

Abstract
Objective
To better characterize the neurologic and cognitive profile of patients with spinocerebellar ataxia 34 (SCA34) caused by ELOVL4 mutations and to demonstrate the presence of ELOVL4 cellular localization and distribution abnormalities in skin-derived fibroblasts.
Methods
We investigated a 5-generation French-Canadian kindred presenting with a late-onset cerebellar ataxia and recruited age- and education-matched controls to evaluate the presence of neurocognitive impairment. Immunohistochemistry of dermal fibroblasts derived from a patient's skin biopsy was performed.
Results
Patients had a late-onset slowly progressive cerebellar syndrome (mean age at onset 47 years; range 32–60 years) characterized by truncal and limb ataxia, dysarthria, hypometric saccades, and saccadic pursuits. No patient had past or current signs of erythrokeratodermia variabilis, which had previously been reported. MRI revealed cerebellar atrophy, with pontine atrophy (4 of 6 patients), and cruciform hypersignal in the pons (2 of 6 patients). Fluorodeoxyglucose-PET showed diffuse cerebellar hypometabolism in all 5 tested patients with subtle parietal hypometabolism in 3. Significant cognitive deficits were found in executive functioning, along with apparent visuospatial, attention, and psychiatric involvement. Immunohistochemistry of dermal fibroblasts showed mislocalization of the ELOVL4 protein, which appeared punctate and aggregated, supporting a dominant negative effect of the mutation on protein localization.
Conclusions
Our findings support the pathogenicity of ELOVL4 mutations in cerebellar dysfunction and provide a detailed characterization of the SCA34 phenotype, with neurocognitive changes typical of the cerebellar cognitive-affective syndrome.
Spinocerebellar ataxia 34 (SCA34), also called ataxia (ATX)-ELOVL4 according to the revised nomenclature,1 was first described in a French-Canadian family with ataxia and erythrokeratodermia variabilis (EKV)2,3 caused by a dominant mutation in ELOVL4, which encodes the elongation of very-long-chain fatty acid (VLCFA) protein 4 (ELOVL4). Since then, 2 Japanese families presenting with a cerebellar disorder and multisystem atrophy–like features on MRI were reported,4 along with 2 sporadic patients with neurocutaneous involvement.5,6 The disease phenotype is still poorly characterized, and little is known on its impact on other neurologic functions, including cognition.
Cognitive and affective dysfunction caused by cerebellar disease has been labeled the cerebellar cognitive-affective syndrome (CCAS), which is characterized by impairment in executive function, visuospatial organization, language, and psychiatric features.7,8 Neurocognitive impairment has been reported in selected hereditary ataxias,9–11 but previous reports of patients with SCA34 only reported normal cognition.3–6
To date, the biochemical validation of ELOVL4 dysfunction in SCA34 remains elusive. Previous reports have demonstrated that serum levels and ratios of VLCFAs (C22 to C26), corresponding to the ELOVL4 substrates and products, remained within normal limits.3,4
Our primary objective was to characterize the neurologic and neurocognitive phenotype of patients with SCA34 from a large family to better define disease manifestations. The secondary objective was to demonstrate that skin fibroblasts of patients with the mutated ELOVL4 allele had subcellular localization or distribution anomalies supporting ELOVL4 dysfunction as the cause of SCA34.
Methods
Clinical and cognitive assessment
We investigated a large 5-generation French-Canadian kindred presenting with a late-onset pure cerebellar ataxia with pontocerebellar atrophy on MRI. A total of 19 affected and unaffected individuals provided written informed consent to participate in the study. We collected genomic DNA and conducted clinical interviews and neurologic evaluations for all participants. All participants were evaluated in December 2013, and 6 affected patients had regular clinical follow-up until July 2018 when they were evaluated by an ophthalmologist to search for retinal abnormalities and were formally rated according to the scale for the assessment and rating of ataxia (SARA). MRI was performed in 6 affected individuals, and 3 underwent nerve conduction studies. All patients were specifically questioned regarding the presence of cutaneous involvement, and one self-reporting patient had a thorough evaluation by a dermatologist.
