=139) Timing of administration: Based on an algorithm of individual cycle length. Women with 28 day cycle had first dose on day 14 before onset of menses Summary measures: Data based on LOCF data |
|
| Outcomes | Hamilton Rating Scale for Depression (HRS‐D); Daily Record Severity of Problems (DRSP); Clinical Global Impression Severity scale (CGI‐S); Clinical Global Impression Improvement scale (CGI‐I); Patient Global Evaluation; Social Adjustment scale (SES); Quality of Life Enjoyment and Satisfaction Questionnaire | |
| Notes | Daily symptom rating Direct expenses funded by Pfizer |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind, identical medication in blister packs containing placebo or sertraline |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 281 randomised, 229 included in intention to‐treat analysis with (81%), with last‐observation‐carried‐forward Twenty‐seven withdrew from sertraline group (protocol violation =3, lost to follow up/ other reason = 13, adverse events = 11) and thirty three from the placebo group (insufficient clinical response = 4, protocol violation =7, lost to follow up / other reason = 21 and adverse events = 1) |
| Selective reporting (reporting bias) | Unclear risk | Adverse event data collected by spontaneous self‐report |
| Other bias | Unclear risk | Only women with a history of response to antidepressants randomised. Placebo responders excluded |
Jermain 1999.
| Methods | Randomised, double‐blind, crossover trial | |
| Participants | Country: USA Site: Research centre in large multi‐speciality clinic Recruitment: Women recruited via advertisements and from referral from affiliated psychiatric and gynaecological clinics.189 women screened, of these 57 had increases in COPE score to be randomised.57 randomised, 40 completed. Refer to Table of bias for details of attrition.Mean age Sertraline first arm 35±7 (25‐47) years, Placebo first arm 38±5 (23‐48) years Inclusion: women aged 19 to 49 years with regular menstrual cycles (23 to 35 days) meeting DSM ‐IV criteria for PMDD. Pre treatment luteal phase score (using COPE) >41 and double follicular phase score for two consecutive menstrual cycles. Follicular phase score < 40 and follicular to luteal phase increase to increase by 30% for at least five pre menstrual symptoms Exclusion: Current Axis 1 disorder, pregnant, significant medical or gynaecologic disorders, taking psychotropic drugs or hormonal medications including the oral contraceptive |
|
| Interventions | Screening: Two cycles Placebo run in: None Intervention: Two cycles treatment with 50mg sertraline luteal phase only increasing to 100mg for non‐improvers versus placebo followed by crossover for two cycles. No washout Timing of administration: Drug was commenced fourteen days before the expected onset of menses and discontinued when menses began Summary data: Data was summarised for luteal phase as a sum of the last seven days of the cycle and averaged over two cycles in the paper. No details of average drug received |
|
| Outcomes | Calendar of Premenstrual Experiences (COPE) patient rated daily assessment; Beck Depression Inventory (BDI) patient rated; Adverse events | |
| Notes | Some data such as adverse events and BDI was not extractable from the first arm of the crossover Funded by Pfizer |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States randomised. Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further details |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 40 completed study out of 57 women randomised completed study (70%) Seventeen women withdrew or data not available. Five withdrew while taking placebo (two due to adverse events) and nine withdrew while taking sertraline (5 due to adverse events) |
| Selective reporting (reporting bias) | Unclear risk | Adverse events data collected by retrospectively by open‐ended enquiry at follow‐up visits |
| Other bias | Unclear risk | Crossover study with no washout period (though used in luteal phase only) |
Kornstein 2006.
| Methods | Randomised double‐blind placebo‐controlled 3‐arm study with parallel groups | |
| Participants | Country: USA Site: Conducted in 22 psychiatric and gynaecological outpatient clinics in the US Recruitment: Women recruited through advertisements in the media. 314 women randomised and allocated to sertraline 25 mg (n=98), sertraline 50.0 mg (n=97) or placebo (N=101) Inclusion: Aged between 24 and 45 years, having regular menstrual cycles (24 to 36 days), and meeting criteria for PMS based on Daily Symptom Report (DSR) for 2 cycles. A total score >80 for luteal phase, along with at least 3 DSR items showing at least moderate severity for 2 out of 6 premenstrual days, moderate distress for at least 2 out of 6 premenstrual days and minimal to no symptoms during the follicular phase (days 5‐10). Exclusion: Decrease of 30% or more in DSR total score for the 6 premenstrual days during the single blind placebo cycle, use of oral contraceptives or other hormonal preparations within six months prior to screening, pregnant, lactating or planning pregnancy, LH levels > 38 or FSH > 20 in patients aged >38 years, post‐hysterectomy or failure to demonstrate ovulation in the two screening cycles, failure to respond to an adequate trial of 2 or more antidepressants to treat premenstrual symptoms, symptomatic endometriosis (or treatment in the past 3 months), history of major depressive episode or other mental disorder or substance misuse in past year, history of eating disorder in previous 2 years, current or lifetime history of psychiatric disorders, current use of any psychotropic medication, positive urine drug screen, current suicide risk, any acute or unstable medical illness or clinically significant laboratory abnormality. |
|
| Interventions | Screening: Two cycles Placebo run in: None Intervention: Two cycles treatment with 25 mg or 50 mg sertraline luteal phase only versus placebo followed by one cycle of continuous dosing followed by one cycle of symptom‐onset dosing; compared to placebo Timing of administration: as above. No details of average drug received Summary measures: Data were summarised for luteal phase as a sum of the last seven days of the cycle and averaged over two cycles in the paper |
|
| Outcomes | Daily Symptom Report; Clinical Global Impression‐ Severity of Illness (CGI‐ S); Clinical Global Improvement (CGI‐I); Patient Global Evaluation (PGE); Quality of Life Enjoyment and Satisfaction Scale (Q‐LES‐Q); Social Adjustment Scale Self Report (SAS‐SR) | |
| Notes | Study funded by Pfizer Data relating to luteal phase SSRI versus placebo comparison (2 cycles) included in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States randomised but method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blinded, no further details |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 269 women analysed out of 314 randomised (86%) 98 allocated to sertraline 25mg and 74 completed (76%) (adverse events = 7, protocol violation = 5, withdrew consent = 4, loss to follow up = 2, other/administrative = 6) 97 allocated to sertraline 50mg 77 completed (79%) (adverse events = 10, protocol violation= 1, withdrew consent = 1, loss to follow up = 5, other/administrative = 3) 101 allocated to placebo and 79 completed (78%) (adverse events = 8, protocol violation = 2, withdrew consent = 6, loss to follow up = 2, other administrative = 4) |
| Selective reporting (reporting bias) | Unclear risk | Adverse effects data collected retrospectively at follow‐up visits |
| Other bias | Unclear risk | Only included women who had responded to an adequate trial of 2 or more antidepressants to treat premenstrual symptoms |
Landen 2007.
| Methods | Randomised, double‐blind placebo‐controlled trial with three parallel arms (intermittent versus continuous versus placebo) | |
| Participants | Country: Sweden Site: Multi‐centre study by four investigators at four centres. Unclear as to what or where these were Recruitment: Mean age placebo group 37±7.1 and intermittent group was 37±5.9 years.Women responding to advertisements, interviewed by telephone and then invited to a screening visit. Inclusion: >18 years, reporting regular menstrual cycles (22‐35 days) and meeting DSM‐IV criteria A‐C for PMDD. Women had to display a 50% increase in irritability and/or depressed mood during the luteal phase as compared to the follicular phase during two screening cycles using a VAS. Mean luteal phase rating of the symptom >/25mm. Exclusion: Meeting DSM‐IV criteria for any Axis 1 disorder other than PMDD during previous 12 months before screening as assessed by Mini International Neuropsychiatric Interview; display a baseline score >10 on Montgomery Asperg Depression Rating Scale in the follicular phase, having tried a SRI for PMDD, taking oral contraceptives or reporting any regular use of any psychoactive drug or any other kind of medication motivating exclusion due to safety reasons. 186 women randomised to intermittent (n=59), continuous (n=60) or placebo (n=59) arms |
|
| Interventions | Screening: Two screening cycles Placebo run in: None Intervention: a) Luteal arm: Placebo until estimated time of ovulation followed by paroxetine 10mg/day given orally for four days and 20mg for the rest of the luteal phase. During the first four days of the follicular phases they received 10mg daily paroxetine for three treatment cycles (n=59). Time of ovulation was estimated on the basis of a normal cycle length for the woman b) Continuous arm: treatment administered daily (n=60) c) Placebo: administered orally throughout the study for three treatment cycles (n=59) Summary measures: LOCF data presented |
|
| Outcomes | VAS scales; Premenstrual Tension Scale (observer and self rated); Clinical Global Improvement (CGI‐I); Patient Global Evaluation (PGE); Sheehan Disability Scale (SDS); Adverse events | |
| Notes | Symptoms rated daily Authors contacted, no response Novo Nordisk and Glaxo SmithKline |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | States double‐blinded, no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 167 women analysed out of 186 randomised (90%), using last‐observation‐carried‐forward Eight randomised women withdrew before taking study med (allocation not stated). Of 59 randomised to intermittent dosing, 50 completed the trial (adverse event = 3, other reason = 6). Of 60 randomised to continuous dosing, 51 completed the trial (adverse event = 5, and other reason = 4). Of 59 randomised to placebo, 51 completed the trial (adverse events = 1, other reason = 7) |
| Selective reporting (reporting bias) | Low risk | Adverse events recorded prospectively and followed up at clinic appointments. No potential selective reporting bias identified |
| Other bias | Unclear risk | No potential for other bias identified |
Menkes 1992.
| Methods | Randomised, double‐blind, placebo‐controlled, crossover trial 2x3 cycles | |
| Participants | Country: New Zealand Site: No details Recruitment: Community sample of volunteers ‐ 23 women screened. After two cycles of screening 21 women had PMS confirmed by psychiatric interview and agreed to be randomised. 21 randomised, 16 completed. Mean age 37.8±4.7 years. 23 women met provisional criteria, Daily Symptom Scores and ratings of PAF Inclusion: Age 18 to 48 years. Confirmation of PMS by psychiatric evaluation Exclusion: Taking regular psychotropics, diuretics, or using hormonal contraception. Any appreciable menstrual irregularity, psychiatric or substance use disorder |
|
| Interventions | Screening: Three cycles of screening Placebo run‐in: None Intervention: Crossover trial of 20mg fluoxetine PO every day for 3 cycles versus placebo PO every day for 3 cycles with 12 day washout period between arms Timing of administration: Medication commenced on twelfth day of menstrual cycle and continued through three cycles stopping at the onset of menses Summary measures: Mean data over three months of treatment presented in paper |
|
| Outcomes | Premenstrual Assessment Form Side effects |
|
| Notes | No data extracted as unable to distinguish first and second arm of study Same patient group as excluded study, Menkes 1992 Daily symptom rating Fluoxetine provided by Eli‐Lilly |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind (patient and rater) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Five women excluded from analysis (23%), two for protocol violation and three due to adverse effects ‐ no details as to which arm of the crossover this occurred in |
| Selective reporting (reporting bias) | Unclear risk | Tolerability assessed at end of each treatment phase, not prospectively. Adverse events not reported by treatment arm |
| Other bias | Unclear risk | Crossover trial with 12‐day washout period |
Miner 2002.
| Methods | Randomised, double‐blind, placebo‐controlled, 3‐arm parallel‐group study (luteal phase weekly dose (LPWD) x one versus LPWD x2 versus placebo) | |
| Participants | Country: Australia, Europe and Mexico Site: Multi‐centre in 30 centres. No details as to what or where these centres were Recruitment: Mean age LPWDX1 37.4±5.8 years, LPWDX2 36.1±5.2 years and placebo 37.4±5.4 years.Not stated where women were recruited from. Refer to table of bias for details of attrition Inclusion: Women aged 18‐45 years, regular menstrual cycles(23‐35 days) were eligible. Meeting PMDD criteria in two screening cycles. Luteal scores average >/3.0 for each of the 5 DRSP symptoms corresponding to the items in DSM‐IV criterion A, with 1>/ symptom corresponding to a DSM IV mood item; if follicular phase scores averaged <2.5 for each of these 5 DRSP symptoms; if mean scores for these 5 DRSP symptoms increased by >/50% from the follicular to the luteal phase; and if scores on the three functional DRSP items were >/4 on >/2 occasions during the luteal phase. Exclusion: Axis 1 psychiatric disorder within previous 6 months (with exception of phobias). Women using hormonal contraceptives or who had used them in previous 3 months. Placebo responders in the single blind placebo run‐in |
|
| Interventions | Screening: Two cycles screening Placebo run‐in: Single blind placebo run‐in Intervention: 3 cycles of treatment or placebo followed by another single blind placebo run out. LPWD X 1 versus LPWD x2 Fluoxetine 90mg PO (n=86) versus Group 3 PLC placebo administered two times during luteal phase at 14 and 7 days before expected menses (n=85) Timing of administration: administered at 14 and 7 days before expected menses Summary measures: Mean data presented |
|
| Outcomes | Self‐completed electronic diary using the Daily Record of Severity of problems Scale (DRSP) for recording daily PMDD symptoms.Mood, physical and social functioning subtotals. Two clinician rated and one patient rated scale. Rating Scale for PreMenstrual Tension (PMTS‐ C) CGI Severity score | |
| Notes | Study was supported by a grant from Forest Laboratories, New York Symptoms rated daily |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation schedule |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind (patient and other not stated) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 257 women randomised, 247 analysed by ITT (96%). (Dropouts: LPWDx1: 3 dropouts of whom 2 were for adverse events; LPWDX2: 2 dropouts, both for adverse events; placebo arm: 5 dropouts, one for adverse events) |
| Selective reporting (reporting bias) | Unclear risk | Does not state how adverse events data were collected |
| Other bias | Unclear risk | Placebo responders excluded |
Ozeren 1997.
| Methods | Randomised, double‐blind, placebo‐controlled 2‐arm parallel trial | |
| Participants | Country: Turkey Site: No details Recruitment: Self‐referred factory workers. 440 women screened. 35 met diagnostic criteria. 35 randomised and 30 completed. Mean age 30.6 ± 7.48 years for treatment group and 31.7 ± 7.42 years for placebo group Inclusion: Women aged 18‐45. Meeting criteria for PMS being a luteal phase score at least twice that of the follicular phase score, and the luteal phase score to be at least 42 and the follicular phase score to be less than 40. Diagnosis confirmed using DSM‐IV and DSM‐3‐R diagnostic criteria Exclusion: Those taking psychotropics, diuretics, antidepressants, anxiolytics, oral contraceptives, hormonal medications and those having major psychiatric disorders, pelvic pathology and irregular menstrual cycles |
|
| Interventions | Screening: Three screening cycles Placebo run‐in: None Intervention: 20mg fluoxetine (n=18) daily PO for 3 cycles versus placebo (n=17) PO for 3 cycles Timing of administration: Medication taken in the morning. No details as to when in the menstrual cycle medication was commenced Summary measures: Mean data presented |
|
| Outcomes | Calendar of Premenstrual Experiences | |
| Notes | Daily symptom rating No details of ITT or power calculation No funding source stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no details as to whom was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Five of 35 women (14%) excluded from efficacy analysis, two from the placebo group and three from the treatment group due to protocol violation or through intolerable adverse effects of fluoxetine |
| Selective reporting (reporting bias) | Unclear risk | Unclear whether data on adverse events collected prospectively. Adverse events data not clearly reported by comparison group |
| Other bias | Low risk | No potential for other bias identified |
Pearlstein 1997.
| Methods | Randomised placebo‐controlled, double‐blind, 3 parallel arms study (fluoxetine versus bupropion versus placebo) | |
| Participants | Country: USA Site: Women treated in two PMS clinics Recruitment: 44 women enrolled, 37 women randomised, 34 completed. Refer to Table of bias for details of attrition. Mean age 36.5±5 years Inclusion: Age 18 ‐ 45 years, regular menstrual cycles (24 to 35 days), in good physical health in 6 months preceding study. A 30% increase in the premenstrual symptom average was required in at least 5 symptom categories as specified by PMDD criteria. Absence of significant follicular phase symptoms and a 30% or greater premenstrual increase in impairment of occupational, social, or interpersonal functioning was required on the basis of daily assessment. Exclusion: Pregnancy, irregular menstruation, serious health problems, use of psychoactive or hormonal medications including oral contraceptive, current Axis 1 disorder (DSM‐III‐R), substance abuse or suicide risk in prior 6 months. |
|
| Interventions | Screening: Two cycles of screening Placebo run‐in: Single‐blind placebo for one cycle Intervention: 20mg fluoxetine orally daily for 2 cycles (n=10) versus placebo administered orally daily for 2 cycles (n=12) Timing of administration: Medication taken daily throughout cycle. Summary measures: Data presented for cycle 3 |
|
| Outcomes | Hamilton Scale for Depression (HAM‐D) Clinical Global Impression (CGI) Global Assessment Score (GAS) Daily Assessment Form (designed by group) | |
| Notes | Daily symptom rating Funded by Eli‐Lilly | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation at separate sites not centrally. Methods not reported |
| Allocation concealment (selection bias) | Unclear risk | Methods not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind. Subjects all took one white capsule in the morning and three red tablets each day in three‐times‐daily dosing. One of the physicians who rated patients was blind to medication but aware of the study design |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3/37 women randomised not analysed for efficacy (8%) ‐ all three withdrew due to mild agitation (one in each group) |
| Selective reporting (reporting bias) | Unclear risk | Unclear how adverse effects data collected |
| Other bias | Unclear risk | Placebo responders excluded |
Pearlstein 2005.
| Methods | Randomised, double‐blind, placebo‐controlled 3‐arm parallel trial. Paroxetine CR 12.5 mg versus 25 mg versus placebo | |
| Participants | Country: USA and Canada Site: 47 outpatient centres Recruitment: 1974 women screened, 371 randomised to Pearlstein. 125 randomised to paroxetine 25mg and 82 completed; 125 randomised to placebo, 96 completed.Mean age paroxetine 25mg 36.5±4.87 years, mean age of placebo 35.8±5.79 years Inclusion: Women aged 18‐45 years with regular menstrual cycles (22‐35 days) and confirmed PMDD based on DSM‐IV. Symptoms to have been present for at least nine out of previous twelve cycles over the previous year. Onset of severe premenstrual symptoms (as recorded on a daily basis) during the luteal phase was followed by symptom subsidence during the follicular phase based on four core symptoms (irritability, tension, affective lability and depressed mood). Required to demonstrate a 200% worsening on one core symptom or a 100% worsening on two or more symptoms during the luteal phase relative to the follicular phase. Baseline Clinical Global Impressions of Severity scale score >/3. Exclusion: Meeting DSM‐IV criteria for other Axis 1 disorder except specific phobias in the previous six months, diagnosed with gynaecological or other clinically significant disease, clinically significant depressive symptomology during the follicular phase, suicide risk, taking medication for PMD, received previous adequate treatment or participated in a clinical trial for PMDD, breastfeeding or pregnant. Using oral or systemic contraception. |
|
| Interventions | Screening: Two screening cycles Placebo run in: One cycle Intervention: Paroxetine 25mg taken orally in the morning throughout the cycle for three cycles versus placebo taken orally in the morning throughout the cycle for three cycles Timing of administration: No details as to which day of the cycle medication commenced. Placebo taken orally in the morning throughout the cycle for three cycles Summary measures: Treatment cycle 3 LOCF data |
|
| Outcomes | Visual Analogue Scale (VAS) recorded daily; Clinical Global Impressions of Severity scale (CGI‐S); Sheehan Disability Scale (SDS) | |
| Notes | Symptoms rated daily Mean VAS score calculated by averaging the item score over the last five days of the luteal phase prior to menstruation Authors contacted, no response Funding GlaxoSmithKline |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, similar appearing medication, no details as to whom was blinded. Evidence that patients were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 371 randomised and 325 analysed by ITT (87%). 12.5 mg group: 105/121 analysed for response (6 excluded, 12 adverse events, 4 loss to follow up, 2 protocol deviation, 5 other reason, 3 lack of efficacy) 25 mg group: 102/125 analysed for response (5 excluded, 20 adverse events, 10 loss to follow up, 5 protocol deviation, 3 other reason) Placebo:118/125 analysed for response (1 excluded, 9 adverse events, 3 loss to follow up, 5 protocol deviation, 8 other reason, 3 lack of efficacy) |
| Selective reporting (reporting bias) | Unclear risk | Does not state how adverse events data were collected |
| Other bias | Unclear risk | Placebo responders apparently excluded |
Steiner 1995.