Five patients and as many cognitively healthy controls underwent formal neurocognitive and neuropsychiatric assessment at the CHU de Québec Memory Clinic (cliniquedememoire.ca). The other affected participants did not wish to collaborate beyond the standard clinical evaluation. Controls were family caregivers and were age- and education-matched to participants. The Mini-Mental State Examination was performed with each participant. Data derived from the Montreal Cognitive Assessment12 and additional standardized and local cognitive measures were summarized according to composite scores for the main cognitive domains: attention (digit span and numeric substitution), executive functions (abstract thinking, lexical verbal fluency, Go/No-Go, Luria, Trail Making B, and Rey Complex Figure Test [RCFT] copy), visuospatial skills (block copy and clock drawing), memory (5-word list free and cued recall), and language (naming, comprehension, and sentence repetition tasks) (appendix e-1, links.lww.com/NXG/A228, for detailed tasks included in each domain). Affective symptoms, including mood alterations and anxiety, along with behavioral changes were assessed by asking patients and caregivers about relevant neuropsychiatric symptoms and by observation of affect and appropriateness of behavior during the cognitive evaluation. Patients' scores were compared with controls' using the paired sample t test. The cognitive involvement appeared milder in the youngest patient (V-1), so she was referred for a detailed neuropsychological assessment (California Verbal Learning Test-II, Delis-Kaplan Executive Function System, DO-80, Taylor complex figure, Minnesota Multiphasic Personality Inventory, Ruff 2 and 7, Tower of London test, and Wechsler Adult Intelligence Scale 4th edition). Functional imaging with [18F]-fluorodeoxyglucose PET (FDG-PET) was performed in the 5 patients who underwent cognitive evaluation.
Whole-exome sequencing and genetic analysis
To identify the genetic mutation underlying ataxia in this family, whole-exome sequencing was performed on 5 affected members. Genomic DNA was extracted from peripheral blood leukocytes following the manufacturer's protocol (Puregene; Gentra Systems, Inc). Whole-exome sequencing (WES) was performed on 5 affected members of the family. Exome capture was performed using SeqCap EZ Exome Library v3 kit (Nimblegen, Roche) with 1 μg of genomic DNA. Exon-enriched DNA libraries from these 5 individuals were sequenced on the Illumina HiSeq 2000 with a paired-end 100-bp length configuration. Reads were mapped against the Human Reference Genome hg19 using the Burrows-Wheeler Aligner (v0.7.5). Single nucleotide variations and copy number variations, including insertions/deletions (indels), were called using the Genome Analysis Toolkit (v2.6)13 and annotated using ANNOVAR13.14 The exomes were analyzed using step-by-step filtering strategies. Basic filters were used to remove low-quality variants: minimum position coverage >6, minimum reads supporting the variant >3, and minimum mutation frequency >0.15. Genetic variants were then filtered to retain only variants respecting the following criteria: (1) shared by all 5 affected family members sequenced by WES, (2) nonsynonymous single nucleotide variants, splicing site variants, or coding indels, (3) with a minor allele frequency <0.1% in Exome Aggregation Consortium, and (4) not present in 186 controls from our in-house data set. To validate our results and examine segregation of the identified ELOVL4 DNA variations with the disease in the family, we used specific PCR primer pairs (forward: CATTGCTTTCCACTGAACACA; reverse CATGCCTTGTACATTTTGTGC) to amplify DNA from 18 family members and sequenced them by Sanger sequencing (Applied Biosystem's 3730xl DNA Analyzer technology). One additional family member was tested with commercially available Sanger sequencing of ELOVL4 from Prevention Genetics.
Fibroblast cell isolation and culture
A skin biopsy was performed on the patient who reported cutaneous involvement. The fibroblast cells were isolated from skin biopsies and kept in liquid nitrogen until used as previously described.15 Briefly, fibroblasts were cultivated in Dulbecco's Modified Eagle Medium with 10% fetal bovin serum (Seradigm) with 100 IU/mL penicillin G (Sigma-Aldrich) and 25 μg/mL gentamicin (Gemini Bio-Products) in 8% CO2 at 37°C. They were cultured in flasks until confluency (100%) prior to be split and seeded in a 6-well plate containing sterile glass coverslips.