| Methods | 3‐arm parallel trial: 20 mg fluoxetine, 60 mg fluoxetine or placebo | |
| Participants | Country: Canada Site: Multi‐centre ‐ 7 university affiliated clinics Recruitment: 405 women screened. 313 randomised to three conditions, 277 completed cycle 1 of phase 2 and were eligible for analysis. 60mg: 106 randomised 20mg: 102 randomised, placebo: 96 105 randomised. Refer to Table of bias for details of attrition. Mean age 36+/‐5 years Inclusion: Women ages 18 to 45 years meeting diagnostic criteria for LLPDD with at least one year history of 5+ symptoms attributable to the disorder that began premenstrually and remitted post‐menstrually. Severe enough to affect activities of daily living as assessed in screening cycles. Menstrual cycles ranging from 24 to 35 days. Exclusion: Pregnant or lactating, taking oral contraceptive, had irregular menstrual cycles, had an unstable medical condition, a seizure disorder with a seizure within the last year, a record of multiple adverse drug reactions, known allergies to inhibitors of the reuptake of serotonin or a history of fluoxetine use. Other major psychiatric syndrome, expressed suicide ideation or intent, had used psychoactive medication or investigational drugs within two months prior to the study or were taking any other medication to treat premenstrual symptoms. |
|
| Interventions | Screening: Two screening cycles Placebo run in: single‐blind placebo Intervention:
Timing of administration: Treatment began on day 1 of the third menstrual cycle Summary measures: Efficacy data used for all women completing at least one cycle of treatment |
|
| Outcomes | Observer and subject assessed VAS Prospective Record of the Impact and Severity of Menstrual Symptomology Side effects |
|
| Notes | Withdrawals are number withdrawn after 6 cycles Analysable data for 1st cycle only Funded by Eli‐Lilly |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind (no details as to whom was blinded) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 313 women randomised; 277 completed cycle 1 of phase 2 and were included in ITT analysis (88%). Only 180/313 completed protocol (58%) 33 withdrawals in fluoxetine (20mg): side effects (11), lack of efficacy (4), loss to follow up (2), personal reasons (9), protocol violation (7); 47 withdrawals in Fluoxetine 60mg group: side effects (35), lack of efficacy (2), loss to follow up (2), personal reasons (4), protocol violation (4); Placebo withdrawals = 53: side effects (8), lack of efficacy (27), loss to follow up (5), personal reasons (6), protocol violations (7) |
| Selective reporting (reporting bias) | Unclear risk | Adverse effects data apparently collected retrospectively |
| Other bias | Unclear risk | Placebo responders excluded |
Steiner 2005.
| Methods | Randomised, double‐blind, placebo‐controlled, fixed dose study | |
| Participants | Country: International multi‐centre study Site: Women attending outpatients department Recruitment: Women recruited from outpatient department. Mean age paroxetine CR 12.5mg 35.9±6.01 years, placebo 36.9±5.51 years. 1615 women screened. 373 randomised Paroxetine12.5 mg: 130/131 analysed, 104/131 completed the trial (1 excluded, 13 adverse events, 4 protocol violation, 1 loss to follow up, 6 other reason, 2 lack of efficacy) Paroxetine 25 mg:116/119 analysed, 87/119 completed trial (3 excluded, 16 adverse events, 2 protocol violation, 3 loss to follow‐up, 6 other reason, 2 lack of efficacy) Placebo: 120/123 analysed, 101 completed the trial (3 excluded, 5 adverse events, 2 protocol violation, 6 other reason, 6 lack of efficacy) Inclusion: Age 18‐45, regular menstrual cycles (22‐35 days), meeting DSM‐IV criteria for PMDD. Having had condition for at least one year during which symptoms needed to have been present for nine out of twelve cycles. Needed to have baseline rating of at least 'mildly ill' according to the Clinical Global Impression severity of illness scale (CGI‐S). Women required to demonstrate a 200% worsening on one core mood symptom or a 100% worsening on two or more of the core mood symptoms during the luteal phases of two or more reference cycles relative to their follicular phases score. Mean follicular phase score </20mm, mean luteal phase score >/40mm. Exclusion: Meeting DSM‐IV criteria for other Axis 1 disorders (except specific phobias) in the six months before screening, diagnosed with gynaecological or other clinically significant disease, had clinically significant depressive symptomatology during the follicular phase, suicide risk, taking medication that could interfere with PMDD symptoms or their assessment, using oral contraceptives, had previous treatment for PMDD, had participated in a trial for PMDD with SSRIs, were pregnant or breastfeeding. |
|
| Interventions | Screening: Two to three screening cycles Placebo run‐in: Single‐blind placebo run‐in taking medication from when they estimated they were 14 days before estimated due date of menses and to continue until start of menses. No medication taken during follicular phase Intervention:
Timing of administration: Requested to take medication once daily in the morning during the luteal phase Summary measures: Data summarised using LOCF for treatment cycle 3, change from baseline |
|
| Outcomes | Visual Analogue Scale (VAS);Observer rated Premenstrual Tension Scale (PMTS‐O); Global Assessment of Disease Severity (CGI‐S); Global Assessment of Disease Improvement (CGI‐I); Patient Global Evaluation (PGE); Sheehan Disability Scale (SDS); Adverse events | |
| Notes | Daily symptom rating Author contacted, no response Funded by GlaxoSmithKline study no 29060/717 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not stated |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind (no details as to whom was blinded) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 366/373 women (98%) analysed by ITT for efficacy after at least one post‐dose assessment |
| Selective reporting (reporting bias) | Unclear risk | Does not state how adverse events data collected |
| Other bias | Unclear risk | Placebo responders excluded |
Steiner 2008.
| Methods | Randomised, double‐blind, placebo‐controlled study. 103 randomised, 99 analysed | |
| Participants | Country: Canada Site: 4 health centres Recruitment: via outpatient clinic Inclusion: aged 18‐45 presenting with PMDD meeting DSM IV criteria, with regular menstrual periods, reliable non hormonal contraception, at least one of four core symptoms prominent (irritability, depressed mood, tension or affective lability), baseline Clinical Global Impressions Severity of Illness Scale score over 3. Exclusion criteria: taking oral contraception, breastfeeding, pregnant or planning pregnancy, any Axis I disorder, suicidal risk, SSRI use for premenstrual symptoms, on medication that could affect PMDD symptoms, clinically significant abnormality on screening blood tests, baseline score of over 10 on Montgomery Asberg Depression Rating Scale. |
|
| Interventions | Screening: 2 cycles Placebo run in: none Intervention: Paroxetine 10 mg or 20 mg versus placebo Timing of administration: luteal Summary measures: change from baseline |
|
| Outcomes | Total symptoms (clinician‐rated PMTS‐O), VAS irritability scale, premenstrual tension scale, response rate using CGI‐S score (1 or 2), response rate (at least 50% reduction in each of VAS mood items), Sheehan disability Scale (SDs not reported) | |
| Notes | Funded by Glaxo. Primary author emailed to request data for total symptoms, but no reply received | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double‐blinded ‐ no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Last‐observation‐carried‐forward analysis used for 99/103 women (96%) |
| Selective reporting (reporting bias) | High risk | Does not state how adverse effects data collected. Data were collected on total symptoms and response rate but reported in published article as p values only, and findings for these total symptoms are marked as "not available" on the sponsor's web site. Findings for severe adverse events (not a review outcome) differ on published and unpublished reports |
| Other bias | Unclear risk | 30% (30/99) of participants had protocol violations. Groups well balanced for demographic characteristics |
Stone 1991.
| Methods | Randomised, double‐blind, placebo‐controlled 2‐arm parallel trial | |
| Participants | Country: USA Site: Based in PMS clinic Recruitment: Women enrolled via self‐referral from newspaper advertisements. 152 completed two cycles of daily symptom rating (42 did not meet criteria of LLPDD). 110 women underwent further psychiatric evaluation. 71 of these were eligible to participate and 25 of these elected to participate and were randomised. The 46 who declined were unwilling to take medication or to be involved in a placebo controlled study. 25 entered first cycle, 5 were eliminated from the study and the remaining 20 were randomised. Mean age 36 years (27 to 45). Mean age fluoxetine 36.6 years, mean age placebo group 35.4 years. Inclusion: Met criteria of DSM‐III‐R diagnosis of LLPDD, physically healthy, and normal gynaecological examination. 30% increase in symptoms during the luteal phase of two cycles in at least five of the ten symptom categories listed in DSM‐III‐R. Average score of premenstrual week had to show 30% increase in severity over average score of postmenstrual week. At least one of the five positive symptoms had to be 'affective'. Exclusion: No current major psychiatric disorder, pregnant, be receiving anti‐depressants, anxiolytics, diuretics, hormones, neuroleptics or have irregular menstrual cycles. |
|
| Interventions | Screening: Two screening cycles Placebo run in: Single blind placebo for first cycle Intervention: 20mg fluoxetine (n=10) every day for 2 cycles versus placebo (n=10) taken daily for two cycles Timing of administration: Medication taken in the morning. No details as to what stage of the menstrual cycle medication commenced Summary measures: Mean final scores |
|
| Outcomes | Daily Assessment Form (DAF); Global Assessment Scale (GAS); Adverse events | |
| Notes | Daily symptom rating Funded by Eli‐Lilly | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated |
| Allocation concealment (selection bias) | Unclear risk | Method not stated |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no details as to whom was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 20 women randomised, all analysed |
| Selective reporting (reporting bias) | Unclear risk | Adverse effects data collected retrospectively at clinic visits |
| Other bias | Unclear risk | Placebo responders excluded |
Su 1997.
| Methods | Randomised, double‐blind, placebo‐controlled, crossover trial | |
| Participants | Country: USA Site: No details Recruitment: Self referred in response to local advertisements or referred by physicians. Nineteen randomised (17 completed). Refer to Table of bias for details of attrition. Mean age 36.5±5.4 years Inclusion: Absence of significant medical illness, absence of significant Axis 1 psychiatric illness, including alcohol and substance misuse. Not taking psychoactive medications, hormonal preparations (including oral contraceptives), mineral supplements, or non‐steroidal anti‐inflammatory medications within the past 6 months. Regular menstrual cycles (23‐35 days). Confirmed diagnosis of PMS via a prospective daily 3 item VAS. >30% increase in mean negative mood symptoms, relative to the actual range of the analogue scale used, in the week before menses compared with the week after menses in at least two out of three cycles. Required to use barrier methods of contraception. Exclusion: Appearance of significant mood symptoms during the follicular phase. |
|
| Interventions | Screening: Three cycles of screening Placebo run in: None Intervention: Crossover trial of 20mg fluoxetine every day cycle 1, 20‐60mg every day for 2 further cycles (mean drug dose during cycle 3 was 29.9±10.6mg) versus placebo. At end of first arm there was one cycle washout period Timing of administration: Medication started on the first day of menses and continued for a full menstrual cycle Summary measures: Results of pre and post menstrual weeks were averaged for the three cycles in each condition in the paper. Composite scores calculated for mood symptoms and social impairment symptoms in paper |
|
| Outcomes | Weekly means of 16 item visual analogue scale (VAS) and 21 item daily rating form (DRF) during seven days before and seven days after menses. Beck Depression Inventory (BDI); State Trait Anxiety Inventory ‐ State form (STAI); Rating Scale for Pre Menstrual Tension (PMTS self and PMTS observer); Physical symptom checklist; Adverse events | |
| Notes | 10 women given m‐CPP (serotonin agonist) during follicular and luteal phase. Women who responded to m‐CPP challenge responded to fluoxetine Unable to extract first arm data from paper Daily symptom rating Fluoxetine provided by Eli‐Lilly |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind (patient and rater) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 17/19 women randomised were included in analysis (89%) . Nineteen were enrolled in study and seventeen completed. Two women in treatment arm dropped out due to intolerable migraine headaches (n=1) and unrelated irregular menstrual bleeding (n=1) no details as to which arm of the crossover this occurred in |
| Selective reporting (reporting bias) | Unclear risk | Does not state how adverse effects data collected |
| Other bias | Low risk | No potential source of other bias identified. Crossover study with one month washout period |
Wikander 1998.
| Methods | Randomised double‐blind placebo‐controlled 4‐arm parallel trial | |
| Participants | Country: Sweden Site: not stated Recruitment: 123 women recruited in total to 4 arm study 78 received medication. 39 randomised, 35 completed this comparison of intermittent versus placebo arm. Women recruited via newspaper advertisements, followed by telephone and then structured interview. Mean age of citalopram intermittent was 37±1 and for placebo was 37±5 years. Refer to Table of bias for details of attrition. Inclusion: Marked premenstrual irritability and/or dysphoria appearing regularly during the two weeks preceding menstrual bleeding and terminating a few days after the onset of menstruation. Criteria A, B, C and D for LLPDD in DSM‐IV‐R. Displaying cyclicity with respect to irritability and depressed mood during at least two cycles of prospective rating. >100% increase in either irritability or dysphoria (or both) during the premenstrual week as compared with the postmenstrual week, a mean premenstrual rating of irritability or dysphoria exceeding 20mm. Exclusion: Previous or ongoing mental illness (apart from major depressive or dysthymic disorders), ongoing major depressive or dysthymic disorders, major depressive disorder or dysthymic disorder < 2 years from the time of the interview, ongoing medication for somatic or mental illness (with the exception of casual analgesics), use of oral contraceptives, ongoing alcohol abuse, ongoing somatic illness, irregular menstrual bleeding or a normal cycle length <25 or > 35 days, ongoing or planned pregnancy and <18 years of age, having previous treatment with antidepressants for premenstrual complaints. |
|
| Interventions | Screening: Screening for 2 cycles Placebo run‐in: None
Timing of administration: Medication commenced on the first day of menses Summary measures: Data was analysed for endpoint data using LOCF. Reports median rather than mean scores, so data not included in meta‐analysis |
|
| Outcomes | Visual analogue scale for irritability, depression, tension, anxiety, appetite, bloating, mastalgia Side effects |
|
| Notes | Data on adverse events could not be extracted Funding by Swedish Medical Research Council, Soderstrom Konigska Nursing Home Foundation, Frederrik and Ingrid Thuring's Foundation, Knut and Alice Wallenberg's Foundation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clearly stated how many randomised. 78 received medication, of whom 69 analysed (88%). Of those who received medication, in the continuous group 2/19 dropped out for side effects, in the semi‐intermittent group 3/20 dropped out, two for side effects and 1 protocol violation; in the intermittent group 1/19 dropped out for side effects and in the placebo group 3/20 dropped out for side effects |
| Selective reporting (reporting bias) | Low risk | Data on adverse effects prospectively collected. No potential source of reporting bias identified |
| Other bias | Unclear risk | No other potential source of bias identified |
Wood 1992.
| Methods | Randomised, placebo‐controlled, crossover trial 2X3 cycles | |
| Participants | Country: USA Site: Women recruited from a PMS clinic Recruitment: Women were between the age of 33‐42 years. 8 women randomised, 8 completed Inclusion: Regular menstrual cycles (26‐32 days), ovulating, onset of premenstrual symptoms during the second half of the menstrual cycle with resolution within the first four days after the onset of menstruation Exclusion: Past or present psychiatric disorder, family history of depression in a first degree relative, significant medical or gynaecological disorders |
|
| Interventions | Screening: Screening for two to three cycles Placebo run‐in: No Intervention: 20mg fluoxetine every day for 3 cycles then crossover to placebo taken daily for 3 cycles Timing of administration: No details as to when in the cycle medication commenced Summary measures: Data pooled, unable to separate first‐arm data |
|
| Outcomes | Calendar premenstrual Experiences, Profile of Mood States, Beck Depression Inventory, STATE‐TRAIT Anxiety Inventory | |
| Notes | Daily symptom rating Funding by National Institute of child health and human development |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, crossover design. "The order in which the subjects received the study drugs was determined according to a preselected randomized list based on the sequential assignment of subject numbers" |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 8/8 women randomised were analysed |
| Selective reporting (reporting bias) | Unclear risk | Data on adverse events extracted from symptom questionnaire, not solicited separately |
| Other bias | Unclear risk | Crossover study, no washout period mentioned |
Yonkers 1997.
| Methods | Randomised, double‐blind, placebo‐controlled, 2‐arm parallel trial. | |
| Participants | Country: USA Site: 12 university affiliated psychiatric and gynaecological departments Recruitment: Women recruited by advertisement and referral. 447 screened, 243 randomised, 200 completed (99 treatment, 101 placebo). 121 received sertraline, there were 22 withdrawals (ineffective n=2, unavailable for follow up n=3, adverse event n=10, laboratory abnormality n=1, intercurrent illness n=2, poor compliance n=4), 122 women received placebo, there were 21 withdrawn (ineffective medication n= 6, unavailable for follow up n= 6, adverse event n=2, laboratory abnormality n=0, intercurrent illness n=1, poor compliance n=6). Mean age of sertraline group 36.8±4.8 (23‐45), mean age of placebo group 36.5±5.0 (25‐45) years. Inclusion: Age range 24 to 45 years, regular menstrual cycles (24 to 36 days), more than two year history of PMDD. Exclusion: Failure to confirm isolated luteal phase symptoms for at least two cycles based on daily symptom ratings, those meeting criteria for other, mood anxiety or eating disorder within previous 6 months, those with alcohol or other drug use or dependence within 12 months and those with a lifetime history of organic mental syndrome, psychotic disorder, or antisocial, schizotypal, or severe borderline personality disorder. Clinically symptomatic endometriosis, hysterectomy, perimenopausal status as determined by FSH >/20U/L, neurological disease or any severe or unstable general medical illness. |
|
| Interventions | Screening: Two screening cycles Placebo run in: One single blind placebo treatment Intervention: 3 cycles of 50‐150mg sertraline every day administered orally (n=121). Mean dosage across the three cycles was 79.1mg versus placebo administered orally daily for three cycles (n=122) Timing of administration: Treatment commenced on day 1 of menses and continued throughout cycle Summary measures: Review has used 'end point' data from paper which is the final visit for each patient |
|
| Outcomes | Daily Record of Severity of Problems (DRSP); Hamilton rating Scale for Depression (HRSD); Clinical Global Impression Scale (CGI‐S) (CGI‐I); Social Adjustment Scale (SAS); Patient Global Evaluation (PGE); Adverse events | |
| Notes | Daily symptom rating Funded by Pfizer | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation in blocks of four |
| Allocation concealment (selection bias) | Low risk | Randomisation occurred at a central location |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double blind with 'matching' placebo ‐ no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 200/243 completed trial (82%). All participants included in analysed by intention to treat, with last value carried forward. Analyses with >20% data missing not reported |
| Selective reporting (reporting bias) | Unclear risk | Data on adverse events collected retrospectively at clinic appointments. No data on rates of individual adverse events in the two groups |
| Other bias | Unclear risk | Placebo responders excluded |
Young 1998.
| Methods | Randomised double‐blind crossover trial | |
| Participants | Country: USA Site: Walter Reed Army Medical Centre Recruitment: 50 women screened, 31 selected. Following two screening cycles 17 women randomised, 11 completed. Three women failed to complete study due to medication side effects (2x sertraline and 1x placebo phase). One subject moved out of area and two discontinued for undetermined reasons. Women recruited from medical centre who had responded to advertisements in local military newspapers and gynaecology clinics. Inclusion: Age between 18 and 45 years. Meeting DSM‐IV criteria. After screening cycles to have overall COPE score 30% greater during late luteal phase compared with follicular phase. Exclusion: Any history of mental health treatment in previous 18 months, taking psychotropic medication. Diagnosis of active disease or pregnancy. |
|
| Interventions | Screening: Two cycles screening with no medication Placebo run in: None Intervention: 50mg sertraline day 15 to menses for 2 cycles versus placebo with one cycle washout Timing of administration: Commencing day 15 to the first day of menses Summary measures: Data from both arms combined |
|
| Outcomes | Calendar of Premenstrual Experiences (COPE) patient completed. Total scores and physical and behavioural subscores | |
| Notes | Unable to extract data as not distinguishable by arm of study. Authors not contacted Independently funded |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind (physicians assessing women were blinded to treatment) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 11/17 included in efficacy analysis (65%) |
| Selective reporting (reporting bias) | Unclear risk | Adverse events not reported |
| Other bias | Low risk | No other potential source of bias identified |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alpay 2001 | Not placebo controlled |
| Brandenburg 1993 | Open trial. Not placebo controlled |
| De la Gandara 1997 | Open trial. Not placebo controlled |
| Diegoli 1998 | Quasi‐randomised (by date of presentation) |
| Elks 1993 | Open trial. Not placebo controlled |
| Flores Ramos 2003 | Randomised trial of intermittent and continuous citalopram. Not placebo controlled |
| Freeman 1996 | Open trial. Not placebo controlled |
| Freeman 1999b | Open trial. Not placebo controlled |
| Freeman 2000 | No extractable symptom data presented; only data on baseline postmenstrual symptoms |
| Freeman 2002 | Open trial. Not placebo controlled |
| Freeman 2005 | Randomised trial but not placebo controlled |
| Glaxo 2002 | Extension study, including only women who completed one of three RCTs and who not experience clinically significant adverse effects |
| Landen 2009 | Unrandomised follow‐up of responders to Landen 2007 |
| Miller 2008 | Participants had premenstrual exacerbation of depression and did not have with a prospective diagnosis of PMS or PMDD |
| Pearlstein 1994 | Open trial. Not placebo controlled |
| Pearlstein 2000 | No extractable symptom data presented; only data on psychosocial functioning |
| Rickels 1990 | Not blind, not randomised, placebo group from parallel trial |
| Steiner 1997 | Open trial. Not placebo controlled |
| Sundblad 1992 | Intervention (clomipramine) not an SSRI |
| Sundblad 1993 | Intervention (clomipramine) not an SSRI |
| Sundblad 1997 | Open trial. Not placebo controlled |
| Veeninga 1990 | RCT. Participants were not screened at study entry as to whether they met diagnostic criteria for PMS or PMDD |
| Wu 2008 | No placebo group |
| Yonkers 1996 | Open trial. Not placebo controlled |
| Yonkers 2002 | Not randomised |
| Yonkers 2006 | Randomised crossover trial using paroxetine. However treatment was only administered for one cycle before crossover and one cycle after crossover. Study did not therefore meet review entry criteria |
Characteristics of ongoing studies [ordered by study ID]
Yonkers 2007.