Immunofluorescence analyses
Fibroblasts cells, grown on glass coverslips, were fixed in 4% (wt/vol) paraformaldehyde in phosphate-buffered saline for 20 minutes and blocked for 1 hour in 5% (vol/vol) goat serum 0.1% Triton X-100 in phosphate-buffered saline at room temperature. Immunofluorescence was performed using antibodies recognizing ELOVL4 (rabbit polyclonal, Abcam ab224608) and calnexin (mouse monoclonal, Invitrogen MA3-027) and used at 1:200 diluted in blocking solution overnight at 4°C. Fluorescent signal distribution was visualized by epifluorescence microscopy after incubation with secondary antibodies, anti-mouse or anti-rabbit IgG conjugated to Alexa Fluor 488 (green) or 594 (red) (Invitrogen mouse 488 #A11001 and rabbit 594 #A21207) diluted 1:500 for 2 hours at room temperature, and mounted in Prolong (Fluoromount-G with 4',6-Diamidino-2-phenylindole, Invitrogen #00-4959-52).
Standard protocol approvals, registrations, and patient consents
The research ethics committee of the CHU de Québec approved the participation of human subjects to this study (PEJ-299, 2012-1311), and written informed consent was obtained from all participants. The participants received no financial compensation.
Data availability
Anonymized data not published in the article will be shared by request from any qualified investigator.
Results
Genetic results
A total of 19 individuals (9 affected and 10 unaffected) underwent a complete neurologic evaluation and genetic testing (table 1). Routine biochemical investigations were normal, and initial genetic screening had excluded SCA1, 2, 3, 6, 7, 8, 17, and fragile X syndrome for this family. WES in 5 affected individuals revealed 426,715 DNA variations. Only 1 variant, a heterozygous missense mutation in the ELOVL4 gene (NM_022726.3 c.504G>C, p.L168F), fulfilled the filtering criteria. The L168F amino acid substitution was located at a highly conserved residue in the third transmembrane domain of the protein. This mutation was predicted to be possibly damaging with a score of 0.867 according to the PolyPhen-2 web site (genetics.bwh.harvard.edu/pph2/).16 This mutation was confirmed in 4 additional affected family members, but was absent in the 10 unaffected relatives. Hence, perfect segregation of the identified c.504G>C mutation with the disease status in the family was observed (figure e-1, links.lww.com/NXG/A228).
Table 1.
Detailed neurologic findings in affected individuals with ELOVL4 mutation

This variant had already been reported to cause SCA34 in another French-Canadian family,3 but no common ancestor could be identified by family history and the haplotype was not reconstructed from the exome data. With agreement of the authors, exomes from the previously described family were retrieved, and kinship coefficients were calculated using KING.17 All pairs of sample between the 2 studies had a phi score of <0.03, which corresponds to a third-degree relationship or second-degree cousins. Although calculating relatedness coefficients using exome sequencing data can be biased due to sparse and unevenly distributed data, this suggested that the patients from the 2 families were not close relatives. Considering the close geographic proximity and common French-Canadian ethnical background, it remains likely that both families have a distant common ancestor that was not identified by family history or relatedness coefficient.
Clinical assessment
The disorder was characterized by a late-onset cerebellar syndrome (mean age at onset 47 years; range 32–60 years) with slow progression. Patients generally required a walker in their 70s and became a wheelchair user in their 80s. Truncal and gait ataxia, along with dysmetria and dysarthria, were remarkable in all patients (table 1). The median SARA score was 16 at last evaluation (range 8–21.5), and there was a correlation between SARA score and age with a Pearson correlation coefficient of 0.84, suggesting disease progression with advancing age. Hypometric saccades and saccadic pursuits were remarkable on examination. Most patients denied current or previous dermatologic involvement, but 1 patient (IV-18) reported erythematous dry skin lesions on the lower limbs during winter beginning in her fifth decade (figure e-2, links.lww.com/NXG/A228). Nummular dermatitis was diagnosed, but there was no typical EKV lesion. Two other patients reported occasional dry skin on the lower limbs in winter, but none had erythematous lesions, and there were no active lesions at the time of evaluation. Moreover, 3 patients reported a history of severe gingivitis that occurred in the second decade and caused a generalized edentation that required dentures. One patient (V-1) had bilateral pale pisciform perimacular retinal lesions on ophthalmologic examination. She had no complains of visual loss and was examined by a retinal disease specialist who concluded that there was no evidence of Stargardt disease or retinitis pigmentosa. All other evaluated patients had a normal ophthalmologic examination. Three patients had mild sensory deficits on evaluation, 2 of whom also had decreased deep tendon reflexes (DTRs). Polyneuropathy was confirmed in 2 of 3 tested patients (table 1).
MRI was performed in 6 patients and revealed mild (1/6) to severe (5/6) vermian and hemispheric cerebellar atrophy in all patients along with pontine atrophy in 4 (figure 1A). Cruciform hypersignal in the pons, also called the hot cross bun sign, was present in 2 patients (III-27 and IV-3) (figure 1B). Other findings were reported only in selected patients, notably mild diffuse cerebral cortical (2/6) or subcortical (2/6) atrophy and leukoencephalopathy (2/6). FDG-PET revealed diffuse cerebellar hypometabolism in all 5 patients. Three patients also had a very subtle bilateral (2/5) or right (1/5) parietal hypometabolism (figure 2C). One other patient had heterogeneous cerebral hypometabolism with preserved basal ganglia, brainstem, and mesiotemporal regions, which was possibly secondary to diffuse vascular involvement. In this patient, MRI had shown subcortical atrophy, but no evidence of previous stroke.
Figure 1. Neuroimaging findings in patients with the c.504G>C mutation in ELOVL4.

(A) Sagittal MRI of a 68-year-old man (IV-3) showing severe cerebellar atrophy with pontine atrophy. (B) Axial MRI from the same patient showing T2 cruciform hypersignal in the pons (indicated by red arrow). (C) Fluorodeoxyglucose-PET of a 70-year-old woman (IV-6) showing severe cerebellar hypometabolism and minimal biparietal hypometabolism. Views are indicated in the top row: Lat. = lateral; Sup. = Superior; Inf. = Inferior; Ant = Anterior; Post. = Posterior; Med. = Medial. GLB indicates normalized to the global metabolism of the brain.
Figure 2. Impaired copy of the RCFT in 3 patients with the c.504G>C mutation in ELOVL4.

(A) RCFT model. (B) Sixty-four-year-old woman with 12 years of education (IV-18). (C) Seventy-two-year-old man with 11 years of education (IV-3) shows significant juxtaposition of details. (D) Eighty-year-old man with 21 years of education (III-3) with a severe dysexecutive syndrome. RCFT = Rey Complex Figure Test.
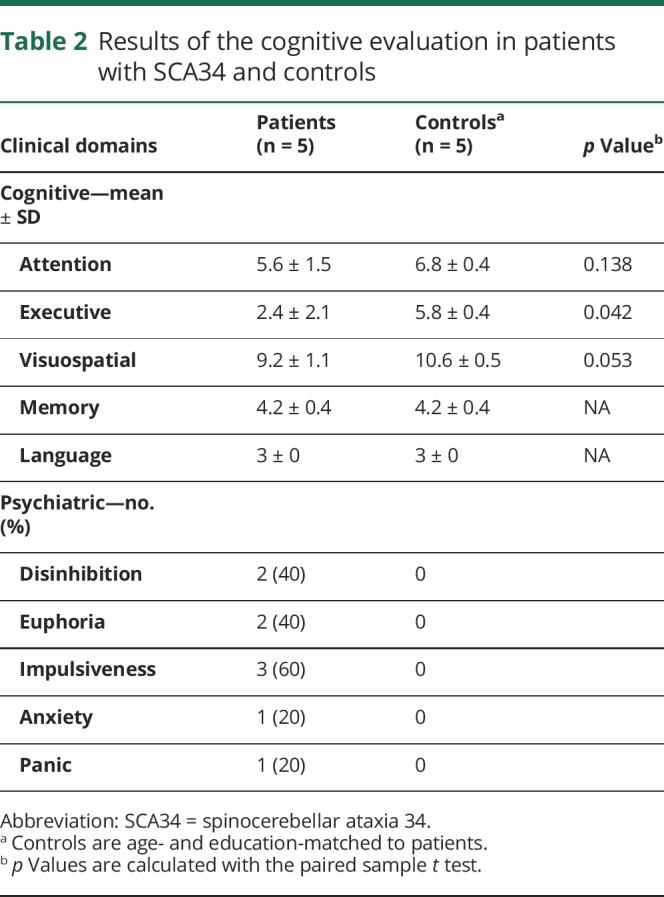
Cognitive assessment
Five patients were evaluated at the Clinique Interdisciplinaire de Mémoire du CHU de Québec. Their performance was compared with 5 age- and education-matched controls (mean education for patients = 12.4 ± 1.51 years vs 13.4 ± 2.07 for controls). The cognitive involvement was homogeneous within affected patients with significant deficits in executive functioning and apparent impairment in visuospatial skills and attention that did not reach statistical significance (table 2). Working memory, psychomotor speed, set shifting, inhibition, and planning skills were particularly affected as revealed by failure to perform backward digit span, decreased lexical verbal fluency, and impaired trail making, Luria sequence, Go/No-Go, and RCFT (figure 2). Visuoconstructive skills and attention were also impaired in all patients, but this did not reach statistical significance. Patients did not present deficits in memory and language. Detailed neuropsychological testing in the youngest patient (V-1) revealed a milder impairment in selective attention and working memory. Psychiatric features were remarkable in 3 patients, 2 of whom exhibited frank disinhibition and euphoria, whereas impulsiveness and anxiety were noted in a third patient (see appendix e-2, links.lww.com/NXG/A228, for detailed evaluations).