| Trial name or title | Evaluating the Effectiveness of Sertraline in Treating Women With Premenstrual Dysphoric Disorder NCT00536198 |
| Methods | RCT |
| Participants | Women with premenstrual dysphoric disorder (n=300) |
| Interventions | Sertraline versus placebo |
| Outcomes | PMTS scale, IDS scale, DRSP scale, SSRI withdrawal checklist |
| Starting date | 2007 to 2012 |
| Contact information | http://clinicaltrials.gov/ct2/show/NCT00536198 |
| Notes |
Yonkers 2010.
| Trial name or title | Comparison of Fluoxetine, Calcium and Placebo for the Treatment of Moderate to Severe Premenstrual Syndrome (PMS) |
| Methods | RCT |
| Participants | Women with moderate to severe PMS |
| Interventions | Fluoxetine, calcium, placebo |
| Outcomes | Efficacy of calcium carbonate to fluoxetine in the treatment of moderate to severe PMS |
| Starting date | 2000 to 2010 |
| Contact information | http://clinicaltrials.gov/ct2/show/NCT00965562 |
| Notes | Principal investigator stated in personal email July 2012 that study had been submitted |
Differences between protocol and review
2013 update:
updated methods section to current Cochrane recommendations;
designated adverse events a primary outcome (previously a secondary outcome);
amalgamated each included study under a single study ID, with all references included (abstracts of included studies formally listed under excluded studies);
reformatted analyses by dose;
unpooled SMDs of change scores and end scores (because the Cochrane Handbook for Systematic Reviews of Interventions recommends that these are not pooled).
Contributions of authors
2013 update
Jane Marjoribanks (JM) led the 2013 update; performed searches; selected included and excluded trials; performed data extraction, quality assessment of trials and statistical analysis; drafted the text and incorporated changes suggested by other authors.
Katrina Wyatt (KW) checked the study selection and read the draft.
Shaughn O'Brien (SO) commented on the draft from a clinical perspective and suggested changes and additions.
Julie Brown (JB) checked the risk of bias assessment and data extraction.
Earlier versions of the review
JB took the lead in writing the 2009 update review, performed searches, selected included and excluded trials, performed data extraction, quality assessment of trials and statistical analysis, drafted the text and incorporated changes suggested by other authors. JM assisted in data extraction in the 2009 update. SO and KW commented on the review update
SO conceptualised the original review, commented on drafts of the protocol and the original review, and provided clinical interpretation of the data. He also assisted in providing comments on draft versions of the update. KW took the lead in writing the original protocol and review, performed initial searches for trials, was involved in selection of included trials, performed data extraction and quality assessment of trials, and assisted in statistical analysis and interpretation of the data. PWD assisted in writing the original protocol and review, contacted authors for additional information, was involved in selection of included trials, performed data extraction and quality assessment of trials, and conducted the statistical analysis and interpretation of the data.
Sources of support
Internal sources
No source provided, Not specified.
External sources
None provided, Not specified.
Declarations of interest
Julie Brown and Jane Marjoribanks have no conflict of interest.
PMS O'Brien has been funded over many years for research personnel, consultancies, lectures and conferences by the following companies: Bayer Schering, Hoechst Marion Roussel Ltd, Shire Pharmaceuticals, Smith Kline Beecham, Eli Lilly, Searle, Sanofi Winthrop, Zeneca, Organon, Solvay Pharmaceuticals and Novo Nordisk.
KM Wyatt has no conflict of interest.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Arrendondo 1997 {published data only}
- Arredondo‐Soberon F, Freeman EW, Sondheimer SJ. Relationship and response of food cravings and depression to sertraline in patients with premenstrual syndrome. Fertility and Sterility 1997;October:S28. [Google Scholar]
Cohen 2002 {published data only}
- Cohen L, Miner C, Brown E, Dillon J. Efficacy of intermittent fluoxetine dosing in premenstrual dysphoric disorder (PMDD). European Neuropsychopharmacology 2001;11 Suppl 3:210. [Google Scholar]
- Cohen L, Miner C, Brown E, Freeman E, Halbreich U, Sundell K, McCray S. Premenstrual daily fluoxetine for premenstrual dysphoric disorder: A placebo controlled, clinical trial using computerized diaries. Obstetrics and Gynecology 2002;100(3):435‐44. [DOI] [PubMed] [Google Scholar]
- Cohen L, Soares C, Yonkers K, et al. Paroxetine controlled release (CR) is effective in the treatment of premenstrual dysphoric disorder (PMDD): Results from a randomized placebo controlled trial. 15th Annual US Psychiatric and Mental Health Congress. October 28‐ November 3rd. Las Vegas, Nevada. 2002:Abstract 39.
- Judge R, Brown E, Miner C, Dillon J. Intermittent fluoxetine dosing in premenstrual dysphoric syndrome. World Journal of Biological Psychiatry 2001;2 Suppl:204. [Google Scholar]
- Miner C, Cohen LS, O'Brien PMS, Davis S, Brown E, Jacobson J. Predictors of response to luteal phase fluoxetine treatment of PMDD. The International Journal of Neuropsychopharmacology 2002;5 Suppl:87. [Google Scholar]
Cohen 2004 {published data only}
- Cohen L, Soares C, Yonkers K, Bellew K, Bridges I, Heller V. Paroxetine controlled release is effective in treating premenstrual dysphoric disorder. Obstetrics and Gynecology 2003;101 Suppl 4:111. [Google Scholar]
- Cohen L, Soares C, Yonkers K, Bellew K, Bridges I, Steiner M. Paroxetine controlled release for premenstrual dysphoric disorder: A double blind, placebo controlled trial. Psychosomatic Medicine 2004;66:707‐13. [DOI] [PubMed] [Google Scholar]
- GlaxoSmithKline. A Double‐Blind, Placebo‐Controlled, Three‐Arm Fixed‐Dose Study of Paroxetine CR Continuous Treatment (12.5 mg/day and 25 mg/day) for Premenstrual Dysphoric Disorder. http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F677&studyId=3742A8A5‐2C3C‐4A40‐B9B5‐7975DF70CF53&compound=paroxetine Accessed 16 August 2012.
- GlaxoSmithKline NZ. Phone call from a representative of GlaxoSmithKline New Zealand (in response to an email sent by J Marjoribanks) confirming that 29060/677 (Cohen 2004) and 29060/689 (Pearlstein 2005) were separate studies July 2012.
- Pearlstein 2012. Email to Jane Marjoribanks from Teri Pearlstein advising that participants in Pearlstein 2005 and Cohen 2004 did not, to her knowledge, overlap. 22.6.12.
Crnobaric 1998 {published data only}
- Crnobaric C, Jasovic‐Gasic M, Milanovanic S, Miljevic C. Treatment of premenstrual dysphoric disorder with fluoxetine during the luteal phase. 9th Congress of the Association of European Psychiatrists. Copenhagen, Denmark, 20‐24 September 1998.
Eriksson 1995 {published data only}
- Eriksson E, Hedberg MA, Andersch B, Sunblad C. The serotonin reuptake inhibitor paroxetin is superior to the noradrenaline reuptake inhibitor maprotiline in the treatment of premenstrual syndrome. Neuropsychopharmacology 1995;12:167‐76. [DOI] [PubMed] [Google Scholar]
Eriksson 2008 {published data only}
- Eriksson E, Ekman A, Sinclair S, Sorvik K, Ysandeer C, Mattson U‐B, Nissbrandt H. Escitalopram administered in the luteal phase exerts a marked and dose‐dependent effect in premenstrual dysphoric disorder. Journal of Clinical Psychopharmacology 2008;28(2):195‐202. [DOI] [PubMed] [Google Scholar]
Freeman 1999 {published data only}
- Freeman EW, Rickels K, Sondheimer S, Polansky M. Differential response to antidepressants in women with premenstrual syndrome. Archives of General Psychiatry 1999;56:932‐9. [DOI] [PubMed] [Google Scholar]
Freeman 2004 {published data only}
- Freeman E, Rickels K, Sondheimer S, Polansky M, Xiao S. Continuous or intermittent dosing with sertraline for patients with severe premenstrual syndrome or premenstrual dysphoric disorder. American Journal of Psychiatry 2004;161(2):343‐51. [DOI] [PubMed] [Google Scholar]
- Freeman EW, Rickels K, Sammel MD, Lin H, Sondheimer SS. Time to relapse after short‐term or long‐term sertraline treatment for severe premenstrual syndromes. Archives of General Psychiatry 2009;66(5):537‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Freeman 2010 {unpublished data only}
- Freeman E. Escitalopram for Premenstrual Syndrome (PMS) in Teens: A Pilot Study NCT00523705. http://clinicaltrials.gov/ct2/show/NCT00523705.
Glaxo 1996 {unpublished data only}
- GlaxoSmithKline. A Double‐Blind, Placebo Controlled Study to Investigate the Efficacy, Safety and Tolerability of Paroxetine in Patients with Premenstrual Dysphoric Disorder. [Study No PAR 29060.400]. http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F400&studyId=80268FE4‐8DB0‐4A5A‐8CF4‐51162DA5EE83&compound=paroxetine Accessed 16 August 2012.
Glaxo 1996a {unpublished data only}
- GlaxoSmithKline. A Multicentre, Double‐Blind, Placebo‐Controlled Study to Investigate the Effects of Paroxetine (5mg OD Vs 10mg OD Vs 20mg OD) in Patients with Premenstrual Dysphoric Disorder (PMDD) [Study No PAR 29060.427]. http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F427&studyId=46B83DBA‐A3AD‐44FB‐84F7‐51BC66679F35&compound=paroxetine Accessed 16 August 2012.
Glaxo 2001 {unpublished data only}
- GlaxoSmithKline. A Double‐Blind, Placebo‐Controlled, 3‐Arm, Fixed‐Dose Study of Paroxetine CR Continuous Treatment (12.5 mg/day and 25 mg/day) for Premenstrual Dysphoric Disorder [Study No 29060/688]. http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F688&studyId=4EAE38F8‐954D‐453B‐BA8C‐32CA4FFAD277&compound=paroxetine Accessed 16 August 2012.
Halbreich 1997 {published data only}
- Halbreich U, Smoller JW. Intermittent luteal phase sertraline treatment of dysphoric premenstrual syndrome. Journal of Clinical Psychiatry 1997;58:399‐402. [DOI] [PubMed] [Google Scholar]
Halbreich 2002 {published data only}
- Halbreich U, Bergeron R, Freeman E, Stout A, Cohen L. Intermittent (luteal phase) dosing of sertraline effective in PMDD. International Journal of Neuropsychopharmacology 2000;3 Suppl 1:248. [Google Scholar]
- Halbreich U, Bergeron R, Stout A, Freeman E, Yonkers K, Pearlstein T, et al. Dosing of sertraline is effective in premenstrual dysphoric disorder. 155th Annual Meeting of the American Psychiatric Association. 13th‐18th May 2000.