Table 2.
Results of the cognitive evaluation in patients with SCA34 and controls
Aberrant ELOVL4 localization in patient fibroblasts
Immunofluorescence analysis of fibroblasts from a healthy control showed homogeneous staining of ELOVL4 seemingly colocalizing with the endoplasmic reticulum marker calnexin at the perinuclear space (figure 3A). However, mislocalization of the ELOVL4 protein beyond the perinuclear region and abnormal punctates and aggregates can be observed in patient-derived fibroblasts carrying the c.504G>C mutation (figure 3B).
Figure 3. Immunofluorescence staining of dermal fibroblasts from a healthy control and a patient with SCA34.

(A) ELOVL4 staining in healthy control's fibroblasts showed homogeneous staining that appears to colocalize with the endoplasmic reticulum marker calnexin at the perinuclear space. The nucleus marker DAPI is shown in blue. Scale bar is 50 μm. (B) ELOVL4 staining in the patient's fibroblasts showed mislocalization of the protein beyond the perinuclear region with a punctate and aggregated appearance. DAPI = 4',6-Diamidino-2-phenylindole; SCA34 = spinocerebellar ataxia 34.
Discussion
We report a large family with a c.504G>C mutation in ELOVL4 associated with cerebellar ataxia and cognitive impairment with prominent executive dysfunction. Our findings provide additional support to the putative role of ELOVL4 mutations in cerebellar dysfunction. Moreover, we report evidence of ELOVL4 cellular abnormalities in SCA34 by demonstrating mislocalization and abnormal aggregation of the protein in a patient's dermal fibroblasts.
In our cohort, all patients presented with a prominent cerebellar syndrome of late onset and slow progression. So far, ELOVL4-linked SCA34 has been reported in patients of Canadian, Japanese, and Brazilian origins, and clinical features vary significantly among described patients (table e-1, links.lww.com/NXG/A228). Notably, decreased DTR and peripheral neuropathy were both observed in 2 patients from the present study, whereas 4 of 8 patients from the previously reported French-Canadian family had a mild peripheral neuropathy.3 However, another study reported increased DTR and positive Babinski signs, upper motor neuron findings that may be associated with the multiple system atrophy–like findings on imagery in these patients.4 Of interest, 2 patients from the present study also presented with a hot cross bun sign, but DTR and plantar reflexes were normal. Hence, it appears that the cerebellar syndrome may be associated with peripheral polyneuropathy or pyramidal signs in patients with SCA34.