- Halbreich U, Bergeron R, Yonkers K, Freeman E, Stout A, Cohen L. Efficacy of intermittent, luteal phase sertraline treatment of premenstrual dysphoric disorder. Obstetrics and Gynecology 2002;100(6):1219‐29. [DOI] [PubMed] [Google Scholar]
- Pearlstein T, Yonkers K, Fayyad R, Gillespie J. Pretreatment pattern of symptom expression in premenstrual dysphoric disorder. Journal of Affective Disorders 2005;85:275‐82. [DOI] [PubMed] [Google Scholar]
Jermain 1999 {published data only}
- Jermain DM, Preece CK, Sykes RL, Kuehl TJ, Sulak PJ. Luteal phase sertraline for the treatment of premenstrual dysphoric disorder. Archives of Family Medicine 1999;8:328‐32. [DOI] [PubMed] [Google Scholar]
Kornstein 2006 {published data only}
- Kornstein S, Gillespie J. Double blind placebo controlled study of sertraline in premenstrual syndrome. 155th Annual Meeting of the American Psychiatric Association. May 18‐23, Philadelphia PA. 2002:NR248.
- Kornstein S, Pearlstein T, Fayyad R, Farfel G, Gillespie J. Low dose sertraline in the treatment of moderate to severe premenstrual syndrome: Efficacy of 3 dosing strategies. Journal of Clinical Psychiatry 2006;67(10):1624‐32. [DOI] [PubMed] [Google Scholar]
Landen 2007 {published data only}
- Bellew KM, Landen M, Hunter B, McCafferty JP. Social functioning improves with paroxetine treatment. 155th Annual Meeting of the American Psychiatric Association. May 18‐23, Philadelphia. 2002:NR282.
- GlaxoSmithKline. A Placebo‐Controlled Study to Investigate the Efficacy of Intermittent and Continuous Treatment With Paroxetine in Patients With Premenstrual Dysphoric Disorder (PMDD). http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F658&studyId=186F6412‐CDE1‐4505‐8ACD‐FBB1858393EB&compound=paroxetine Accessed 16 August 2012.
- Landen M, Sorvik K, Ysander C, Allgulander C, Nissbrandt H, Gezelius B, et al. A placebo controlled trial exploring the efficacy of paroxetine for the treatment of premenstrual dysphoria. American Psychiatric Association, 155th annual meeting. May 18‐23. Philadelphia, PA., 2002.
- Landen M, Ysander K, Sorvik K, Nissbrandt H, Allgulander C, Hunter B, et al. A placebo controlled study of the efficacy of intermittent and continuous treatment with paroxetine for premenstrual dysphoric disorder (PMDD). European Neuropsychopharmacology 2001;11 Suppl 3:308‐9. [Google Scholar]
- Landen MSG. A Placebo‐Controlled Study to Investigate the Onset of Action of Paroxetine in Premenstrual Dysphoria. http://clinicaltrials.gov/ct2/show/NCT00516113 2007.
- Länden M, Nissbrandt H, Allgulander C, Sorvik K, Ysander C, Eriksson E. Placebo controlled trial comparing intermittent and continuous paroxetine in premenstrual dysphoric disorder. Neuropsychopharmacology 2007;32:153‐61. [DOI] [PubMed] [Google Scholar]
Menkes 1992 {published data only}
- Menkes DB, Taghavi E, Mason PA, Spears GF, Howard RC. Fluoxetine treatment of severe premenstrual syndrome. BMJ 1992;305(6489):346‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menkes DB, Taghavi E, Mason PA, Spears GF, Howard RC. Fluoxetine treatment of severe premenstrual syndrome. International Clinical Psychopharmacology 1993;8:95‐102. [DOI] [PubMed] [Google Scholar]
Miner 2002 {published data only}
- Miner C, Brown E, McCray S, Gonzales J, Wohlreich M. Weekly luteal phase dosing with enteric coated fluoxetine 90mg in premenstrual dysphoric disorder: A randomised, double blind, placebo controlled clinical study. Clinical Therapeutics 2002;24(3):417‐33. [DOI] [PubMed] [Google Scholar]
- Miner C, Cohen LS, O'Brien PMS, Davis S, Brown E, Jacobson J. Predictors of response to luteal phase fluoxetine treatment of PMDD. The International Journal of Neuropsychopharmacology 2002;5 Suppl:87. [Google Scholar]
Ozeren 1997 {published data only}
- Ozeren S, Corakci A, Yucesoy I, Mercan R, Erhan G. Fluoxetine in the treatment of premenstrual syndrome. European Journal of Obstetrics, Gynecology, and Reproductive Biology 1997;73:167‐70. [DOI] [PubMed] [Google Scholar]
Pearlstein 1997 {published data only}
- Pearlstein TB, Stone AB, Lund SA, Scheft H, Zlotnick C, Brown WA. Comparison of fluoxetine, bupropion and placebo in the treatment of premenstrual dsyphoric disorder. Journal of Clinical Psychopharmacology 1997;17:261‐6. [DOI] [PubMed] [Google Scholar]
Pearlstein 2005 {published data only}
- GlaxoSmithKline. A Double‐Blind, Placebo‐Controlled, 3‐Arm, Fixed‐Dose Study of Paroxetine CR Continuous Treatment (12.5 mg/day and 25 mg/day) for Premenstrual Dysphoric Disorder. http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F689&studyId=6F9A5C75‐99C3‐4F11‐AED7‐A122263D1746&compound=paroxetine Accessed 16 August 2012.
- GlaxoSmithKline NZ. Phone call from a representative of GlaxoSmithKline New Zealand (in response to an email sent by J. Marjoribanks) confirming that 29060/677 (Cohen 2004) and 29060/689 (Pearlstein 2005) were separate studies July 2012.
- Pearlstein 2012. Email to Jane Marjoribanks from Teri Pearlstein advising that participants in Pearlstein 2005 and Cohen 2004 did not, to her knowledge, overlap. 22.6.12.
- Pearlstein T, Bellew K, Endicott J, Steiner M. Paroxetine controlled release for premenstrual dysphoric disorder: remission analysis following a randomized, double blind, placebo controlled trial. 41st Annual Meeting of the American College of Neuropsychopharmacology (ACNP), San Juan, Puerto Rico. Dec 8‐12 2002.
Steiner 1995 {published data only}
- Dillon J, Steiner M, Judge R, Brown E. Fluoxetine improves social functioning in women with premenstrual dysphoria. International Journal of Gynecology and Obstetrics 2000;70 Suppl 3:99. [Google Scholar]
- Koke S, Steiner M, Judge R, Babcock S, Dillon J. Efficacy of fluoxetine in improving mood symptoms and social impairments in patients with PMDD. Psychosomatics 2001;42(2):178. [Google Scholar]
- Nilsson M Judge R, Brown E, Schuler C. Fluoxetine's efficacy in improving mood. physical and social impairment symptoms associated with PMDD. International Journal of Gynecology and Obstetrics 2000;Abstract Book 3:96. [Google Scholar]
- Steiner M, Babcock S, McCray SD, Romano S. Fluoxetine's efficacy in improving premenstrual dysphoric disorder. 152nd Annual Meeting of the American Psychiatric Association, Washington DC. USA. 15‐20th May 1999.
- Steiner M, Babcock S, Steinberg SI, Stewart DE, Carter D, Berger C. Fluoxetine's efficacy in improving physical symptoms associated with pdd: results from a multisite, randomized placebo controlled trial. 152nd Annual Meeting of the American Psychiatric Association. Washington DC USA. 15‐20 May 1999.
- Steiner M, Brown E, Trzepacz P, Dillon J, Berger C, Carter D, et al. Fluoxetine improves functional work capacity in women with premenstrual dysphoric disorder. Archives of Women's Mental Health 2003;6:71‐7. [DOI] [PubMed] [Google Scholar]
- Steiner M, Judge R, Brown E, Dillon J. Fluoxetine improves social functioning in women with premenstrual dysphoric disorder (PMDD). International Journal of Neuropsychopharmacology. 2000; Vol. 3, issue suppl1:247.
- Steiner M, Judge R, Brown E, Dillon J. Fluoxetine's effect on food craving during the luteal phase in women with premenstrual dysphoric disorder (PMDD). International Journal of Gynecology and Obstetrics 2000;70(4):D147. [Google Scholar]
- Steiner M, Romano S, Babcock S. Fluoxetine's efficacy in improving physical symptoms associated with PMDD. Journal of the European College of Neuropsychopharmacology 1999;9 Suppl 5:208. [Google Scholar]
- Steiner M, Romano S, Babcock S. Fluoxetine's efficacy in improving physical symptoms associated with PMDD. Journal of the European College of Neuropsychopharmacology 2001;Suppl:208. [Google Scholar]
- Steiner M, Romano S, Babcock, Dillon J, Shuler C, Berger C, et al. The efficacy of fluoxetine in improving physical symptoms associated with premenstrual dysphoric disorder. British Journal of Obstetrics and Gynaecology 2001;108:462‐8. [DOI] [PubMed] [Google Scholar]
- Steiner M, Romano SJ, Babcock S, McCray S, Dillon JA. Efficacy of fluoxetine in improving mood symptoms, physical symptoms and social impairment in patients with PMDD. Journal of the European College of Neuropsychopharmacology 2001;Suppl:208. [Google Scholar]
- Steiner M, Steinberg S, Stewart D, Carter D, Berger C, Reid R, et al. Fluoxetine in the treatment of premenstrual dysphoric disorder. The New England Journal of Medicine 1995;332:1529‐34. [DOI] [PubMed] [Google Scholar]
- Steiner M, Trezepacz PT, Brown E. Premenstrual dysphoric disorder (PMDD) and work efficiency: Response to fluoxetine in a randomized clinical trial. European Neuropsychopharmacology 2000;10(3):226‐7. [Google Scholar]
- Steiner M, Trzepacz PT, Brown E. Premenstrual dysphoric disorder (PMDD) and work efficiency: response to fluoxetine in a randomized clinical trial. Journal of European College of Neuropsychopharmacology 2001;Suppl:226. [Google Scholar]
- Trzepacz PT, Steiner M, Brown E. Premenstrual dysphoric disorder and work efficiency: Response to fluoxetine in a randomized clinical trial. Psychosomatics 2001;42(2):184. [Google Scholar]
- Trzepacz PT, Steiner M, Brown E, Dillon J. Premenstrual dysphoric disorder and work efficiency: Response to fluoxetine in a randomized clinical trial. World Journal of Biological Psychiatry 2001;2 Suppl:340S. [Google Scholar]
Steiner 2005 {published data only}
- Gee M, Bellew K, Holland F, Erp E, Perera P, McCafferty J. Luteal phase dosing of paroxetine controlled release is effective in treating PMDD. 156th Annual Meeting American Psychiatric Association, May 17‐22 San Francisco, CA. 2003:NR760.