We report patients with SCA34 with deficits in executive functioning that were statistically different from age- and education-matched controls, along with apparent visuospatial and attention deficits. Although some of the cognitive tasks could have been affected by reduced hand coordination, such as the RCFT and Trail Making Test, the deficits observed went beyond what would be expected in a pure cerebellar motor syndrome. This was associated with psychiatric features in 3 patients. These findings appear typical of the CCAS7,8 and provide additional evidence of neurocognitive and neurobehavioral impairment in hereditary degenerative ataxias. This is consistent with the results of recent functional imaging studies showing that the cerebellum is composed of several functional areas: the sensorimotor region in the anterior lobe and lobule VIII, the cognitive region in the posterior lobes, and the affective region in the posterior vermis.18–20 The CCAS is expected to result from disruption of cerebro-cerebellar loops, particularly cerebellar projections to association cortices in the dorsolateral prefrontal and parietal lobes and to the limbic regions.19–21 In our patients, functional imaging showed cerebellar hypometabolism, which was associated with mild parietal hypometabolism in 3 patients. This could be attributable to a diaschisis phenomenon, wherein severe damage to the cerebellar hemispheres could result in reduced cerebral metabolic activity through deactivation of the cerebello-thalamic-cortical projections.22 Indeed, the parietal lobe has important afferent and efferent connections with the cerebellum. These appear to be involved in the pathophysiology of parietal ataxia, or crossed cerebellar diaschisis, a phenomenon in which patients with a primary parietal lesion develop secondary cerebellar dysfunction.23 The parietal hypometabolism observed here could reflect a similar phenomenon, i.e., parietal dysfunction secondary to a primary cerebellar lesion. A previous report of parietal hypometabolism in a pure recessive cerebellar ataxia phenotype caused by SYNE1 recessive mutations supports this hypothesis.9 This phenomenon may contribute to the visuospatial and attention deficits observed in our patients and may be amenable to potential treatments targeting diaschisis.22
As opposed to previously reported patients with the same mutation,3 our patients did not report any history of skin lesions associated with EKV. All patients were thoroughly questioned regarding past or current erythematous or hyperkeratotic lesions, but only the patient reporting active lesions underwent a complete skin examination. It is possible that subtle skin lesions may have been overlooked by patients themselves, but this is unlikely to explain alone the absence of EKV in our patients because the cutaneous findings reported in previous studies were pronounced and symptomatic.3,6 Absence of EKV may reflect the role of modifier genes or environmental factors involved in cutaneous disease or possibly a milder disease severity that would have gone unnoticed. The late-onset skin dryness and nummular dermatitis reported by a few patients, along with the early gingivitis episodes in 3 patients, may represent milder mucocutaneous involvement or coincidental findings. In the previously reported patients with the same mutation, skin disease was shown to have incomplete penetrance,3 and absence of EKV in SCA34 families has been reported in patients of Japanese origin.4
Immunofluorescence analyses using dermal fibroblasts from an affected patient revealed mislocalization and aggregation of the ELOVL4 protein. Previous studies did not demonstrate significant serum lipid anomalies in patients with SCA34, and cellular abnormalities in human tissue had not been reported.3,4 ELOVL4 is involved in the elongation of VLCFA with carbon chains of 24 atoms or longer and catalyzes the rate-limiting reaction in the production of 2 distinct products: very-long-chain saturated fatty acids (VLC-SFAs), which are involved in skin barrier formation and sphingolipid production, and VLC polyunsaturated fatty acids, which are essential for photoreceptor cells of the macula and also for production of sphingolipids.24 In the cerebellum, the protein is located in all parts of the cerebellar cortex and deep cerebellar nuclei, and it is mainly expressed in neurons and oligodendrocytes,25 which is compatible with the role of VLC-SFA in myelin production. Skin appears as a relevant tissue to investigate the biochemical anomalies associated with ELOVL4 mutations, considering that the brain and skin are the 2 human organs with the highest concentrations of sphingolipids, some of which are dependent on the presence of ELOVL4 for synthesis.26 The mislocalization and aggregation of the ELOVL4 protein observed in the patient's fibroblast cells appear similar to those found in previous studies of Stargardt-like macular dystrophy type 3 (STGD3), which is associated with ELOVL4 dominant mutations.27–29 More specifically, Logan et al.27 demonstrated that ELOVL4 mutations associated with STGD3 cause mislocalization of the protein beyond the endoplasmic reticulum in a punctate and aggregated appearance very similar to what we observed. In this study, the mutant protein was shown to interact with wild-type ELOVL4 to alter the localization of the protein and to exert a dominant negative effect on enzymatic activity that worsened if the protein was redirected to the endoplasmic reticulum.27 Therefore, it is likely that the c.504G>C ELOVL4 mutation may also exert its pathogenic effect through a dominant negative mechanism that involves interaction with wild-type ELOVL4, mislocalization, and possibly reduction in the production of VLCFA. Although the skin biopsy was performed in the patient that reported cutaneous involvement, we expect that the ELOVL4 mislocalization and aggregation observed in this patient's skin fibroblasts would be similar in other patients carrying the same mutation and underlies the pathophysiologic mechanism of the disorder in other tissues as well, including the cerebellum.