- GlaxoSmithKline. A Double‐Blind, Placebo‐Controlled, 3‐Arm, Fixed‐Dose Study of Paroxetine CR Intermittent Dosing (12.5 mg and 25 mg) for Premenstrual Dysphoric Disorder [Study No 29060/717]. http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F717&studyId=CA9A3538‐05F2‐43D8‐A1E2‐342281DDD178&compound=paroxetine Accessed 16 August 2012.
- Steiner M, Hirschberg AL, Bergeron R, Holland F, Gee MD, Erp E. Luteal phase dosing with paroxetine controlled release (CR) in the treatment of premenstrual dysphoric disorder. American Journal of Obstetrics and Gynecology 2005;193:352‐60. [DOI] [PubMed] [Google Scholar]
Steiner 2008 {published data only}
- GlaxoSmithKline. A randomized, double‐blind, placebo‐controlled trial of intermittent treatment with paroxetine 10mg/day and 20mg/day versus placebo in Canadian women with Premenstrual Dysphoric Disorder. http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F621&studyId=8696C1BE‐93D7‐4883‐AD54‐2BC6CF7740BB&compound=paroxetine Accessed 16 August 2012.
- Hamilton Health Sciences Corporation. Luteal Phase Administration of Paroxetine for the Treatment of PMDD: A Randomized, Double‐Blind, Placebo‐Controlled Trial in Canadian Women NCT00620581. http://clinicaltrials.gov/ct2/show/NCT00620581 2008. [DOI] [PubMed]
- Steiner M, Ravindran AV, LeMedello J‐M, Carter D, Huang JO, Anonychuk AM, Simpson SD. Luteal phase administration of paroxetine for the treatment of premenstrual dysphoric disorder: a randomized, double‐blind, placebo‐controlled trial in Canadian women. Journal of Clinical Psychiatry 2008;69:991‐8. [DOI] [PubMed] [Google Scholar]
Stone 1991 {published data only}
- Stone AB, Pearlstein TB, Brown WA. Fluoxetine in the treatment of late luteal phase dysphoric disorder. Journal of Clinical Psychiatry 1991;52:290‐3. [PubMed] [Google Scholar]
- Stone AB, Pearlstein TB, Brown WA. Fluoxetine in the treatment of premenstrual syndrome. Psychopharmacology Bulletin 1990;26:331‐5. [PubMed] [Google Scholar]
- Yonkers KA, Halbreich U, Freeman E, Brown C, Pearlstein T. Sertraline in the treatment of premenstrual dysphoric disorder. Psychopharmacology Bulletin 1996;32(1):41‐6. [PubMed] [Google Scholar]
Su 1997 {published data only}
- Su TP, Schmidt PJ, Danaceau MA, Tobin MB, Rosenstein RL, Murphy DL, et al. Fluoxetine in the treatment of premenstrual dysphoria. Neuropsychopharmacology 1997;16(5):346‐56. [DOI] [PubMed] [Google Scholar]
Wikander 1998 {published data only}
- Wikander I, Sunblad C, Andersch B, Dagnell I, Zylberstein D, Bengtsson F, et al. Citalopram in premenstrual dysphoria: Is intermittent treatment during luteal phases more effective than continuous medication throughout the menstrual cycle?. Journal of Clinical Psychopharmacology 1998;18:390‐8. [DOI] [PubMed] [Google Scholar]
Wood 1992 {published data only}
- Wood SH, Mortola JF, Chan YF, Moossazadeh F, Yen SS. Treatment of premenstrual syndrome with fluoxetine: a double‐blind, placebo‐controlled, crossover study. Obstetrics and Gynecology 1992;80:339‐44. [PubMed] [Google Scholar]
Yonkers 1997 {published data only}
- Yonkers KA, Halbriech U, Freeman E, Brown C, Endicott J, Frank E, et al. Symptomatic improvement of premenstrual dysphoric disorder with sertraline treatment. JAMA 1997;278(12):983‐8. [PubMed] [Google Scholar]
Young 1998 {published data only}
- Young SA, Hurt PH, Benedeck DM, Howard RS. Treatment of PDD with sertraline during the luteal phase: a randomised, double‐blind, placebo controlled crossover trial. Journal of Clinical Psychiatry 1998;59:76‐80. [DOI] [PubMed] [Google Scholar]
- Young SA, Hurt PH, Benedek DM, Howard RS. Treatment of premenstrual dysphoric disorder with sertraline during the luteal phase: A randomized, double blind placebo controlled crossover trial. American Psychiatric Association 150th Annual Meeting, San Diego USA. 1997. [DOI] [PubMed]
- Young SA, Hurt PH, Benedek DM, Howard RS. Treatment of premenstrual dysphoric disorder with sertraline during the luteal phase: A randomized, double blind placebo controlled crossover trial. Journal of Clinical Psychiatry 1998;59:76‐80. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Alpay 2001 {published data only}
- Alpay F, Turhan N. Intermittant versus continuous sertraline therapy in the treatment of premenstrual dysphoric disorders. International Journal of Fertility 2001;46:228‐31. [PubMed] [Google Scholar]
Brandenburg 1993 {published data only}
- Brandenburg S, Tuynman‐Qua H, Verheij R, Pepplinkhuizen L. Treatment of premenstrual syndrome with fluoxetine: an open study. International Clinical Psychopharmacology 1993;8:315‐7. [DOI] [PubMed] [Google Scholar]
De la Gandara 1997 {published data only}
- Gandara Martin JJ. Trastorno disforico premenstrual: Tratamiento a largo plazo con fluoxetina y discontinuacion [Premenstrual dysphoric disorder:long term treatment with fluoxetine and discontinuation]. Actas Luso‐Espanolas de Neurologia, Psiquiatria y Ciencias Afines 1997;25:235‐42. [PubMed] [Google Scholar]
Diegoli 1998 {published data only}
- Diegoli M, DaFonseca A, Diegoli CA, Pinotti JA. A double blind trial of four medications to treat severe premenstrual syndrome. International Journal of Gynecology and Obstetrics 1998;62:63‐7. [DOI] [PubMed] [Google Scholar]
Elks 1993 {published data only}
- Elks ML. Open trial of fluoxetine therapy for premenstrual syndrome. Southern Medical Journal 1993;86:503‐7. [DOI] [PubMed] [Google Scholar]
Flores Ramos 2003 {published data only}
- Fores Ramos M, Ontiveros Uribe M, Cortes Sotres J. Comparison between continuous and intermittent treatment with citalopram..... [Comparacion entre el tratamiento continuo y el intermitente con citalopram para el trastorno disforico premenstrual]. Salud Mental 2003;26(003):37‐45. [Google Scholar]
Freeman 1996 {published data only}
- Freeman EW, Rickels K, Sondheimer SJ. Comparison of serotonergic and noradrenergic antidepressant medications in treatment of premenstrual syndrome (PMS). XXIst Collegium Internationale Neuro‐psychopharmacologicum, Glasgow, Scotland. 12‐16th July 1998.
- Freeman EW, Rickels K, Sondheimer SJ. Sertraline versus desipramine in PMS treatment. 151st Annual Meeting of the American Psychiatric Association, Toronto, Canada. 30th May‐4th June 1998, issue 118C.
- Freeman EW, Rickels K, Sondheimer SJ, Wittmaack FM. Sertraline versus desipramine in the treatment of premenstrual syndrome: an open label trial. Journal of Clinical Psychiatry 1996;57:7‐11. [PubMed] [Google Scholar]
Freeman 1999b {published data only}
- Freeman EW, Rickels K, Arredono F, Lee‐Chuan K, Pollack S, Sondheimer S. Full or half cycle treatment of severe premenstrual syndrome with a serotonergic antidepressant. Journal of Cliniical Psychopharmacology 1999;19(1):3‐9. [DOI] [PubMed] [Google Scholar]
Freeman 2000 {published data only}
- Freeman EW, Sondheimer SJ, Polansky M, Garcia‐Espagna B. Predictors of response to sertraline treatment of severe premenstrual syndromes. Journal of Clinical Psychiatry 2000;61(8):579‐84. [DOI] [PubMed] [Google Scholar]
Freeman 2002 {published data only}
- Freeman E, Jabara S, Sondheimer S, Auletto R. Citalopram in PMS patients with prior SSRI treatment failure: A preliminary study. Journal of Womens Health & Gender‐based Medicine 2002;11(5):459‐64. [DOI] [PubMed] [Google Scholar]
Freeman 2005 {published data only}
- Freeman E, Sondheimer S, Sammel M, Ferdousi T, Lin H. A preliminary study of luteal phase versus symptom‐onset dosing with escitalopram for premenstrual dysphoric disorder. Journal of Clinical Psychiatry 2005;66(6):769‐73. [DOI] [PubMed] [Google Scholar]
Glaxo 2002 {unpublished data only}
- GlaxoSmithKline. A 3‐Month, Double‐blind, Placebo‐controlled, Fixed‐dose, Extension Study of Paroxetine CR (12.5 mg and 25 mg/day) Continuous Treatment for PMDD Patients Completing Studies 29060/677, 688 or 689 [Study no 29060/711]. http://www.gsk‐clinicalstudyregister.com/result_detail.jsp?protocolId=29060%2F711&studyId=4A384A1A‐46F7‐4E8B‐915B‐4B324BB64491&compound=paroxetine Accessed 16 August 2012.
Landen 2009 {published data only}
- Landen M, Erlandsson H, Bengtsson F, Andersch B, Eriksson E. Short onset of action of a serotonin reuptake inhibitor when used to reduce premenstrual irritability. Neuropsychopharmacology 2009;34:585‐92. [DOI] [PubMed] [Google Scholar]
Miller 2008 {published data only}
- Miller MN, Newell CL, Miller BE, Frizzell PG, Kayser RA, Ferslew KE. Variable dosing of sertraline for premenstrual exacerbation of depression: a pilot study. Journal of Women's Health 2008;17(6):993‐7. [DOI] [PubMed] [Google Scholar]
Pearlstein 1994 {published data only}
- Pearlstein TB, Stone AB. Long term fluoxetine treatment of late luteal phase dysphoric disorder. Journal of Clinical Psychiatry 1994;55:332‐5. [PubMed] [Google Scholar]
Pearlstein 2000 {published data only}
- Pearlstein T, Haskett R, Stout A, Frank E, Endicott J. Sertraline improves psychosocial functioning in premenstrual dysphoric disorder. 151st Annual Meeting of the American Psychiatric Association, 30th May‐ 4th June Toronto, Canada. 1998.