As expected in the evaluation of a rare disease, this study is limited by the small number of patients who originated from a single family such that the clinical features described here may not reflect the whole range of clinical involvement in patients with different mutations. This was highlighted by the comparison between the findings from the present and previous studies (table e-1, links.lww.com/NXG/A228), which showed the broad range of clinical findings in patients with SCA34. Nevertheless, it is likely that the neurocognitive impairment described here is not mutation-specific and would be present in all patients with a cerebellar motor syndrome because the cognitive deficits are also a direct consequence of cerebellar degeneration. Similarly, the skin biopsy findings would have to be confirmed in tissue specimens from additional patients with different mutations to confirm that the dominant negative effect on protein localization observed here is a ubiquitous mechanism in this disorder.
Our findings support the role of ELOVL4 in cerebellar function and present a more precise and exhaustive characterization of the SCA34 phenotype. Clinicians should consider the diagnosis of SCA34 (ATX-ELOVL4) even in the absence of EKV and systematically assess cognitive and psychiatric features with specific emphasis on executive, visuospatial, and attention deficits. Analysis of dermal fibroblasts from a patient with SCA34 supports a dominant negative effect on ELOVL4 localization. More research is needed to understand the specific role of ELOVL4 and VLCFA in the cerebellum to better understand the specific pathologic mechanisms of selective cerebellar dysfunction in SCA34.
Acknowledgment
The authors thank Bastien Paré for his contribution to the fibroblast cell isolation and culture. Affiliation: Laval University Experimental Organogenesis Research Center/LOEX, Division of Regenerative Medicine, CHU de Québec Research Center – Enfant-Jésus Hospital, Québec, Canada.
Glossary
- CCAS
cerebellar cognitive-affective syndrome
- DTR
deep tendon reflex
- EKV
erythrokeratodermia variabilis
- FBS
fetal bovin serum
- FDG-PET
fluorodeoxyglucose-PET
- MoCA
Montreal Cognitive Assessment
- RCFT
Rey Complex Figure Test
- SCA34
spinocerebellar ataxia 34
- STGD3
Stargardt-like macular dystrophy type 3
- VLCFA
very long chain fatty acid
- VLC-SFA
very-long-chain saturated fatty acid
Appendix. Authors
Study funding
No targeted funding reported.
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Rossi M, Anheim M, Durr A, et al. The genetic nomenclature of recessive cerebellar ataxias. Mov Disord 2018;33:1056–1076. [DOI] [PubMed] [Google Scholar]
- 2.Giroux JM, Barbeau A. Erythrokeratodermia with ataxia. Arch Dermatol 1972;106:183–188. [PubMed] [Google Scholar]
- 3.Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, et al. Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol 2014;71:470–475. [DOI] [PubMed] [Google Scholar]
- 4.Ozaki K, Doi H, Mitsui J, et al. A novel mutation in ELOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a broadened spectrum of SCA34. JAMA Neurol 2015;72:797–805. [DOI] [PubMed] [Google Scholar]
- 5.Bourassa CV, Raskin S, Serafini S, Teive HA, Dion PA, Rouleau GA. A new ELOVL4 mutation in a case of spinocerebellar ataxia with erythrokeratodermia. JAMA Neurol 2015;72:942–943. [DOI] [PubMed] [Google Scholar]
- 6.Bourque PR, Warman-Chardon J, Lelli DA, et al. Novel ELOVL4 mutation associated with erythrokeratodermia and spinocerebellar ataxia (SCA 34). Neurol Genet 2018;4:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain 1998;121(pt 4):561–579. [DOI] [PubMed] [Google Scholar]
- 8.Manto M, Marien P. Schmahmann's syndrome—identification of the third cornerstone of clinical ataxiology. Cerebellum Ataxias 2015;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laforce R Jr, Buteau JP, Bouchard JP, Rouleau GA, Bouchard RW, Dupre N. Cognitive impairment in ARCA-1, a newly discovered pure cerebellar ataxia syndrome. Cerebellum 2010;9:443–453. [DOI] [PubMed] [Google Scholar]
- 10.Pedroso JL, Franca MC Jr, Braga-Neto P, et al. Nonmotor and extracerebellar features in Machado-Joseph disease: a review. Mov Disord 2013;28:1200–1208. [DOI] [PubMed] [Google Scholar]
- 11.Fancellu R, Paridi D, Tomasello C, et al. Longitudinal study of cognitive and psychiatric functions in spinocerebellar ataxia types 1 and 2. J Neurol 2013;260:3134–3143. [DOI] [PubMed] [Google Scholar]
- 12.Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699. [DOI] [PubMed] [Google Scholar]
- 13.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pare B, Touzel-Deschenes L, Lamontagne R, et al. Early detection of structural abnormalities and cytoplasmic accumulation of TDP-43 in tissue-engineered skins derived from ALS patients. Acta Neuropathol Commun 2015;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics 2010;26:2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kansal K, Yang Z, Fishman AM, et al. Structural cerebellar correlates of cognitive and motor dysfunctions in cerebellar degeneration. Brain 2017;140:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoodley CJ, Valera EM, Schmahmann JD. Functional topography of the cerebellum for motor and cognitive tasks: an fMRI study. Neuroimage 2012;59:1560–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoodley CJ, Schmahmann JD. Evidence for topographic organization in the cerebellum of motor control versus cognitive and affective processing. Cortex 2010;46:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salmi J, Pallesen KJ, Neuvonen T, et al. Cognitive and motor loops of the human cerebro-cerebellar system. J Cogn Neurosci 2010;22:2663–2676. [DOI] [PubMed] [Google Scholar]
- 22.Carrera E, Tononi G. Diaschisis: past, present, future. Brain 2014;137:2408–2422. [DOI] [PubMed] [Google Scholar]
- 23.Morihara R, Yamashita T, Deguchi K, et al. Familial and sporadic chronic progressive degenerative parietal ataxia. J Neurol Sci 2018;387:70–74. [DOI] [PubMed] [Google Scholar]
- 24.Agbaga MP. Different mutations in ELOVL4 affect very long chain fatty acid biosynthesis to cause variable neurological disorders in humans. Adv Exp Med Biol 2016;854:129–135. [DOI] [PubMed] [Google Scholar]
- 25.Sherry DM, Hopiavuori BR, Stiles MA, et al. Distribution of ELOVL4 in the developing and adult mouse brain. Front Neuroanat 2017;11:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brush RS, Tran JT, Henry KR, McClellan ME, Elliott MH, Mandal MN. Retinal sphingolipids and their very-long-chain fatty acid-containing species. Invest Ophthalmol Vis Sci 2010;51:4422–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Logan S, Agbaga MP, Chan MD, et al. Deciphering mutant ELOVL4 activity in autosomal-dominant Stargardt macular dystrophy. Proc Natl Acad Sci U S A 2013;110:5446–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grayson C, Molday RS. Dominant negative mechanism underlies autosomal dominant Stargardt-like macular dystrophy linked to mutations in ELOVL4. J Biol Chem 2005;280:32521–32530. [DOI] [PubMed] [Google Scholar]
- 29.Maugeri A, Meire F, Hoyng CB, et al. A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Invest Ophthalmol Vis Sci 2004;45:4263–4267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published in the article will be shared by request from any qualified investigator.