- Pearlstein TB, Halbreich U, Batzar ED, Brown CS, Endicott J, Frank E, et al. Psychosocial functioning in women with premenstrual dysphoric disorder before and after treatment with sertraline or placebo. Journal of Clinical Psychiatry 2000;61(2):101‐9. [DOI] [PubMed] [Google Scholar]
Rickels 1990 {published data only}
- Rickels K. Fluoxetine in the treatment of PMS. Current Therapeutic Research 1990;48:161‐8. [Google Scholar]
Steiner 1997 {published data only}
- Steiner M, Korzekwa MI, Lamont J, Wilkins A. Intermittent fluoxetine dosing in the treatment of women with premenstrual dysphoria. Psychopharmacology Bulletin 1997;33:771‐4. [PubMed] [Google Scholar]
Sundblad 1992 {published data only}
- Sundblad C, Modigh K, Andersch B, Eriksson E. Clomipramine effectively reduces irritability and dysphoria: a placebo controlled trial. Acta Psychiatrica Scandinavia 1992;85:39‐47. [DOI] [PubMed] [Google Scholar]
Sundblad 1993 {published data only}
- Sundblad C, Hedberg M, Eriksson E. Clomipramine administered during the luteal phase reduces the symptoms of premenstrual syndrome: A placebo controlled trial. Neuropsychopharmacology 1993;9(2):133‐45. [DOI] [PubMed] [Google Scholar]
Sundblad 1997 {published data only}
- Sundblad C Wikander I, Andersch B, Eriksson E. A naturalistic study of paroxetine in premenstrual syndrome: efficacy and side‐effects during ten cycles of treatment. European Neuropsychopharmacology 1997;7(3):201‐6. [DOI] [PubMed] [Google Scholar]
Veeninga 1990 {published data only}
- Veeninga AT, Westenberg HG, Weusten JT. Fluvoxamine in the treatment of menstrually related disorders. Psychopharmacology 1990;102:414‐6. [DOI] [PubMed] [Google Scholar]
Wu 2008 {published data only}
- Wu K‐Y, Liu C‐Y, Hsiao M‐C. Six‐month paroxetine treatment of premenstrual dysphoric disorder: continuous versus intermittent treatment protocols. Psychiatry and Clinical Neurosciences 2008;62:109‐14. [DOI] [PubMed] [Google Scholar]
Yonkers 1996 {published data only}
- Yonkers KA, Gullion C, Williams A, Novak K, Rush AJ. Paroxetine as a treatment for premenstrual dysphoric disorder. Journal of Clinical Psychopharmacology 1996;16(1):3‐8. [DOI] [PubMed] [Google Scholar]
Yonkers 2002 {unpublished data only}
- Yonkers KA. Antidepressant Treatment for Premenstrual Syndrome and Premenstrual Dysphoric Disorder. http://clinicaltrials.gov/ct2/show/NCT00048854.
Yonkers 2006 {published data only}
- Yonkers K, Holthausen G, Poschman K, Howell H. Symptom‐onset treatment for women with premenstrual dysphoric disorder. Journal of Clinical Psychopharmacology 2006;26(2):198‐202. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
Yonkers 2007 {unpublished data only}
- Yonkers KA, Altemus M, Kornstein S. Evaluating the Effectiveness of Sertraline in Treating Women With Premenstrual Dysphoric Disorder. http://www.clinicaltrials.gov/ct2/show/NCT00536198 2007.
Yonkers 2010 {unpublished data only}
- Yonkers KA. Comparison of Fluoxetine, Calcium and Placebo for the Treatment of Moderate to Severe Premenstrual Syndrome (PMS). http://clinicaltrials.gov/ct2/show/NCT00965562 2010.
Additional references
ACOG 2000
- American College of Obstetricians and Gynecologists. Premenstrual syndrome. ACOG PracticeBulletin No. 15. Washington, DC: ACOG 2000.
APA 2000
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington, DC:American Psychiatric Association; 2000. p. 774. Diagnostic and statistical manual of mental disorders. 4th Edition. Washington, DC: American Psychiatric Association, 2000:774. [Google Scholar]
Baker 2012
- Baker LJ, O'Brien PMS. Premenstrual syndrome (PMS): A peri‐menopausal perspective. Maturitas 72 2012;72:121‐5. [DOI] [PubMed] [Google Scholar]
Cohen 1988a
- Cohen J. Statistical Power Analysis in the Behavioral Sciences (2nd edition). Statistical Power Analysis in the Behavioral Sciences. 2nd Edition. Hillsdale (NJ): Lawrence Erlbaum Associates, Inc., 1988. [Google Scholar]
Epperson 2012
- Epperson CN, Steiner M, Hartlage SA, Erikkson E, Schmidt PJ, Jones I, Yonkers KA. Premenstrual Dysphoric Disorder: Evidence for a New Category for DSM‐5. American Journal of Psychiatry 2012;169(5):465‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ford 2012
- Ford O, Lethaby A, Roberts H, Mol BWJ. Progesterone for premenstrual syndrome. Cochrane Database of Systematic Reviews 2012, Issue 3. [DOI: 10.1002/14651858.CD003415.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Freeman 2009
- Freeman EW, Rickels K, Sammel MD, Lin H, Sondheimer SS. Time to relapse after short‐term or long‐term sertraline treatment for severe premenstrual syndromes. Archives of General Psychiatry 2009;66(5):537‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Freeman 2012
- Freeman EW, Sammel MD, Lin H, Rickels K, Sondheimer SJ. Clinical subtypes of premenstrual syndrome and responses to sertraline treatment. Obstetrics and Gynecology 2011;118(6):1293‐1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Glaxo 2012
- GlaxoSmithKline Government Affairs, Public Policy, Patient Advocacy. Public Disclosure of Clinical Research. Downloaded from: http://www.google.co.nz/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&ved=0CDAQFjAA&url=http%3A%2F%2Fwww.gsk.com%2Fpolicies%2FGSK‐on‐disclosure‐of‐clinical‐trial‐information.pdf&ei=lqIIUee4GuTDmQX9hoD4Ag&usg=AFQjCNEcA2G3XGrrh9BkqK1RedmQ53VgQg&bvm=bv.41642243,d.dGY.
Halbreich 2008
- Halbreich U. Selective serotonin reuptake inhibitors and initial oral contraceptives for the treatment of PMDD: effective but not enough. CNS Spectrum 2008;13(7):566,569‐72. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011 ]. Available from www.cochrane‐handbook.org.
Ismail 2006
- Ismail KMK, Crome IB, O'Brien PMS. In: Higham J editor(s). Psychological disorders in obstetrics and gynaecology for the MRCOG and beyond. London: The Royal College of Obstetricicians and Gynaecologists, 2006:37. [Google Scholar]
Jing 2009
- Jing Z, Yang X, Ismail KMK, Chen XY, Wu T Cochrane Database of Systematic Reviews 2009, Issue 1. Chinese herbal medicine for premenstrual syndrome. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD006414.pub2] [DOI] [PubMed] [Google Scholar]
Kleinstauber 2012
- Kleinstauber M, Witthoft M, Hiller W. Cognitive‐behavioral and pharmacological interventions for premenstrual syndrome or premenstrual dysphoric disorder: a meta‐analysis. Journal of Clinical Psychology in Medical Settings 2012;19:308‐19. [DOI] [PubMed] [Google Scholar]
Lopez 2012
- Lopez LM, Kaptein AA, Helmerhorst FM. Oral contraceptives containing drospirenone for premenstrual syndrome. Cochrane Database of Systematic Reviews 2012, Issue 2. [DOI: 10.1002/14651858.CD006586.pub4] [DOI] [PubMed] [Google Scholar]
Magos 1986
- Magos AL, Brincat M, Studd JW. Trend analysis of the symptoms of 150 women with a history of th premenstrual syndrome. American Journal of Obstetrics and Gynecology 1986;155(2):277‐82. [DOI] [PubMed] [Google Scholar]
Micromedex 2013
- DRUGDEX® Evaluations. Micromedex.2 database Accessed 2.5.13 via University of Auckland library.
Naheed 2013
- Naheed B, O'Brien PMS, Uthman OA, O'Mahony F. Non‐contraceptive oestrogen‐containing preparations for controlling symptoms of premenstrual syndrome. Cochrane Database of Systematic Reviews 2013, Issue 4. [DOI: 10.1002/14651858.CD010503] [DOI] [PMC free article] [PubMed] [Google Scholar]
O'Brien 2011
- O'Brien PMS, Backstrom T, Brown C, Dennerstein L, Endicott J, Epperson CN, et al. Towards a consensus on diagnostic criteria, measurement and trial design of the premenstrual disorders: the ISPMD Montreal consensus. Archives of Womens Mental Health 2011;14:13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pearlstein 2002
- Pearlstein T. Selective serotonin reuptake inhibitors for premenstrual dysphoric disorder. Drugs 2002;62(13):1869‐85. [DOI] [PubMed] [Google Scholar]
Pearlstein 2007
- Pearlstein T. Prevalence, impact, on morbidity and burden of disease. In: O’Brien PMS, Rapkin A, Schmidt P editor(s). The premenstrual syndromes: PMS and PMDD. London: Informa Healthcare, 2007:37‐47. [Google Scholar]
Plouffe 1993
- Plouffe L, Stewart K, Craft KS, Maddox MS, Rausch MD. Diagnostic and treatment results from a south‐eastern academic center‐based premenstrual syndrome clinic: The first year. Americal Journal of Obstetrics and Gynecology 1993;169(2):295‐307. [DOI] [PubMed] [Google Scholar]
Rapkin 2008
- Rapkin AJ, Winer SA. The pharmacologic management of premenstrual dysphoric disorder. Expert Opinion in Pharmacotherapy 2008;9(3):429‐45. [DOI] [PubMed] [Google Scholar]
Shah 2008
- Shah NJ, Jones JB, Aperi J, Shemtov R, Karne A. Selective serotonin reuptake inhibitors for premenstrual syndrome and premenstrual dysphoric disorder. A meta‐analysis. Obstetrics and Gynecology 2008;111(5):1175‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shaw 2003
- Shaw S, Wyatt K, Campbell J, Ernst E, Thompson‐Coon J. Vitex agnus castus for premenstrual syndrome. Cochrane Database of Systematic Reviews 2003, Issue 4. [DOI: 10.1002/14651858.CD004632] [DOI] [Google Scholar]
Yu 2005
- Yu J, Liu B, Liu Z, Welch V, Wu T, Clarke J, Smith CA. Acupuncture for premenstrual syndrome. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD005290] [DOI] [Google Scholar]
References to other published versions of this review
Dimmock 2000
- Dimmock PW, Wyatt KM, Jones PW, O'Brien PMS. Efficacy of selective serotonin‐reuptake inhibitors in premenstrual syndrome: a systematic review. Lancet 2000;356:1131‐6. [DOI] [PubMed] [Google Scholar]